

Abstract
Treatment options are limited for esophageal carcinoma (EC). G47Δ, a triple-mutated, conditionally replicating herpes simplex virus type 1 (HSV-1), exhibits enhanced killing of tumor cells with high safety features. Here, we studied the efficacy of G47Δ using preclinical models of human EC. In vitro, G47Δ showed efficient cytopathic effects and replication capabilities in all eight human esophageal cancer cell lines tested. In athymic mice harboring subcutaneous tumors of human EC (KYSE180, TE8, and OE19), two intratumoral injections with G47Δ significantly inhibited the tumor growth. To mimic the clinical treatment situations, we established an orthotopic EC model using luciferase-expressing TE8 cells (TE8-luc). An intratumoral injection with G47Δ markedly inhibited the growth of orthotopic TE8-luc tumors in athymic mice. Furthermore, we evaluated the safety of applying G47Δ to the esophagus in mice. A/J mice inoculated intraesophageally or administered orally with G47Δ (107 plaque-forming units [pfu]) survived for more than 2 months without remarkable symptoms, whereas the majority with wild-type HSV-1 (106 pfu) deteriorated within 10 days. PCR analyses showed that the G47Δ DNA was confined to the esophagus after intraesophageal inoculation and was not detected in major organs after oral administration. Our results provide a rationale for the clinical use of G47Δ for treating EC.
Keywords: oncolytic virus therapy, G47Δ, esophageal cancer, preclinical safety, orthotopic tumor model, herpes simplex virus, teserpaturev, biodistribution, intraesophageal inoculation, esophageal squamous cell carcinoma
Graphical abstract
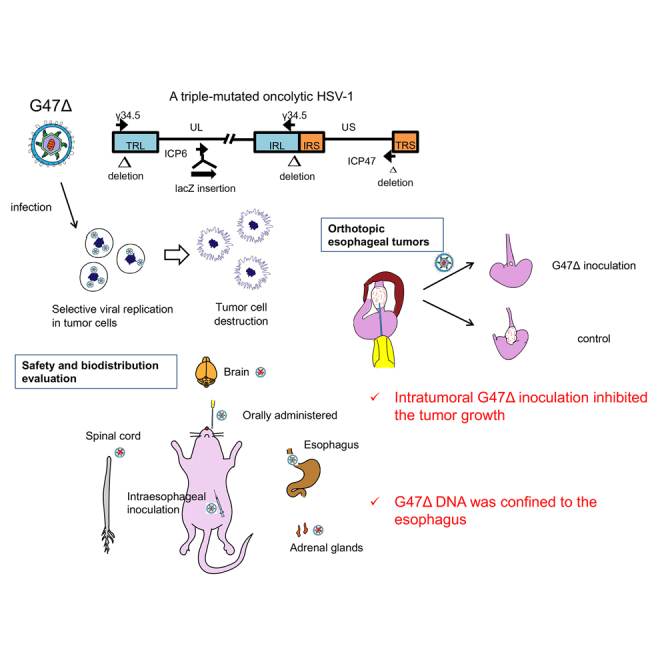
An intratumoral injection with oncolytic herpes virus G47Δ is efficacious in a clinically relevant orthotopic mouse model of esophageal cancer. G47Δ is safe when inoculated intraesophageally in mice, and G47Δ DNA distribution is confined to the esophagus. Intratumoral G47Δ treatment can be a potential therapeutic approach for esophageal cancer patients.
Introduction
Esophageal carcinoma (EC) remains a substantial cause of cancer-related mortality worldwide.1 Despite recent advances in diagnosis and multimodal treatments, the survival outcome of EC patients has improved only modestly over the past decades, with the 5-year survival ranging from 15% to 25%.2,3 Esophageal squamous cell carcinoma accounts for the majority of the incident esophageal cancer cases each year, particularly in the high-incidence regions of eastern Asia and Africa, while esophageal adenocarcinoma is more prevalent in North America and Western Europe.4
Radical esophagectomy with pre- or post-operative chemo-/chemoradiotherapy is the mainstay of treatment for resectable, locally advanced EC, and chemotherapy for unresectable or relapsed EC.5, 6, 7 New modalities, such as taxane-based triplet regimens8 and immune checkpoint inhibitors,9, 10, 11 have recently been applied for treating advanced EC. Unfortunately, immune checkpoint inhibitors show low response rates in patients with gastroesophageal cancer.12 EC treatment options are limited when the tumor progresses or the patient is intolerant to first-line standard chemotherapy. Therefore, there is a high need for developing novel therapeutic strategies for refractory EC.
Oncolytic viruses exhibit antitumor activity through selective killing of cancer cells and induction of specific antitumor immunity,13 and have emerged as promising immunotherapeutic agents for currently incurable malignancies.14 Many oncolytic viruses have been tested in clinical trials, of which talimogene laherparepvec (T-VEC) was the first to be approved by the US Food and Drug Administration (FDA) for patients with malignant melanoma in 2015.15,16 The clinical implementation of oncolytic viruses has been rather slow for patients with gastroesophageal cancer.17, 18, 19 Oncolytic adenovirus, alone20 or in combination with chemotherapeutic agents,21,22 has been investigated in mouse models with subcutaneous EC tumors. Phase I and II clinical trials are ongoing using a telomerase-specific oncolytic adenovirus in combination with pembrolizumab or with radiotherapy.23,24
G47Δ is a triple-mutated, third-generation oncolytic herpes simplex virus type 1 (HSV-1), which was developed by an additional deletion of the α47 gene from the genome of G207, a second-generation oncolytic HSV-1, to enhance the oncolytic activity and retain the immunogenicity of infected cancer cells.25,26 In the course of selective cancer cell destruction, G47Δ facilitates the priming of the immune system with cancer neoantigens, serving as a platform to revert immunologically “cold” tumors into “hot” tumors.25,27,28 G47Δ can be armed with therapeutic genes of interest to increase its antitumor potency.29, 30, 31 The immunomodulatory features of G47Δ can potentially overcome tumor resistance to immune checkpoint inhibitors.32
Notably, stereotactic injections with G47Δ provided excellent survival benefit for patients with glioblastoma in a phase II clinical trial (manuscript under preparation), and G47Δ awaits a governmental approval as a new drug in Japan. In preclinical studies, G47Δ has shown efficacy not only for brain tumors but also for a variety of cancers.33, 34, 35, 36, 37 G47Δ has also been used in clinical trials for prostate cancer, olfactory neuroblastoma, and malignant mesothelioma.38 Because of its versatile efficacy and the proven safety in the human brain, the clinical application of G47Δ should further expand in the near future.
In this study, we explore the usefulness of G47Δ as a therapeutic agent for EC in preclinical models, including a clinically relevant orthotopic mouse EC model. Furthermore, we evaluate the safety of G47Δ for treating EC by direct inoculation or by oral administration in the esophagus of HSV-1 sensitive A/J mice.
Results
Cytopathic effect and replication capability of G47Δ in vitro
To characterize the oncolytic activities of G47Δ in esophageal cancer, we studied its cytopathic effects and replication capabilities in eight human esophageal cancer cell lines in vitro. In all cell lines examined, G47Δ at a multiplicity of infection (MOI) of 0.1 caused >60% cell destruction within 4 days after infection (Figure 1A). TE8 cells (esophageal squamous cell carcinoma) and OE19 cells (esophageal adenocarcinoma) were especially susceptible to G47Δ, with >80% cell destruction by day 4 at an MOI of 0.01 (Figure 1A). We further examined the replication capabilities of G47Δ in human esophageal cancer cells. Virus yields increased by 48 h after infection at an MOI of 0.01 in all cell lines tested, although the extent of replication varied among cell lines (Figure 1B). These observations indicate that G47Δ can replicate well and exert efficient cell killing in human esophageal cancer cells, irrespective of tissue types.
Figure 1.

Cytopathic effects and virus yields of G47Δ in human EC cell lines
(A) Cells were seeded onto six-well plates at 2 × 105 cells/well or onto 96-well plates at optimal cell density. After an overnight incubation, the cells were infected with G47Δ (MOI of 0.01 or 0.1 or mock). The cell viability was determined daily either by counting surviving cells with a Coulter Counter or by the CellTiter96 Aqueous Non-Radioactive Cell Proliferation Assay. The percentage of surviving cells is expressed as a percentage of mock-infected controls. G47Δ exhibited good cytopathic effects at an MOI of 0.1 in all human esophageal cancer cell lines examined. Data are presented as the mean of triplicates ±SD. One-way ANOVA followed by Dunnett's test was used to determine statistical significance (∗∗∗p < 0.001; ns, not significant; versus mock-infected controls). (B) Cells were seeded onto six-well plates at 3 × 105 cells/well. Triplicate wells were infected with G47Δ at an MOI of 0.01. At 24 or 48 h after infection, cells were collected and progeny virus was titered on Vero cells. The dotted line shows the assumed titer of administered G47Δ per well. Vero cell line was used as a control. In all cell lines tested, G47Δ showed good replication capabilities by 48 h after infection. The results presented are the mean of triplicates ±SD.
Efficacy of G47Δ in subcutaneous EC tumor models
Next, we studied the in vivo efficacy of G47Δ using three mouse models with subcutaneous tumors of human esophageal cancer. Athymic mice harboring KYSE180, TE8, or OE19 tumors of approximately 6 mm in diameter were inoculated intratumorally with G47Δ (4 × 104, 2 × 105, or 1 × 106 plaque-forming units [pfu]) or mock twice (days 0 and 3). Intratumoral inoculations with G47Δ caused a significant inhibition of tumor growth in all subcutaneous tumor models even at the lowest dose tested (p < 0.01 versus mock on day 30 [KYSE180], day 22 [TE8], and day 27 [OE19]; Figure 2). In all models, there was no significant difference among the three doses used, although there seemed to be a tendency for dose dependency.
Figure 2.

Efficacy of G47Δ in subcutaneous EC tumor models
Subcutaneous tumors of a human esophageal cancer cell line (KYSE180 (upper left), TE8 (upper right), or OE19 (lower left)) were generated in 6-week-old female athymic mice. Established tumors, 5–6 mm in diameter, were inoculated with G47Δ (4 × 104, 2 × 105, or 1 × 106 pfu) or mock on days 0 and 3 (n = 9–10 per group). G47Δ treatment caused a significant inhibition of tumor growth in all subcutaneous tumor models irrespective of the dose used. Each experiment was conducted at least twice with similar results. The results presented are the mean ± SEM (n = 9–10). One-way ANOVA followed by Dunnett's test was used to determine statistical significance (∗∗p < 0.01; ∗∗∗p < 0.001).
Generation of an orthotopic EC tumor model
Subcutaneous xenograft tumor models do not fully reflect the clinical features of the tumors.39 To evaluate the efficacy of G47Δ under a more clinically relevant condition, we attempted to generate a reproducible orthotopic EC tumor model that mimics the clinical situation of treating a tumor in its organ of origin.40
We generated orthotopic tumors in athymic mice using eight human esophageal cancer cell lines (Figure S1), and evaluated the change in body weight, the engraftment rate, and the survival rate. Most of the mice harboring orthotopic tumors did not show marked loss in body weight, irrespective of the type of implanted tumor cells (Figure S2A). All tumor cell lines showed a high engraftment rate, four of eight cell lines being 100% (Figure S2B). Most tumor models showed a rather long survival, except for the OE19 model, in which half the animals died within 90 days due to the progression of peritoneal dissemination (Figure S2B). The orthotopic tumor model with TE8 cells (esophageal squamous cell carcinoma) exhibited a 100% engraftment rate and a consistent survival course without peritoneal dissemination, and therefore was considered the best suited among the tumor models evaluated for assessing the antitumor efficacy of G47Δ.
To monitor the tumor growth using a noninvasive, semiquantitative bioluminescent imaging method,41 we established TE8 cells that stably express luciferase (TE8-luc). When the TE8-luc cells were subcutaneously implanted into athymic mice, a significant and strong correlation was observed between tumor volume and the luciferase emission level (r = 0.945, p < 0.001, data not shown).
Intratumoral G47Δ treatment inhibited the growth of orthotopic tumors
The potential of G47Δ as a therapeutic means for esophageal cancer was studied using the orthotopic tumor model of TE8-luc. The experimental design is depicted in Figure 3A. Female athymic mice were inoculated with TE8-luc (2 × 106 cells) into the abdominal esophagus wall (on day −18). The mice were randomly divided into two groups 3 days later (n = 10 per group), at which time point there was no significant difference in the total photon counts of orthotopic tumors between the two groups. Eighteen days after the tumor implantation (on day 0), the established orthotopic tumors were intratumorally inoculated with G47Δ (1 × 106 pfu) or mock. In vivo imaging system (IVIS) for tumor-encoded luciferase revealed a significant reduction in tumor burden in mice treated with G47Δ compared with those treated with mock (p < 0.01 on day 13; Figure 3B). The changes of IVIS images of two treatment groups with time are shown in Figure 3C. The weight of harvested tumors from the G47Δ-treated mice on day 34 was significantly lower than that from the mock-treated surviving mice (median 32.1 mg versus 76.4 mg, p < 0.001).
Figure 3.

Efficacy of G47Δ in an orthotopic EC model
(A) Experimental design. Female athymic mice were inoculated with TE8-luc cells (1 × 106 cells) into the esophageal wall (day −18). After confirmation of the orthotopic tumors with IVIS 3 days after the implantation (on day −15), the mice were randomly divided into two groups (n = 10 per group), and were treated with an intratumoral inoculation with G47Δ (1.0 × 106 pfu) or mock on day 0. The photon counts of the tumors were calculated with IVIS every 3–4 days. On day 34, all surviving mice were euthanized and the tumor weight was compared between the groups. (B) The average of the logarithmic photon flux was plotted on a chart. G47Δ treatment significantly decreased the total bioluminescence from tumors (p < 0.01 on day 13). Each experiment was conducted at least twice with similar results. The results presented are the mean ± SEM (n = 10). The Welch's t test was used to determine statistical significance (∗∗p < 0.01). (C) The changes of IVIS images with time of both treatment groups are shown.
Hematoxylin-eosin (HE) staining of cross sections of the abdominal esophagus of mice with orthotopic tumors revealed that tumor cells invade extensively across the layers and into the lumen of the esophagus in mock-treated mice (Figure 4A), whereas the layered structure of the esophagus was well preserved in G47Δ-treated mice (Figure 4B). Immunohistochemical staining for HSV-1 in the same cross section indicated HSV-1 positivity, presumably depicting replicating G47Δ, localized within the tumor at 34 days after treatment (Figure 4C).
Figure 4.

Histology and immunohistochemistry of orthotopic EC tumors treated with G47Δ
Orthotopic EC tumors in athymic mice were treated with an intratumoral inoculation with G47Δ (1 × 106 pfu) or mock or G47Δ. Mice were euthanized 34 days after the treatments, orthotopic tumors excised, and the paraffin-embedded tissue sections stained with HE or immunostained with an anti-HSV-1 antibody. The red dotted circles indicate the extent of local tumor invasion (T, tumor; L, lumen; E, esophageal wall). Representative HE staining images of orthotopic EC tumors inoculated with mock (A) and G47Δ (B). (A) In orthotopic EC treated with mock, tumor cells invade extensively across the layers of the esophageal wall, narrowing the lumen of the esophagus. (B) In orthotopic EC treated with G47Δ, the tumor size remains small and the original structure of the esophageal wall is preserved. (C) The same tumor from (B) immunostained for HSV-1. HSV-1 positivity, presumably indicating replicating G47Δ, localizes within the tumor. Original magnification, ×30 (A) and ×40 (B, C), as indicated. Scale bars: 1 mm (A) and 500 μm (B, C).
Safety evaluation of intraesophageal inoculation and oral administration of G47Δ
To evaluate the safety of applying G47Δ in the esophagus, A/J mice were intraesophageally inoculated with mock, G47Δ (1 × 107 pfu), or wild-type HSV-1 strain F (1 × 106 pfu) (n = 5 per group). In another set of experiments, A/J mice were orally administered with mock, G47Δ (1 × 107 pfu), or strain F (1 × 106 pfu) (n = 5 per group). A/J is known to be one of the most susceptible mouse strains to HSV-1 infection.42 Each mouse was monitored twice a week for clinical manifestations based on three parameters (Figure 5A) and the body weight for 2 months.
Figure 5.

Safety evaluation of G47Δ applied to the esophagus of A/J mice
A/J mice were intraesophageally inoculated or orally administered with mock, G47Δ (1 × 107 pfu) or wild-type HSV-1 strain F (1 × 106 pfu) (n = 5 per group). Cages were then blinded, and each mouse was monitored twice a week for clinical manifestations and body weight for 2 months. (A) Clinical manifestation scores were estimated based on three parameters: appearance, activity, and response. Each parameter has a response on a five-point (0–4 points) ranging from normal (4) to death (0). Score 0 means the death of the mouse. (B) Time course changes in clinical manifestations (top) and body weight ratio based on the body weight on day 0 (bottom) in mice inoculated intraesophageally with mock (left), G47Δ (middle), or strain F (right). (C) Time course changes in clinical manifestations (top) and body weight ratio (bottom) in mice administered orally with mock (left), G47Δ (middle), or strain F (right). In both experiments (B and C), all mice treated with G47Δ survived without remarkable manifestations, but many of the mice treated with strain F deteriorated rapidly and became moribund.
In both experiments, intraesophageal inoculation and oral administration, mice given either mock or G47Δ all survived, with only a few showing a transient and minor decrease in clinical scores in the beginning and without weight loss (mock, Figures 5B and 5C [left]; G47Δ, Figures 5B and 5C [middle]). In contrast, many of the mice given strain F deteriorated within 10 days of administration and became moribund. With strain F, three out of five died by direct intraesophageal inoculation (Figure 5B [right]) and four out of five died by oral administration (Figure 5C [right]). These observations indicate that G47Δ is at least 10 times less toxic than strain F when applied to the esophagus in HSV-1-susceptible A/J mice.
Biodistribution of G47Δ administered to the esophagus
Before investigating the biodistribution of G47Δ, we sought to obtain data on the tendency of biodistribution using wild-type HSV-1. Strain F (1 × 107 pfu) was administered either intraperitoneally or orally to A/J mice, one mouse each was sacrificed daily until day 5, and the amount of viral DNA in nine organs was detected by qPCR. When strain F was administered intraperitoneally, the virus tended to spread widely to the organs of the peritoneum, then replicated in the adrenal glands, followed by replication in the spinal cord and the brain (Figure S3A). When strain F was administered orally, the virus was detected from organs of initial infection, the gastro-esophagus, and the lung (presumably from aspiration), then tended to enter and replicate in the brain (Figure S3B). Two potential pathways have been suggested for HSV-1 to spread after an intraperitoneal administration in mice: (1) from the adrenal glands via the spinal cord to the brainstem, or (2) from the myenteric plexus of the gut via the vagal nerves to the brainstem,43 which supports the results of our preliminary study. Therefore, for the main studies with G47Δ, we chose the esophagus, the adrenal glands, the spinal cord, and the brain as major organs to investigate.
To investigate the biodistribution of G47Δ after application in the esophagus, A/J mice were intraesophageally inoculated with G47Δ (1.0 × 107 pfu) or strain F (1 × 107 pfu) (n = 12 per group). Again, in another set of experiments, A/J mice were orally administered with G47Δ (1.0 × 107 pfu) or strain F (1.0 × 107 pfu) (n = 12 per group). Three mice per group were sacrificed on days 1, 3, 5, and 7, and the amount of G47Δ DNA in major organs was detected by qPCR.
In A/J mice receiving a direct inoculation with G47Δ into the subserosa of the esophagus, G47Δ DNA was detected at high levels in the esophagus on day 1 (three out of three), then gradually decreased, but could still be detected in all three mice on day 7 (Figure 6A, top). G47Δ DNA was also detected in the adrenal glands (two out of three) on day 1, but not detected by day 7 (Figure 6A, top). G47Δ DNA was undetectable in the spinal cord and detected only in one out of three on two occasions in the brain (Figure 6A, top). In marked contrast, in mice receiving a direct inoculation with strain F into the subserosa of the esophagus, the copy numbers of strain F DNA remained high throughout the time course in the esophagus and increased over time in the adrenal glands, the spinal cord, and the brain (Figure 6A, bottom), indicating the replication of strain F in the nervous system. In A/J mice receiving an oral administration with G47Δ, G47Δ DNA was not detected in major organs throughout the time course, except for one out of three in the esophagus on day 1 (Figure 6B, top). In contrast, in mice receiving an oral administration with strain F, strain F DNA was detected at high levels throughout the time course in the esophagus and markedly increased over time in the spinal cord and brain, indicating that orally administered strain F can enter and replicate in the central nervous system (Figure 6B, bottom).
Figure 6.

Biodistribution of G47Δ applied to the esophagus of A/J mice
A/J mice were intraesophageally inoculated or orally administered with G47Δ (1 × 107 pfu) or wild-type HSV-1 strain F (1 × 107 pfu) (n = 12 per group). Three mice per group were sacrificed on days 1, 3, 5, and 7, and the amount of G47Δ DNA in major organs (esophagus, adrenal glands, spinal cord, and brain) was detected by qPCR. Each dot represents the viral DNA copy number of the specimen (n = 3). (A) In mice receiving a direct inoculation with G47Δ into the subserosa of the esophagus (top), high levels of G47Δ DNA were detected from the esophagus throughout the time course. G47Δ DNA was also detected in the adrenal glands early after inoculation, but not detected by day 7. In mice receiving a direct inoculation with strain F into the subserosa of the esophagus (bottom), the copy numbers of strain F DNA remained high throughout the time course in the esophagus and increased over time in the adrenal glands, spinal cord, and brain. (B) In mice receiving an oral administration with G47Δ, G47Δ DNA was not detected in major organs throughout the time course, except for one in the esophagus on day 1 (top). In mice receiving an oral administration with strain F, strain F DNA was detected at high levels throughout the time course in the esophagus and markedly increased over time in the spinal cord and brain (bottom).
To summarize, G47Δ DNA distribution was confined mainly to the esophagus, the inoculated site, after a direct inoculation. G47Δ DNA was almost undetectable in the esophagus, and there was none in other major organs, after an oral administration. These observations further confirm the safety of G47Δ when applied to the esophagus.
Discussion
Currently available therapies provide unsatisfactory outcomes for patients with EC, underscoring the need for innovative treatment strategies.44 We demonstrate that all human esophageal cancer cell lines tested are highly susceptible to G47Δ, irrespective of tumor histology. G47Δ exhibits remarkable efficacy in subcutaneous EC xenograft models and in a clinically relevant orthotopic EC tumor model. G47Δ proves to be safe when applied to the esophagus of HSV-1 sensitive A/J mice, either intraesophageally or orally. These findings indicate that G47Δ could be useful for improving the clinical outcome of EC patients.
To our knowledge, the efficacy of an oncolytic virus has not been investigated in an orthotopic EC tumor model. Orthotopic tumor models reportedly characterize the cellular and biological features of tumors in a more clinically relevant way than subcutaneous tumor models, and therefore allow the study of tumor-stromal interaction, which is important to understand tumorigenesis and tumor responses to treatments.39,40,45 In addition, our orthotopic tumor model enables accurate monitoring of tumor growth, without invasive procedures, through bioluminescence imaging.41,46 Using the orthotopic tumor model, we find that an intratumoral inoculation with G47Δ is highly efficacious. Unlike in the mouse model, a direct intratumoral injection into esophageal cancer in patients is easily doable via endoscope, so it could become a principal means of treating EC in clinical settings.47
We further show that application of G47Δ to the esophagus is safe, using HSV-1-sensitive A/J mice. An intraesophageal inoculation with a high dose of G47Δ does not cause a substantial clinical manifestation, and G47Δ DNA is detectable only locally within the esophagus. The safety studies with oral administration were performed on supposition of a leakage of G47Δ into the esophageal lumen or spraying of G47Δ to the tumor surface as a potential route of administration. The results support that G47Δ is safe even if it was administered orally. The safety features of G47Δ are not at all natural features of HSV-1 but are acquired by the well-designed, triple mutations within the genome.25 In fact, we show that a wild-type HSV-1 strain F, not only when inoculated directly into the subserosa of the esophagus but also when administered orally, can enter the central nervous system and replicate, resulting in death of the majority of animals even at one-tenth of a dose of G47Δ. G47Δ DNA was detected in the brain of one mouse out of three that received a direct inoculation with G47Δ into the subserosa of the esophagus. The finding is presumably due to the neurotropic nature of HSV-1.48 Although it is unknown whether G47Δ can become latent in sensory neurons, because G47Δ can replicate in cancer cells only, G47Δ DNA in normal cells in the brain should stay harmlessly as DNA without ever producing a virus.
The replication capability of HSV-1 in normal cells is best demonstrated in brain tissue in vivo. A previous study showed that all of the A/J mice inoculated with strain F (2 × 103 pfu) in the brain deteriorated rapidly and became moribund within 7 days, clearly demonstrating the replication capability of strain F in the normal brains of HSV-1-susceptible A/J mice.25 On the other hand, G47Δ did not cause any manifestation when inoculated into the brain of A/J mice at 2 × 106 pfu. Furthermore, the first-in-human clinical trial in patients with recurrent glioblastoma (UMIN000002661) proved the safety of G47Δ when injected into the human brain tumor. The subsequent phase II clinical trial in patients with glioblastoma (UMIN000015995) further confirmed the safety of G47Δ when injected repeatedly in the brain tumor for the maximum of six doses. This phase II trial led to recent approval of G47Δ as a new drug for malignant glioma in Japan. Besides glioblastoma, a phase I study in patients with castration-resistant prostate cancer (UMIN000010463) demonstrated the safety of G47Δ when injected into the prostate. The results from the present study and these preclinical and clinical studies indicate that G47Δ does not replicate in normal tissues.
Because the biodistribution study was performed in normal A/J mice without tumors, the actual biodistribution of G47Δ in humans can only be observed in patients with esophageal cancer and therefore awaits a clinical trial. Furthermore, because the only natural host of HSV-1 is human, it is difficult to extrapolate the biodistribution data of mice to those in humans. However, previous preclinical studies demonstrated that intracerebral inoculation with high-titer G207 resulted in no viral distribution beyond the central nervous system at any time point after inoculation in non-human primates49,50 and BALB/c mice.51 The biodistribution of G207 in non-human primates did not differ from that in mice. The safety and biodistribution evaluation of G47Δ in HSV-1-susceptible A/J mice therefore at least suggests the safety and biodistribution profile in humans. As observed in preclinical safety studies in A/J mice, G47Δ was in fact safe when used in the human brain.
Neoadjuvant therapy followed by surgery is a clinical paradigm for patients with advanced EC. Recent preclinical studies in mouse models have shown that a neoadjuvant use of oncolytic viruses has a potential of eradicating the microscopic presence of cancer cells. For example, replicating pox virus administered systemically prior to surgery could reverse surgical stress-induced natural killer cell suppression in metastatic breast cancer and melanoma.52 Preoperative uses of Maraba virus and vaccinia virus proved to be useful for controlling postsurgical cancer recurrence via activation of innate immunity and specific antitumor immunity.53,54 Furthermore, we recently demonstrated that G47Δ can be used as a neoadjuvant therapy to suppress tumor relapse after radiofrequency ablation.34 Since neoadjuvant chemo-/chemoradiotherapy is standardly used for resectable EC patients, a neoadjuvant use of G47Δ for EC is a possibility that merits testing preclinically and clinically. A direct oncolysis with G47Δ alone may not be sufficient for a cure. We recently showed that G47Δ in combination with systemic administration of immune checkpoint inhibitors can cure half of the mice bearing murine EC tumors.55 Arming of G47Δ with immunostimulatory genes has been shown to result in enhanced efficacy.29 Human interleukin-12-expressing oncolytic HSV-1 with a G47Δ backbone (T-hIL12) is currently being tested in patients with melanoma (jRCT2033190086).
In conclusion, G47Δ is efficacious in both subcutaneous and orthotopic EC tumors in mice. G47Δ is safe when a high dose is administered intraesophageally and orally. Intratumoral injections with G47Δ thus constitute a practical and useful therapeutic approach for human esophageal cancer.
Materials and methods
Cell lines
Human esophageal squamous cell carcinoma cell lines KYSE70, KYSE180, KYSE220, T.Tn, and TE8, and human esophageal adenocarcinoma cell lines OE19, OE33, SKGT-4, and Vero (African green monkey kidney) cells were used. KYSE70, KYSE180, KYSE220, and T.Tn were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). OE19, OE33, and SKGT-4 were bought from the European Collection of Authenticated Cell Cultures (Porton Down, Salisbury, UK). TE8 and Vero cells were purchased from the Institute of Physical and Chemical Research (Riken, Saitama, Japan) and the American Type Culture Collection (Rockville, MD), respectively. Cells were cultured according to the directions provided by the suppliers. Luciferase-expressing TE8 (TE8-luc) cells were established with pre-made luciferase lentiviral particles expressing the firefly luciferase3 gene (#LVP326, GenTarget) according to the manufacturer's protocol. Clonal selection was performed in a medium containing 5 μg/mL blasticidin (Wako, Japan). All cell lines were used within 10 passages and confirmed to be mycoplasma free using the MycoAlert Mycoplasma Detection Kit.
Viruses
G47Δ and a wild-type HSV-1, strain F, were grown, purified, and titered by plaque assay on Vero cells as described previously.25,26
Cytotoxicity assays
In vitro cytotoxicity studies were performed as previously described.56 Briefly, cells were seeded onto six-well plates at 2 × 105 cells/well and incubated overnight at 37°C. The following day, the cells were infected with G47Δ (MOI of 0.01 or 0.1) or mock, and further incubated at 34.5°C. The number of surviving cells was counted daily with a Coulter Counter (Beckman Coulter, Fullerton, CA) and expressed as a percentage of mock-infected controls.
Virus yield studies
Cells were seeded onto six-well plates at 5 × 105 cells/well and incubated overnight at 37°C. The following day, triplicate wells were infected with G47Δ at an MOI of 0.01. At 24 and 48 h after infection, the cells were scraped into the medium and lysed by three cycles of freezing and thawing. The extracted viruses were titrated in Vero cells, and viral plaques were counted 3 days after infection as described previously.56
Animal experiments
All animal experiment protocols conformed to all relevant regulatory standards and were approved by the Committee for Ethics of Animal Experimentation and were in accordance with the Guideline for Animal Experiments in the University of Tokyo. Six-week-old female athymic (BALB/c nu/nu) mice were purchased from Clea Japan (Tokyo, Japan). Mice were maintained under specific pathogen-free conditions and provided with sterile food, water, and cages.
Subcutaneous tumor models
Subcutaneous tumors of esophageal cancer were generated by inoculating 3 × 106 cells (KYSE180), 4 × 106 cells (OE19), or 5 × 106 cells (TE8) into the right flank of 6-week-old female athymic mice (BALB/c nu/nu). When tumors reached approximately 5–7 mm in diameter, the animals were randomized, and mock or G47Δ (4 × 104, 2 × 105, or 1 × 106 pfu) in 20 μL of phosphate-buffered saline (PBS) containing 10% glycerol was inoculated into the right-flank tumors twice (n = 9–10 for each group). Mock-infected extract was prepared from virus buffer-infected cells.28 The tumor size was measured using a digital caliper (Mitutoyo) twice per week and the tumor volume was calculated using the following formula: tumor volume (mm3) = length × width × height. Mice were euthanized when the maximum diameter of the tumor reached 24 mm. Each experiment was conducted at least twice with similar results.
An orthotopic tumor model of esophageal squamous cell carcinoma
Orthotopic implantation of TE8-luc cells (2 × 106 cells) was performed in 6-week-old female athymic mice as previously reported.41 Briefly, a slanting 10-mm skin incision was made in the left upper abdomen under anesthesia. The stomach was pulled down with tweezers so that the abdominal esophagus could be seen. A 1-mL syringe with a 30-gauge needle (Nipro, Tokyo, Japan), in which TE8-luc cells were suspended in a 10-μL volume of RPMI medium and Matrigel (2.0 × 106 cells/20 μL), was inserted into the anterior wall of the abdominal esophagus. The tumor cells were then slowly injected so that they were not spilt on the peritoneal cavity. The appearance of a small swelling at the injection site indicated successful cell injection. The esophagus was then returned to the peritoneal cavity, and the abdominal wall and skin were closed with 5-0 polydioxanone (PDS).
Three days after the tumor cell inoculation, orthotopic tumor volume was determined by bioluminescence imaging with an IVIS (IVIS Lumina Series III, SPI, Japan) and the mice were randomly divided into two groups (n = 10). Eighteen days after the tumor implantation (on day 0), the mice were inoculated intratumorally with mock or G47Δ (1 × 106 pfu) in 20 μL of PBS containing 10% glycerol. The total photon counts of orthotopic tumors were observed. On day 34, all mice were euthanized, the tumors were harvested, and the weight measurement and the histological examinations were performed.
In vivo photon counting analysis was conducted twice a week on the IVIS with Living Image acquisition and analysis software (Living Image 4.4., Xenogen, United States) as previously described.57 For detection of bioluminescence, the mice were anesthetized and intraperitoneally injected with D-luciferin (150 mg/kg; PerkinElmer, Waltham, MA). Each experiment was conducted at least twice with similar results.
Histological and immunohistochemical analysis of orthotopic tumors
Mice harboring orthotopic TE8-luc tumors were treated and euthanized 34 days after the treatment as scheduled in Figure 3A. Orthotopic tumors were harvested, fixed in 10% formaldehyde neutral buffer solution (Sigma-Aldrich, St Louis, MOSA) for 72 h, and embedded in paraffin. Sections (5-μm thick) were rehydrated using an alcohol gradient and subjected to heat-mediated antigen retrieval using target retrieval solution S1700 (Dako, Santa Clara, CA).
Sections were mounted on silanized slides (Dako Cytomation, Glostrup, Denmark) and stained with HE. Sequential sections were subjected to immunohistochemical analysis to detect HSV-1. The sections were treated with peroxidase blocking solution (Dako) and Blocking One (Nacalai Tesque, Kyoto, Japan), incubated with a rabbit polyclonal anti-HSV-1 antibody (1:2,000 dilution, 3 μg/mL) (Dako Cytomation), rinsed, and incubated with an HRP-conjugated goat anti-rabbit IgG antibody (Nichirei Bioscience, Tokyo, Japan). The sections were developed with 3,3′-diaminobenzidine (DAB) Peroxidase Substrate kit (Vector laboratories, Burlingame, CA), and then counterstained with hematoxylin. A NanoZoomer Digital Pathology slide scanner (Hamamatsu Photonics K.K., Hamamatsu, Japan) was used to view slides.
Safety evaluation of G47Δ applied to the esophagus
G47Δ (1 × 107 pfu), strain F (1 × 106 pfu), or mock (PBS containing 10% glycerol) in a volume of 10 μL was injected into the subserosa of esophagus, or orally administrated into the lumen of esophagus using a feeding needle, in 6-week-old female A/J mice (n = 5 for each group). Cages were then blinded and mice monitored for changes in clinical manifestations and body weight twice a week for 2 months. Animal care was in accordance with institution guidelines.
Virus biodistribution studies
Six-week-old female A/J mice were given G47Δ (1 × 107 pfu) or strain F (1 × 106 pfu) by two different administration routes as described above; i.e., intraesophageal inoculation (n = 12 per group) and oral administration (n = 12 per group). Mice were sacrificed at 1, 3, 5, and 7 days after the administration (n = 3 for each day), and the amount of viral DNA in major organs (esophagus, adrenal glands, spinal cord, and brain) was quantified using semiquantitative real-time PCR. As a preliminary study, strain F (1 × 107 pfu) was administered either intraperitoneally or orally to 6-week-old female A/J mice, one mouse each was sacrificed daily until day 5, and the amount of viral DNA in nine organs (brain, spinal cord, gastro-esophagus, small intestine, colon, liver, lungs, kidneys, and adrenal glands) was quantified using semiquantitative real-time PCR.
The organs were homogenized, and genomic DNA was extracted using the QIAamp DNA mini kit (Qiagen) according to the manufacturer's instructions. Absolute quantification of viral DNA was conducted using real-time TaqMan PCR (the 7500 Fast Real-Time PCR System, Applied Biosystems) using HSV-1 gB primers (forward primer, 5′-GGCACGCGGCAGTACTTT-3′; reverse primer, 5′-CCATGCGCTCGCAGAGA-3′; TaqMan probe, 5′-GGTCGACAGGCACCTACAATGCCG-3′). The standard plasmid containing the glycoprotein B sequence of strain F and G47Δ served as positive control, and the values were used to generate a standard curve from 10 to 1.0 × 106 copies.
Statistical analysis
All data were expressed as the mean ± standard deviation (SD) or the mean ± standard error of the mean (SEM). Correlations were analyzed graphically using scatterplots and by the Pearson correlation coefficient, with application of the appropriate significance test (t test). For cytopathic effect studies and subcutaneous tumor studies, one-way ANOVA followed by Dunnett's test was used, and, for others, Student's t test or the Welch's t test was used to analyze the statistical significance of differences. The survival curves were analyzed by the Kaplan-Meier method, and p < 0.05 was considered as statistically significant using the log rank test. In the figures, standard symbols are used: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant. Statistical analyses were carried out with JMP 14.0.0 (SAS Institute, Cary, NC).
Acknowledgments
This research is supported in part by grants to T.T. from Practical Research for Innovative Cancer Control, Japan Agency for Medical Research and Development (AMED; grant number JP18ck0106416), and Translational Research Program, AMED (grant number JP20lm0203140).
Author contributions
S.Y., K.S., and T.T. were involved with the conception and performance of experiments, statistical analysis, and writing the manuscript. M.I. assisted with some of the experiments. S.Y. and M.T. were involved with the conception and design of experiments. K.S. and T.T. participated in manuscript preparation. All authors reviewed and edited the manuscript.
Declaration of interests
T.T. owns the patent right for G47Δ in multiple countries, including Japan.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.10.012.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Lagergren J., Smyth E., Cunningham D., Lagergren P. Oesophageal cancer. Lancet. 2017;390:2383–2396. doi: 10.1016/S0140-6736(17)31462-9. [DOI] [PubMed] [Google Scholar]
- 3.Wang V.E., Grandis J.R., Ko A.H. New strategies in esophageal carcinoma: translational insights from signaling pathways and immune checkpoints. Clin. Cancer Res. 2016;22:4283–4290. doi: 10.1158/1078-0432.CCR-16-0292. [DOI] [PubMed] [Google Scholar]
- 4.Abnet C.C., Arnold M., Wei W.Q. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360–373. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ajani J.A., D'Amico T.A., Almhanna K., Bentrem D.J., Besh S., Chao J., Das P., Denlinger C., Fanta P., Fuchs C.S., et al. Esophageal and esophagogastric junction cancers, version 1.2015. J. Natl. Compr. Canc. Netw. 2015;13:194–227. doi: 10.6004/jnccn.2015.0028. [DOI] [PubMed] [Google Scholar]
- 6.Lordick F., Mariette C., Haustermans K., Obermannová R., Arnold D. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016;27:v50–v57. doi: 10.1093/annonc/mdw329. [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa Y., Uno T., Oyama T., Kato K., Kato H., Kawakubo H., Kawamura O., Kusano M., Kuwano H., Takeuchi H., et al. Esophageal cancer practice guidelines 2017 edited by the Japan Esophageal Society: part 2. Esophagus. 2019;16:25–43. doi: 10.1007/s10388-018-0642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamasaki M., Yasuda T., Yano M., Hirao M., Kobayashi K., Fujitani K., Tamura S., Kimura Y., Miyata H., Motoori M., et al. Multicenter randomized phase II study of cisplatin and fluorouracil plus docetaxel (DCF) compared with cisplatin and fluorouracil plus Adriamycin (ACF) as preoperative chemotherapy for resectable esophageal squamous cell carcinoma (OGSG1003) Ann. Oncol. 2017;28:116–120. doi: 10.1093/annonc/mdw439. [DOI] [PubMed] [Google Scholar]
- 9.Janjigian Y.Y., Bendell J., Calvo E., Kim J.W., Ascierto P.A., Sharma P., Ott P.A., Peltola K., Jaeger D., Evans J., et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J. Clin. Oncol. 2018;36:2836–2844. doi: 10.1200/JCO.2017.76.6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang Y.K., Boku N., Satoh T., Ryu M.H., Chao Y., Kato K., Chung H.C., Chen J.S., Muro K., Kang W.K., et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:2461–2471. doi: 10.1016/S0140-6736(17)31827-5. [DOI] [PubMed] [Google Scholar]
- 11.Kato K., Cho B.C., Takahashi M., Okada M., Lin C.Y., Chin K., Kadowaki S., Ahn M.J., Hamamoto Y., Doki Y., et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:1506–1517. doi: 10.1016/S1470-2045(19)30626-6. [DOI] [PubMed] [Google Scholar]
- 12.Schoenfeld A.J., Hellmann M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443–455. doi: 10.1016/j.ccell.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuhara H., Ino Y., Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi: 10.1111/cas.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoda R., Nagalo B.M., Arora M., Egan J.B., Bogenberger J.M., DeLeon T.T., Zhou Y., Ahn D.H., Borad M.J. Oncolytic virotherapy in upper gastrointestinal tract cancers. Oncolytic Virother. 2017;7:13–24. doi: 10.2147/OV.S161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Quintanilla J., Seah I., Chua M., Shah K. Oncolytic viruses: overcoming translational challenges. J. Clin. Invest. 2019;129:1407–1418. doi: 10.1172/JCI122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 17.Macedo N., Miller D.M., Haq R., Kaufman H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer. 2020;8:e001486. doi: 10.1136/jitc-2020-001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terrero G., Lockhart A.C. Role of immunotherapy in advanced gastroesophageal cancer. Curr. Oncol. Rep. 2020;22:112. doi: 10.1007/s11912-020-00975-y. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara T. Multidisciplinary oncolytic virotherapy for gastrointestinal cancer. Ann. Gastroenterol. Surg. 2019;3:396–404. doi: 10.1002/ags3.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Komaki R., Wang L., Fang B., Chang J.Y. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin. Cancer Res. 2008;14:2813–2823. doi: 10.1158/1078-0432.CCR-07-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma G., Kawamura K., Li Q., Okamoto S., Suzuki N., Kobayashi H., Liang M., Tada Y., Tatsumi K., Hiroshima K., et al. Combinatory cytotoxic effects produced by E1B-55kDa-deleted adenoviruses and chemotherapeutic agents are dependent on the agents in esophageal carcinoma. Cancer Gene Ther. 2010;17:803–813. doi: 10.1038/cgt.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J., Li N., Zhao J., Lu J., Ma Y., Zhu Q., Dong Z., Liu K., Ming L. Histone deacetylase inhibitor trichostatin A enhances the antitumor effect of the oncolytic adenovirus H101 on esophageal squamous cell carcinoma in vitro and in vivo. Oncol. Lett. 2017;13:4868–4874. doi: 10.3892/ol.2017.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemunaitis J., Tong A.W., Nemunaitis M., Senzer N., Phadke A.P., Bedell C., Adams N., Zhang Y.A., Maples P.B., Chen S., et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. 2010;18:429–434. doi: 10.1038/mt.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan U., Biran T., Ocean A.J., Popa E.C., Ruggiero J.T., Paul D., Garcia C., Carr-Locke D., Sharaiha R., Urata Y., et al. Phase II study of a telomerase-specific oncolytic adenovirus (OBP-301, Telomelysin) in combination with pembrolizumab in gastric and gastroesophageal junction adenocarcinoma. J. Clin. Oncol. 2019;37:TPS4145. [Google Scholar]
- 25.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. U S A. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mineta T., Rabkin S.D., Yazaki T., Hunter W.D., Martuza R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 27.Fukuhara H., Martuza R.L., Rabkin S.D., Ito Y., Todo T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin. Cancer Res. 2005;11:7886–7890. doi: 10.1158/1078-0432.CCR-05-1090. [DOI] [PubMed] [Google Scholar]
- 28.Todo T., Martuza R.L., Dallman M.J., Rabkin S.D. In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity. Cancer Res. 2001;61:153–161. [PubMed] [Google Scholar]
- 29.Fukuhara H., Ino Y., Kuroda T., Martuza R.L., Todo T. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65:10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]
- 30.Ino Y., Saeki Y., Fukuhara H., Todo T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin. Cancer Res. 2006;12:643–652. doi: 10.1158/1078-0432.CCR-05-1494. [DOI] [PubMed] [Google Scholar]
- 31.Tsuji T., Nakamori M., Iwahashi M., Nakamura M., Ojima T., Iida T., Katsuda M., Hayata K., Ino Y., Todo T., et al. An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumor effect against human gastric cancer. Int. J. Cancer. 2013;132:485–494. doi: 10.1002/ijc.27681. [DOI] [PubMed] [Google Scholar]
- 32.Saha D., Martuza R.L., Rabkin S.D. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32:253–267.e5. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakatake R., Kaibori M., Nakamura Y., Tanaka Y., Matushima H., Okumura T., Murakami T., Ino Y., Todo T., Kon M. Third-generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018;109:600–610. doi: 10.1111/cas.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamada T., Tateishi R., Iwai M., Koike K., Todo T. Neoadjuvant use of oncolytic herpes virus G47Δ enhances the antitumor efficacy of radiofrequency ablation. Mol. Ther. Oncolytics. 2020;18:535–545. doi: 10.1016/j.omto.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugawara K., Iwai M., Yajima S., Tanaka M., Yanagihara K., Seto Y., Todo T. Efficacy of a third-generation oncolytic herpes virus G47Δ in advanced stage models of human gastric cancer. Mol. Ther. Oncolytics. 2020;17:205–215. doi: 10.1016/j.omto.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishino R., Kawase Y., Kitawaki T., Sugimoto N., Oku M., Uchida S., Imataki O., Matsuoka A., Taoka T., Kawakami K., et al. Oncolytic virus therapy with HSV-1 for hematological malignancies. Mol. Ther. 2021;29:762–774. doi: 10.1016/j.ymthe.2020.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oku M., Ishino R., Uchida S., Imataki O., Sugimoto N., Todo T., Kadowaki N. Oncolytic herpes simplex virus type 1 (HSV-1) in combination with lenalidomide for plasma cell neoplasms. Br. J. Haematol. 2021;192:343–353. doi: 10.1111/bjh.17173. [DOI] [PubMed] [Google Scholar]
- 38.Taguchi S., Fukuhara H., Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019;49:201–209. doi: 10.1093/jjco/hyy170. [DOI] [PubMed] [Google Scholar]
- 39.Tung L.N., Song S., Chan K.T., Choi M.Y., Lam H.Y., Chan C.M., Chen Z., Wang H.K., Leung H.T., Law S., et al. Preclinical study of novel curcumin analogue SSC-5 using orthotopic tumor xenograft model for esophageal squamous cell carcinoma. Cancer Res. Treat. 2018;50:1362–1377. doi: 10.4143/crt.2017.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bibby M.C. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur. J. Cancer. 2004;40:852–857. doi: 10.1016/j.ejca.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 41.Kuroda S., Kubota T., Aoyama K., Kikuchi S., Tazawa H., Nishizaki M., Kagawa S., Fujiwara T. Establishment of a non-invasive semi-quantitative bioluminescent imaging method for monitoring of an orthotopic esophageal cancer mouse model. PLoS One. 2014;9:e114562. doi: 10.1371/journal.pone.0114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kastrukoff L.F., Lau A.S., Thomas E.E. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS) Herpesviridae. 2012;3:4. doi: 10.1186/2042-4280-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irie H., Harada Y., Yoshihashi H., Kimura T., Kojima M., Kataoka M., Saito M., Sugawara Y., Mori W. Spread of herpes simplex virus type-1 (Miyama +GC strain) to the central nervous system after intraperitoneal inoculation: the role of the myenteric plexus of the gut. Arch. Virol. 1989;105:247–257. doi: 10.1007/BF01311361. [DOI] [PubMed] [Google Scholar]
- 44.Ilson D.H., van Hillegersberg R. Management of patients with adenocarcinoma or squamous cancer of the esophagus. Gastroenterology. 2018;154:437–451. doi: 10.1053/j.gastro.2017.09.048. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y., Toneri M., Ma H., Yang Z., Bouvet M., Goto Y., Seki N., Hoffman R.M. Real-time GFP intravital imaging of the differences in cellular and angiogenic behavior of subcutaneous and orthotopic nude-mouse models of human PC-3 prostate cancer. J. Cell. Biochem. 2016;117:2546–2551. doi: 10.1002/jcb.25547. [DOI] [PubMed] [Google Scholar]
- 46.Yanagihara K., Takigahira M., Takeshita F., Komatsu T., Nishio K., Hasegawa F., Ochiya T. A photon counting technique for quantitatively evaluating progression of peritoneal tumor dissemination. Cancer Res. 2006;66:7532–7539. doi: 10.1158/0008-5472.CAN-05-3259. [DOI] [PubMed] [Google Scholar]
- 47.Chahlavi A., Rabkin S., Todo T., Sundaresan P., Martuza R. Effect of prior exposure to herpes simplex virus 1 on viral vector-mediated tumor therapy in immunocompetent mice. Gene Ther. 1999;6:1751–1758. doi: 10.1038/sj.gt.3301003. [DOI] [PubMed] [Google Scholar]
- 48.Connolly S.A., Jardetzky T.S., Longnecker R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021;19:110–121. doi: 10.1038/s41579-020-00448-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Todo T., Feigenbaum F., Rabkin S.D., Lakeman F., Newsome J.T., Johnson P.A., Mitchell E., Belliveau D., Ostrove J.M., Martuza R.L. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in Aotus. Mol. Ther. 2000;2:588–595. doi: 10.1006/mthe.2000.0200. [DOI] [PubMed] [Google Scholar]
- 50.Hunter W.D., Martuza R.L., Feigenbaum F., Todo T., Mineta T., Yazaki T., Toda M., Newsome J.T., Platenberg R.C., Manz H.J., et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J. Virol. 1999;73:6319–6326. doi: 10.1128/jvi.73.8.6319-6326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sundaresan P., Hunter W.D., Martuza R.L., Rabkin S.D. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J. Virol. 2000;74:3832–3841. doi: 10.1128/jvi.74.8.3832-3841.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tai L.H., de Souza C.T., Bélanger S., Ly L., Alkayyal A.A., Zhang J., Rintoul J.L., Ananth A.A., Lam T., Breitbach C.J., et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013;73:97–107. doi: 10.1158/0008-5472.CAN-12-1993. [DOI] [PubMed] [Google Scholar]
- 53.Bourgeois-Daigneault M.C., Roy D.G., Aitken A.S., El Sayes N., Martin N.T., Varette O., Falls T., St-Germain L.E., Pelin A., Lichty B.D., et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 54.Ahmed J., Chard L.S., Yuan M., Wang J., Howells A., Li Y., Li H., Zhang Z., Lu S., Gao D., et al. A new oncolytic Vaccinia virus augments antitumor immune responses to prevent tumor recurrence and metastasis after surgery. J. Immunother. Cancer. 2020;8:e000415. doi: 10.1136/jitc-2019-000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugawara K., Iwai M., Ito H., Tanaka M., Seto Y., Todo T. Oncolytic herpes virus G47Δ works synergistically with CTLA-4 inhibition through dynamic intratumoral immune modulation. Mol. Ther. Oncolytics. 2021;22:129–142. doi: 10.1016/j.omto.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Todo T., Rabkin S.D., Sundaresan P., Wu A., Meehan K.R., Herscowitz H.B., Martuza R.L. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum. Gene Ther. 1999;10:2741–2755. doi: 10.1089/10430349950016483. [DOI] [PubMed] [Google Scholar]
- 57.Toyoshima M., Tanaka Y., Matumoto M., Yamazaki M., Nagase S., Sugamura K., Yaegashi N. Generation of a syngeneic mouse model to study the intraperitoneal dissemination of ovarian cancer with in vivo luciferase imaging. Luminescence. 2009;24:324–331. doi: 10.1002/bio.1112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.