

Abstract
Background & Aims:
Intratumor molecular heterogeneity is a key feature of tumorigenesis and is linked to treatment failure and patient prognosis. Here, we aimed to determine what drives tumor cell evolution by performing single-cell transcriptomic analysis.
Methods:
We analyzed 46 hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA) biopsies from 37 patients enrolled for interventional studies at the NIH Clinical Center, with 16 biopsies collected before and after treatment from 7 patients. We developed a novel machine learning-based consensus clustering approach to track cellular states of 57,000 malignant and non-malignant cells including tumor cell transcriptome-based functional clonality analysis. We determined tumor cell relationships using RNA velocity and reverse graph embedding. We also studied longitudinal samples from 4 patients to determine tumor cellular state and its evolution. We validated our findings in bulk transcriptomic data from 488 patients with HCC and 277 patients with iCCA.
Results:
Using transcriptomic clusters as a surrogate for functional clonality, we observed an increase in tumor cell state heterogeneity which was tightly linked to patient prognosis. Furthermore, increased functional clonality was accompanied by a polarized immune cell landscape which included an increase in pre-exhausted T-cells. We found that SPP1 expression was tightly associated with tumor cell evolution and microenvironmental reprogramming. Finally, we developed a user-friendly online interface as a knowledge base for a single-cell atlas of liver cancer.
Conclusions:
Our study offers insight into the collective behavior of tumor cell communities in liver cancer as well as potential drivers of tumor evolution in response to therapy.
Keywords: Tumor cell state, Functional clonality, Tumor evolution, Liver cancer, Single cell, Tumor transcriptomic heterogeneity, Tumor microenvironments, T cells, Osteopontin
Graphical Abstract

Lay Summary
Intratumor molecular heterogeneity is a key feature of tumorigenesis, and is linked to treatment failure and patient prognosis. In this study, we present a single-cell atlas of liver tumors from patients treated with immunotherapy and describe intratumoral cell states and their hierarchical relationship. We suggest osteopontin, encoded by the gene SPP1, as a candidate regulator of tumor evolution in response to treatment.
INTRODUCTION
Tumorigenesis is a consequence of an evolutionary process by which somatic cells acquire genetic and epigenetic alterations when exposed over time to extrinsic or intrinsic factors such as adverse tumor microenvironments (TME)[1]. It is known that tumor cells within a solid lesion exist as a social community where each tumor cell interacts with one another, as well as their TME which includes the extracellular matrix, tumor vasculature and immune cells. This TME provides a setting in which tumor cells sense and adapt to environmental cues such as available nutrients, oxygen, as well as toxic agents such as chemotherapeutics for its survival and growth[2]. As such, a tumor lesion is under the constant pressure of Darwinian evolutionary selection reminiscent to the collective behavior of the animal kingdom[3]. Consequently, such an intrinsic ‘survival of the fittest’ trait, found in most if not all malignant solid tumors, can result in vast heterogeneity within tumor cell populations known as intratumor heterogeneity (ITH)[1], a feature universally linked to tumor aggressiveness[4]. In the same way that biodiversity within a defined ecosystem promotes fitness and survival of an organism, ITH has a similar role in sustaining tumor survival. Evidence of ITH has been observed in most solid tumor types by both tumor bulk and single cell analyses[5]. However, these studies have a limitation in interpreting tumor clonality and its evolution since a majority of studies utilize tumor bulk, which eliminates individuality among cells. Our recent single cell analysis, together with many studies in the literature, indicate a tight link between tumor aggressiveness and ITH[4, 6]. Thus, presence of ITH creates unique challenges for targeted therapy, leading to drug resistance, treatment failure and poor outcomes[7]. This begs the questions of what are the cellular factors driving the collective behaviors of a tumor cell community? Are there intrinsic drivers regulating tumor cell evolution, similar to the proposed model of ITH as a reflection of the inherent biology of a given tumor type[7, 8]? Understanding the biodiversity and clonality of solid cancers on a cellular or lesion-specific basis may provide conceptual knowledge about carcinogenesis with practical implications for diagnosis and treatment.
Traditionally, tracing tumor cell clonality relies on somatic mutations found in tumor cells as cancer is viewed as a genetic disease[9]. However, it is increasingly recognized that a majority of somatic mutations are known as passenger mutations as they are not causative of cancer development and thus are not involved in positive selection during tumor evolution[10]. This makes it less desirable to use genetic tracing with tumoral mutational data to reconstruct clonality, especially from the current single-cell DNA sequencing platform due to a high false positivity. A recent elegant study of mouse and human lung tumorigenesis revealed that a high plasticity cell state common to mouse and human lung tumors drives cellular heterogeneity and that cell state heterogeneity arises largely independently of genetic variation during lung cancer evolution[11]. In contrast, describing cellular clonality in normal cells such as defining subclones and clonal expansion in immune cells has commonly been used to define immune cell states. Given a high-plasticity cell state during tumor evolution, defining tumor cell functional clonality and its cell state heterogeneity is conceptually better achieved by determining tumor cell clusters at single-cell levels with transcriptomic similarities as well as their cell lineage and evolutional trajectories[12].
Primary liver cancer is among the top five deadliest cancers in the world, of which the most common types are hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA)[13]. Both tumor types have been shown to have extensive intertumor and intratumor heterogeneity[14, 15]. This is unsurprising given the presence of multiple etiological factors, extensive genomic complexity and underling tumor biology in HCC and iCCA[16]. While most HCC and iCCA are refractory to systemic therapeutics that target tumor cells[17], some HCC patients show remarkable and durable responses to immune checkpoint inhibitors either alone or in combination with ablation or anti-VEGF treatment[18, 19]. Our recent study using single-cell transcriptome analysis revealed a strong link between ITH and liver cancer prognosis[6], which offered a mechanistic explanation for the differential treatment responses and provided a rationale for the use of a combination therapy of immune checkpoint inhibitors and anti-vascular treatment to improve therapeutic efficacies[19]. Thus, our single-cell transcriptome analysis provides sufficient resolution to faithfully determine liver tumor cell communities. To extend our initial study, we aimed to determine what drives ITH and to delineate tumor cell evolution in HCC and iCCA biospecimens from 37 patients enrolled in various treatment protocols at the National Institutes of Health (NIH) Clinical Center. Moreover, we developed an online interface as a knowledge base for a single-cell atlas in liver cancer (scAtlasLC, https://scatlaslc.ccr.cancer.gov). In conclusion, our study provides rich resources and valuable insights into the understanding of a complex liver cancer ecosystem and identifies osteopontin (OPN) as a potential player driving ITH.
MATERIALS AND METHODS
Human sample collection.
Fresh liver tumor biopsies were collected with preoperative informed consent from patients participating at NIH Clinical Center for interventional studies, following approval by the ethics committee of the National Institutes of Health (ClinicalTrials.gov Identifier: NCT01313442). A total of 37 liver cancer patients (HCC, 25; iCCA, 12) were involved in this study, with 46 tumor biopsies collected before or after treatment. Among the tumor biopsies, 19 samples were collected and analyzed in our previous study[6]. The ages of the patients ranged from 35 to 81, with a median age of 65. Fourteen patients were tested positive for HCV, four patients positive for HBV, two with fatty liver and one positive for both HBV and HDV. A majority of the patients were treated with immune checkpoint inhibitors of durvalumab, tremelimumab, pembrolizumab to target PD-L1, CTLA-4 as well as PD-1. The detailed clinical data of the patients was summarized in Table S1.
RESULTS
Single-cell atlas of tumor ecosystem in liver cancer
We performed single-cell RNA sequencing (scRNA-seq) of liver tumors to study tumor cell states, cellular hierarchy and its evolution that contribute to ITH (Figure 1A and Table S1). In a subset of seven patients, liver tumor biopsies were available from multiple time points, including before and after treatment. We obtained single-cell transcriptomic profiles of 56,721 single cells (Figure 1B and Table S2). Epithelial marker genes were highly expressed in some of the cells, suggesting potential tumoral origins (Figure 1C). We analyzed three biopsies collected at different time points from one patient (H34) and found a relatively similar tumor ecosystem makeup among all three biopsies such as T cells, B cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), tumor-associated endothelial cells (TECs) and epithelial cells, revealing relatively stable tumor stromal compositions over time (Figures 1D and S1A). We identified 17,164 malignant cells using a method previously described[6] (Figures S1B–S1D). We found malignant cells formed patient-specific clusters, each with a heterogeneous population (Figure 1E, top panel), consistent with previous studies[6]. Consistently, pairwise correlation analysis revealed a stronger association of single cells within a tumor than between tumors (Figure 1E, bottom panel). In contrast, non-malignant cells mainly grouped according to cell types, as annotated according to known cell lineage specific marker genes unique to T cells, B cells, CAFs, TAMs, TECs, hepatocytes and cholangiocytes (Figure 1F). We confirmed the histology of liver tumors in our study by histopathology analysis (Figure 1G).
Figure 1. Single-cell transcriptomic profiling of primary liver tumor.

(A) Workflow of liver tumor biopsy collection, processing, sequencing, and computational analysis.
(B) t-SNE plot of 56,721 single-cells from 46 liver tumor samples (indicated by colors). Case ID was named according to histological subtypes of liver cancer as well as biopsy timing. H, HCC; C, iCCA; B, baseline (the first biopsy); F, follow-up biopsy after treatment.
(C) t-SNE plot of single-cells in (B) colored by epithelial score. Epithelial score was determined by the average expression of epithelial marker genes.
(D) t-SNE plot of a representative case (H34) with biopsies collected at baseline and follow-up study during treatment.
(E) t-SNE plot (top) and tumor heterogeneity (bottom) of malignant cells from 30 tumors with > 15 malignant cells in each tumor.
(F) t-SNE plot (top) and known lineage-specific marker genes (bottom) of non-malignant cells. Cell types were indicated by colors.
(G) Histopathology of the eight representative tumors. Scale bars, 100 μm.
Tumor cell functional clonality and tumor branching evolution
We determined tumor cell states as a functional clonality of malignant cells within each tumor by developing a consensus clustering algorithm (see supplementary text) (Figures S2A and S2B). We searched similarities among tumor sub-clusters based on a hierarchical clustering method (Figure 2A, top two panels). Bootstrap analysis[20] was applied to measure the confidence of the hierarchical relationship of the tumor clusters sharing similar cellular states (Figure S2C, top panel). We found three major branches in the phylogenetic tree and referred the branches using branching index (BI) as BI-A, BI-B, and BI-C. All tumors in BI-A are from HCC cases, while BI-B and BI-C contain tumors from both HCC and iCCA. In all three branches, tumor cell clusters within a tumor tend to be grouped together forming a hierarchical architecture. To further determine cluster-to-cluster relationship within a tumor, we applied two independent methods: reverse graph embedding method[21] and RNA velocity[22], to learn the single-cell trajectory and potential cell-lineage among malignant cells (Figure 2A, middle panels rows 3–5). Both methods revealed similar tumor cell trajectories, which largely agree with the cluster relationship within each tumor using a hierarchical clustering method (Figure 2A, top panel). We developed a strategy to search for conserved genes of different branches (Figure 2B). We reasoned that a gene that is elevated in tumor cells and expressed ubiquitously at the branch or sample level, is likely to drive the function of the corresponding branch or sample. This strategy revealed SPP1 as the top conserved gene in BI-B and BI-C (Figure S2C, bottom panel). Noticeably, SPP1 expression was often enriched in tumor cells at the beginning of a cell lineage (Figure 2A, panel row 6), and was much higher in BI-B/C than BI-A (Figure 2A, bottom panel). Moreover, SPP1 was mainly expressed in malignant cells rather than non-malignant cells (Figure 2C). Thus, SPP1 could act as a driver of tumor evolution.
Figure 2. Tumor cell functional clonality and tumor branching evolution.
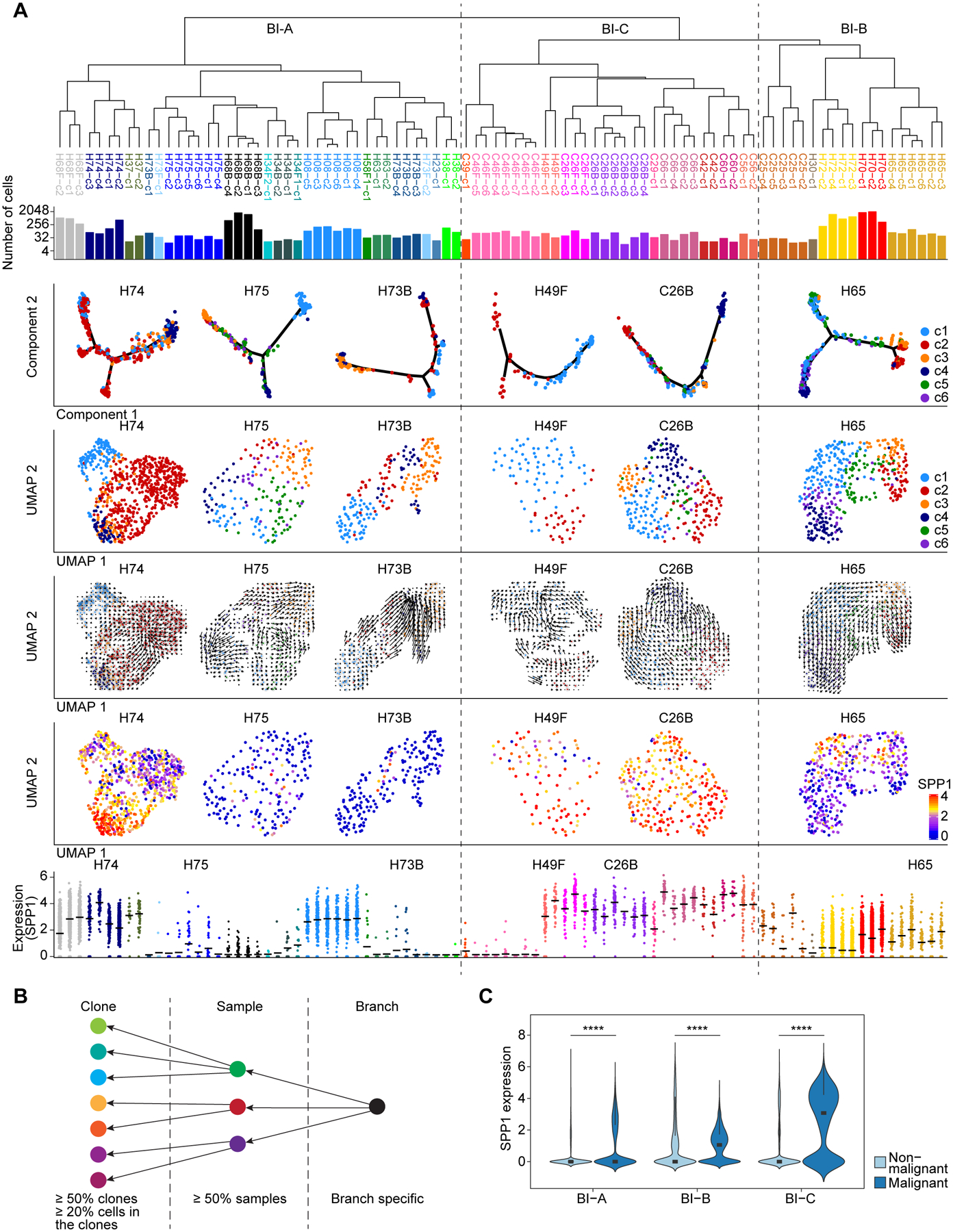
(A) Tumor phylogenetic tree constructed by hierarchical clustering of all the clusters from 30 tumors (indicated by colors) with > 15 malignant cells. BI-A, BI-B and BI-C were defined according to the hierarchical relationship. Panels from top to bottom: phylogenetic tree (top); the number of cells in each sub-cluster (row 2); single-cell trajectory (row 3), tumor cell functional clonality (row 4), RNA velocity-based cell lineage (row 5), and SPP1 expression (row 6) of several representative tumors; SPP1 expression in each sub-cluster of the 30 tumors, with representative tumors labeled on top of the jitter plot (bottom). Line segments in the jitter plot indicate the mean values.
(B) Strategy used for searching conserved genes of tumor branches.
(C) SPP1 expression in malignant cells and non-malignant cells of different branches. Wilcoxon test was performed to indicate statistical significance. The width of a violin plot represents the density of gene expression values. Box spans the first quartile to the third quartile of the values while segment inside a box indicates the median value. **** p-value < 0.0001.
Tumor functional clonality is associated with patient prognosis
We found that there was an increasing number of sub-clusters in tumors from BI-A, BI-B, and BI-C (Figure S3A), suggesting that tumors in BI-B and BI-C were more diverse and thus likely to be more aggressive than tumors in BI-A, a feature described in previous studies[4, 6, 23]. Consistently, patients whose tumor had a higher cluster number had a much shorter survival than patients with a lower cluster number (Figure 3A). In addition, there was a significant trend of overall survival of patients in BI-A, BI-B and BI-C (Figure 3B). Due to a small number of cases in BI-B, we combined BI-B and BI-C as BI-B&C to increase the statistical power. Consistently, patients in BI-B&C had a significantly shorter survival than patients in BI-A (Figure 3C). We also performed survival analysis of HCC patients separately and found similar results (Figure 3D). We did not perform separate analysis for iCCA as they were found only in BI-B&C.
Figure 3. Tumor cell state heterogeneity is associated with patient prognosis.

(A) Overall survival of all patients with low number of clusters (Clonality-Low) and high number of clusters (Clonality-High) separated by the median value.
(B and C) Overall survival of all the patients from different branches, by using three-group comparison (B) and two-group comparison (C).
(D) Overall survival of HCC patients from different branches.
(E) The enriched pathways of tumors from BI-A, BI-B and BI-C. Only tumors with more than two clusters detected as well as at least one enriched pathway with FDR adjusted q value < 0.05 were involved in the heatmap. NES, normalized enrichment score.
(F) Functional analysis of clusters within each tumor. Top three panels show the expression of hypoxia, EMT and angiogenesis related genes in each sub-cluster of the tumors from BI-A, BI-B and BI-C. Bottom three panels show the expression of hypoxia, EMT and angiogenesis related genes on the UMAP of several representative tumors. Line segments in the jitter plot indicate the mean values.
Log-rank test and log-rank test for trend were preformed to show the statistical significance in (A-D).
We carried out gene enrichment analysis using differential expression genes among the functional clusters within each tumor to determine common cluster-related features. We found that tumors in BI-B and BI-C were enriched in tumor aggressiveness-related pathways, such as epithelial–mesenchymal transition (EMT), hypoxia, TNF alpha signaling as well as glycolysis (Figure 3E and Figure S3B). We found that genes related to hypoxia, EMT, and angiogenesis defined by GSEA (Table S3) were elevated in BI-B and BI-C compared with BI-A (Figure 3F, top three panels). While EMT was significantly associated with patient outcomes, hypoxia and angiogenesis only showed a trend (Figure S3C). Moreover, these genes were heterogeneously expressed among sub-clusters within a tumor, suggesting that different clusters may serve different roles in a tumor ecosystem (Figure 3F, bottom three panels). These results are consistent with the survival analysis described above, indicating that patients in BI-B and BI-C tend to gain aggressive tumor features.
Our single-cell results described above were based on a limited number of cases. To further validate the above findings, we analyzed bulk transcriptomic data from two HCC cohorts of 488 patients (i.e., Liver Cancer Institute, LCI, cohort and TCGA HCC cohort) and two iCCA cohorts of 277 patients (i.e., International Cancer Genomics Consortium, ICGC, cohort and Japan cohort). We conducted differential gene expression analysis among sub-clusters of malignant cells from BI-B&C and BI-A to generate tumor functional clonality surrogate gene signatures. The derived signatures from BI-A and BI-B&C were then applied in the four cohorts for survival risk prediction, respectively. We obtained remarkably consistent results from the four cohorts, with a better separation of high-risk and low-risk groups of patients as well as a higher hazard ratio by using the surrogate signature from BI-B&C than the signature from BI-A (Figures S3D and S3E). We also found consistent results using the functional clonality surrogate signatures of only HCC cases (Figures S3D and S3F). To determine the relationship between tumor branches identified in this study and known molecular subtypes defined by bulk transcriptome data, we generated pseudo-bulk transcriptome of individual tumors using average gene expression of all single cells within each tumor. We then performed a comparison of pseudo-bulk transcriptome data to bulk transcriptome linked to molecular subtypes of HCC and iCCA [14]. We found that the prognostic subtypes of patients in BI-A and BI-B&C are mostly consistent with that of HCC and iCCA molecular subtypes although tumor cell states identified in our single-cell study was reconstructed using only malignant cells (Figure S3G). Taken together, our results indicate that tumor functional clonality is associated with patient prognosis, independent of tumor types.
TME polarization
When analyzing T cells, B cells, TAMs, CAFs and TECs separately, we found that these cells differed in their transcriptomic profiles between BI-A and BI-B&C, suggesting a potential reprogramming of non-malignant cells by malignant cells (Figure S4A). We focused mainly on T cells (Figure S4B; see Methods for details) as most of the patients in our study were treated with immune checkpoint inhibitors. We first extracted CD4+ or CD8+ T cells by using marker genes of CD4, CD8A or CD8B, respectively. A graph-based clustering method was then applied to define T-cell subtypes according to known marker genes (Figures S4C–S4H), following the approach described in recent studies[24–26]. We finally remapped the determined T cell subtypes to all the T cells based on clustering analysis (Figures 4A and S5A). In total, we identified 20 T-cell subtypes, including 10 CD4+ and 10 CD8+ T-cell subtypes using the same criteria previously described[24–27]. We found a nice trajectory of both CD4+ and CD8+ T cells initiated from naïve T cells and further branching into cytotoxic T cells and exhausted T cells (Figures 4A, S4E, and S4H). Moreover, immune checkpoint molecules were mainly expressed in regulatory T cells (CD4-c10-FOXP3), pre-exhaustion T cells (CD8-c6-CXCL13), and exhaustion T cells (CD8-c5-PDCD1) as shown in Figure 4B.
Figure 4. Landscape of T cells in liver cancer.
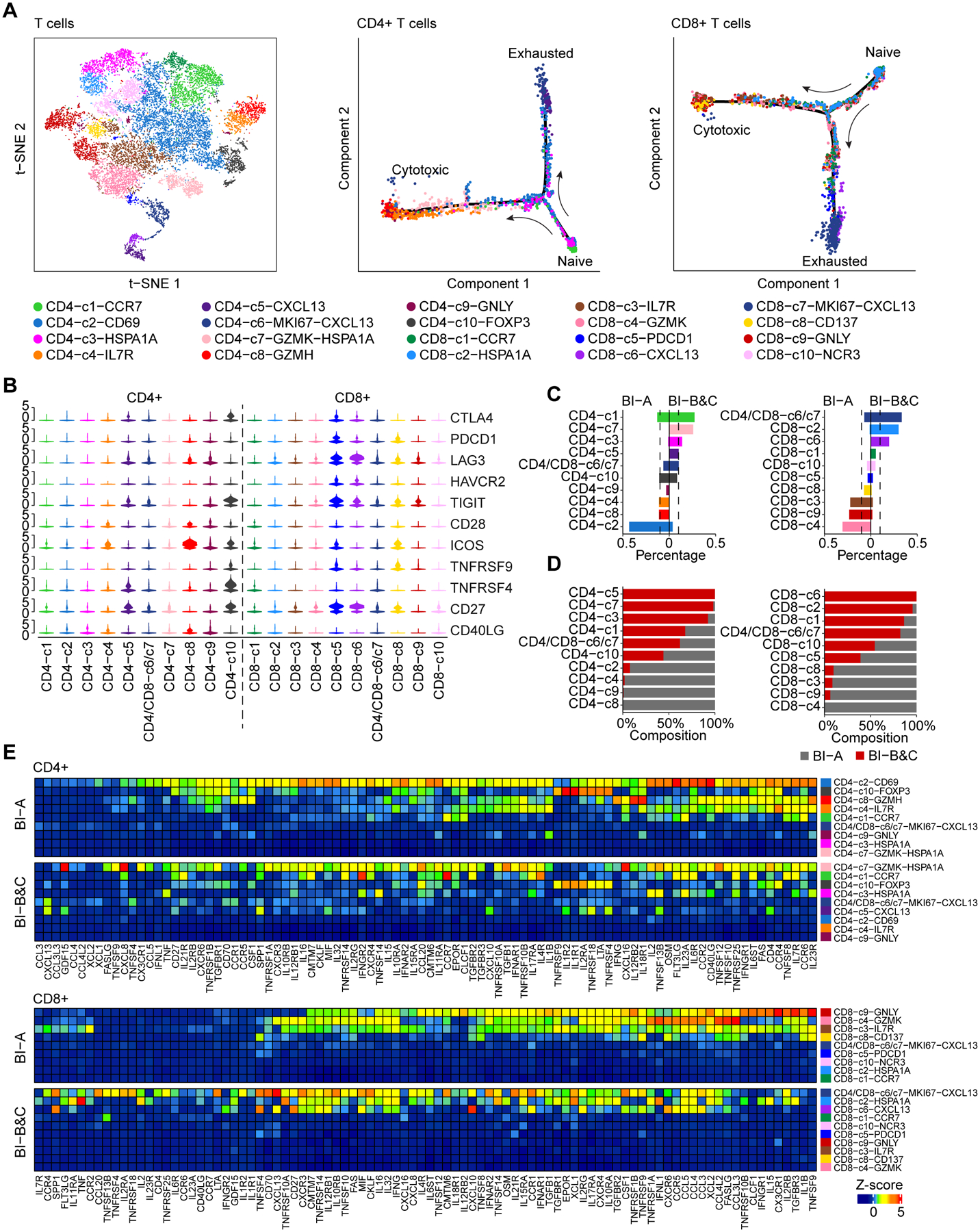
(A) t-SNE plot of all the 19,587 T cells (left), as well as single cell trajectory of CD4+ (middle) and CD8+ (right) T cells. T-cell subtypes are indicated by colors.
(B) Violin plot of immune checkpoint molecules in all T-cell subtypes. The width of a violin plot represents the density of gene expression values.
(C) The percentage of each CD4+ (left) and CD8+ (right) T-cell subtype in BI-A and BI-B&C. Dashed lines indicate 10%.
(D) Composition of each CD4+ (left) and CD8+ (right) T-cell subtype from BI-A and BI-B&C.
(E) Cytokines and chemokines produced by each CD4+ (top) and CD8+ (bottom) T-cell subtype from BI-A and BI-B&C. Gene expression was weighted by the percentage of each T-cell subtype, followed by z-score transformation.
We compared the proportion of T-cell subtypes between BI-A and BI-B&C. We found a polarization of both CD4+ and CD8+ T-cell subtypes among the two branches (Figures 4C and 4D). Large proportions of memory T cells of CD4-c2-CD69, CD8-c4-GZMK, and CD8-c3-IL7R, as well as a group of cytotoxic T cells of CD8-c9-GNLY were observed in BI-A. In contrast, proliferative pre-exhaustion T cells of CD4/CD8-c6/c7-MKI67-CXCL13 and pre-exhaustion T cells of CD8-c6-CXCL13 were enriched in BI-B&C. In general, the levels of cytokines and chemokines were much higher in CD4+ and CD8+ T-cells from BI-A than that of BI-B&C (Figure 4E). Cytotoxic T cells of CD8-c9-GNLY were a major source of cytokines and chemokines in BI-A while proliferative pre-exhaustion T cells of CD4/CD8-c6/c7-MKI67-CXCL13 mainly produced those cellular factors in BI-B&C. We found similar results in branching analysis of granzymes and perforin, which may reflect the cytotoxicity of T cells (Figure S5B). In addition, both CD4+ and CD8+ T cells from BI-A were enriched in immune response related pathways, which were not found in T cells from BI-B&C (Figures S5C and S5D). We found similar results when HCC cases were analyzed separately (data not shown). Taken together, these results support a polarization of T cells landscape between BI-A and BI-B&C.
Next, we used CellPhoneDB[28] to analyze cell communications by searching for ligand-receptor interactions between malignant cells (sources of ligands) and T cells (sources of receptors). We observed much stronger interactions between malignant cells and T cells in BI-B&C than that of BI-A for both CD4+ and CD8+ T cells (Figure 5), suggesting that tumor cells from BI-B&C have an elevated activity of ligand-receptor interactions. Strikingly, SPP1-CD44 was a top interaction pair between malignant cells and T cells, further supporting the key role of SPP1 in the tumor ecosystem.
Figure 5. Communications of malignant cells and T cells.

Ligand-receptor interactions of malignant cells and CD4+ T cells (left) as well as malignant cells and CD8+ T cells (right). Dashed lines separate interactions in tumor ecosystems from BI-A and BI-B&C. Row stands for ligand and receptor pairs. red, ligand from malignant cells; blue, receptor from T cells. Column represents pair of malignant cells (red) and T-cell subtype (blue).
We also determined cell states of CAFs, TECs, TAMs and B cells using a shared nearest neighbor modularity optimization based clustering algorithm (Figure S6A) and found that the clusters within each cell type were polarized between BI-A and BI-B&C (Figures S6B–S6D). For instance, we found four clusters within the CAF population, with HSPA1A+ (cluster b), ID4+ (cluster c) and THBS2+ (cluster d) cells distributed at the terminal of three different branches of the trajectory while THY1+ (cluster a) cells along the trajectory were in between. We observed enrichment of HSPA1A+ and ID4+ cells in BI-A tumors, THBS2+ cells in BI-B&C tumors, and THY1+ cells in both BI-A and BI-B&C, indicating potential gradual reprogramming of CAFs by BI-B&C tumor. Collectively, our results suggest a polarization of TME with tumor branching evolution.
Tumor evolution in response to treatment
Tumor evolutionary trajectories may change over time in response to treatment. Among 37 patients, we obtained at least two core biopsies from 7 patients at different time points (Table S1). Among them, 4 cases with biopsies having at least 15 malignant cells in each biopsy before (baseline) and after treatment were used for evolutionary trajectory analysis by RNA velocity method (Figures 6A and 6B). For two cases of C26 and H68, we found remarkably different lineage of malignant cells before and after treatment, with a weak connection of the clusters among two biopsies. In contrast. the clusters from before and after treatment in H34 and H73 were mixed, indicating a relatively stable population of malignant cells from these patients during treatment (Figures 6A and 6B). Consistent with the RNA velocity analysis, hierarchical clustering analysis revealed that the clusters from baseline and treatment clustered separately for C26 and H68 but were more homogenous in H34 and H73 (Figure 6C). To study the genomic similarities between sub-clusters within each case, we calculated a pairwise correlation of CNVs between malignant cells before and after treatment (intra-patient) as well as among tumors from different patients (inter-patient). We also performed a randomization of CNV segments to establish a background correlation value distribution as a reference. We found a much higher genomic similarity in paired biopsies compared to inter-patient biopsies (Figure 6D), supporting the hypothesis that paired tumors are more likely from the same cell of origin. Interestingly, we found relatively higher degrees of genomic similarity in paired biopsies from C26, H34 and H73, compared to H68 (Figure 6D), suggesting that paired tumor biopsies of H68 are much more genomically unique. It is plausible that the follow-up biopsy of H68 may be a de novo tumor rather than disseminated tumor cells from its baseline, as its pairwise correlation values are much closer to that of the intertumor-derived values (Figure 6D). Strikingly, SPP1 was significantly elevated in malignant cells of C26 and H68 after treatment, while no significant changes of SPP1 expression was observed in H34 and H73 with similar malignant cell populations before and after treatment (Figure 6E). These results once again highlight the role of SPP1 in tumor evolution. Collectively, these data may explain why H34, H68 and H73 responded to immunotherapy while C26 didn’t. While the small number of cases with longitudinal biopsies limit our ability to provide a conclusive statement of the mechanisms for varying patient responses, we are collecting samples from more cases and may decode mechanisms of diverse patient responses to immunotherapy in the future. To help researchers who are interested in exploring our single cell data for hypothesis development and validation, we developed a user-friendly online interface of single-cell atlas in liver cancer as scAtlasLC (https://scatlaslc.ccr.cancer.gov). We will continuously update new single-cell data derived from our NCI CCR CLARITY study.
Figure 6. Analysis of liver tumor collected at different time points.
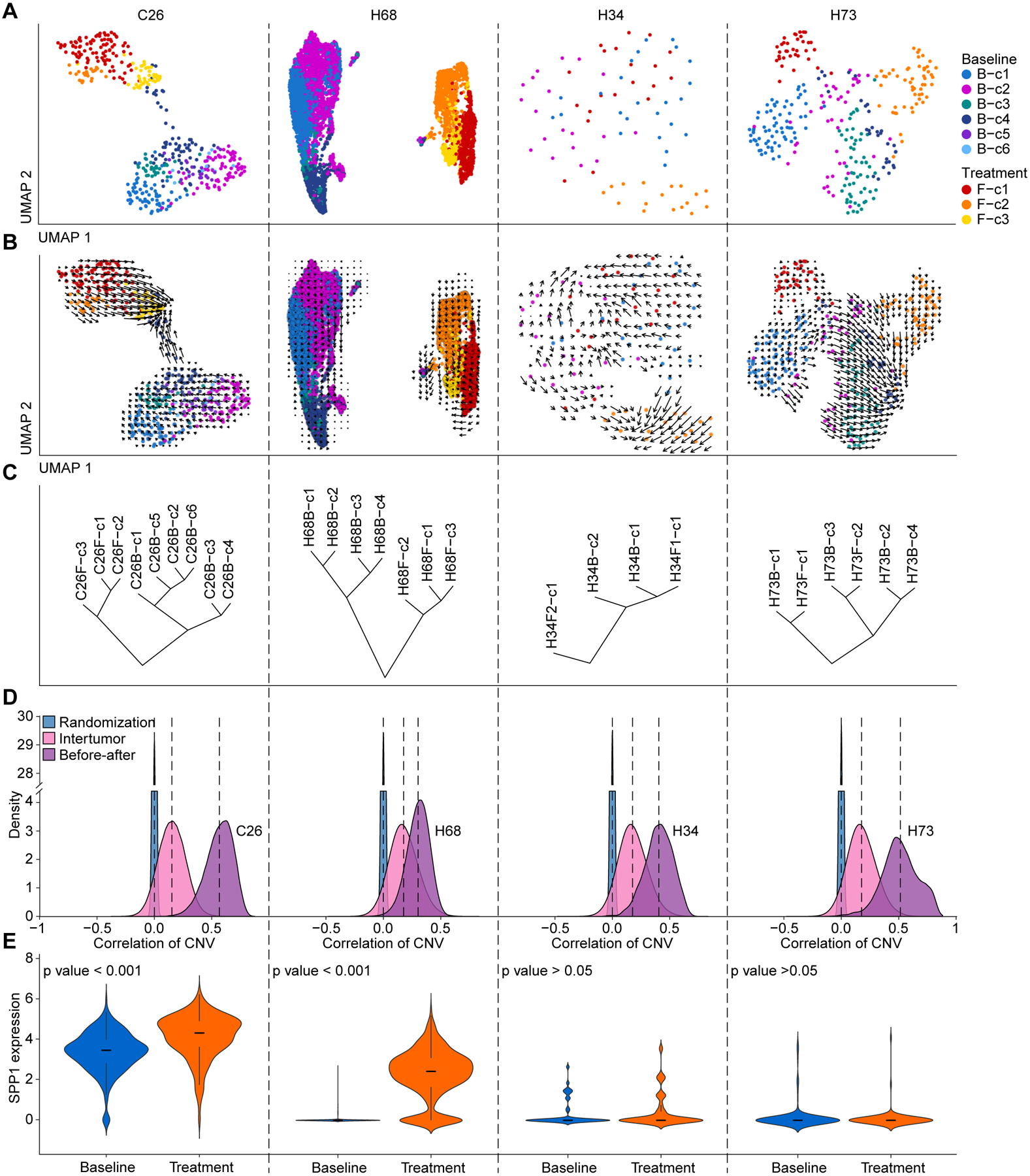
(A and B) Tumor cell functional clonality (A) and RNA velocity-based cell lineage (B) of malignant cells from 4 cases with > 15 malignant cells obtained from each biopsy at baseline or after treatment. Clusters are indicated by colors.
(C) Hierarchical clustering of the tumor cell clusters from baseline and after-treatment biopsies within each case.
(D) Pairwise correlation of CNVs between malignant cells at baseline and after treatment (light purple), between tumors (pink) as well as the correlation of malignant cells with a randomization of CNV segments (blue).
(E) SPP1 expression in malignant cells from baseline and treatment. The width of a violin plot represents the density of gene expression values. Box spans the first quartile to the third quartile of the values while segment inside a box indicates the median value.
We also compared the landscape of T cells in biopsies collected between the first biopsy (baseline) and the second biopsy (after treatment) (Figure S7). There was a smaller difference of CD4+ T-cell subtypes among the first biopsy but a much bigger difference among the second biopsy between BI-A and BI-B&C, with a large proportion of memory T cells of CD4-c2-CD69 in BI-A while CD4-c3-HSPA1A was enriched in BI-B&C. For CD8+ T cells, cytotoxic T cells of CD8-c9-GNLY and proliferative pre-exhaustion T cells of CD4/CD8-c6/c7-MKI67-CXCL13 were enriched in the first biopsy from BI-A and BI-B&C, whereas effector memory T cells of CD8-c4-GZMK were largely in BI-A while CD8-c2-HSPA1A was mainly in BI-B&C from the second biopsy. Consistent with the differential expression patterns of cytokines, chemokines, granzymes and perforin in T cells between BI-A and BI-B&C described above, we also observed similar differential expression patterns between the first biopsy (baseline) and the second biopsy (after treatment) linked to BI-A and BI-B&C (Figure S8). However, it is unclear what the functional significance of the T-cell subtypes that express HSPA1A is as this subtype has not been described previously[24–27]. Further studies are needed to determine the functional roles of CD4-c3-HSPA1A and CD8-c2-HSPA1A T cells in BI-B&C tumors.
DISCUSSION
Biodiversity is a common trait in solid-organ malignancy and is thought to be evolutionarily selected for due to its ability to increase tumor cell fitness and survival, possibly via a common molecular mechanism(s). As such, faithfully defining tumor clonality and its evolutionary trajectory is paramount in understanding the mechanistic collective behavior of tumor cells within a solid-organ malignancy and defining its potential drivers. While genome sequencing of bulk tumor has provided compelling evidence for the presence of ITH[29], this approach is limited due to its lack of sufficient resolution to define tumor cell functional clonality within diverse subclones and their distinct functionality-based evolutionary trajectories. The current methods to define driver mutations are also limited due to their inability to define missense mutations, somatic copy number alterations, structural variants and epigenetic alterations. This complexity fuels the ongoing debate about the definitions of tumor clonality and its drivers in the context of tumor heterogeneity[30]. Improved methodologies are being developed to analyze bulk tumor samples with the hopes to better define and evaluate tumor clonality[31]. However, given the lack of optimal resolution, it is warranted that understanding tumor evolution should be sought out at the single-cell resolution in a collaborative effort such as through the NCI Cancer Moonshot Initiative[32].
So how do we better define tumor cell functional clonality at single-cell resolution, given that the current primitive state of single-cell sequencing technologies at the genomic level has a high error rate for mutational information? Thus, we sought to determine the clonality of liver tumors using single-cell transcriptomics, an approach that has matured in recent years. Our rationale to use transcriptome to define tumor subclones is as follows: first, while genetic and epigenetic alterations drive tumorigenesis, the vast majority of these alterations may not be linked to phenotypic consequences[30]. Thus, tracing genetic information with a low degree of confidence to their functional impact could be misleading for monitoring tumor evolution. Second, single-cell transcriptomic analysis has been used successfully to define distinct cell types with defined functionality-based clones reflecting cellular states and phenotypic features in various organs such as the brain and liver[33–35]. Third, each tumor is comprised of cell populations with diverse transcriptional programs. As tumor subclones evolve through phenotypic selection to adapt to various environments, functional diversity may be the consequence of genetic heterogeneity. Thus, tracing subclones based on their transcriptional program may represent a closer cellular state in regards to their phenotypic features, a strategy that has been increasingly recognized as tumor functional heterogeneity[12]. Using this strategy, we have determined tumor cell clonality and its hierarchical relationship in HCC and iCCA. We found that patients whose tumors have a higher cluster number have a much shorter survival compared to those with a lower cluster number. Phylogenetic analysis of subclusters reveals strong evidence of functionality-based tumor branching evolution in HCC and iCCA, consistent with the Darwinian tumor evolution model proposed by Peter Nowell[36]. CNV analysis revealed similarities in clonality between transcriptome-based subclones and CNV-based subclones, supporting the hypothesis that functional diversity is the consequence of genetic heterogeneity and thus better represents functionality-based evolutionary trajectories. Moreover, we validated our findings using independent and publicly available tumor bulk data from 488 HCC patients and 277 iCCA patients. Interestingly, iCCA patients generally have a much higher clonality and refractory treatment response than HCC patients enrolled in our interventional studies. It is plausible that tumor cell evolution may drive an increase in biodiversity, making treatment ineffective.
Identifying players of tumor cell evolution is of significant importance as it can facilitate the development of better approaches for cancer intervention. Using two independent strategies, we identified SPP1 as the top candidate in our liver cancer patients. Consistently, we found that SPP1 expression patterns follow hierarchical relationships of tumor cell branching evolution with a much higher expression level in BI-B/C clusters than BI-A clusters which are associated with treatment response and patient survival. SPP1 encodes osteopontin (OPN), a phosphorylated glycoprotein expressed in various tissues and cell types linked to human diseases[37]. The role of OPN in HCC progression is well documented[38]. Its ability to induce tumor cell invasion and metastasis in HCC is likely to be linked to its ability to drive tumor cell evolution that can adapt the tumor microenvironment for its survival. Furthermore, OPN may regulate different immune cell types to modulate host immunity. OPN exists as several isoforms that exert different functions by interacting differentially with its receptors CD44 and integrins[39, 40]. It is plausible that SPP1 may drive tumor cell biodiversity and further in vitro and in vivo experimental models are warranted to functionally prove this observation.
Supplementary Material
Highlights.
Single cell landscape of liver cancer in response to immunotherapy was determined.
Functional clonality as a surrogate of tumor cell state for liver cancer prognosis.
Liver tumor cell evolution linked to polarized immune cell landscape.
Osteopontin as a potential player of tumor cell evolution.
ACKNOWLEDGEMENTS:
We thank members of the Wang laboratory for critical discussions; the Greten laboratory for managing clinical programs; Sean P. Martin for clinical data collection; Wei Tang for advice on data analysis; Zachary Rae for additional laboratory work; the patients, families and nurses for contribution to this study.
Financial support statement:
This work was supported by grants (ZIA BC 010877, ZIA BC 010876, ZIA BC 010313 and ZIA BC 011870) from the intramural research program of the Center for Cancer Research, National Cancer Institute of the United States.
ABBREVIATIONS:
- BI
branching index
- CAF
cancer-associated fibroblast
- CNV
chromosomal copy number variation
- EMT
epithelial–mesenchymal transition
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- ICGC
International Cancer Genomics Consortium
- ITH
intratumor heterogeneity
- iCCA
intrahepatic cholangiocarcinoma
- LCI
Liver Cancer Institute
- OPN
osteopontin
- scAtlasLC
single-cell atlas in liver cancer
- scRNA-seq
single-cell RNA sequencing
- TAM
tumor-associated macrophage
- TEC
tumor-associated endothelial cell
- TME
tumor microenvironment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: The authors declare no competing interests.
Data availability:
Single-cell transcriptomic data are available at the Gene Expression Omnibus (accession number GSE151530).
ADDITIONAL INFORMATION
Supplementary information is available for this paper at https://
REFERENCES
- [1].Maley CC, Aktipis A, Graham TA, Sottoriva A, Boddy AM, Janiszewska M, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer 2017;17:605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501:346–354. [DOI] [PubMed] [Google Scholar]
- [3].Gordon DM. The ecology of collective behavior. PLoS Biol 2014;12:e1001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016;22:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 2021;27:212–224. [DOI] [PubMed] [Google Scholar]
- [6].Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019;36:418–430.e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Khatib S, Pomyen Y, Dang H, Wang XW. Understanding the cause and consequence of tumor heterogeneity. Trends Cancer 2020;6:267–271. [DOI] [PubMed] [Google Scholar]
- [8].Iacobuzio-Donahue CA, Litchfield K, Swanton C. Intratumor heterogeneity reflects clinical disease course. Nature Cancer 2020;1:3–6. [DOI] [PubMed] [Google Scholar]
- [9].Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nature reviews Genetics 2019;20:404–416. [DOI] [PubMed] [Google Scholar]
- [10].Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics 2014;15:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020;38:229–246 e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gonzalez-Silva L, Quevedo L, Varela I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020;6:13–19. [DOI] [PubMed] [Google Scholar]
- [13].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- [14].Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM, et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017;32:57–70 e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].TheCancerGenomeAtlasResearchNetwork. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang XW, Thorgeirsson SS. The biological and clinical challenge of liver cancer heterogeneity. Hepat Oncol 2014;1:349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Worns MA, Galle PR. HCC therapies--lessons learned. Nat Rev Gastroenterol Hepatol 2014;11:447–452. [DOI] [PubMed] [Google Scholar]
- [18].Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382:1894–1905. [DOI] [PubMed] [Google Scholar]
- [20].Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006;22:1540–1542. [DOI] [PubMed] [Google Scholar]
- [21].Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nature methods 2017;14:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature 2018;560:494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kwon SM, Budhu A, Woo HG, Chaisaingmongkol J, Dang H, Forgues M, et al. Functional genomic complexity defines intratumor heterogeneity and tumor aggressiveness in liver cancer. Scientific Reports 2019;9:16930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017;169:1342–1356 e1316. [DOI] [PubMed] [Google Scholar]
- [25].Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, et al. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 2019;38:2451–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018;24:978–985. [DOI] [PubMed] [Google Scholar]
- [27].Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564:268–272. [DOI] [PubMed] [Google Scholar]
- [28].Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 2018;563:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med 2015;21:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Salcedo A, Tarabichi M, Espiritu SMG, Deshwar AG, David M, Wilson NM, et al. A community effort to create standards for evaluating tumor subclonal reconstruction. Nat Biotechnol 2020;38:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 2020;181:236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 2018;563:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018;555:524–528. [DOI] [PubMed] [Google Scholar]
- [36].Nowell PC. The clonal evolution of tumor cell populations. Science (New York, NY) 1976;194:23–28. [DOI] [PubMed] [Google Scholar]
- [37].Icer MA, Gezmen-Karadag M. The multiple functions and mechanisms of osteopontin. Clin Biochem 2018;59:17–24. [DOI] [PubMed] [Google Scholar]
- [38].Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. NatMed 2003;9:416–423. [DOI] [PubMed] [Google Scholar]
- [39].Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 2007;26:6361–6371. [DOI] [PubMed] [Google Scholar]
- [40].Kanayama M, Xu S, Danzaki K, Gibson JR, Inoue M, Gregory SG, et al. Skewing of the population balance of lymphoid and myeloid cells by secreted and intracellular osteopontin. Nat Immunol 2017;18:973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.