

Abstract
N-acetylglutamate synthase deficiency (NAGSD, MIM #237310) is an autosomal recessive urea cycle disorder caused either by decreased expression of the NAGS gene or defective NAGS enzyme resulting in decreased production of N-acetylglutamate (NAG), an allosteric activator of carbamylphosphate synthetase 1 (CPS1). NAGSD is the only urea cycle disorder that can be effectively treated with a single drug, N-carbamylglutamate (NCG), a stable NAG analog, which activates CPS1 to restore ureagenesis. We describe three patients with NAGSD due to four novel non-coding sequence variants in the NAGS regulatory regions. All three patients had hyperammonemia that resolved upon treatment with NCG. Sequence variants NM_153006.2:c.427–222G>A and NM_153006.2:c.427–218A>C reside in the 547 bp long first intron of NAGS and define a novel NAGS regulatory element that binds retinoic X receptor α. Sequence variants NC_000017.10:g.42078967A>T (NM_153006.2:c.−3065A>T) and NC_000017.10:g.42078934C>T (NM_153006.2:c.−3098C>T) reside in the NAGS enhancer, within known HNF1 and predicted glucocorticoid receptor binding sites, respectively. Reporter gene assays in HepG2 and HuH-7 cells demonstrated that all four substitutions could result in reduced expression of NAGS. These findings show that analyzing non-coding regions of NAGS and other urea cycle genes can reveal molecular causes of disease and identify novel regulators of ureagenesis.
Keywords: N-acetylglutamate synthase, N-acetylglutamate, non-coding sequence variants, urea cycle, urea cycle disorders, N-acetylglutamate synthase deficiency, mutation analysis, regulatory element, intron
Introduction
N-acetylglutamate synthase deficiency (NAGSD, MIM# 237310) is an autosomal recessive urea cycle disorder caused by pathogenic variants in the N-acetylglutamate synthase (NAGS) gene. Its product, the NAGS enzyme (EC 2.3.1.1) catalyzes formation of N-acetylglutamate (NAG), which is essential for the activity of carbamylphosphate synthetase 1 (CPS1, EC 6.3.4.16), the rate-limiting enzyme of the urea cycle (Waterlow, 1999). NAGSD is the only urea cycle disorder that can be effectively treated with a drug N-carbamylglutamate (NCG), a stable NAG analog, which binds to and activates CPS1 to completely restore ureagenesis in NAGS deficient patients (Caldovic et al., 2004; Grisolia & Cohen, 1952, 1953; Haberle, 2011). NAGSD can manifest either within hours after birth if there is complete loss of NAGS function or at any time thereafter in patients with residual NAGS activity. Symptoms of NAGSD include lethargy progressing to coma and death with laboratory studies showing elevated blood ammonia and glutamine, and low citrulline concentrations (Ah Mew & Caldovic, 2011; Haberle, 2011).
The human NAGS gene, located on chromosome 17, has seven exons that encode a 534 amino acid protein and a regulatory region that extends at least 3 kb upstream of the first exon (Caldovic et al., 2002; Elpeleg, Shaag, Ben-Shalom, Schmid, & Bachmann, 2002; Haberle et al., 2003; Heibel et al., 2011). Expression of the NAGS gene is controlled by the promoter and −3kb enhancer, which were identified based on their conservation in mammalian NAGS genes (Heibel et al., 2011; Heibel et al., 2012). The NAGS promoter binds the transcription factors specificity protein 1 (Sp1), cAMP response element binding (CREB) and farnesoid X receptor (FXR) (Heibel et al., 2012; Renga et al., 2011). The −3 kb NAGS enhancer binds transcription factors hepatocyte nuclear factor 1 (HNF1) and nuclear factor Y (NF-Y) and directs liver-specific expression of the NAGS gene (Heibel et al., 2012).
The establishment of accurate molecular diagnostic methods for NAGSD is clinically important because the biochemical phenotype cannot be easily differentiated from other proximal urea cycle disorders, but unlike these other disorders, NAGSD can be effectively treated with NCG (Ah Mew & Caldovic, 2011; Haberle, 2011). Although most of the sequence variants causing NAGSD have been found in the NAGS coding region (Al Kaabi & El-Hattab, 2016; Caldovic, Morizono, & Tuchman, 2007; Kim et al., 2015; Sancho-Vaello et al., 2016; van de Logt, Kluijtmans, Huigen, & Janssen, 2017), three pathogenic variants have been found in the non-coding regions (Heibel et al., 2012; Sonaimuthu et al., 2021; Williams et al., 2018). Therefore, understanding of the NAGS gene regulation and the function of its regulatory elements is essential for confirming the pathological nature of sequence variants found in patients. In this study, we identify a novel regulatory element in the 547 bp long first intron of the NAGS gene based on the sequence variants found in patients with NAGSD and describe two new pathogenic sequence variants in the −3 kb enhancer of the NAGS gene.
Materials and Methods
Case Descriptions
All methods were performed in accordance with the relevant guidelines and regulations of the participating institutions. Mutation analysis was conducted with the approval of the Institutional Review Boards of the University of Utah School of Medicine and the Zurich University Children’s Hospital. Informed consent was obtained either from all study subjects or their parents/guardians.
Case 1
The patient, a male, was hospitalized at 25 months of age for vomiting, dehydration, pneumonia and progressive lethargy. He was found to have metabolic acidosis with mildly elevated plasma lactate (2.2–2.4 mmol/L, normal 0.7–2.1 mmol/L) and elevated ammonia (hyperammonemia at 120 μmol/L on presentation, normal 21–50). Toxicology testing was negative as well as blood and urine cultures. He was admitted to the intensive care unit (ICU) where his ammonia decreased to 43 μmol/L upon administration of intravenous fluids containing glucose and, eventually lipids, with clinical improvements of becoming more alert and engaged. During the initial hospitalization peak ammonia was 185 μmol/L. Newborn screening was reviewed and was normal at 1 and 8 days of age, with normal citrulline and glutamine/citrulline ratio. He had history of developmental delays (unable to walk, no words at 25 months of age) and had a previous hospitalization for altered mental status and dehydration with metabolic acidosis at 17 months of age at which time intussusception was suspected (ammonia was not measured at that time). A head computed tomography (CT) was normal. Otherwise, the child was growing normally and had a normal head size.
This child was the product of in vitro fertilization from sperm donor to a healthy mother. He had a 4-year-old half-sister (different sperm donor) with mild speech delay, improving with therapy.
Plasma amino acids showed elevated glutamine (peak 1448 μmol/L on admission, normal 410–700) with mildly low citrulline (9 μmol/L before supplements, normal 10–60) and arginine at low end of normal (44 μmol/L, normal 40–160). Orotic acid in the urine was mildly increased at 6.2–7.3 mmol/mol creatinine (normal 0.7–5.1) with normal orotidine (1.7–2.4 mmol/mol creatinine, normal 0.7–4.2). Urine organic acids had markedly elevated 3-hydroxybutyric and acetoacetic acids, mild lactic/pyruvic aciduria and dicarboxylic aciduria suggesting catabolic state that could explain the metabolic acidosis. Plasma acylcarnitine profile had elevated concentrations of acetylcarnitine and C4OH-(3-hydroxybutyryl-) carnitine (0.98µmol/L; normal range: < 0.19), suggestive of ketosis. Neither the urine organic acids, nor the plasma acylcarnitine profile were suggestive of an organic acidemia. The child was discharged home on a low protein diet with medical food, citrulline supplements, and glycerol phenylbutyrate. Whole exome sequencing was denied by insurance. Standard genetic testing covering the exons of the OTC, CPS1, NAGS, and CA5A genes did not reveal pathogenic sequence variants.
Despite adherence to therapy, the child continued to have swings in ammonia and elevated glutamine in the plasma amino acids. The child was admitted again to the hospital at 26 months of age because of croup and poor intake leading to hyperammonemia (179 µmol/L). Labs and clinical status normalized with breathing treatment, dextrose containing intravenous fluids and intralipids. In view of the persistent instability, the child was tried on NCG (100 mg/kg per day) that resulted in normalization of ammonia and plasma amino acids. These remained normal even after sequentially stopping glycerol phenylbutyrate, citrulline supplements, amino acid modified medical food and the low protein diet. Clinically, his development started to improve with the initial therapies, including protein restricted diet. At 26 months, he had his first independent step and first word. He was walking independently at 31 months. By 39 months he had >100 words which he began to place in sentences.
Case 2
The patient was the first son of unrelated Dutch parents, born at 41 weeks of gestation. At day 4, he had a remarkable tachypnea and lethargy that prompted blood gas analysis. The results showed a respiratory alkalosis (pH 7.6, CO2 2.5 kPa) and elevated ammonia concentration (270–309 µmol/L). Physical examination revealed no additional abnormalities. A urea cycle defect was suspected; therefore, the patient was referred to a metabolic center and treated with sodium benzoate, an ammonia scavenger medication, dietary protein restriction and later with NCG. Cerebral ultrasound was normal with no signs of edema or intracranial bleeding.
Metabolic investigations revealed highly elevated plasma glutamine (1043 µmol/L, normal 322–733) and low-to-normal plasma citrulline whereas the excretion of orotic acid was normal. This led to the possible diagnosis of NAGSD. Treatment with protein restriction, citrulline supplementation and NCG resulted in normal metabolic profile and clinical condition. Sequencing of CPS1 cDNA identified four common SNPs that could not explain patient’s clinical manifestation and prompted sequencing of the NAGS gene.
The child developed normally except for an impaired vision due to Leber congenital amaurosis. At present, 7 years old, he attends regular school with the necessary adaptations because of his blindness. His growth is unremarkable. His diet is normal and with adequate NCG medication his amino acid profile is normal.
Case 3
This patient is a boy, born at term after uneventful pregnancy and delivery. His parents declared non-consanguinity. The boy’s development was normal, although he presented aversion to meat and dairy. At the age of eight years, after a week of dairy-rich diet, he presented to a hospital with symptoms of gastroenteritis, balance problems and qualitative disturbances of consciousness. CNS infection and brain tumor were ruled out and further diagnostic tests revealed rapidly increasing hyperammonemia (198 µmol/L, followed by 281 µmol/L; normal <60), with high concentrations of glutamine in the serum and CSF (1173 µmol/L and 3033 µmol/L, respectively; normal ranges 152 – 700 and 365 – 700), low-normal citrulline concentrations (11.1 µmol/L in serum and 3.6 µmol/L in CSF; normal ranges 1 – 55 and 0 – 8, respectively), normal serum but decreased CSF arginine concentrations (30.9 µmol/L and 13.4 µmol/L, respectively; normal ranges 6 – 187 and 14.2 – 25.8) and normal ornithine concentrations (12.8 µmol/L in serum and 16.9 µmol/L in CSF). Urinary organic acids revealed elevated orotic acid (9.5 mmol/mol creatinine; normal range <1.1). The treatment with hemodiafiltration was started immediately but the ammonia level remained high (291 µmol/L) and the patient’s neurologic status worsened. Administration of sodium benzoate, sodium phenylbutyrate and NCG led to decrease of ammonia concentration to normal levels (< 60 µmol/L) within hours and patient status improved rapidly. Biochemical tests done on the third day of treatment revealed almost normal serum glutamine concentration (530 µmol/L) and orotic acid (4.80 mmol/mol creatinine). The low protein diet was implemented, but the patient still required high doses of sodium benzoate, L-arginine (initially intravenously and then orally) and hypercaloric diet to keep ammonia at acceptable levels (70 – 100 µmol/L). Because laboratory investigations and quick response to treatment with sodium benzoate, sodium phenylbutyrate and NCG suggested NAGSD, therapeutic trial with NCG at a dose of 100 mg/kg was performed after withdrawal of ammonia scavengers. Ammonia normalized rapidly from 120 µmol/L to 44 µmol/L after administration of NCG. Following the start of treatment with NCG at a dose of 15 mg/kg, the patient had no hyperammonemic episodes and is doing well clinically, although he still prefers low-protein diet. Next generation sequencing of the urea cycle gene panel did not identify pathogenic variants in coding sequences and splice sites of urea cycle genes. This and patient’s response to NCG treatment prompted sequencing of the NAGS gene regulatory regions.
Identification of Patient Sequence Variants
Case 1
DNA was extracted from proband’s whole blood and prepared using TruSeq DNA PCR-free libraries (Illumina). The Illumina HiSeq X Ten System (Illumina) was used to sequence the proband’s DNA to a median 84x whole-genome coverage. Sequence reads were aligned to the GRCh37 reference genome (including decoy sequences from the GATK resource bundle) using the BWA-MEM (Li, 2013). The resulting BAM files were de-duplicated, INDEL realignment and base quality recalibration was performed using Picard tools (Toolkit, 2019) followed by the GATK variant discovery toolkit using a best practices workflow (DePristo et al., 2011). Quality control metrics indicated high quality sequence, alignment and variant calling. Sequence variant data for the proband in VCF file format was loaded into the Fabric Genomics (formerly Omicia) platform (Genomics), and genes involved in the urea cycle were examined in detail within this framework to identify variants that may have an impact on gene function. This analysis identified 12 non-coding sequence variants in CPS1, CA5A and NAGS genes (Supp Table S1). Ten of the variants were excluded from consideration because they affect low to moderately conserved base pairs that do not reside in conserved elements.
Cases 2 and 3
Sequence analysis of coding exons, including flanking intronic regions, as well as the promoter and enhancer region were carried out as described before (Williams et al., 2018). Sequencing of the regulatory element in the NAGS intron 1 was performed using the BigDye Terminator Cycle Sequencing kit version 1.1 and an ABI 3130 genetic analyzer (Applied Biosystems by Life Technologies Europe BV, Zug, Switzerland) (Williams et al., 2018) after amplification with 5’-gca gga tac gct gcg ggc tc-3’ and 5’-gtg ggc cag acg tgg tgc tc-3’ primers and following amplification conditions: 15 min initial denaturation at 95°C; 38 cycles of denaturation (20 s at 95°C), annealing (20 s at 58°C) and extension (1 min at 72°C); and a final extension at 72°C for 7 min. If not provided, genomic DNA was extracted from peripheral blood leukocytes.
Bioinformatic Analysis
Regulatory element in the first intron of human NAGS gene was identified by querying UCSC Genome Browser for sequence conservation and predicted regulatory elements in the NAGS gene (chr17:42,078,702–42,086,453 of the GRCh37/hg19 human genome assembly). UCSC Genome Browser conservation track has a 200 bp-long conserved region (chr17: 42,082,680–42,082,880) within first intron of the NAGS gene, which is 547 bp long (chr17: 42,082,458–42,083,004). This region of conservation is contained in a cis-acting conserved regulatory element (chr17:42,082,631–42,082,888) of the cCRE track (Consortium, 2012) of the UCSC Genome Browser. The In Other Genomes (Convert) tool in the View menu of the UCSC Genome Browser was used to download genomic sequences from 20 mammalian species (Supp. Table S2) that correspond to conserved element in NAGS intron 1. These sequences were aligned using Clustal Omega (Sievers et al., 2011) and the alignment was visualized with WebLogo 3 (Crooks, Hon, Chandonia, & Brenner, 2004). Transcription factors that bind to the conserved element within first intron of the NAGS gene were identified by data mining the ENCODE project database (Ljubica Caldovic (July 20, 2020)) (Caldovic, 2018). The following filters were applied to experiment matrix in the ENCODE database: TF ChIP-seq, organism (Homo sapiens), and biosample term name (liver); results were visualized in the UCSC genome browser for the chr17:42,078,702–42,086,453 genomic region. The hepatocyte nuclear factor 4α (HNF4α) binding site (chr17:42,082,640–42,082,899 in the GRCh37/hg19 human genome assembly) was identified from the Transcription Factor ChIP-seq Clusters track of the UCSC Genome Browser. The retinoic X receptor α (RXR α) and Sp1 binding sites were identified by comparing Logo alignment of NAGS intronic elements with the respective MA0115.1 and MA0079.3 position matrices from the JASPAR database of transcription binding sites (Fornes et al., 2020).
The In Other Genomes(Convert) tool in the View menu of the UCSC Genome Browser was used to download genomic sequences from 24 mammalian species (Supp. Table S3) that correspond to the −3kb enhancer (Heibel et al., 2012) of the human NAGS gene (chr17:42,078,635–42,079,129 of the GRCh37/hg19 human genome assembly). Downloaded sequences were aligned using Clustal Omega (Sievers et al., 2011) and the alignment was visualized with WebLogo 3 (Crooks et al., 2004). Histone modifications of the −3 kb enhancer and transcription factors that bind to it were identified by data mining the ENCODE project database (Ljubica Caldovic (July 20, 2020)) (Caldovic, 2018). The following filters were applied to experiment matrix in the ENCODE database: TF ChIP-seq, Histone ChIP-seq, organism (Homo sapiens), and biosample term name (liver); results were visualized in the UCSC genome browser for the chr17:42,078,635–42,079,129 genomic region. The glucocorticoid receptor (GR) binding site (chr17:42,078,920–42,078,938 in the GRCh37/hg19 human genome assembly) was identified from the HMR Conserved Transcription Factor Binding Sites track of the UCSC Genome Browser. The HNF4a, RXRα, Sp1 and Yin Yang 1 (YY1) transcription factor binding sites were identified by comparing Logo alignment of NAGS −3 kb enhancers with the respective MA0114.1, MA1147.1, MA0079.1 and MA0095.2 position matrices from the JASPAR database of transcription binding sites (Fornes et al., 2020).
Table browser of the UCSC Genome Browser and genomic coordinates of the four base pairs altered in patients with NAGSD were used to query GERP (Cooper et al., 2010; Goode et al., 2010), phyloP (Siepel et al., 2005), and PhastCons (Siepel et al., 2005) tracks and determine conservation in vertebrate genomes. Precomputed CADD scores (Kircher et al., 2014; Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019) for the four sequence variants were downloaded from the CADD web site (https://cadd.gs.washington.edu/). MutationTaster2 (Schwarz, Rodelsperger, Schuelke, & Seelow, 2010) was used to determine the likelihood that each of the four sequence variants causes NAGSD.
Plasmid Construction and Expression Studies
Names and inserts of plasmids used in this study are listed in Table 1. Plasmid p4.23hE (Heibel et al., 2011), renamed minP-E in this study (Table 1), was the template for introduction of the NC_000017.10:g.42078967A>T (NAGS:c.−3065A>T) and NC_000017.10:g.42078934C>T (NAGS:c-3098C>T) sequence changes using mutagenic primers 5’-tcc ctc cga cct ggg act ccg gga c-3’ and 5’-gtc ccg gag tcc cag gtc gga ggg a-3’ for the c.−3065A>T variant and 5’-cat tga ccc tgg gac cac tgt gtc ccc-3’ and 5’-ggg gac aca gtg gtc cca ggg tca atg-3’ for the c.−3098C>T variant. Site directed mutagenesis was performed using QuickChange Site-Directed Mutagenesis kit (Agilent Technologies) according to manufacturer’s instructions. Resulting plasmids were called c.−3065A>C and c.−3098C>T (Supp. Table S4), and the presence of the correct sequence changes were verified with DNA sequencing.
Table 1.
Conservation scores and predicted effects of sequence variants found in patients with NAGS deficiency.
| Variant | Conservation Score | Predicted Effect | |||
|---|---|---|---|---|---|
| GERP† | PhyloP 100Way‡ |
PhastCons 100Way§ |
CADD C-score |
Mutation Taster |
|
| c.−3098C>T¶ | 3.98 | 0.22 | 0.998 | 16.2 | Disease Causing |
| c.−3065A>T# | 4.51 | 3.53 | 0.984 | 20.2 | Disease Causing |
| c.427–222G>A | 4.17 | 4.85 | 1.000 | 19.7 | Disease Causing |
| c.427–218A>G | 4.15 | 2.98 | 1.000 | 19.1 | Disease Causing |
GERP scores range between −12.36 and 6.18. Higher score indicates higher sequence conservation.
PhyloP100Way scores range between −20 and 7.532. Higher score indicates higher sequence conservation.
PhastCons100Way scores range between 0 and 1. Higher score indicates higher sequence conservation.
Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence NM_153006.2. The HGVS name for this variant is NC_000017.10:g.42078934C>T.
Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence NM_153006.2. The HGVS name for this variant is NC_000017.10:g.42078967A>T.
Plasmid Prom-Ex1-Int1-Luc (Sonaimuthu et al., 2021) was a template for introduction of the NM_153006.2:c.427–222G>A and NM_153006.2:c.427–218A>C sequence changes. Mutagenic primers 5’-ccg acc ttg gat tcc ggg aca tct ttg ggg g-3’ and 5’-ccc cca aag atg tcc cgg aat cca agg tcg g-3’ were used to generate the c.427–222G>A sequence change while primers 5’-tcc ctc cga cct ggg act ccg gga c-3’ and 5’-gtc ccg gag tcc cag gtc gga ggg a-3’ were used for the c.427–218A>C sequence change. The QuickChange Site-Directed Mutagenesis kit (Agilent Technologies) was used to introduce the mutations according to manufacturer’s instructions; resulting plasmids were called c.427–222G>A and c.427–218A>C (Table 1) and their correct sequence was verified with DNA sequencing.
Culturing of the HepG2 cells (ATCC) and the HuH-7 cells (Charles Rice laboratory, Rockefeller University), their transfections and reporter assays were carried out as described previously (Sonaimuthu et al., 2021; Williams et al., 2018).
Results
Non-coding sequence variants found in patients with NAGSD
We have identified three subjects with NAGSD and pathogenic sequence variants in the non-coding regions of the NAGS. Subject 1 is a compound heterozygote for two non-coding sequence variants in the NAGS gene: the NAGS:c.427–222G>A located in the first intron of the NAGS gene and NAGS:c.−3065A>T, located in the −3kb NAGS enhancer and adjacent to the previously identified NAGS:c-3064C>A pathogenic sequence variant (Heibel et al., 2012). Subject 2 is a compound heterozygote for sequence variants NAGS:c.427–218A>C, located in the NAGS intron 1, and NAGS:c.1494G>A or p.Trp498Ter in exon 7, leading to a premature termination of translation at codon 498 (Supp. Figure S1). Subject 3 is a homozygote for the sequence variant NAGS:c.−3098C>T (Supp. Figure S2), located in the −3kb NAGS enhancer (Heibel et al 2012). Subjects 1 and 3 had milder, late onset disease which is consistent with partial NAGSD. The more severe, neonatal NAGSD in subject 2 is consistent with the presence of one loss of function NAGS allele.
Query of the gnomAD (Karczewski et al., 2020), dbSNP153 (Sherry et al., 2001) and 1000 Genomes Project (Genomes Project et al., 2015) databases indicated that none of the four non-coding sequence variants have been previously reported. The GERP (Cooper et al., 2010; Goode et al., 2010) and phyloP (Siepel et al., 2005) scores of the c.−3065A>T, c.427–222G>A, and c.427–218A>C sequence variants indicated that they affected highly conserved base pairs while the base pair affected by the c.−3098C>T is not as conserved in mammalian genomes (Table 1). However, the phastCons (Siepel et al., 2005) scores of all four sequence variants indicated that they reside within conserved elements of the human genome (Table 2). Functional effect prediction programs Combined Annotation Dependent Depletion (CADD) (Kircher et al., 2014; Rentzsch et al., 2019) and MutationTaster2 (Schwarz et al., 2010) indicated that all four sequence variants should be disease causing.
Table 2.
VariantValidator has been used to verify HGVS nomenclature of the 4 variants. VariantValidator has been used to verify HGVS nomenclature of the 4 variants. The table below lists variant names in HGVS nomenclature and the corresponding designations that were used in the manuscript. Correct HGVS names of all 4 variants have been added in the abstract and Materials and Methods section.
| Position on Chromosome 17 | HGVS Nomenclature | Variant designation in the manuscript |
|---|---|---|
| 42,078,934 | NC_000017.10:g.42078934C>T | NAGS:c-3098C>T and c-3098C>T |
| 42,078,967 | NC_000017.10:g.42078967A>T | NAGS:c.−3065A>T and c.−3065A>T |
| 42,082,783 | NM_153006.2:c.427–222G>A | NAGS:c.427–222G>A and c.427–222G>A |
| 42,082,787 | NM_153006.2:c.427–218A>G | NAGS:c.427–218A>G and c.427–218A>G |
Because phastCons scores of the two intronic sequence variants indicated that they reside in a region of high sequence conservation we used data mining approaches to further characterize this region (Caldovic, 2018). Query of the phastCons conservation track of the UCSC Genome Browser revealed a 200 bp conserved element with genomic coordinates chr17:42,082,680–42,082,799 (GRCh37/hg19 human genome assembly) in the first intron of the NAGS gene, which is 547 bp long (Figure 1A). Moreover, based on its biochemical signatures this region was identified as a candidate cis-regulatory element by ENCODE data analysis center (Figures 1A-B and (Consortium, 2012)). The ENCODE Project database was also queried for transcription factors that bind to this region in the human liver. This revealed that the NAGS intronic element binds RXRα, HNF4α and Sp1 transcription factors (Figure 1B). Transcription factor Chip-Seq track of the UCSC Genome Browser was then used to locate more precisely the HNF4α binding site, while comparisons with the Sp1 and RXRα position similarity matrices from the JASPAR database of transcription factor binding sites (Fornes et al., 2020) were used to locate Sp1 and RXRα transcription factor binding sites (Figure 1C). The c.427–222G>A and c.427–218A>C sequence variants are located within the RXRα binding site and affect base pairs that are highly conserved in mammalian NAGS genes and almost invariant in its canonical recognition sequence (Yang, Subauste, & Koenig, 1995).
Figure 1. Sequence variants in the novel regulatory element in the NAGS intron 1.
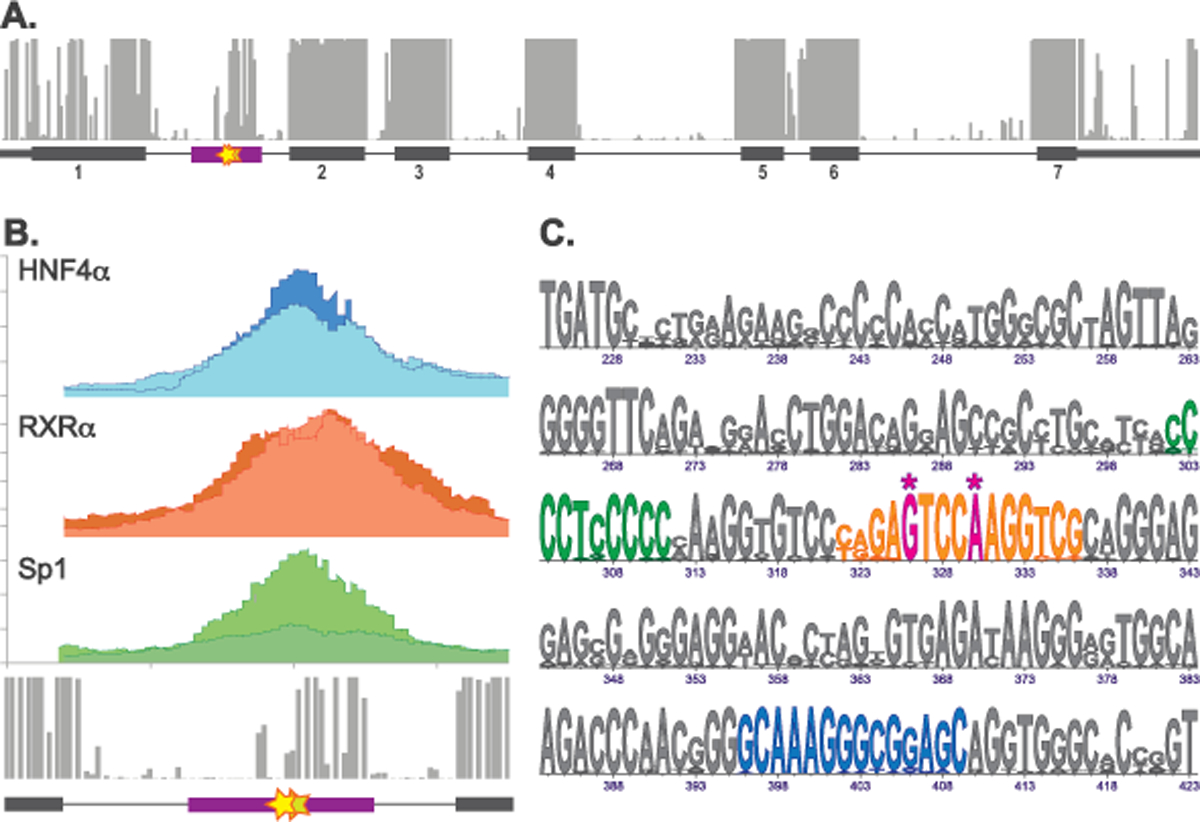
A. The phastCons track of the UCSC Genome Browser showing conserved regions in the NAGS gene (top) and map of the NAGS gene (bottom) showing genomic region chr17:42,081,993 – 42,086,412 of the GRCh37/hg19 human genome assembly with exons as gray boxes, introns as gray lines and predicted cis-acting conserved regulatory element in the NAGS intron 1 as purple box. Numbers below gray boxes indicate exon numbers. B. Genomic region chr17:42,082,368–42,083,068 of the GRCh37/hg19 human genome assembly. Results of the ENCODE ChIP-Seq experiments showing binding of transcription factors HNF4α (blue), RXRα (orange) and Sp1 (green) to NAGS intron 1 in the human liver tissue (top) and phastCons track of the UCSC Genome Browser showing conserved region within NAGS intron 1 (bottom). Two shades of blue, orange and green indicate results of the two ChIP-Seq biological replicates in the ENCODE database. C. LOGO alignment of the conserved intronic sequences from 20 mammals. Numbers indicate bp positions in the NAGS intron 1 where G of the donor splice site is +1. Magenta – pathogenic sequence variants. Blue – HNF4α binding site. Orange – RXRα binding site. Green – Sp1 binding site. Asterisks indicate positions of pathogenic sequence variants NM_153006.2:c.427–222G>A and NM_153006.2:c.427–218A>C.
Functional testing of sequence variants in the first intron of the NAGS gene
The pathogenic nature of the two sequence variants found in the first intron of the NAGS gene was confirmed using reporter gene assays in the HuH-7 and HepG2 cell lines. The c.427–222G>A and c.427–218A>C sequence variants were introduced into Prom-Ex1-Int1-Luc construct, which harbors luciferase gene fused to the promoter, exon 1 and intron 1 of the human NAGS gene (Supp. Table S3). Construct with the wild-type sequence of NAGS intron 1 was used as a control; constructs that contain NAGS promoter controlling expression of either luciferase gene (Prom-Luc; Supp. Table S3) or luciferase gene fused to the NAGS exon 1 (Prom-Ex1-Luc; Supp. Table S3) were additional controls while construct harboring only luciferase reporter gene was used as a negative control. Luciferase activity was lower in the HuH-7 cells transfected with the Prom-Ex1-Int1 construct harboring the c.427–218A>C variant while there was a trend (p=0.06, n=9) towards lower luciferase activity in in the HuH-7 cells transfected with the construct containing the c.427–222G>A sequence variant (Figure 2B). Luciferase activity was lower in the HuH-7 cells transfected with the Prom-Luc and Prom-Luc-Ex1 constructs compared to the HuH-7 cells transfected with the Prom-Ex1-Int1-Luc construct suggesting that presence of the NAGS intron 1 enhances gene expression in this cell line (Figure 2B). Reporter gene assays in the HepG2 cells revealed that presence of either c.427–222G>A or c.427–218A>C sequence variants caused reduced luciferase activity compared to the HepG2 cells transfected with the Prom-Ex1-Int1-Luc plasmid (Figure 2C). As in the HuH-7 cells, luciferase activity was higher in the HepG2 cells transfected with the Prom-Ex1-Int1-Luc plasmid than in the cells transfected with the Prom-Ex1-Luc construct (Figure 2C). However, luciferase activity was lower in the HepG2 cells transfected with plasmids that contain the NAGS exon 1 coding sequence suggesting that its translation led to decreased expression of the reporter gene in this cell line (Figure 2C). Taken together these data strongly suggest that the conserved element in the first intron of the NAGS gene can enhance its expression and that the c.427–222G>A and c.427–218A>C sequence variants cause NAGSD by reducing expression of the NAGS gene.
Figure 2. Functional testing of the sequence variants found in NAGS intron 1 from patients with NAGSD.
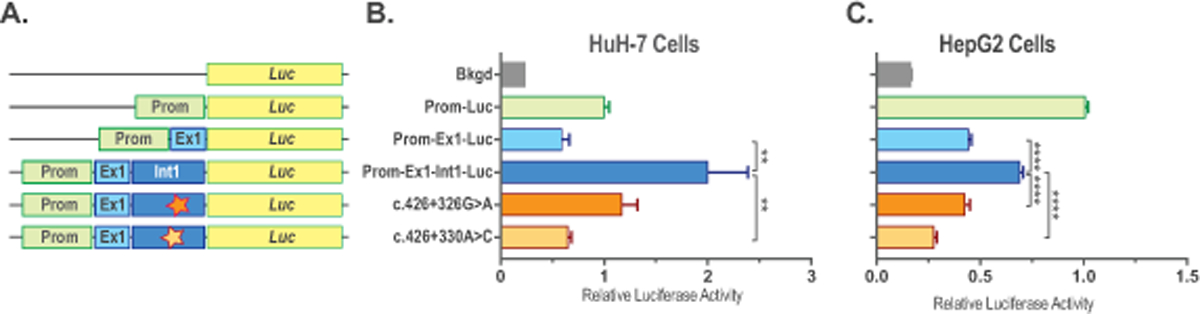
Expression constructs (A) and relative luciferase activity in HuH-7 (B) and HepG2 (C) cells transfected with the promoter-less plasmid pGL4.10[luc2] (Bkgd), plasmid with NAGS promoter controlling luciferase expression (Prom-Luc), plasmid with NAGS promoter controlling luciferase fused to the coding sequence of NAGS exon 1 (Prom-Ex1-Luc), plasmid with NAGS promoter controlling luciferase fused to the NAGS exon and intron 1 (Prom-Ex1-Int1-Luc) and plasmids with intronic mutations found in patients with NAGS deficiency (NM_153006.2:c.427–222G>A and NM_153006.2:c.427–218A>C). Data represent mean ± SEM of n=9 measurements. ** indicate p<0.01, **** indicates p<0.0001.
Functional testing of sequence variants in the −3kb enhancer of the NAGS gene
Subjects 1 and 3 had deleterious sequence variants in the first intron and coding sequence of their NAGS genes; we used functional testing of the sequence variants found in the −3 kb enhancer of their NAGS genes to establish molecular diagnosis of NAGSD. Epigenetic mark H3K27Ac of the NAGS −3 kb enhancer in the human liver indicate active enhancer status while H3K4me3 epigenetic mark, characteristic of active promoters (Kouzarides, 2007), of the −3 kb enhancer suggests that it may regulate NAGS gene expression through initiation of enhancer mRNA (Figure 3A) (Natoli & Andrau, 2012). Query of the ENCODE database revealed that −3 kb enhancer harbors conserved GR binding site and binds transcription factors HNF4α, RXR α, Sp1, and YY1 in addition to previously identified HNF1, NF-Y binding sites (Heibel et al., 2012) and NF1C conserved site (Williams et al., 2018) (Figure 3). The c.−3065A>T sequence variant is located within HNF1 binding site (Figure 3B), one base pair upstream of the previously identified pathogenic NAGS sequence variant (Heibel et al., 2011) while the c.−3098C>T sequence variant resides in the predicted GR binding site (Figure 3B).
Figure 3. Sequence variants in the −3kb enhancer of the NAGS gene.
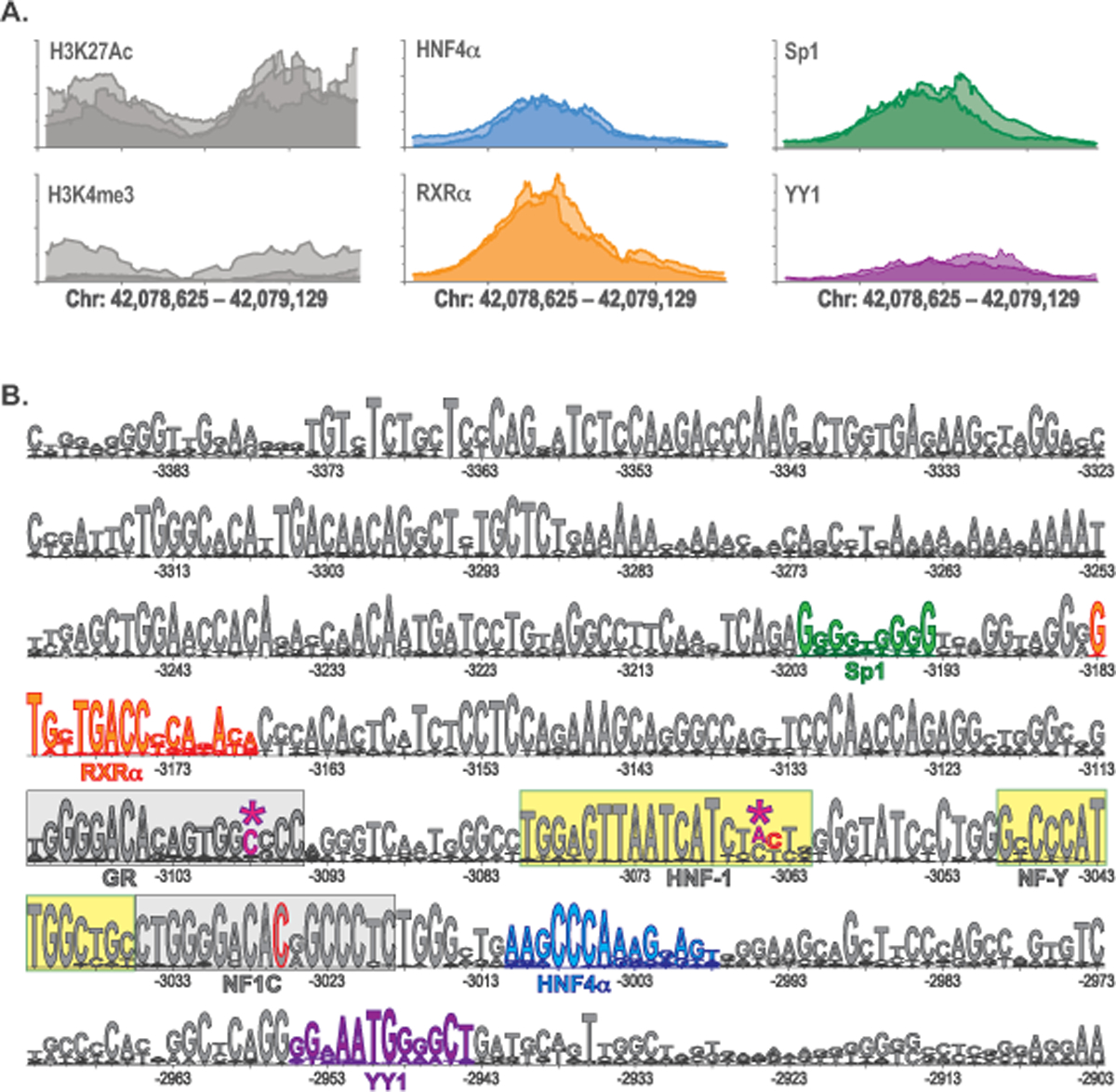
A. Genomic region chr17:42,078,635–42,079,129 of the GRCh37/hg19 human genome assembly. Results of the ENCODE ChIP-Seq experiments showing H3K27Ac and H3K4me3 histone modifications (gray) and binding of transcription factors HNF4α (blue), RXRα (orange), Sp1 (green) and YY1 (purple) to −3kb enhancer in the human liver tissue. Shades of gray, blue, orange, green, and purple indicate results of ChIP-Seq biological replicates in the ENCODE database. B. LOGO alignment of the −3kb enhancer sequences from 25 mammals. Confirmed HNF1 and NF-Y binding sites are highlighted in yellow. Conserved GR and NF1C binding sites are highlighted in gray. Magenta and asterisks – pathogenic sequence variants reported here: NC_000017.10:g.42078934C>T (c-3098C>T) and NC_000017.10:g.42078967A>T (c.−3065A>T). Gray with red outline – location of the previously reported sequence variants: NC_000017.10:g.42079006C>T (c.−3026C>T) and NC_000017.10:g.42078968C>A (c.−3064C>A). Blue – HNF4α binding site. Orange – RXRα binding site. Green – Sp1 binding site. Purple – YY1 binding site. Numbers indicate the distance of base pairs in the NAGS −3 kb enhancer from translation initiation codon; its A is +1.
Reporter gene assays were used to determine whether c.−3065A>T and c.−3098C>T sequence variants can affect gene expression in HuH-7 and HepG2 cells. Base pair changes that correspond to the two −3 kb enhancer variants found in subjects 1 and 3 were introduced into minP-E plasmid (Table S3) followed by transfection of the plasmids into either HuH-7 or HepG2 cells. The minP-E plasmid was used as a positive control; cells transfected with plasmid containing either only minimal promoter or promoter-less plasmid were used as negative controls. Depending on the experiment, luciferase activity increased approximately 8 to 10-fold in the HuH-7 cells and 19 to 29-fold in the HepG2 cells when −3 kb enhancer was placed upstream of the minimal promoter (Figure 4). Luciferase activity was slightly above background level in the presence of the c.−3065A>T sequence variant in both HuH-7 and HepG2 cells (Figure 4A-B) strongly suggesting that this sequence variant can have deleterious effect on the NAGS gene expression. In the presence of the c.−3098C>T sequence variant luciferase activity was reduced by approximately one half in the HuH-7 cells and by about two thirds in the HepG2 cells compared to cells expressing plasmid with the wild-type −3 kb enhancer (Figure 4C-D). This suggests that the c.−3098C>T sequence variant likely has a negative effect on the NAGS gene expression.
Figure 4. Functional testing of the sequence variants found in the NAGS −3kb enhancer from patients with NAGSD.
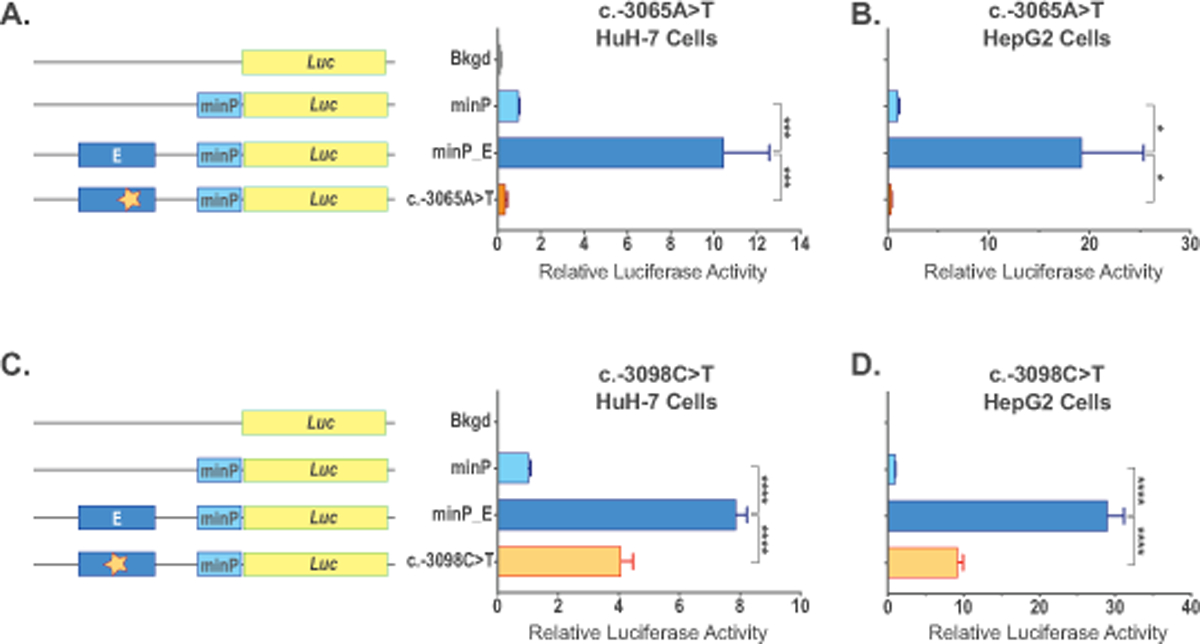
Expression constructs and effect of the NC_000017.10:g.42078967A>T (c.−3065A>T) sequence variant on luciferase gene expression was tested in HuH-7 (A) and HepG2 (B) cells. Expression constructs and effect of the NC_000017.10:g.42078934C>T (c-3098C>T) sequence variant on luciferase gene expression was tested in HuH-7 (C) and HepG2 (D) cells. Cells were transfected with the promoter-less plasmid pGL4.10[luc2] (Bkgd), plasmid with minimal eukaryotic promoter controlling luciferase expression (minP), plasmid harboring −3kb NAGS enhancer and minimal promoter controlling luciferase expression (minP_E), and plasmids with enhancer mutations found in patients with NAGS deficiency (c.−3065A>T and c.−3098C>T). Data represent mean ± SEM of n=9 measurements. * indicate p<0.05, *** indicate p<0.001, **** indicate p<0.0001.
Discussion
This report describes four new non-coding sequence variants in the NAGS gene that can cause reduced NAGS expression and NAGSD. None of the four sequence variants have been previously reported and all four reduced luciferase activity in reporter gene assays. Two of the sequence variants are located in a conserved region of the first NAGS intron and define a novel regulatory element of the NAGS gene. This novel regulatory element binds transcription factors HNF4α, RXR α and Sp1 in human liver based on the data from the ENCODE project. First introns of many human genes often harbor regulatory elements based on their conservation in mammalian genomes, presence of DNase hypersensitive sites and epigenetic histone modifications indicative of active regulatory elements (Jo & Choi, 2019; Park, Hannenhalli, & Choi, 2014). Several regulatory elements found within first introns bind Sp1 transcription factor (Beaulieu et al., 2011; Bornstein, McKay, Morishima, Devarayalu, & Gelinas, 1987; Guerin, Leclerc, Verreault, Labrie, & Luu-The, 1995; Liska, Robinson, & Bornstein, 1992), similar to the regulatory element within first intron of human NAGS gene identified in this study.
The c.427–222G>A and c.427–218A>C sequence variants both affect highly conserved base pairs in the RXRα binding site suggesting a role for this transcription factor in the regulation of NAGS expression. Decreased reporter gene activity in cells transfected with constructs containing sequence variants within RXRα binding site, which is consistent with decreased NAGS expression in patients with the two sequence variants, suggests that RXRα acts as transcriptional activator of NAGS. RXRα transcription factor is a nuclear receptor that binds vitamin A metabolites 9-cis-retinoic acid (Evans & Mangelsdorf, 2014) and 9-cis-13,14-dihydroretinoic acid; the latter is a better candidate for physiological RXRα ligand because it has been detected in the liver (Krezel, Ruhl, & de Lera, 2019). RXRα regulates transcription either as a homodimer or heterodimer with retinoic acid receptor, thyroid receptor or vitamin D receptor. Ongoing efforts of the ENCODE project may reveal whether RXRα binds as a homo- or heterodimer to regulatory element in the first intron of human NAGS gene. There are no reports of regulation of urea cycle enzymes by vitamin D. Both vitamin A and thyroid hormone play a role in the protein metabolism of rats. Vitamin A deficiency results in increased protein catabolism and higher expression of urea cycle genes and enzymes in adult and growing rats (Esteban-Pretel et al., 2010; McClintick et al., 2006). This effect of vitamin A deficiency on expression of urea cycle genes is likely an indirect consequence of increased protein catabolism. This does not exclude activation of NAGS expression by RXRα and can be consistent with decreased expression of NAGS due to sequence variants that may decrease binding of receptors for vitamin A and its metabolites. Manipulation of the thyroid hormone levels in rats affect abundance of urea cycle enzymes, but direction of the change depends on the duration of hypothyroidism and control of food intake by experimental animals. Prolonged hypothyroidism in rats, lasting 4–7 weeks, resulted in increased abundance of urea cycle enzymes and capacity to produce urea probably due to decreased food intake and weight loss in the hypothyroid animals (Marti, Portoles, Jimenez-Nacher, Cabo, & Jorda, 1988; Silvestri et al., 2006). In a different set of studies, hypothyroidism lasting two weeks led to increased production of urea and urea cycle intermediates, including NAG. However, neither hypo- nor hyperthyroidism led to changes in expression of urea cycle genes in mouse liver (Feng, Jiang, Meltzer, & Yen, 2000; Flores-Morales et al., 2002) as well as abundance and activity of urea cycle enzymes in rat liver (Hayase, Naganuma, Koie, & Yoshida, 1998; Hayase, Yonekawa, Yokogoshi, & Yoshida, 1991; Hayase, Yonekawa, & Yoshida, 1992, 1993; Hayase & Yoshida, 1995). This suggests that thyroid hormone receptors are unlikely to regulate expression of NAGS by forming heterodimers with RXRα.
Two of the sequence variants were found in the −3 kb enhancer of the NAGS gene. We queried ENCODE project database for epigenetic marks found in this region in the human liver. Acetylation of the lysine 27 of the histone H3 in this region indicates that it is an active enhancer of the human NAGS gene. The lysine 4 of the histone H3 is tri-methylated in the −3 kb enhancer. Although this epigenetic mark indicates active promoters, many enhancers can bind RNA polymerase II and initiate transcription of enhancer RNA (eRNA) (Natoli & Andrau, 2012). Closer inspection of the Transcription Factor ChIP-Seq track of the UCSC Genome Browser revealed that RNA polymerase II binds to the −3 kb NAGS enhancer in HepG2 cells and likely initiates transcription of an eRNA from this region. Unidirectional transcription of eRNA from the −3 kb NAGS enhancer may explain its inability to act in the orientation independent manner (Heibel et al., 2012), while decreased transcription and/or stability of the eRNA could be a novel molecular mechanism of NAGSD.
The c.−3065A>T sequence variant affects a base pair that is highly conserved in mammals and located in the HNF1 transcription factor binding site. Negative effect of the c.−3065A>T sequence variant on HNF1 binding to the −3kb enhancer is a likely explanation for the deleterious effect of this variant. A sequence variant that reduces binding of HNF1 to −3 kb enhancer and located immediately downstream of the c.−3065A>T was found in a patient with NAGSD (Heibel et al., 2011). Two pathogenic sequence variants found in the HNF1 binding site of the −3 kb enhancer stress the importance of this transcription factor for expression of the NAGS gene and normal ureagenesis.
The second variant found in the −3 kb enhancer is located in the predicted GR binding site. This variant reduced luciferase activity presumably through reduced binding of GR to its binding site in the −3 kb enhancer. Circadian fluctuations of glucocorticoid secretion regulate expression of urea cycle genes and enzymes during feeding and fasting periods to accommodate removal of excess ammonia that is released as amino acids enter gluconeogenesis (Luna-Moreno, Garcia-Ayala, & Diaz-Munoz, 2012). The role of glucocorticoids in regulation of ureagenesis was revealed through decreased abundance and activity of urea cycle enzymes in adrenalectomized rats (Hazra, DuBois, Almon, Snyder, & Jusko, 2008; McLean & Gurney, 1963). GR binds to regulatory elements and activates expression of the rat Cps1 gene (Christoffels et al., 1998; Christoffels et al., 2000; Christoffels, van den Hoff, Moorman, & Lamers, 1995; Schoneveld, Gaemers, Hoogenkamp, & Lamers, 2005). Regulation of other urea cycle genes by GR is indirect and requires ongoing protein synthesis of transcription factors that directly regulate rat ornithine transcarbamylase, argininosuccinate synthetase 1, argininosuccinate lyase and arginase 1 (Gebhardt & Mecke, 1979; Lin, Snodgrass, & Rabier, 1982; Morris & Kepka-Lenhart, 2002; Nebes & Morris, 1988; Ulbright & Snodgrass, 1993). A role for GR in regulation of ureagenesis in humans is supported by the observation of abnormal concentrations of urea cycle intermediates and low urea concentration in the blood of patients with Addison’s disease (Okun et al., 2015) and in patients receiving prednisolone treatment (Wolthers, Hamberg, Grofte, & Vilstrup, 2000). NAGSD in one of our patients and functional tests of the c.−3098C>T variant, located in the predicted GR binding site, suggest that GR might directly regulate NAGS expression. Unfortunately, the data about GR binding to DNA in human liver are not yet available in the ENCODE database.
ENCODE project and database enabled discovery of the new regulatory element and transcription factors that regulate NAGS gene expression. Our analysis focused on the first intron of the NAGS gene and −3 kb NAGS enhancer. This does not exclude existence of additional regulatory elements further upstream of the NAGS gene, including long-range enhancers that could be located millions of base pairs away from NAGS. Until recently, limited ability to sequence DNA combined with small number of patients with urea cycle disorders precluded identification of such long-range enhancers and silencers that regulate NAGS and other urea cycle genes in model organisms and/or patients with urea cycle disorders. Such long-range regulatory elements can be identified through integrated analysis of the ENCODE project data (Consortium et al., 2020; Fishilevich et al., 2017; Stelzer et al., 2016). Increasing efficiency and decreasing price of whole genome sequencing will enable detecting pathogenic sequence variants in the distant regulatory elements in patients with NAGSD and other urea cycle disorders, which will confirm their role in the regulation of expression of urea cycle genes.
Molecular diagnosis of NAGSD is important because it is the only urea cycle disorder that can be effectively treated with a drug (Caldovic et al., 2004; Haberle, 2011). Two sequence variants found in the first intron define a new NAGS regulatory element that binds and implicates transcription factors HNF4α and RXRα in the regulation of NAGS expression and ureagenesis. The four non-coding sequence variants that cause NAGSD reported here bring the total number of non-coding, disease-causing NAGS variants to seven, which is almost 14% of deleterious NAGS sequence variants (Al Kaabi & El-Hattab, 2016; Bijarnia-Mahay et al., 2018; Cartagena et al., 2013; Cavicchi et al., 2018; Heibel et al., 2011; van de Logt et al., 2017; Williams et al., 2018). This underscores the importance of analyzing both coding and non-coding regions of the NAGS gene, which is amenable to both Sanger and next generation sequencing, for the presence of disease-causing sequence variants.
Supplementary Material
Acknowledgements
This work was supported by Public Health Service Grant R01DK064913 from the National Institutes of Health, Recordati Rare Diseases, Inc, and the Rashid Family Foundation. Work on urea cycle disorders is supported by the Swiss National Science Foundation (grant 320030_176088 to JH). Mutation analysis for NAGSD at the Zurich laboratory is supported by Recordati Rare Diseases. Whole genome sequencing of DNA from case 1 was supported by the Utah Genome project. We are grateful for the support and resources from the Center for High Performance Computing at the University of Utah for computational analysis of case 1.
Grant numbers: R01DK064913 and 320030_176088
Footnotes
Conflicts of Interest
Mutation analysis (Dr. Häberle and Ms. Rüfenacht) for cases 2 and 3 and functional testing of the four non-coding variants (Dr. Caldovic and Ms. Haskins) were supported by the Recordati Rare Diseases, Inc. that manufactures and sells NCG as Carbaglu® (carglumic acid), which is used for treatment of NAGS deficiency.
Data Availability
Data that support the findings of this study are available in the supplementary material of this article
All 4 sequence variants have been deposited to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) with following accession numbers:
SCV001733621 for NM_153006.2:c.427–222G>A
SCV001733623 for NC_000017.10:g.42078967A>T
SCV001733619 for NM_153006.2:c.427–218A>C
SCV001759970 for NC_000017.10:g.42078934C>T
Web Resources
UCSC Genome Browser: https://genome.ucsc.edu
gnomAD database: https://gnomad.broadinstitute.org/
ENCODE Project: https://www.encodeproject.org/
WebLogo 3: http://weblogo.threeplusone.com/
JASPAR Database: http://jaspar.genereg.net/
Fabric Genomics: https://fabricgenomics.com/
Illumina: https://www.illumina.com/
Picard Toolkit: http://broadinstitute.github.io/picard/
References
- Ah Mew N, & Caldovic L (2011). N-acetylglutamate synthase deficiency: an insight into the genetics, epidemiology, pathophysiology, and treatment. Appl Clin Genet, 4, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Kaabi EH, & El-Hattab AW (2016). N-acetylglutamate synthase deficiency: Novel mutation associated with neonatal presentation and literature review of molecular and phenotypic spectra. Mol Genet Metab Rep, 8, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu E, Green L, Elsby L, Alourfi Z, Morand EF, Ray DW, & Donn R (2011). Identification of a novel cell type-specific intronic enhancer of macrophage migration inhibitory factor (MIF) and its regulation by mithramycin. Clin Exp Immunol, 163(2), 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijarnia-Mahay S, Haberle J, Jalan AB, Puri RD, Kohli S, Kudalkar K, . . . Verma IC (2018). Urea cycle disorders in India: clinical course, biochemical and genetic investigations, and prenatal testing. Orphanet J Rare Dis, 13(1), 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P, McKay J, Morishima JK, Devarayalu S, & Gelinas RE (1987). Regulatory elements in the first intron contribute to transcriptional control of the human alpha 1(I) collagen gene. Proc Natl Acad Sci U S A, 84(24), 8869–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldovic L (2018). Data mining approaches for understanding of regulation of expression of the urea cycle genes. In Uchiumi F (Ed.), Gene Expression and Control Croatia: IntechOen. [Google Scholar]
- Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, & Tuchman M (2004). Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr, 145(4), 552–554. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Gracia Panglao M, Gallegos R, Yu X, Shi D, . . . Tuchman M (2002). Cloning and expression of the human N-acetylglutamate synthase gene. Biochem Biophys Res Commun, 299(4), 581–586. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, & Tuchman M (2007). Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat, 28(8), 754–759. [DOI] [PubMed] [Google Scholar]
- Cartagena A, Prasad AN, Rupar CA, Strong M, Tuchman M, Ah Mew N, & Prasad C (2013). Recurrent encephalopathy: NAGS (N-acetylglutamate synthase) deficiency in adults. Can J Neurol Sci, 40(1), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavicchi C, Chilleri C, Fioravanti A, Ferri L, Ripandelli F, Costa C, . . . Morrone A (2018). Late-Onset N-Acetylglutamate Synthase Deficiency: Report of a Paradigmatic Adult Case Presenting with Headaches and Review of the Literature. Int J Mol Sci, 19(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Grange T, Kaestner KH, Cole TJ, Darlington GJ, Croniger CM, & Lamers WH (1998). Glucocorticoid receptor, C/EBP, HNF3, and protein kinase A coordinately activate the glucocorticoid response unit of the carbamoylphosphate synthetase I gene. Mol Cell Biol, 18(11), 6305–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Habets PE, Das AT, Clout DE, van Roon MA, Moorman AF, & Lamers WH (2000). A single regulatory module of the carbamoylphosphate synthetase I gene executes its hepatic program of expression. J Biol Chem, 275(51), 40020–40027. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, van den Hoff MJ, Moorman AF, & Lamers WH (1995). The far-upstream enhancer of the carbamoyl-phosphate synthetase I gene is responsible for the tissue specificity and hormone inducibility of its expression. J Biol Chem, 270(42), 24932–24940. [DOI] [PubMed] [Google Scholar]
- Consortium, Encode Project. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, Encode Project, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, . . . Weng Z (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature, 583(7818), 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, & Nickerson DA (2010). Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods, 7(4), 250–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G , Chandonia JM , & Brenner S, E. (2004). WebLogo: a sequence logo generator. Genome Res, 14(6), 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, . . . Daly MJ (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 43(5), 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elpeleg O, Shaag A, Ben-Shalom E, Schmid T, & Bachmann C (2002). N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy. Ann Neurol, 52(6), 845–849. [DOI] [PubMed] [Google Scholar]
- Esteban-Pretel G, Marin MP, Cabezuelo F, Moreno V, Renau-Piqueras J, Timoneda J, & Barber T (2010). Vitamin A deficiency increases protein catabolism and induces urea cycle enzymes in rats. J Nutr, 140(4), 792–798. [DOI] [PubMed] [Google Scholar]
- Evans RM, & Mangelsdorf DJ (2014). Nuclear Receptors, RXR, and the Big Bang. Cell, 157(1), 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Jiang Y, Meltzer P, & Yen PM (2000). Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol, 14(7), 947–955. [DOI] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, . . . Cohen D (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford), 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Morales A, Gullberg H, Fernandez L, Stahlberg N, Lee NH, Vennstrom B, & Norstedt G (2002). Patterns of liver gene expression governed by TRbeta. Mol Endocrinol, 16(6), 1257–1268. [DOI] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, . . . Mathelier A (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res, 48(D1), D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt R, & Mecke D (1979). Permissive effect of dexamethasone on glucagon induction of urea-cycle enzymes in perifused primary monolayer cultures of rat hepatocytes. Eur J Biochem, 97(1), 29–35. [DOI] [PubMed] [Google Scholar]
- Genomes Project, Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, . . . Abecasis GR (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomics, Fabric. Retrieved from https://fabricgenomics.com/)
- Goode DL, Cooper GM, Schmutz J, Dickson M, Gonzales E, Tsai M, . . . Sidow A (2010). Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res, 20(3), 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisolia S, & Cohen PP (1952). The catalytic role of carbamyl glutamate in citrulline biosynthesis. J Biol Chem, 198(2), 561–571. [PubMed] [Google Scholar]
- Grisolia S, & Cohen PP (1953). Catalytic role of of glutamate derivatives in citrulline biosynthesis. J Biol Chem, 204(2), 753–757. [PubMed] [Google Scholar]
- Guerin SL, Leclerc S, Verreault H, Labrie F, & Luu-The V (1995). Overlapping cis-acting elements located in the first intron of the gene for type I 3 beta-hydroxysteroid dehydrogenase modulate its transcriptional activity. Mol Endocrinol, 9(11), 1583–1597. [DOI] [PubMed] [Google Scholar]
- Haberle J (2011). Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther Clin Risk Manag, 7, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle J, Schmidt E , Pauli S , Kreuder JG , Plecko B , Galler A , . . . Koch HG (2003). Mutation analysis in patients with N-acetylglutamate synthase deficiency. Human Mutation, 21(6), 593–597. [DOI] [PubMed] [Google Scholar]
- Hayase K, Naganuma Y, Koie M, & Yoshida A (1998). Role of N-acetylglutamate turnover in urea synthesis by rats treated with the thyroid hormone. Biosci Biotechnol Biochem, 62(3), 535–539. [DOI] [PubMed] [Google Scholar]
- Hayase K, Yonekawa G, Yokogoshi H, & Yoshida A (1991). Triiodothyronine administration affects urea synthesis in rats. J Nutr, 121(7), 970–978. [DOI] [PubMed] [Google Scholar]
- Hayase K, Yonekawa G, & Yoshida A (1992). Changes in liver concentration of N-acetylglutamate and ornithine are involved in regulating urea synthesis in rats treated with thyroid hormone. J Nutr, 122(5), 1143–1148. [DOI] [PubMed] [Google Scholar]
- Hayase K, Yonekawa G, & Yoshida A (1993). Arginine affects urea synthesis in rats treated with thyroid hormone. J Nutr, 123(2), 253–258. [DOI] [PubMed] [Google Scholar]
- Hayase K, & Yoshida A (1995). Role of ornithine in urea synthesis in rats treated with thyroid hormone. Biosci Biotechnol Biochem, 59(5), 801–804. [DOI] [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Snyder GH, & Jusko WJ (2008). Pharmacodynamic modeling of acute and chronic effects of methylprednisolone on hepatic urea cycle genes in rats. Gene Regul Syst Bio, 2, 1–19. [PMC free article] [PubMed] [Google Scholar]
- Heibel SK, Ah Mew N, Caldovic L, Daikhin Y, Yudkoff M, & Tuchman M (2011). N-carbamylglutamate enhancement of ureagenesis leads to discovery of a novel deleterious mutation in a newly defined enhancer of the NAGS gene and to effective therapy. Hum Mutat, 32(10), 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heibel SK, Lopez GY, Panglao M, Sodha S, Marino-Ramirez L, Tuchman M, & Caldovic L (2012). Transcriptional regulation of N-acetylglutamate synthase. PLoS One, 7(2), e29527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illumina. Illumina Retrieved from https://www.illumina.com/
- Jo SS, & Choi SS (2019). Analysis of the Functional Relevance of Epigenetic Chromatin Marks in the First Intron Associated with Specific Gene Expression Patterns. Genome Biol Evol, 11(3), 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, . . . MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim YM, Lee BH, Cho JH, Kim GH, Choi JH, & Yoo HW (2015). Short-term efficacy of N-carbamylglutamate in a patient with N-acetylglutamate synthase deficiency. J Hum Genet, 60(7), 395–397. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet, 46(3), 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell, 128(4), 693–705. [DOI] [PubMed] [Google Scholar]
- Krezel W, Ruhl R, & de Lera AR (2019). Alternative retinoid X receptor (RXR) ligands. Mol Cell Endocrinol, 491, 110436. [DOI] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Retrieved from https://arxiv.org/abs/1303.3997 [Google Scholar]
- Lin RC, Snodgrass PJ, & Rabier D (1982). Induction of urea cycle enzymes by glucagon and dexamethasone in monolayer cultures of adult rat hepatocytes. J Biol Chem, 257(9), 5061–5067. [PubMed] [Google Scholar]
- Liska DJ, Robinson VR, & Bornstein P (1992). Elements in the first intron of the alpha 1(I) collagen gene interact with Sp1 to regulate gene expression. Gene Expr, 2(4), 379–389. [PMC free article] [PubMed] [Google Scholar]
- Luna-Moreno D, Garcia-Ayala B, & Diaz-Munoz M (2012). Daytime restricted feeding modifies 24 h rhythmicity and subcellular distribution of liver glucocorticoid receptor and the urea cycle in rat liver. Br J Nutr, 108(11), 2002–2013. [DOI] [PubMed] [Google Scholar]
- Marti J, Portoles M, Jimenez-Nacher I, Cabo J, & Jorda A (1988). Effect of thyroid hormones on urea biosynthesis and related processes in rat liver. Endocrinology, 123(5), 2167–2174. [DOI] [PubMed] [Google Scholar]
- McClintick JN, Crabb DW, Tian H, Pinaire J, Smith JR, Jerome RE, & Edenberg HJ (2006). Global effects of vitamin A deficiency on gene expression in rat liver: evidence for hypoandrogenism. J Nutr Biochem, 17(5), 345–355. [DOI] [PubMed] [Google Scholar]
- McLean P, & Gurney MW (1963). Effect of adrenalectomy and of growth hormone on enzymes concerned with urea synthesis in rat liver. Biochem J, 87, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM Jr., & Kepka-Lenhart D (2002). Hormonal induction of hepatic mitochondrial ornithine/citrulline transporter mRNA. Biochem Biophys Res Commun, 294(4), 749–752. [DOI] [PubMed] [Google Scholar]
- Natoli G, & Andrau JC (2012). Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet, 46, 1–19. [DOI] [PubMed] [Google Scholar]
- Nebes VL, & Morris SM Jr. (1988). Regulation of messenger ribonucleic acid levels for five urea cycle enzymes in cultured rat hepatocytes. Requirements for cyclic adenosine monophosphate, glucocorticoids, and ongoing protein synthesis. Mol Endocrinol, 2(5), 444–451. [DOI] [PubMed] [Google Scholar]
- Okun JG, Conway S, Schmidt KV, Schumacher J, Wang X, de Guia R, . . . Rose AJ (2015). Molecular regulation of urea cycle function by the liver glucocorticoid receptor. Mol Metab, 4(10), 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Hannenhalli S, & Choi SS (2014). Conservation in first introns is positively associated with the number of exons within genes and the presence of regulatory epigenetic signals. BMC Genomics, 15, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Cipriani S, D’Amore C, Zampella A, Monti MC, . . . Fiorucci S (2011). The nuclear receptor FXR regulates hepatic transport and metabolism of glutamine and glutamate. Biochim Biophys Acta, 1812(11), 1522–1531. [DOI] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res, 47(D1), D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Vaello E, Marco-Marin C, Gougeard N, Fernandez-Murga L, Rufenacht V, Mustedanagic M, . . . Haberle J (2016). Understanding N-Acetyl-L-Glutamate Synthase Deficiency: Mutational Spectrum, Impact of Clinical Mutations on Enzyme Functionality, and Structural Considerations. Hum Mutat, 37(7), 679–694. [DOI] [PubMed] [Google Scholar]
- Schoneveld OJ, Gaemers IC, Hoogenkamp M, & Lamers WH (2005). The role of proximal-enhancer elements in the glucocorticoid regulation of carbamoylphosphate synthetase gene transcription from the upstream response unit. Biochimie, 87(11), 1033–1040. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, & Seelow D (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods, 7(8), 575–576. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, & Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res, 29(1), 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, . . . Haussler D (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res, 15(8), 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, . . . Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri E, Moreno M, Schiavo L, de Lange P, Lombardi A, Chambery A, . . . Goglia F (2006). A proteomics approach to identify protein expression changes in rat liver following administration of 3,5,3’-triiodo-L-thyronine. J Proteome Res, 5(9), 2317–2327. [DOI] [PubMed] [Google Scholar]
- Sonaimuthu P, Senkevitch E, Haskins N, Uapinyoying P, McNutt M, Morizono H, . . . Caldovic, L. (2021). Gene delivery corrects N-acetylglutamate synthase deficiency and enables insights in the physiological impact of L-arginine activation of N-acetylglutamate synthase. Sci Rep, 11(1), 3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, . . . Lancet D (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics, 54, 1 30 31–31 30 33. [DOI] [PubMed] [Google Scholar]
- Toolkit, Picard. (2019). Picard Toolkit. Broad Institute, GitHub Repository Retrieved from http://broadinstitute.github.io/picard/ [Google Scholar]
- Ulbright C, & Snodgrass PJ (1993). Coordinate induction of the urea cycle enzymes by glucagon and dexamethasone is accomplished by three different mechanisms. Arch Biochem Biophys, 301(2), 237–243. [DOI] [PubMed] [Google Scholar]
- van de Logt AE, Kluijtmans LA, Huigen MC, & Janssen MC (2017). Hyperammonemia due to Adult-Onset N-Acetylglutamate Synthase Deficiency. JIMD Rep, 31, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterlow JC (1999). The mysteries of nitrogen balance. Nutrition Research Reviews, 12, 25–54. [DOI] [PubMed] [Google Scholar]
- Williams M, Burlina A, Rubert L, Polo G, Ruijter GJG, van den Born M, . . . Caldovic L (2018). N-Acetylglutamate Synthase Deficiency Due to a Recurrent Sequence Variant in the N-acetylglutamate Synthase Enhancer Region. Sci Rep, 8(1), 15436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthers T, Hamberg O, Grofte T, & Vilstrup H (2000). Effects of budesonide and prednisolone on hepatic kinetics for urea synthesis. J Hepatol, 33(4), 549–554. [DOI] [PubMed] [Google Scholar]
- Yang YZ, Subauste JS, & Koenig RJ (1995). Retinoid X receptor alpha binds with the highest affinity to an imperfect direct repeat response element. Endocrinology, 136(7), 2896–2903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.