

Abstract
We examined safety, tolerability, and efficacy of SGS-742, a GABA-B receptor antagonist, in patients with succinic semialdehyde dehydrogenase deficiency (SSADH-D). This was a single-center randomized, double-blind cross-over phase II clinical trial of SGS-742 versus placebo in patients with SSADH-D. Procedures included transcranial magnetic stimulation (TMS) and the Adaptive Behavior Assessment Scale (ABAS). Nineteen subjects were consented and enrolled; mean age was 14.0 +/− 7.5 years; 11 (58%) were female. We did not find a significant effect of SGS-742 on the ABAS score, motor threshold, and paired-pulse stimulation. The difference in recruitment curve slopes between treatment groups was 0.003 (p = 0.09). There was no significant difference in incidence of adverse effects between drug and placebo arms. SGS-742 failed to produce improved cognition and normalization of cortical excitability as measured by the ABAS and TMS. Our data do not support the current use of SGS-742 in SSADH-D.
Keywords: cognition, efficacy, inborn errors of metabolism, neurodevelopment, treatment, epilepsy
INTRODUCTION
Succinic semialdehyde dehydrogenase deficiency (SSADH-D) (also termed 4-hydroxybutyric aciduria) is a rare autosomal recessive disorder due to pathogenic variants in ALDH5A1. Loss of function in SSADH results in reduced GABA (gamma-aminobutyric acid) catabolism, leading to elevated levels of GABA and gamma-hydroxybutyric acid (GHB) and additional metabolites in body fluids and brain parenchyma1,2. Over 450 cases have been identified worldwide3, making this the most prevalent pediatric neurotransmitter disorder. Diagnosis is primarily based on detection of elevated GHB in the urine organic acid profile4 but can also be made by detecting low SSADH levels or the pathogenic ALDH5A1 gene variant. Most patients present with developmental delay noted in the first two years of life, in addition to hypotonia, ataxia, speech disturbance, and intellectual disability. At least half develop epilepsy. As patients age, they exhibit more compulsive behavior, sleep disturbances, and seizures5.
The signs and symptoms of SSADH-D result from various metabolic derangements, primarily the elevations in GHB and GABA which affect GABAergic neurotransmission, disrupt the glial-neuronal glutamate/GABA-glutamine shuttle, and alter dopamine and serotonin homeostasis. At physiologic levels, GHB acts primarily at high affinity GHB and α4β1δ-GABA-A receptors, and other yet-identified sites6. In SSADH-D, where GHB concentrations typically reach 100-500 times normal levels, GHB acts as a weak GABA-B receptor agonist7. This GABA-B receptor activation modulates neurotransmission at GABA-A synapses8,9, disproportionately impacting inhibitory interneurons, and leads to disinhibition of excitatory neurons. These changes result in downregulation of GABA-A receptors evident on flumazenil-PET10 and decreased GABA-B function as measured by transcranial magnetic stimulation (TMS) in patients with SSADH-D11.
Despite an expanding literature on the pathophysiology and diverse metabolic disruptions in SSADH-D, there are no successful targeted therapies. Pharmacologic treatment is generally aimed at ameliorating symptoms of the disease, primarily seizures and psychiatric sequelae. No one anti-seizure medication (ASM) has emerged as the treatment of choice for SSADH-D. Vigabatrin, an irreversible inhibitor of GABA-transaminase, has been proposed as a logical intervention since it inhibits the conversion of GABA to GHB. However, multiple reports detail lack of efficacy12, or worsening of specific symptoms such as seizures or level of alertness13. While vigabatrin will lead to at least transient decreases in CSF GHB levels14, there may be a deleterious effect related to increases in CSF (and brain) GABA levels15. Similarly, clinicians may wish to avoid valproic acid, which inhibits residual SSADH activity, though there are reports of efficacy16. Enzyme replacement therapy is challenging, in part because SSADH is a mitochondrial enzyme whose delivery into the mitochondria is coupled with its ribosomal biosynthesis17.
One hurdle in developing effective therapies is the nature of the disorder. As mentioned above, SSADH-D is extremely rare. The variability in phenotypic severity makes selection of standardized assessments able to capture the range of neurocognitive symptoms rather difficult. And since only about half develop seizures (which may be infrequent), changes in seizure frequency is unlikely to prove useful. Only TMS has detected a significant change in a therapeutic trial of taurine for SSADH-D18.
Preliminary animal work in the SSADH mutant model has suggested benefit from treatment with SGS-742 (3-aminopropyl-n-butyl phosphinic acid), a GABA-B receptor antagonist. SGS-742 is orally absorbed, with peak plasma concentration achieved after four hours and elimination half life of approximately four hours, >99% being excreted unchanged in the urine. Early reports showed improved attention, reaction time, visual information processing, and working memory in mice, rats and monkeys19. It may yield a preferential effect at pre-synaptic GABA-B receptors which generally act to reduce neurotransmitter release20. In SSADH deficient (Aldh5a1−/−) mice, SGS-742 showed a dramatic dose-dependent improvement in spike-wave discharges and absence seizures identified by electrocorticography, while topiramate was ineffective21. In a phase II double blind, placebo-controlled study in 110 adults with mild cognitive impairment, oral administration of SGS742 600 mg three times daily for eight weeks significantly improved attention, reaction time, visual information processing, and working memory22,23. No clear drug-related serious adverse events or drug related effects on cardiovascular or laboratory variables were reported22. The drug has not yet been used in children.
We aimed to examine safety, tolerability, and efficacy of SGS-742 on the neuropsychological function and cortical excitability in a small group of patients with SSADH deficiency. We hypothesized that: a. patients would show improvements in activities of daily living based on parent questionnaire, b: patients would have lengthening toward normal values of the cortical silent period, and return of long interval intracortical inhibition, and c. patients would show improvement on global assessment ratings.
METHODS
Patients
Eligible patients were age four years and older with SSADH-D as determined by documented 4-hydroxybutyric aciduria on two separate tests, documented succinic semialdehyde dehydrogenase quantitative enzyme deficiency or presence of two pathogenic ALDH5A1 gene variants, and clinical features consistent with SSADH-D. We excluded patients with current alcohol use (>14 drinks/wk in men and >7 drinks/wk in women) or recreational drug use for the 16 month period of this study, patients with a history of other major medical disorders with clinical fluctuations, or requiring therapy that might affect study participation or drug response such as severe depression or psychoses, renal or hepatic disease, and patients requiring treatment with drugs known to affect the GABAergic system, including vigabatrin and benzodiazepines (except for acute seizure treatment). Pregnant and lactating women were also excluded, and women of child-bearing potential were required to use a reliable form of contraception (abstinence included).
Letters were mailed to families of patients with a history of SSADH-D. This letter was posted to the SSADH Association website. We also recruited patients from a cohort followed by, or referred to, Dr. Pearl in the Boston Children’s Hospital Neurology Department. During the course of routine clinical care, patients were informed about the study and, upon their request, were provided the information to contact the NIH personnel on their own initiative. Several patients had participated in previous NIH SSADH-D studies10,11.
Patients were screened in the NINDS Clinical Epilepsy Section (CES) outpatient clinic for inclusion in the protocol by CES physicians or licensed practitioners. Dr. Pearl supervised confirmation of the diagnosis.
Study design
This was a single-center randomized, double-blind cross-over phase II clinical trial of SGS-742 versus placebo. Figure 1 depicts the overall study design. Patients underwent baseline testing as detailed in the assessments section below and were then randomized to first receive either active drug or placebo at a 1:1 ratio. Patients received their first dose of study pills at the NIH, returned 12-24 hours later to be assessed for adverse reactions, then returned home. All patients were given an exact written titration schedule to follow. Patients/families were contacted at least once every two weeks by phone and/or completion of patients surveys on the Clinical Trial Survey System. These surveys included questions regarding the severity and chronicity of symptoms. Patients/families were also asked the following data and safety monitoring questions: Has the patient seen a health care provider since last visit? Has the patient had any seizures since last visit? Has the patient had a change in medications over the past week? Any missed doses of study drug? Any unexpected/unusual symptoms over the past week? Treatment during this first phase continued for six months before tapering and washout. Following a 9 week total washout period (+/− 2 weeks), including drug taper, patients entered the other treatment arm (phase two). Repeat assessments were conducted at the conclusion of each treatment arm. The final visit occurred 4-7 weeks after completion of the final drug taper.
Figure 1.
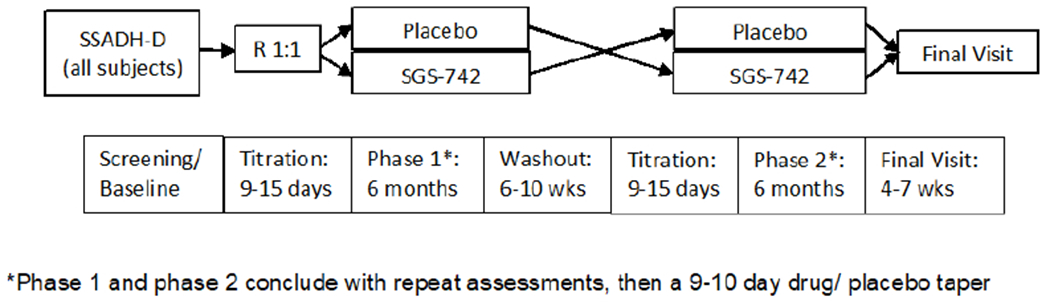
Randomized, double-blinded crossover study design.
SGS-742 was synthesized by IRIX pharmaceuticals in a GCP facility. We recommended that patients take the study drug three times each day prior to meals because administration of food decreases oral systemic availability of SGS-742. The drug and matching placebo were encapsulated by the NIH pharmacy. The pharmacy created the randomization schedule and ensured blinding of patients, family, and investigators. The study was approved by the NIH Combined Neurosciences Institutional Review Board. We obtained written informed consent for all participants, their legal guardian, or legally authorized representative, as appropriate.
During titration of SGS-742, patients received an initial dose of 2.5 mg/kg/dose (up to 150 mg) tid X 3 days, which was then increased in increments of 2.5 mg/kg/dose every 3 days up to the target dose of 10 mg/kg/dose (up to 600 mg) tid. If patients experienced side effects, the next scheduled dose increase was delayed until side effects abated or the dose was reduced in 2.5 mg/kg per day increments until side effects abated. The drug taper schedule exactly mirrored this increase: 7.5 mg/kg/dose (up to 450 mg) tid X 3 days, 5 mg/kg/dose (up to 300 mg) tid X 3 days, 2.5 mg/kg/dose (up to 150 mg) tid X 3 days, then stop. Placebo was titrated and tapered in capsules matching the SGS-742 dosing stages.
Assessments
Assessments were conducted at baseline, end of first treatment arm (phase 1), and end of second treatment arm (phase 2). Procedures included neurological and systemic physical examination, electroencephalogram (EEG), transcranial magnetic stimulation (TMS), neuropsychological testing, and blood and urine tests. TMS was not done within 24 hours of a reported seizure. At each visit during drug treatment, patients had a medical history and physical examination and review of seizure calendars and any adverse effects. Laboratory tests were obtained when indicated for change in clinical status. Patients and families kept a record of seizures and any possible medication side effects.
Neuropsychological Assessments
We planned a comprehensive language and cognitive evaluations using a selection of assessments based on age and ability. Planned language assessments included the Wechsler Nonverbal Scale of Ability24 for patients up to age 22 years, the Wechsler Adult Intelligence Scale25 for patients age 22 years and older, and the Neuropsychological Assessment Battery Language Module Confrontation Naming and Auditory Comprehension Subtests26. Other tools used for cognitive assessment were the Rule Shift from the Behavioral Assessment of Dysexecutive Syndrome27 (a short test of working memory), a computerized test of reaction time and go/no-go to assess reaction time and attention, the Texas Functional Living Scale28 (an assessment of practical reading, math, language comprehension and memory developed for individuals with cognitive limitations), the Adaptive Behavior Assessment Scale (ABAS)29 to be completed by parent/guardian which assesses functional skills, the Wechsler Preschool and Primary Scale of Intelligence-IV30 for patients between the ages of 2.5 and 7 years, and the Wechsler Intelligence Scale for Children-V31 for patients age 6 through 17 years old. The Vineland Adaptive Ability (VABS) Composite: Parent Form32 was given to all parents/guardians. This questionnaire provides an estimate of the patient’s communication, daily living, socialization and motor functioning.
However, the functional level for both child and adult patients was too low to derive statistically analyzable data for any but the ABAS. The ABAS is a widely used parent/guardian questionnaire that assesses adaptive abilities including communication, social adaptability and practical skills regarding their child. Each of the 232 items is rated on a scale of 0 (unable to perform) to 2 (performs independently). Raw scores are converted to age-adjusted normative data (mean=100 +/−15).
Other Procedures
Routine video EEG was performed according to standard procedures using the international 10-20 standard method of electrode placement. Blood was collected for CBC, clinical chemistry, and hepatic panels. We performed urine pregnancy testing on women of child-bearing age.
TMS
TMS was delivered through a round coil (90 mm diameter) connected to 2 Magstim 2002 magnetic stimulators via a BiStim-module (Magstim, Dyfed, UK). The coil was placed over the contralateral motor cortex at the site, and in the orientation, that consistently produced the maximum motor evoked potential (MEP) amplitude from the right first dorsal interosseus (FDI) muscle.
Subjects were seated in a comfortable chair with their hands resting on a pillow. Electrodes were applied to the skin over the right FDI in a belly-tendon montage with the reference electrode placed at least 4 cm distal to the active electrode and a ground electrode positioned over the dorsum of the hand. The electromyogram (EMG) signal was amplified (Coulburn Isolated Bioamplifier, model V75-04), bandpass filtered (90 Hz to 1 kHz), digitized (analog/digital rate 40 kHz), and recorded (Signal version 4.05, Cambridge Electronics, UK) for offline analysis. The EMG was monitored continuously for relaxation by visual inspection. The resting motor threshold (MT) was determined by increasing stimulus intensity in increments of 5 % of maximum output until a MEP was recorded and then adjusting by 1% increments to the lowest stimulator output required to produce MEPs of at least 50 μV on 5 out of 10 consecutive trials.
MEP recruitment curve (RC), cortical silent period (CSP), short and long interval intracortical inhibition (SICI and LICI), and intracortical facilitation (ICF) were measured. Subjects were instructed to rest during the RC and paired pulse paradigms, and to sustain a contraction of the FDI by pinching the thumb and first finger during CSP determination. Individual trials were repeated or later excluded if muscle activity was apparent in the 100 ms before stimulation in the RC and paired pulse paradigms, or if EMG amplitude dropped below baseline, determined visually, in the 100 ms prior to stimulation for CSP. For RC determination, five MEPs were recorded at each of eight percentages (when possible - some percentages of the MT exceeded maximum stimulator output for certain patients) of the MT in the following order: 90%, 130%, 100%, 140%, 110%, 150%, 120%, 160%. The CSP was measured visually from the end of stimulus artifact to the first return of sustained EMG activity for 10 trials at 110, 120, 130, and 140% MT. For SICI and ICF measurements, we set the conditioning stimulus at 70%, and the test stimulus at 120%, of MT. Interstimulus intervals were 2 and 10 ms. For LICI, we used 120% of MT for both stimuli with an interstimulus interval of 100 ms. We used the mean MEP to the conditioning stimulus in the LICI experiment as the control MEP. ICI and ICF were determined by the ratio of the mean conditioned MEP to this control. We delivered and averaged 10 trials for each measurement.
Statistical analysis
The sample size was calculated based on detecting a change in the auditory comprehension subtest of the Neuropsychological Assessment Battery Language Module. Using a two-tailed, paired t-test, an effect size of 0.75 would require 16 patients at 80% power and an alpha-level of 0.05.
Descriptive statistics were provided for demographic variables using mean and standard deviation, as well as number and percent. Linear mixed effects models were used with ABAS, motor threshold, and SICI, ICF, and LICI as outcome variables, and treatment group and baseline effect as predictor variables. Subject was specified as a random effect to account for repeated observations within an individual (one on each treatment). Recruitment curves were analyzed with the outcome as average MEP and percent of motor threshold (90-160%), treatment group, and the interaction of motor threshold and treatment as the main predictors of interest. Order of administration was added into the primary model (ABAS) as a sensitivity analysis to check for order effect, as well as an interaction between treatment and baseline age. Raw changes for each outcome measure were also described using the mean (SD) change between follow-up and baseline for each treatment group, to descriptively supplement the model results.
Adverse events were investigated and described by system and treatment group. Graphical displays were also used to show the relationship between treatment and various outcome measures.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the NIH NINDS Institutional Review Board. Written informed consent was obtained from all participants (or guardians of participants) in the study. The clinical trial identifier number is NCT02019667.
Data Availability Statement
The study protocol, statistical analysis plan, and de-identified data are available for qualified individuals upon reasonable request. Data will be stored for at least three years as per federal guidelines.
RESULTS
Nineteen subjects were consented and enrolled at the NIH between March 2014 and October 2017. Details on recruitment and handling of subjects is found in the CONSORT flow diagram (figure 2). The final follow up visit was in January 2019. Mean age was 14.0 +/− 7.5 years at enrollment (range 5 to 34.5) and 11 (58%) were female. Mean age at diagnosis was 4.7 +/− 4.6 years (range 0-20 years). Table 1 provides details on diagnosis, neuroimaging, and clinical findings for each subject.
Figure 2.
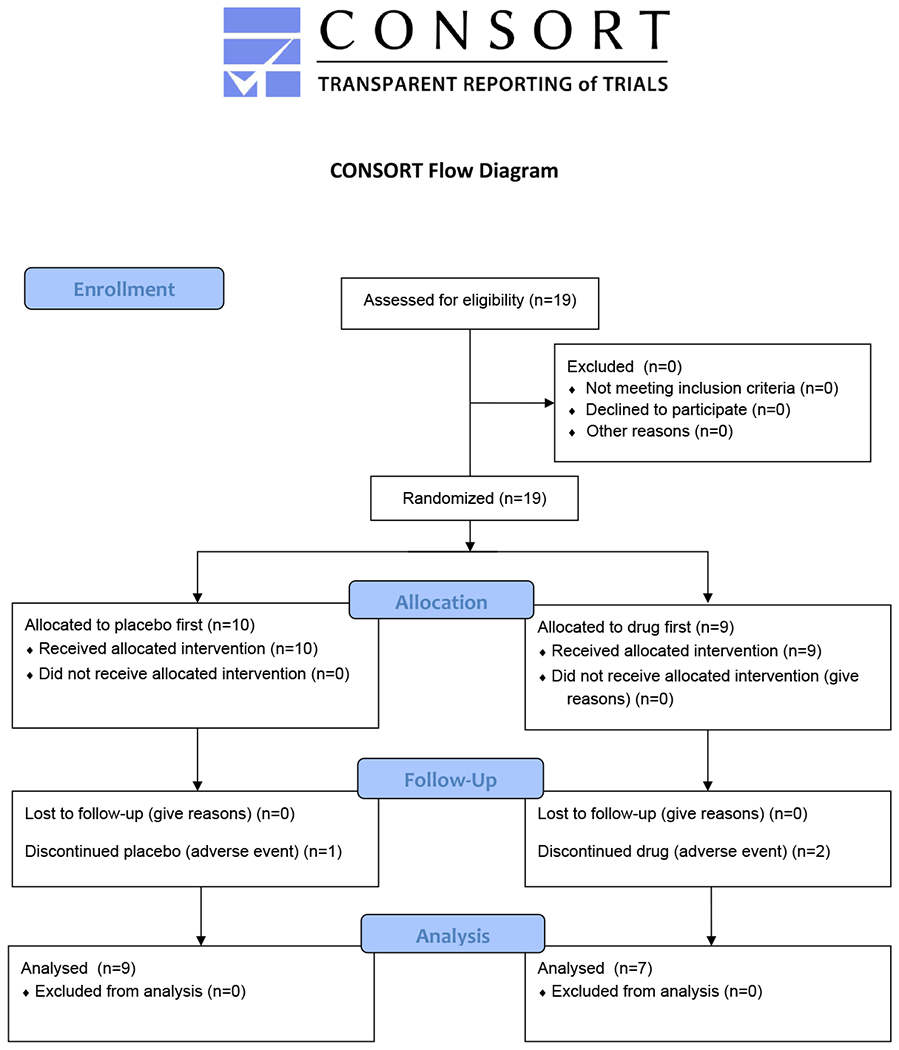
CONSORT flow diagram detailing recruitment and handling of subjects.
Table 1.
Clinical findings, MRI brain findings, and ALDH5A1 variant or other method of SSADH-D diagnosis for each subject. ADHD = attention deficit-hyperactivity disorder, GP = globus pallidus, OCD = obsessive-compulsive disorder.
| Patient | Clinical | MRI brain | ALDH5A1 Variants (NM_001080.3) or other diagnosis |
|---|---|---|---|
| 1 | developmental delay, seizures, depression | bilateral GP increased T2 | c.612G>A (p.Trp204Ter); c.1015-2A>C |
| 2 | global delay, juvenile rheumatoid arthritis | bilateral GP, thalami, subthalamic nuclei increased T2 | c.803G>A (p.Gly268Glu); c.1558G>C (p.Gly520Arg) |
| 3 | global delay, mild ataxia | bilateral GP increased T2 | c.803G>A (p.Gly268Glu); c.1558G>C (p.Gly520Arg) |
| 4 | global delay, Seizures, strabismus, poor fine motor | bilateral GP increased T2 | c.612G>A (p.Trp204Ter); c.1402+2T>C |
| 5 | global delay, strabismus, OCD, ADHD, prediabetes | bilateral GP, dentate nuclei increased T2, deep/subcortical WM gliosis, L>R; left frontal PVWM cavitation (small) | homozygous c.612G>A (p.Trp204Ter) |
| 6 | global delay, ataxia, seizures | bilateral GP, dentate increased T2 | c.104_127del (p.Ser35Ter); c.1015-2A>C |
| 7 | global delay, seizures, ataxia, hypotonia, behavior disorder | bilateral GP increased T2 | c.608C>T (p.Pro203Leu); c.819delT (p.Asp274Ilefs*27) |
| 8 | global delay, ataxia, seizures, OCD, poor fine motor | bilateral GP, subthalmic nn., superior sustantia nigra, caudate, putamen, and dentate increased T2; vermian and paravermian cerebellar atrophy | homozygous c.923G>A (p.Gly308Asp) |
| 9 | global delay, OCD, possible seizures | bilateral GP increase T2, watershed zone ischemia | c.278G>T (p.Cys93Phe); c.566_567insTTGCCCT (p.Val190fs) |
| 10 | global delay, OCD, ataxia | normal | enzymatic quantification |
| 11 | global delay, behavior disorder | not done | enzymatic quantification |
| 12 | mild developmental delay | normal | Elevated urine γ-hydroxybutyrate in multiple samples |
| 13 | global delay | bilateral GP increased T2 | c.612G>A (p.Trp204Ter); c.1234C>T (p.Arg412Ter) |
| 14 | global delay, hypotonia | bilateral GP, cerebellar dentate increased T2 | c.612G>A (p.Trp204Ter); c.1234C>T (p.Arg412Ter) |
| 15 | global delay, ? Movement disorder | bilateral GP, subthalamic nuclei increased T2 | c.517C>T (p.Arg173Cys); c.1015-2A>C |
| 16 | global delay; asthma, seizures | bilateral GP increased T2 | homozygous c.608 C>T (p.Pro203Leu) |
| 17 | global delay, behavior disorder | not done | c.664delG (p.Gly222Alafs*5); c.803G>A (p.Gly268Glu) |
| 18 | global delay | bilateral GP, cerebellar dentate nuclei increased T2 | c.967_968dupCA (p.Gln323Hisfs*4); c.1597G>A (p.Gly533Arg) |
| 19 | global delay, OCD, ataxia, possible Seizures | bilateral GP increased T2 | c.803G>A (p.Gly268Glu); c.851G>A (p.Gly284Asp) |
Summary findings are presented in Table 2. In the mixed model with ABAS score as an outcome variable, we did not find a significant effect of SGS-742 on the ABAS score (baseline-adjusted treatment effect = −0.69, p = 0.80; figure 3). Not surprisingly, baseline score had a significant relation to scores on both drug and placebo, meaning higher follow-up scores in either treatment group came from individuals with higher baseline scores (β = 1.20, p<0.001). Order of treatment administration did not significantly alter the findings, and was not significantly related to the outcome.
Table 2.
Baseline, placebo, and SGS-742 descriptives of primary (ABAS) and secondary (TMS) outcome measures with mean, standard deviation, and number who completed testing. ABAS = Adaptive Behavior Assessment Scale, MT = motor threshold, PPS = paired-pulse stimulation, ICF = intra-cortical facilitation, LICI = long interval intra-cortical inhibition; SICI = short interval intra-cortical inhibition. ABAS data presented are the standardized scores. MT is presented as the percent of maximum stimulator output. PPS are presented as a percentage of the baseline mean motor evoked potential elicited by stimulation at 120% of the MT.
| Variable | Baseline | Placebo | SGS | |
|---|---|---|---|---|
| ABAS | 50.5 (14.0); 17 | 58.1 (18.6); 15 | 58.7 (17.2); 15 | |
| MT | 68.3 (14.9); 18 | 66.6 (14.8); 16 | 68.1 (13.2); 17 | |
| PPS | ||||
| ICF | 1.0 (46.9); 18 | 53.0 (65.6); 16 | 44.0 (61.5); 17 | |
| LICI | −10.9 (79.5); 18 | 37.7 (240); 16 | −8.34 (115); 17 | |
| SICI | −25.2 (52.0); 18 | 15.4 (120); 16 | −34.1 (46.0); 17 | |
Figure 3.
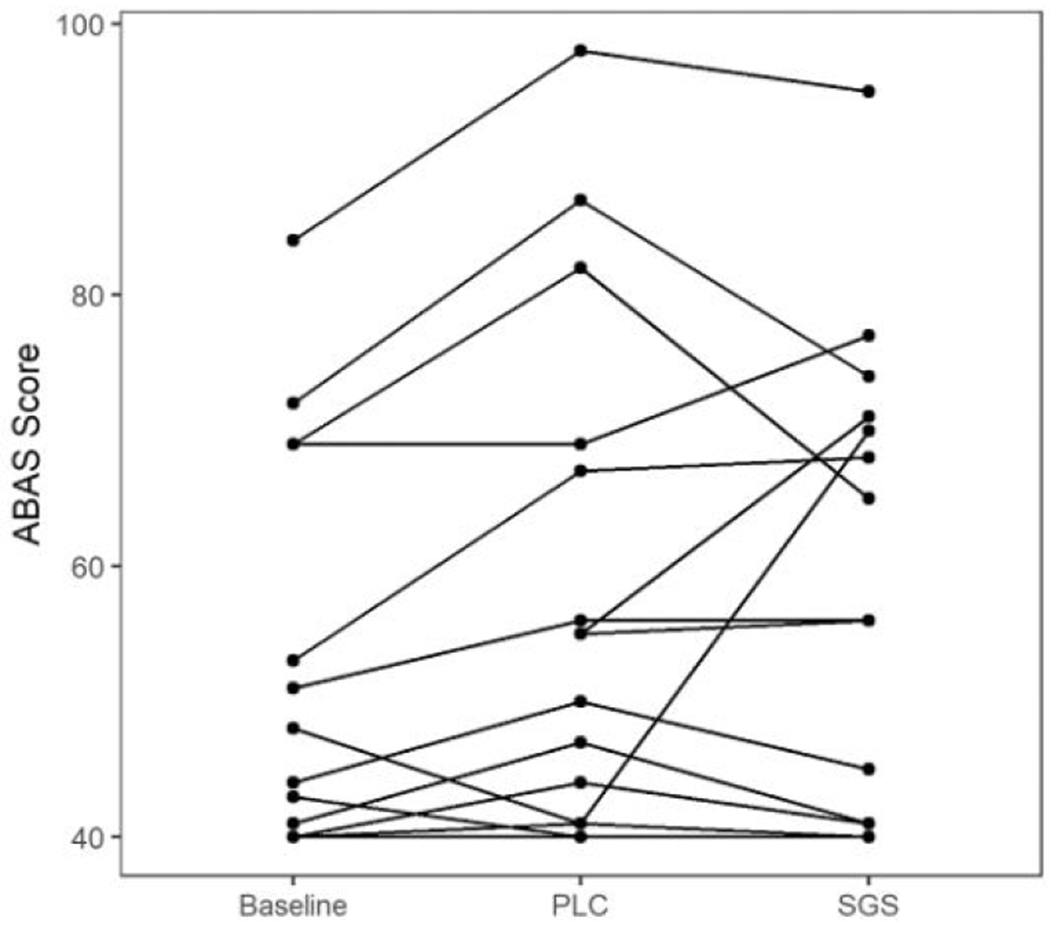
General adaptive composite standardized scores on the Adaptive Behavior Assessment System, Third Edition (ABAS-3) for each subject in each condition.
The data from other neuropsychological tests were too limited by patient performance to allow statistical analysis. Fourteen participants between the ages 10 and 24 were assessed using standardized tests of intellectual ability, word retrieval and attention. The Wechsler Nonverbal Scale of Intelligence (WNV) was attempted at baseline. Five were unable to attend and focus. These individuals (4 children and 1 young adult), were observed by the neuropsychologist while parent/guardian responded to the interview edition of the Vineland Adaptive Behavior Scale. These participants were either too young for individually administered standardized tests of ability or were unable to attend, focus or respond meaningfully.
Cognitive ability was determined by achieved Full Scale standard score (mean 100 +/− 15) on the WNV (mean: 52.7 +/− 18.8). Of the 14 patients, one obtained a Full Scale IQ score WNV in the average range (T1=93, T2=93, T3=97); for the remainder of the subjects (N=9) the mean Full Scale IQ scores ranged from 31 to 61. The results of the Vineland were used for four individuals unable to respond meaningfully either verbally or in writing. The mean of the scores on Vineland Adaptive Composite for those unable to complete the WNV was 66.6 +/− 7.05 (mean = 100 +/− 15). The VABS General Adaptive Composite mean of 66.6 +/− 7 (mean = 100 +/15) is in the deficient range. Examination of the results suggests that individuals generally had more well-developed social skills in the context of less well developed conceptual or practical skills. For those patients able to respond to the individually administered scales of intellectual ability, VABS scores were consistent with ability on the Wechsler tests. Thus the majority (13 of 14 participants) of these subjects meet established criteria for Intellectual Disability (DSM-V).
Results were similar for TMS measures. There was no significant effect of treatment on motor threshold (figure 3) (baseline-adjusted treatment effect = 1.22%, p = 0.47). Paired-pulse stimulation and recruitment curve results also showed no significant drug effects (figure 4). Baseline-adjusted treatment effect was −49.4% (p = 0.13) for SICI, −10.5% (p = 0.53) for ICF, and −41.2% (p = 0.27) for LICI. Results were robust to removal of a single patient outlier. The difference in recruitment curve slopes between treatment groups was 0.003 (p = 0.09).
Figure 4a-c.
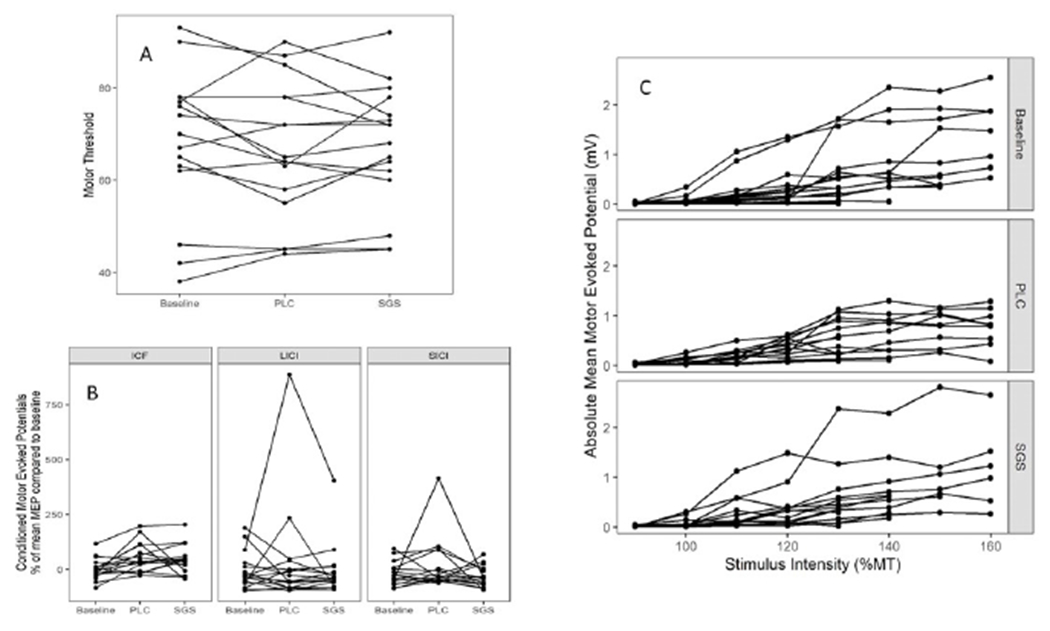
Results of transcranial magnetic stimulation. 4a. Motor threshold, expressed as a percentage of maximal stimulator output for each patient in each condition. 4b. Conditioned motor evoked potentials (MEP) for paired-pulse stimuli including intra-cortical facilitation (ICF), long interval intra-cortical inhibition (LICI), and short interval intra-cortical inhibition (SICI). Values are expressed as a percentage of the mean MEP compared to the baseline (conditioning) MEP in each patient for each study condition.4c. Recruitment curves for each subject across each condition, expressed as absolute mean motor evoked potential amplitude in millivolts at increasing stimulus intensities (expressed as a percentage of the motor threshold).
We attempted to obtain a CSP for all patients, but were often unsuccessful due to lack of patient cooperation. Furthermore, there was no observational difference when we examined the results graphically. Therefore, we did not analyze the CSP formally.
Five patients had a well-documented history of seizures and were taking anti-seizure medications. Three of the five had seizures during the study; there was no difference in frequency or severity between active drug and placebo periods. One, not currently on treatment, had a history of past seizures, and two patients reported episodes of staring and unresponsiveness, or ‘spacing out,’ but had had previous non-diagnostic studies including 24 hour continuous EEG. Neither of these two had any observed or reported episodes during the drug study.
There was no interaction between patient age and treatment; younger patients were not more likely to show improvement in ABAS on active drug.
Adverse Effects
Adverse effects were generally mild, usually requiring either no intervention over-the-counter or transient medical outpatient treatment. The number of events reported per patient varied from 0 to 19. There was no difference in incidence between drug and placebo arms for any body system (Table 3). One subject had a large number of reported side effects on placebo. Two developed molluscum contagiosum. Upper respiratory infections were common, perhaps due to the preponderance of children in the study.
Table 3.
Adverse events, by system, reported on SGS-742 and placebo
| System | AEs on SGS-742 | AEs on Placebo | Total |
|---|---|---|---|
| CNS | 14 | 21 | 35 |
| GI | 20 | 18 | 38 |
| GU | 0 | 1 | 0 |
| HEENT | 2 | 9 | 11 |
| Infection | 7 | 16 | 23 |
| Musculoskeletal | 1 | 0 | 1 |
| skin | 3 | 9 | 12 |
| general | 5 | 9 | 14 |
| total | 52 | 83 | 135 |
CNS and GI systems were most frequently affected. Common CNS complaints included lethargy, fatigue, increased irritability or emotional lability, anxiety, and insomnia. The most common GI AEs were abdominal pain, and decreased appetite; diarrhea and vomiting were reported less frequently. Two patients required transient dose reductions for gastrointestinal complaints, both while taking placebo.
Three patients were withdrawn from the study for an adverse event, two during drug treatment and one during placebo treatment. One patient developed a rash after the fourth dose of study drug. Another patient developed priapism on placebo. He was also taking risperidone. A third patient had exacerbation of a previously existing behavior disorder while on study drug. The first two adverse events resolved spontaneously, and the third after symptomatic treatment. All sixteen study completers achieved and were maintained on the full goal dose of 10 mg/kg (up to 600 mg) three times daily.
Laboratory tests
No consistent changes in blood counts or chemistry parameters were noted in either active drug or placebo periods. Two patients had mildly elevated liver function tests (alanine amino transferase and alkaline phosphatase) on active drug, and one (alkaline phosphatase) on placebo. Eight patients had positive urine urobilinogen (six at baseline) intermittently during the study. One had had a positive test on previous evaluation.
EEG
One patient had only a single EEG, and two had two rather than three. There were no changes on serial records for 13 patients. Three patients had a better organized background/decreased slowing on placebo, and one on active drug, compared to baseline. Spike-wave discharges seen on the baseline EEG in the latter patient were not present on the record at the end of the active drug phase, but reappeared at the end of the placebo phase. One patient’s record at the end of the placebo phase showed increased slowing and occasional interictal epileptiform discharges not present at baseline, but these features were seen as well on the EEG at the end of the active drug phase.
DISCUSSION
Our study failed to find an effect of SGS-742 on our primary outcome measure of the ABAS score, or on the secondary outcome measures of TMS parameters. We enrolled only 19 subjects, with 16 completers. Nevertheless, our data do not support a current role for the drug in SSADH treatment.
The side effects we observed were expected based on previous clinical trial data. Only two patients needed dose adjustment due to GI distress We do not know if the patient who developed a rash on the fourth dose of SGS-742 was reacting to the active drug or to other components of the capsule. Risperdone may have been responsible for the priapism experienced by one patient. Considering reports of increased anxiety and insomnia, SGS-742 may have led to the exacerbation of the behavior disorder leading to withdrawal. The increased urine urobilinogen, present before drug initiation, was not associated with any hematologic abnormalities, and may have been due to a laboratory cross-reaction.
It is possible that the dose we used was too low to show an effect. The lack of increased reported adverse reactions with SGS-742 may lend support to this notion. However, the maximum dose achieved by patients in this study was equivalent to prior studies demonstrating clinical improvement in adults with mild cognitive impairment22,23.
Our underlying hypothesis and strategy of blocking GABA-B receptors may have been inadequate, or wrong. The biochemical perturbations underlying the clinical symptoms of the disorder may be too complex to respond to a single targeted approach. Oxidative damage, mitochondrial dysfunction, and aberrant lipid biosynthesis are a component of various organic acidemias, including SSADH-D33–37 Altered autophagy, mitophagy, and pexophagy, through increased GABA-mediated activation of the mTOR complex38, may further contribute to oxidative stress and impaired mitochondrial function, and are implicated in a variety of neurodegenerative diseases. Perhaps a multi-faceted therapeutic approach addressing these mechanisms would yield positive results.
Blockade of additional receptors may be required to achieve therapeutic effects. There are multiple types of GABA-A and GABA-B receptors, in addition to GHB receptors, with different distribution and function. SSADH-D results in markedly elevated CSF GHB and GABA levels1,18,2. GABA levels are well above elevations that may be seen in other neuropediatric disorders39. At physiologic levels, GHB acts at high-affinity sites including GHB receptor(s), α4β1δ-GABA-A receptors (largely located at extra-synaptic sites), and other unidentified GHB receptors – only about 40% of GHB high–affinity sites have been identified6. However, at the extreme levels of GHB seen in SSADH-D, GHB also activates low-affinity targets: namely GABA-B receptors7,40 and α4β2/3δ-GABA-A receptors. Counterintuitively, activation of these δ-preferring GABA-A receptors induces spike-wave discharges and absence seizures in naïve rats41, speaking to the complexity inherent in the balance of excitation and inhibition exacted through GABA.
In addition to altered GABA receptor activation, SSADH-D has broad effects on various neurotransmitter systems. GHB inhibits presynaptic dopamine release and potentiates dopamine turnover42,43. CSF metabolite profiles from 13 unrelated patients with SSADH-D demonstrated significantly elevated GHB (65- to 230-fold), high free and total GABA (up to threefold), and low glutamine44. These findings, including a drop in glutamine over time, have been documented in the affected animal model45. In addition, there was a linear correlation in both HVA and 5-HIAA levels with increasing GHB concentration, suggesting enhanced dopamine and serotonin turnover44. Elevated GABA combined with low glutamine suggest disruption of the glial-neuronal glutamine/GABA/glutamate shuttle necessary for replenishment of neuronal neurotransmitters.
It is also possible that the patients were already too old to benefit from the therapy, despite our inclusion of patients down to 4 years old. We did not see any age-related trends toward improvement. Multiple lines of evidence from animal models and human subjects demonstrate an early shift in GABAergic receptor activation from depolarizing to hyperpolarizing shortly after birth46. Use-dependent downregulation of GABA-A and GABA-B receptors occurs in the first few weeks of life in the SSADH deficient mouse47,48. An age-dependent decrease in GABA and GHB levels towards more normal values follows49,50. This decrease in GABA and GHB levels, paired with downregulation of GABA-A and GABA-B receptors10,48, may explain the evolving phenotype seen in human subjects, who show more tendency towards compulsive behavior, sleep disturbance, and seizures with age5.
The outcome measures we used, the ABAS and TMS parameters, may not have been sensitive enough to show therapeutic effects. Individuals with SSADH-D have a fairly broad range of phenotypes and cognitive abilities4, generally performing in the moderately impaired range18, making it quite challenging to select appropriate neuropsychological tests that can be compared across subjects. Unfortunately, despite incorporating multiple measures into our neuropsychological evaluation, none except the ABAS allowed for analysis due to poor patient performance. The ABAS is a widely used parent/guardian questionnaire that assesses adaptive abilities including communication, social adaptability and practical skills regarding their child. Items focus on practical, everyday activities required to function, care for oneself, and interact with others effectively and independently. On a four-point response scale, raters indicate whether, and how frequently, the individual performs each activity. It is particularly useful for people with intellectual disability and can provide a measure of change in adaptive skills over time.
TMS provides surrogate measures of neurotransmitter system function, and has previously indicated abnormal GABA-B receptor-mediated inhibition in this population, as evidenced by shortening of the CSP and loss of LICI11. Other measures, including motor threshold, SICI, ICF, and RC, principally reflecting voltage-gated sodium channel activity, GABA-A receptor activity, and excitatory neurotransmitter function51 are no different among patients and controls. Since only disruption in GABA-B function in these patients has been demonstrated by TMS, and because SGS-742 blocks both pre- and post-synaptic GABA-B receptors, the lack of normalization in the CSP and LICI may be due to these competing effects. If one could selectively block presynaptic GABA-B receptors without affecting post-synaptic GABA-B receptors, one might see the desired effect on LICI and CSP.
In conclusion, we found no change in cognition or normalization of cortical excitability as measured by the ABAS and TMS in this randomized, placebo-controlled trial of SGS-742 in patients with SSADH-D. Our data do not support the current use of SGS-742 in SSADH-D.
Supplementary Material
Supplementary File 1. CONSORT checklist
ACKNOWLEDGEMENTS
The monetary and logistical support of the SSADH Association for this study is gratefully acknowledged.
FUNDING
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the SSADH Association, R01 NS082286 (PI: KMG), NINDS, NIH and by the NIH Intramural Research Program.
Footnotes
Trial registry number NCT02019667. Phase 2 Clinical Trial of SGS-742 Therapy in Succinic Semialdehyde Dehydrogenase Deficiency. https://clinicaltrials.gov/ct2/show/NCT02019667
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ETHICAL APPROVAL
The study was approved by the NIH Combined Neurosciences Institutional Review Board.
REFERENCES
- 1.Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology. 2003;60:1413–1417. [DOI] [PubMed] [Google Scholar]
- 2.Malaspina P; Roullet JB; Pearl PL; Ainslie GR; Vogel KR; Gibson KM Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem Int. 2016;99:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearl PL, Wiwattanadittakul N, Roullet JB, Gibson KM. Succinic Semialdehyde Dehydrogenase Deficiency. In: GeneReviews [online]. Available at: www.GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK1195/. Accessed July 16, 2020.
- 4.Gordon N. Succinic semialdehyde dehydrogenase deficiency (SSADH) (4-hydroxybutyric aciduria, gamma-hydroxybutyric aciduria). Eur J Paediatr Neurol. 2004;8:261–265. [DOI] [PubMed] [Google Scholar]
- 5.DiBacco ML, Roullet JB, Kapur K, et al. Age-related phenotype and biomarker changes in SSADH deficiency. Ann Clin Transl Neurol. 2018;6:114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bay T Eghorn LF, Klein AB, Wellendorph P. GHB receptor targets in the CNS: Focus on high-affinity binding sites. Biochem Pharmacol. 2014;87:220–228. [DOI] [PubMed] [Google Scholar]
- 7.Lingenhoehl K, Brom R, Heid J, et al. Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacol. 1999;38:1667–1673. [DOI] [PubMed] [Google Scholar]
- 8.Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balasubramanian S, Teissére JA, Raju DV, Hall RA. Hetero-oligomerization between GABAA and GABAB receptors regulates GABAB receptor trafficking. J Biol Chem. 2004;279:18840–18850. [DOI] [PubMed] [Google Scholar]
- 10.Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology. 2009;73:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reis J, Cohen LG, Pearl PL, et al. GABAB-ergic motor cortex dysfunction in SSADH deficiency. Neurology. 2012;79:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogel KR, Pearl PL, Theodore WH, McCarter RC, Jakobs C, Gibson KM. Thirty years beyond discovery—clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J Inherit Metab Dis. 2013;36:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matern D, Lehnert W, Gibson KM, Korinthenberg R. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (gamma-vinyl-GABA). J Inherit Metab Dis. 1996;19:313–318. [DOI] [PubMed] [Google Scholar]
- 14.Ergezinger K, Jeschke R, Frauendienst-Egger G, Korall H, Gibson KM, Schuster VH. Monitoring of 4-hydroxybutyric acid levels in body fluids during vigabatrin treatment in succinic semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54:686–689. [DOI] [PubMed] [Google Scholar]
- 15.Pearl PL, Gropman A. Monitoring gamma-hydroxybutyric acid levels in succinate-semialdehyde dehydrogenase deficiency. Ann Neurol. 2004;55:599. [DOI] [PubMed] [Google Scholar]
- 16.Shinka T, Ohfu M, Hirose S, Kuhara T. Effect of valproic acid on the urinary metabolic profile of a patient with succinic semialdehyde dehydrogenase deficiency. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;792:99–106. [DOI] [PubMed] [Google Scholar]
- 17.Didiášová M, Banning A, Brennenstuhl H, et al. Succinic semialdehyde dehydrogenase deficiency: an update. Cells. 2020;9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber JM, Pearl PL, Dustin I, et al. Biomarkers in a taurine trial for succinic semialdehyde dehydrogenase deficiency. JIMD Rep. 2016;30:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helm KA, Haberman RP, Dean SL, et al. GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharm. 2005;48:956–964. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Wojtowicz JM. Effect of GABA(B) receptors on synaptic interactions in dentate gyrus granule neurons of the rat. Neuroscience. 1997;79:117–127. [DOI] [PubMed] [Google Scholar]
- 21.Pearl PL, Gibson KM, Cortez MA, et al. SSADH Deficiency: Lessons from Mice and Men. J Inherit Metab Dis. 2009;32:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froestl W, Gallaher M, Jenkins H, et al. SGS742: the first GABA(B) receptor antagonist in clinical trials. Biochem Pharmacol. 2004;68:479–487. [DOI] [PubMed] [Google Scholar]
- 23.Tomlinson J, Cummins H, Wendt J, et al. SGS742, a novel GABAB receptor antagonist, improves cognition in patients with mild cognitive impairment. Meeting of the American Academy of Neurology, San Francisco, April 27, 2004, P02.053. [Google Scholar]
- 24.Wechsler D, Naglieri JA. Wechsler Nonverbal Scale of Ability. Pearson; 2006. [Google Scholar]
- 25.Wechsler D. Wechsler Adult Intelligence Scale. Pearson; 2008. [Google Scholar]
- 26.Stern RA, White T. Neuropsychological Assessment Battery. Par, Inc.; 2003. [Google Scholar]
- 27.Wilson BA, Alderman N, Burgess PW, Emslie H, Evans JJ. Behavioral Assessment of Dysexecutive Syndrome. Pearson; 1996. [Google Scholar]
- 28.Cullum M, Saine K, Weiner MF. Texas Functional Living Scale. Pearson; 2009. [PubMed] [Google Scholar]
- 29.Oakland T, Harrison PL. Adaptive Behavior Assessment System-II: Clinical Use and Interpretation. Elsevier Inc.; 2008. [Google Scholar]
- 30.Wechsler D. Wechsler Preschool and Primary Scale of Intelligence-IV. Pearson; 2012. [Google Scholar]
- 31.Wechsler D. Wechsler Intelligence Scale for Children-V. Pearson; 2003 [Google Scholar]
- 32.Niemi AK, Brown C, Moore T, Enns GM, Cowan TM. Evidence of redox imbalance in a patient with succinic semialdehyde dehydrogenase deficiency. Mol Genet Metab Rep. 2014;1:129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sparrow S, Cicchetti D, & Saulnier C. Vineland Adaptive Behavior Scales—Third Edition (Vineland-3). Circle Pines, MN: American Guidance Service; 2016. [Google Scholar]
- 34.Kim KJ, Pearl PL, Jensen K, et al. Succinic semialdehyde dehydrogenase: Biochemical-molecular-clinical disease mechanisms, redox regulation, and functional significance. Antioxid Redox Signal. 2011;15:691–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sgaravatti AM, Sgarbi MB, Testa CG, et al. Gamma-hydroxybutyric acid induces oxidative stress in cerebral cortex of young rats. Neurochemi Int. 2007;50:564–570. [DOI] [PubMed] [Google Scholar]
- 36.Silva AR, Ruschel C, Helegda C, et al. Inhibition of rat brain lipid synthesis in vitro by 4-hydroxybutyric acid. Metab Brain Dis. 1999;14:157–164. [DOI] [PubMed] [Google Scholar]
- 37.Latini A, Scussiato K, Leipnitz G, Gibson KM, Wajner M. Evidence for oxidative stress in tissues derived from succinate semialdehyde dehydrogenase-deficient mice. J Inherit Metab Dis. 2007;30:800–810. [DOI] [PubMed] [Google Scholar]
- 38.Lakhani R, Vogel KR, Till A, et al. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med. 2014;6: 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cortés-Saladelafont E, Molero-Luis M, Cuadras D, et al. Gamma-aminobutyric acid levels in cerebrospinal fluid in neuropaediatric disorders. Dev Med Child Neurol. 2018;60:780–792. [DOI] [PubMed] [Google Scholar]
- 40.Mathivet P, Bernasconi R, De Barry J, Marescaux C, Bittiger H. Binding characteristics of γ-hydroxybutyric acid as a weak but selective GABAB receptor agonist. Eur J Pharmacol. 1997;321:67–75. [DOI] [PubMed] [Google Scholar]
- 41.Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maitre M. The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Progr Neurobiol. 1997;51:337–361. [DOI] [PubMed] [Google Scholar]
- 43.Wong CG, Bottiglieri T, Snead OC. GABA, gamma-hydroxybutyric acid, and neurological disease. Ann Neurol. 2003;54 Suppl 6:S3–S12. [DOI] [PubMed] [Google Scholar]
- 44.Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria). Biol Psych. 2003;54:763–768. [DOI] [PubMed] [Google Scholar]
- 45.Gupta M, Hogema BM, Gromope M, et al. Murine Succinate Semialdehyde Dehydrogenase. Ann Neurology. 2003;54 (suppl 6):S81–S90. [DOI] [PubMed] [Google Scholar]
- 46.Cherubini E, Gaiars JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. [DOI] [PubMed] [Google Scholar]
- 47.Buzzi A, Wu Y, Frantseva MV. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res. 2006;1090:15–22. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Buzzi A, Frantseva M. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABA(A) receptor-mediated mechanisms. Ann Neurol. 2006;59:42–52. [DOI] [PubMed] [Google Scholar]
- 49.Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol. 2008;8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansen SS, Wang X, Sejer Pedersen D, et al. Gamma-hydroxybutyrate (GHB) content in hair samples correlates negatively with age in succinic semialdehyde dehydrogenase deficiency. JIMD Rep. 2017;36:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ilic TV, Meintzschel F, Cleff U, Ruge D, Kessler KR, Ziemann U. Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J Physiol. 2002;545:153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1. CONSORT checklist
Data Availability Statement
The study protocol, statistical analysis plan, and de-identified data are available for qualified individuals upon reasonable request. Data will be stored for at least three years as per federal guidelines.