

Abstract
Male sexual orientation is a scientifically and socially important trait shown by family and twin studies to be influenced by environmental and complex genetic factors. Individual genome-wide linkage studies (GWLS) have been conducted, but not jointly analyzed. Two main datasets account for > 90% of the published GWLS concordant sibling pairs on the trait and are jointly analyzed here: MGSOSO (Molecular Genetic Study of Sexual Orientation; 409 concordant sibling pairs in 384 families, Sanders et al. (2015)) and Hamer (155 concordant sibling pairs in 145 families, Mustanski et al. (2005)). We conducted multipoint linkage analyses with Merlin on the datasets separately since they were genotyped differently, integrated genetic marker positions, and combined the resultant LOD (logarithm of the odds) scores at each 1 cM grid position. We continue to find the strongest linkage support at pericentromeric chromosome 8 and chromosome Xq28. We also incorporated the remaining published GWLS dataset (on 55 families) by using meta-analytic approaches on published summary statistics. The meta-analysis has maximized the positional information from GWLS of currently available family resources and can help prioritize findings from genome-wide association studies (GWAS) and other approaches. Although increasing evidence highlights genetic contributions to male sexual orientation, our current understanding of contributory loci is still limited, consistent with the complexity of the trait. Further increasing genetic knowledge about male sexual orientation, especially via large GWAS, should help advance our understanding of the biology of this important trait.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10508-021-02035-3.
Keywords: Chromosome 8, Chromosome Xq28, Complex trait, Genome-wide linkage scan, Sexual orientation
Introduction
Male homosexuality runs in families, and twin studies have shown that genetic contributions appear to account for a moderate proportion of the variation in male sexual orientation with heritability estimated at ~ 32% (for review, see Bailey et al., 2016). Three genome-wide linkage studies (GWLS) have been conducted on male sexual orientation, all focusing on concordant sibling pairs (2010homosexual brothers)—we refer here to these GWLS datasets as Hamer (Mustanski et al., 2005), MGSOSO (Molecular Genetic Study of Sexual Orientation) (Sanders et al., 2015), and Canadian (Ramagopalan et al., ). The Hamer GWLS combined samples from two earlier studies (Hamer et al., 1993; Hu et al., 1995) with newly collected families (Mustanski et al., 2005) to total 155 independent concordant sibling pairs in 145 families. While linkage to chromosome Xq28 was prominent in the earlier linkage studies focusing on chromosome X (Hamer et al., 1993; Hu et al., 1995), the Hamer GWLS instead had its strongest finding of suggestive linkage at chromosome 7q36 (Mustanski et al., 2005). Another research group collected 55 families in Canada and performed a GWLS, with the strongest (albeit not significant) linkage reported at chromosome 14q32 (Ramagopalan et al., 2010). The MGSOSO performed a GWLS on 409 independent concordant sibling pairs in 384 families, making its strongest finding of significant (Lander & Kruglyak, 1995) linkage at pericentromeric chromosome 8 and also detecting suggestive (Lander & Kruglyak, 1995) linkage (supportive evidence of previous findings) at chromosome Xq28 (Sanders et al., 2015). In order to extract the maximal positional information from GWLS of currently available family resources, we jointly analyzed the Hamer and MGSOSO datasets (and included the Canadian dataset by meta-analyzing published summary statistics).
Method
Joint Linkage Analyses
The two jointly analyzed datasets used very similar phenotype definitions for homosexual men from their questionnaire data: Hamer used “Kinsey 5–6” for several questions (attraction, fantasy, behavior, and self-identification) (Mustanski et al., 2005), and MGSOSO used “Kinsey 5–6” for fantasy along with homosexual identity (Sanders et al., 2015). The Hamer dataset consisted of 441 individuals in 145 families genotyped with 408 short tandem repeat polymorphism genetic markers (STRPs) (Mustanski et al., 2005), and the MGSOSO dataset consisted of 908 individuals in 384 families and genotyped with 45,387 single-nucleotide polymorphism genetic markers (SNPs) (Sanders et al., 2015). Various quality control steps had already been performed in the respective GWLS as previously detailed (Mustanski et al., 2005; Sanders et al., 2015). After obtaining collaborative access to genotypes for each dataset, we conducted multipoint nonparametric linkage analyses with Merlin v1.1.2 (Abecasis et al., 2002) on the Hamer (Mustanski et al., 2005) and MGSOSO (Sanders et al., 2015) datasets separately since they were genotyped differently (STRPs vs. SNPs). To integrate, we found the genetic positions of the respective markers in the Rutgers Map v.3 (hg19 build) (Nato et al., 2018) and then used the nonparametric S-pairs and grid 1 cM options to perform multipoint linkage on both data sets, followed by combining LOD scores at each grid position across the marker sets.
Meta-Analyses of Summary Statistics
For phenotype definitions for homosexual men, the Canadian dataset used an interview approach based on identity and corroboration by sibling, and on a sub-sample all also had Kinsey 5–6 for several questions (attraction, fantasy, and behavior) (Rice et al., 1999a, b). As we were unable to access genotypes for the Canadian dataset (accounting for < 10% of the families in GWLS on the trait), we were only able to incorporate the Canadian GWLS by meta-analyzing summary statistics. Thus, we used the plotted multipoint Canadian GWLS Fig. 1 (Ramagopalan et al., 2010) and interpolated into cM bins enabling use of GWLS meta-analytic methods not needing genotypes, namely the multi-scan probability (MSP) approach utilizing regional p-values (Badner & Gershon, 2002), and the rank-based genome scan meta-analysis (GSMA) approach (Levinson et al., 2003; Wise & Lewis, 1999).
Fig. 1.

Joint multipoint linkage analysis of the combined Hamer and MGSOSO datasets. Multipoint LOD scores are plotted v. the chromosomal positions for the nonparametric linkage analysis. Adjacent chromosomes are separated by alternating black and gray lines
Results
The multipoint plots for the Hamer and the MGSOSO datasets for the current analyses (Supplementary Figs. 1 and 2, respectively) line up very well with the original GWLS manuscripts’ multipoint plots–Fig. 1a (Mustanski et al., 2005) and Fig. 1 (Sanders et al., 2015), respectively. This overlap of multipoint findings was found despite some differences between the original reports (Mustanski et al., 2005; Sanders et al., 2015) and the current manuscript in statistical analysis software (Aspex vs. Merlin for the Hamer dataset) and genetic map used (deCode vs. Rutgers for both the Hamer and MGSOSO datasets). The joint analysis of the combined Hamer and MGSOSO datasets is shown in Fig. 1, with zoomed-in plots of the top two multipoint linkage peaks from this joint GWLS depicted for chromosomes 8 and X in Fig. 2. The results of the meta-analyses of summary statistics from Hamer, MGSOSO, and Canadian GWLS datasets are presented in Supplementary Tables 1 (MSP) and 2 (GSMA).
Fig. 2.
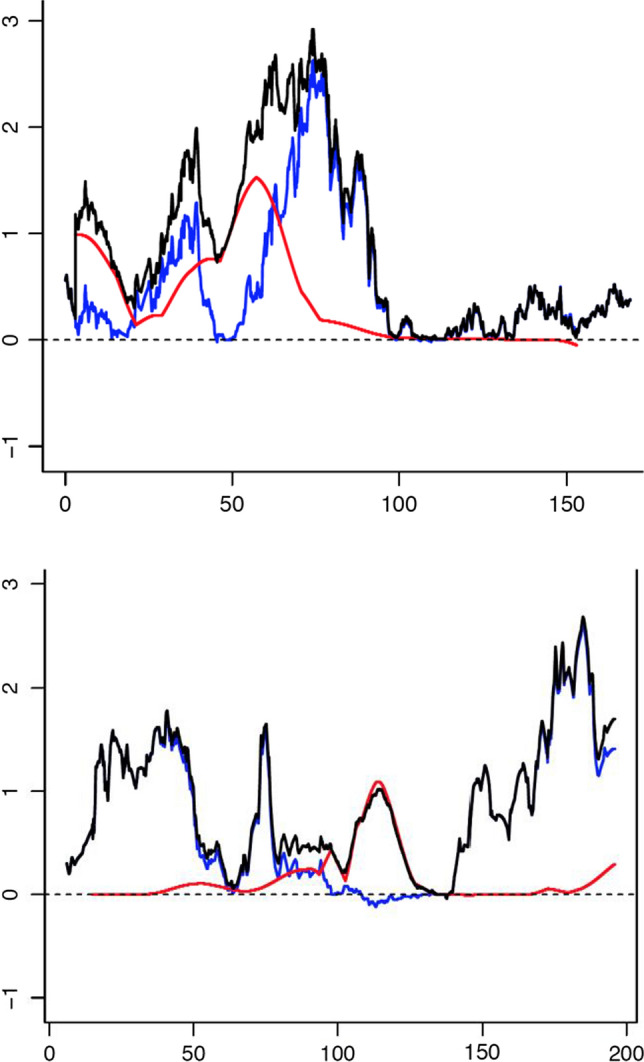
Individual chromosomal plots of multipoint linkage peaks on chromosomes 8 and X. Multipoint LOD scores are plotted v. the chromosomal positions for the nonparametric linkage analysis, the red line indicating the results from the Hamer dataset, the blue line for the MGSOSO dataset, and the black line for the joint analysis. Chromosome 8 (top) has its multipoint linkage peak in the pericentromeric region, and chromosome X (bottom) has its multipoint linkage peak at Xq28
Discussion
Our primary analysis for this investigation was the joint analysis of multipoint linkage from the Hamer and MGSOSO datasets (Mustanski et al., 2005; Sanders et al., 2015), to which each dataset contributed some peaks (Fig. 1, Supplementary Figs. 1 and 2). Overall, the maximum multipoint peaks increased little in height, though the pericentromeric chromosome 8 peak was broadened (Fig. 2). Chromosomes 8 and X retained the highest multipoint peaks genome-wide, mostly arising from the larger (MGSOSO) dataset (Fig. 2). The joint analysis gives a more comprehensive picture of shared and heterogeneous linkage regions (e.g., at pericentromeric chromosome 8), the studies share overlapping peaks (possibly suggesting heterogeneity, perhaps with different genes involved in the different datasets), and the evidence broadens the search. The secondary analyses on summary statistics using MSP and GSMA to incorporate all three (Hamer, MGSOSO, Canadian) GWLS datasets showed no genome-wide significant results though suggestive findings remained present. The joint analysis of multipoint linkage (Fig. 1) extracted the available positional information from collaborating GWLS, though previous GWLS findings were not much further strengthened in these analyses. Nevertheless, this provides information to complement other approaches, such as helping prioritize findings from GWAS. Linkage and association studies measure different genetic properties (i.e., segregation of a region within families, vs. correlation of alleles in a population), both of which provide clues about underlying trait genetics. Thus, since GWLS are different from GWAS, we were unable to directly combine any GWAS (e.g., Ganna et al., 2019) with the studied GWLS in our GWLS meta-analysis. Limitations include those inherent to linkage (as opposed to GWAS) of traits with complex genetics (e.g., their limited utility for phenotypes with contributions from more than one or a few genes); on the other hand, linkage retains some advantages over association approaches, such as being robust to allelic heterogeneity (Lipner & Greenberg, 2018). Accumulating genetic studies of the trait such as by much enlarged GWAS (e.g., Ganna et al., 2019) will be especially useful, given its successful application in the study of other phenotypes manifesting complex genetics (e.g., Fig. 3b in Sullivan et al. (2018)).
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by NICHD, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Award No. R01HD041563 for the linkage sample to Alan R. Sanders, M.D.; and Award No. R21HD080410 for meta-analyses to Alan R. Sanders, M.D. and Eden R. Martin, Ph.D.), and by intramural NIH funds (to Dean H. Hamer, Ph.D.). We thank the men and their families for their participation.
Compliance with Ethical Standards
Human and Animal Rights and Informed Consent
There was no participant contact for the current study, as it was a meta-analysis of previously collected genetic data. Two of the three studied samples (MGSOSO and Hamer) describe ethical aspects in their earlier manuscripts in more detail, but we briefly summarize here. For the third sample, the Canadian one (Ramagopalan et al., 2010), we only meta-analyzed published summary statistics, i.e., we used no individual level data. For MGSOSO, institutional review board (IRB) approval was obtained from NorthShore University HealthSystem, and all participants provided informed consent (Sanders et al., 2015). For the Hamer dataset, IRB approval was obtained from the National Cancer Institute, and all participants provided informed consent (Mustanski et al., 2005).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: Graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genetics. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Badner JA, Gershon ES. Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Molecular Psychiatry. 2002;7:56–66. doi: 10.1038/sj/mp/4000922. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Vasey PL, Diamond LM, Breedlove SM, Vilain E, Epprecht M. Sexual orientation, controversy, and science. Psychological Science in the Public Interest. 2016;17:45–101. doi: 10.1177/1529100616637616. [DOI] [PubMed] [Google Scholar]
- Boyles AL, Scott WK, Martin ER, Schmidt S, Li YJ, Ashley-Koch A, Bass MP, Schmidt M, Pericak-Vance MA, Speer MC, Hauser ER. Linkage disequilibrium inflates type I error rates in multipoint linkage analysis when parental genotypes are missing. Human Heredity. 2005;59:220–227. doi: 10.1159/000087122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganna A, Verweij KJ, Nivard MG, Maier R, Wedow R, Busch AS, Abdellaoui A, Guo S, Sathirapongsasuti JF, Lichtenstein P, Lundström S. Large-scale GWAS reveals insights into the genetic architecture of same-sex sexual behavior. Science. 2019 doi: 10.1126/science.aat7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer DH. Genetics and male sexual orientation. Science. 1999;285:803. doi: 10.1126/science.285.5429.803a. [DOI] [Google Scholar]
- Hamer DH, Hu S, Magnuson VL, Hu N, Pattatucci AM. A linkage between DNA markers on the X chromosome and male sexual orientation. Science. 1993;261:321–327. doi: 10.1126/science.8332896. [DOI] [PubMed] [Google Scholar]
- Hu S, Pattatucci AM, Patterson C, Li L, Fulker DW, Cherny SS, Kruglyak L, Hamer DH. Linkage between sexual orientation and chromosome Xq28 in males but not in females. Nature Genetics. 1995;11:248–256. doi: 10.1038/ng1195-248. [DOI] [PubMed] [Google Scholar]
- Huang Q, Shete S, Amos CI. Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. American Journal of Human Genetics. 2004;75:1106–1112. doi: 10.1086/426000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nature Genetics. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Levinson DF, Levinson MD, Segurado R, Lewis CM. Genome scan meta-analysis of schizophrenia and bipolar disorder, part I: Methods and power analysis. American Journal of Human Genetics. 2003;73:17–33. doi: 10.1086/376548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipner EM, Greenberg DA. The rise and fall and rise of linkage analysis as a technique for finding and characterizing inherited influences on disease expression. Methods in Molecular Biology. 2018;1706:381–397. doi: 10.1007/978-1-4939-7471-9_21. [DOI] [PubMed] [Google Scholar]
- McPeek MS, Sun L. Statistical tests for detection of misspecified relationships by use of genome-screen data. American Journal of Human Genetics. 2000;66:1076–1094. doi: 10.1086/302800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustanski BS, Dupree MG, Nievergelt CM, Bocklandt S, Schork NJ, Hamer DH. A genomewide scan of male sexual orientation. Human Genetics. 2005;116:272–278. doi: 10.1007/s00439-004-1241-4. [DOI] [PubMed] [Google Scholar]
- Nato, A. Q., Buyske, S., & Matise, T. C. (2018). The Rutgers map: A third-generation combined linkage-physical map of the human genome. Retrieved from http://compgen.rutgers.edu/rutgers_maps.shtml
- Ramagopalan SV, Dyment DA, Handunnetthi L, Rice GP, Ebers GC. A genome-wide scan of male sexual orientation. Journal of Human Genetics. 2010;55:131–132. doi: 10.1038/jhg.2009.135. [DOI] [PubMed] [Google Scholar]
- Rice G, Anderson C, Risch N, Ebers G. Male homosexuality: Absence of linkage to microsatellite markers at Xq28. Science. 1999;284:665–667. doi: 10.1126/science.284.5414.665. [DOI] [PubMed] [Google Scholar]
- Rice G, Risch N, Ebers G. Genetics and male sexual orientation. Science. 1999;285:803. doi: 10.1126/science.285.5429.803a. [DOI] [Google Scholar]
- Sanders AR, Martin ER, Beecham GW, Guo S, Dawood K, Rieger G, Badner JA, Gershon ES, Krishnappa RS, Kolundzija AB, Duan J. Genome-wide scan demonstrates significant linkage for male sexual orientation. Psychological Medicine. 2015;45:1379–1388. doi: 10.1017/S0033291714002451. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, Cichon S, Edenberg HJ, Faraone SV, Gelernter J, Mathews CA. Psychiatric genomics: An update and an agenda. American Journal of Psychiatry. 2018;175:15–27. doi: 10.1176/appi.ajp.2017.17030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigginton JE, Abecasis GR. PEDSTATS: Descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- Wise LH, Lewis CM. A method for meta-analysis of genome searches: Application to simulated data. Genetic Epidemiology. 1999;17(Suppl. 1):S767–S771. doi: 10.1002/gepi.13701707126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.