

Summary
Although natural killer (NK) cells have become a promising immune effector cell for chimeric antigen receptor (CAR)-based therapy, generating human CAR-NK cells with high transgene efficiency has been challenging. In this protocol, we describe how to generate CAR-NK cells with transduction efficiencies >15% from healthy donor ex vivo expanded NK cells using third generation lentiviral vectors (LVs). We also show how to assess CAR-NK cell anti-tumor function in vitro using a flow cytometry-based killing assay.
For complete details on the use and execution of this protocol, please refer to Portillo et al. (2021).
Subject areas: Cancer, Cell Biology, Cell culture, Cell-based Assays, Flow Cytometry/Mass Cytometry, Gene Expression, Immunology, Molecular Bio
Graphical abstract
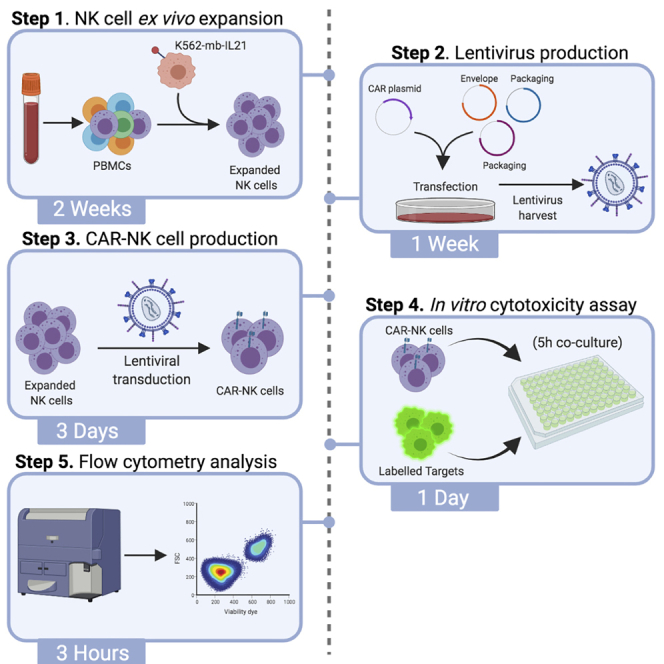
Highlights
-
•
PBMC isolation and expansion of human NK cells from healthy donors
-
•
Production of third generation lentiviral vectors for NK cell transduction
-
•
A protocol for the generation of CAR-modified NK cells from human expanded NK cells
-
•
A flow cytometry-based assay to assess CAR-NK cell cytotoxicity in vitro
Although natural killer (NK) cells have become a promising immune effector cell for chimeric antigen receptor (CAR)-based therapy, generating human CAR-NK cells with high transgene efficiency has been challenging. In this protocol, we describe how to generate CAR-NK cells with transduction efficiencies >15% from healthy donor ex vivo expanded NK cells using third generation lentiviral vectors (LVs). We also show how to assess CAR-NK cell anti-tumor function in vitro using a flow cytometry-based killing assay.
Before you begin
Note: Within each section of the protocol we have indicated when a reagent/equipment can be substituted with a similar product, otherwise use the provided reagent/equipment.
Culture K562-mb-IL21 feeder cells
Timing: Minimum 2 weeks before NK cell expansion from PBMCs
This section describes how to culture the K652 feeder cells expressing membrane bound IL-21 (K562-mb-IL21) that are used to generate expanded NK cells. The K562-mb-IL21 cells were generated by retroviral transduction with truncated CD19, CD64, CD86 and CD137L and by transduction using the Sleeping Beauty transposon expressing mbIL-21 (Denman et al., 2012; Singh et al., 2011). The sequence of mb-IL21 is owned by Kiadis Pharma (https://www.kiadis.com/) and the sequence is expressed from the MNDU3 promoter (Singh et al., 2011). The K562-mb-IL21 cells are available upon request through a Material Transfer Agreement (MTA).
-
1.Thaw cryopreserved K562-mb-IL21 cells.
-
a.Add 10 mL of complete RPMI media to 15 mL falcon tube.
-
b.Remove frozen K562-mb-IL21 cryovial from liquid nitrogen tank.
-
c.Place K562-mb-IL21 cryovial in a 37°C water bath immediately after retrieving from liquid nitrogen tank. As soon as there is one ice crystal remaining, transfer cells drop wise into 10 mL of complete RPMI media.
-
d.Centrifuge tube at 300 × g for 5 min.
-
e.Remove supernatant and count cells with Trypan Blue.
-
a.
-
2.
Resuspend K562-mb-IL21 cells to a final concentration of 0.5 × 106 cells/mL in complete RPMI media. Incubate cells at 37°C and 5% CO2
-
3.
Change media every 2–3 days and maintain cells at 0.5 × 106 cells/mL.
Isolation of peripheral blood mononuclear cells (PBMCs) from whole blood
Obtain approval from the appropriate ethics board prior to conducting research with human samples. All research conducted in this study was approved by the Hamilton Integrated Research Ethics Board in Hamilton, Ontario. Before taking blood ensure that you have obtained written and signed consent from the healthy donors. Follow all appropriate safety guidelines when working with human blood including use of a biological safety cabinet.
-
4.
Prepare 50 mL falcon tubes containing 15 mL of Lymphoprep density gradient medium.
Alternatives: Other density gradient media suitable for PBMC isolation can be used such as Ficoll.
-
5.
Collect 15–30 mL of whole blood from a healthy donor using BD ACD Solution A Vacutainers.
-
6.
Transfer 15 mL of peripheral blood per 50 mL falcon tube.
-
7.
Use 15 mL of sterile 2% FBS-PBS to wash vacutainer tubes and dilute the peripheral blood 1:1 in the 50 mL falcon tube.
-
8.
Set your pipettor to slow speed and begin slowly overlaying the 30 mL of diluted peripheral blood onto the falcon tube pre-filled with Lymphoprep.
CRITICAL: Make sure the diluted blood does not break through the Lymphoprep layer to avoid red blood cell contamination after centrifugation. Tilt the tube and allow the blood to flow down the tube wall.
-
9.
Centrifuge at 20°C–22°C for 20 min at 600 × g with no brakes. See Figure 1 for visual representation of blood component layers after centrifugation.
-
10.
Slowly remove ∼10 mL of the plasma layer without disturbing the buffy coat and discard.
-
11.
Slowly collect the buffy coat and place into a new 50 mL falcon tube being careful to not disturb the red blood cell pellet at the bottom of the tube.
Note: Buffy coats collected from two tubes can be combined into one 50 mL falcon tube.
-
12.
Top up the tube with 2% FBS-PBS and centrifuge at 300 × g for 5 min with the break on to remove remaining Lymphoprep.
-
13.
Discard supernatant and count isolated PBMCs with Trypan Blue.
Pause point: At this point you can proceed directly to the ex vivo NK cell expansion using fresh PBMCs or cryopreserve them for later use.
Figure 1.

Schematic of the different blood layers obtained from the density gradient centrifugation pre- and post-spin.
Ex vivo expansion of human NK cells using K562-mb-IL21 feeder cells
This section describes how to generate highly activated CD3-CD56superbright expanded NK cells from human PBMCs.
-
14.Transfer K562-mb-IL21 cells to a 50 mL falcon tube and centrifuge at 300 × g for 5 min.
-
a.Remove supernatant and count with Trypan Blue.
-
b.Determine total number of K562-mb-IL21 cells needed to replenish desired number of PBMCs at a 2:1 ratio.
-
a.
Example: If expanding a starting number of 2.5 million PBMCs you need to irradiate 5 million K562-mb-IL21 cells. Depending on the number of NK cells you would like to expand, irradiate the corresponding amount of K562-mb-IL21 plus 2–5 million extra cells to account for pipetting error.
-
15.
Resuspend K562-mb-IL21 cells to 5 × 106 cells/mL in complete RPMI media and irradiate cells using a gamma cell irradiator at 100 Gy for 30 min.
-
16.Co-culture irradiated K562-mb-IL21 feeder cells with PBMCs at a 2:1 ratio, respectively.
-
a.Count PBMCs with Trypan Blue.
-
b.Add 2.5 million PBMCs and 5 million irradiated K562-mb-IL21 cells into a 50 mL falcon tube and top up with complete RPMI media to a final volume of 0.5 × 106 cells/mL based on the starting PBMC cell number.
-
c.Supplement with 100 IU/mL of IL-2.
-
a.
-
17.
Incubate the co-culture at 37°C and 5% CO2.
Note: Starting with 2.5 million PBMCs usually yields a sufficient amount of expanded NK cells as this method typically induces a ∼103–104 fold expansion after 21 days (Denman et al., 2012).
-
18.Replace media on co-culture every other day.
-
a.Transfer co-culture to a 50 mL falcon tube and centrifuge at 300 × g for 5 min.
-
b.Remove supernatant and add 5 mL of fresh complete RPMI media, keeping the same total volume used in the replenishing day.
-
c.Supplement with 100 IU/mL of IL-2.
-
d.Place co-culture back into tissue culture flask and incubate at 37°C and 5% CO2.
-
a.
-
19.
Every 7 days, count NK cells in the culture and add freshly irradiated K562-mb-IL21 cells at a 2:1 ratio. See Troubleshooting 1 if low viability or low expansion is observed.
Note: Prior to proceeding with lentiviral transduction, ensure that the expanded NK cells are >75% CD3-CD56superbirhgt (Troubleshooting 2).
Culture SKBR3 breast cancer tumor cells
The following protocol describes how to passage the human breast cancer SKBR3 cell line established from a 43-year old Caucasian female with malignant mammary adenocarcinoma and which naturally overexpresses HER2. Other target cells can be used depending on the specificity of the CAR construct used in your study.
-
20.Thaw cryopreserved SKBR3 cells.
-
a.Add 10 mL of complete DMEM media to 15 mL falcon tube.
-
b.Remove frozen SKBR3 cryovial from liquid nitrogen tank.
-
c.Place SKBR3 cryovial in a 37°C water bath immediately after retrieving from liquid nitrogen tank. As soon as there is one ice crystal remaining, transfer cells drop wise into 10 mL of complete DMEM media.
-
d.Centrifuge tube at 300 × g for 5 min.
-
e.Remove supernatant and count cells with Trypan Blue.
-
a.
-
21.
Resuspend 2.5–5 million SKBR3 cells in 20 mL of complete DMEM and place in a T-150 flask.
-
22.Passage cells every 2–3 days once they reach confluency.
-
a.Discard spent media from flask. Add 10 mL of warm PBS to wash the cells.
-
b.Add 5 mL of 1× Trypsin-EDTA (0.05%) and incubate at 37°C for 3–5 min.
-
c.Lightly tap flask to dislodge all cells from the flask.
-
d.Neutralize Trypsin by adding 3 times the volume of warm complete DMEM media.
-
e.Split confluent flask by 1:4.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE Mouse Anti-Human IgG Fc (1:20) | BioLegend | Cat#409304; Clone HP6017; RRID:AB_10895907 |
| APC Mouse Anti-Human CD271 (NGFR) (1:20) | BioLegend | Cat#345108; Clone ME20.4; RRID:AB_10645515 |
| BV421 Mouse Anti-Human CD56 (1:20) | BD Biosciences | Cat#562751; Clone NCAM16.2; RRID:AB_2732054 |
| APC-H7 Mouse Anti-Human CD3 (1:20) | BD Biosciences | Cat#560275; Clone SK7; RRID:AB_1645475 |
| VioBright FITC Mouse Anti-Human CD271 (LNGFR) (1:50) | Miltenyi Biotec Inc. | Cat#130-113-423; Clone ME20.4-1.H4; RRID:AB_2734064 |
| Bacterial and virus strains | ||
| Third Generation Self-Inactivating Lentivirus | Generated in this study | N/A |
| Biological samples | ||
| Healthy adult PBMCs | Isolated from peripheral blood of healthy volunteers | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lymphoprep | STEMCELL Technologies | Cat#07861 |
| Recombinant Human IL-2 | PeproTech | Cat#200-02; Accession# P60568 |
| Recombinant Human ErbB2/Her2 Fc Chimera Protein | R&D Systems | Cat#1129-ER-050; Accession# NP_004439 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | Ca# H9268; CAS: 28728-55-4 |
| 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE) | Sigma-Aldrich | Cat#21888; CAS 150347-59-4 |
| eBioscience Fixable Viability Dye eFluor 780 (1:1000) | Thermo Fisher Scientific | Cat#65-0865-18 |
| Fixable Viability Stain 510 (1:1000) | BD Biosciences | Cat#564406; RRID: AB_2869572 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668019 |
| Sodium butyrate | Sigma-Aldrich | Cat#B5887; CAS: 156-54-7 |
| Opti-MEM | Thermo Fisher Scientific | Cat#31985070 |
| Fetal Bovine Serum | Gibco | Cat#12484028 |
| Bovine Serum Albumin | Rockland Inc. | Cat#BSA-50 |
| RMPI 1640 Medium, powder | Gibco | Cat#31800105 |
| DMEM Medium, powder | Gibco | Cat#12800017 |
| L-Glutamine | Bioshop Canada Inc | Cat#GLU102; CAS: 56-85-9 |
| HEPES | In house | N/A |
| Penicillin/Streptomycin | Gibco | Cat#15140122 |
| Trypan Blue Solution, 0.4% | Life Technologies | Cat#15250061 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | Cat#D12345 |
| 10× Trypsin-EDTA (0.5%) | Gibco | Cat#15400054 |
| 16% Paraformaldehyde (PFA) | Electron Microscopy Sciences | Cat#15710(EM) |
| Experimental models: cell lines | ||
| Human: K562-mb-IL21 (Clone 9) cells (Female) | Laboratory of Dean A. Lee | Denman et al. (2012) |
| Human: SKBR-3 cells (Female) | Laboratory of Karen Mossman | RRID: CVCL_0033 |
| Human: HEK293T cells (Fetal) | Laboratory of Jonathan L. Bramson | RRID: CVCL_0063 |
| Recombinant DNA | ||
| Plasmid: pCCL-Darpin-hCD8a NGFR | Laboratory of Jonathan L. Bramson | Hammill et al. (2015) |
| Plasmid: pCCL-CMV-NGFR | Laboratory of Jonathan L. Bramson | Hammill et al. (2015) |
| Plasmid: pRSV-REV | Addgene | Addgene #12253 |
| Plasmid: pMD2.G | Addgene | Addgene #12259 |
| Plasmid: pMDLg-pRRE | Addgene | Addgene #12251 |
| Software and algorithms | ||
| FACSDIVA Software | BD Biosciences | https://www.bdbiosciences.com/ |
| FlowJo Software | BD Biosciences | https://www.flowjo.com/ |
| Other | ||
| BD Vacutainer™ Glass Blood Collection Tubes with Acid Citrate Dextrose (ACD) | Thermo Fisher Scientific | Cat#02-684-26 |
| 150 mL 0.45 μM polyenersulfone (PES) filter | Thermo Fisher Scientific | Cat#165-0045 |
| 38.5 mL Ultracentrifuge tubes | Beckman Coulter | Cat#344058 |
| SW-32 Ti Swinging Bucket Rotor | Beckman Coulter | Cat#369694 |
| T-150 tissue culture flasks | Fisher Scientific | Cat#0877248 |
| 15 cm NUNC plates | Thermo Fisher Scientific | Cat#168381 |
| 5 mL polysterene tubes | Fisher Scientific | Cat#149591A |
| 50 mL falcon tubes | Falcon | Cat#352098 |
| 15 mL falcon tubes | Falcon | Cat#352096 |
| 96-well U bottom plate with lid | Fisher Scientific | Cat#0877254 |
| 24-well Tissue culture plate with lid | Falcon | Cat# 353047 |
| 20 μL Sterile filtered pipette tips | DiaMed | Cat#DIATEC530-8001 |
| 100 μL Sterile filtered pipette tips | DiaMed | Cat#DIATEC530-8251 |
| 1000 μL Sterile filtered pipette tips | DiaMed | Cat#DIATEC530-9001 |
| 5 mL serological pipettes | Fisher Scientific | Cat#1367522 |
| 10 mL serological piptettes | Fisher Scientific | Cat# 1367520 |
| 25 mL serological pipettes | Fisher Scientific | Cat# 136682 |
| 1.5 mL microcentrifuge tubes | Fisher Scientific | Cat# 05408129 |
| LSRFortessa Flow Cytometer | BD Biosciences | N/A |
Materials and equipment
Complete RPMI media
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 media | n/a | 435 mL |
| FBS | 10% | 50 mL |
| HEPES (1 M) | 10 mM | 5 mL |
| L-glutamine (0.2 M) | 2 mM | 5 mL |
| Penicillin/Streptomycin | 1% | 5 mL |
| Total | n/a | 500 mL |
[Keep sterile and store at 4°C for up to 1 month]
Complete DMEM media
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM media | n/a | 435 mL |
| FBS | 10% | 50 mL |
| HEPES (1 M) | 10 mM | 5 mL |
| L-glutamine (0.2 M) | 2 mM | 5 mL |
| Penicillin/Streptomycin | 1% | 5 mL |
| Total | n/a | 500 mL |
[Keep sterile and store at 4°C for up to 1 month]
1× Trypsin EDTA (0.05%)
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | n/a | 90 mL |
| 10× Trypsin-EDTA (0.5%) | 1× | 10 mL |
| Total | n/a | 100 mL |
[Sterile filter and store at 4°C for up to 4 months]
Polybrene stock
| Reagent | Final concentration | Amount |
|---|---|---|
| Sterile Water | n/a | 20 mL |
| Hexadimethrine bromide (Polybrene) | 8000 μg/mL | 0.160 g |
| Total | n/a | 20 mL |
[Sterile filter and store at −80°C for up to 4 months]
NK cell transduction media
| Reagent | Final concentration | Amount |
|---|---|---|
| Complete RPMI media | n/a | 10 mL |
| Hexadimethrine bromide (Polybrene; 8000 μg/mL) | 8 μg/mL | 10 μL |
| IL-2 (100 IU/uL) | 500 IU/mL | 50 μL |
| Total | n/a | 10 mL |
[Make fresh each time]
5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE) stock
| Reagent | Final concentration | Amount |
|---|---|---|
| DMSO | n/a | 896.9 μL |
| CFSE | 50 mM | 25 mg |
| Total | n/a | 896.9 μL |
[Store aliquots at −20°C for up to 12 months]
Recombinant ErbB2/HER2 Fc chimeric protein stock
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | n/a | 500 μL |
| ErbB2/HER2 Fc chimeric protein | 100 μg/mL | 50 μg |
| Total | n/a | 500 μL |
[Keep sterile and store aliquots at −20°C for up to 3 months]
FACS buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | n/a | 500 mL |
| Bovine Serum Albumin | 0.2% | 1 g |
| Total | n/a | 500 mL |
[Sterile filter and store at 4°C for up to 2–3 months]
2% PFA
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | n/a | 7 mL |
| 16% Paraformaldehyde (PFA) | 2% | 1 mL |
| Total | n/a | 8 mL |
[Store at 4°C for up to 2 weeks protected from light]
Step-by-step method details
Third generation lentivirus production and harvest by ultracentrifugation
These steps describe how to generate 3rd generation LVs and has been adapted with some modifications from Hammill et al. (2016).
-
1.Thaw low passage (P16 or less) cryopreserved HEK293T cells in a 37°C water bath.
-
a.Add 10 mL of complete DMEM media to 15 mL falcon tube.
-
b.Remove frozen HEK293T cryovial from liquid nitrogen tank.
-
c.Place HEK293T cryovial in a 37°C water bath immediately after retrieving from liquid nitrogen tank. As soon as there is one ice crystal remaining, transfer cells drop wise into the 15 mL falcon tube.
-
d.Centrifuge tube at 300 × g for 5 min.
-
e.Remove supernatant and count cells with Trypan Blue.
-
a.
-
2.
Resuspend 1.5–2 million HEK293T cells in 20 mL of complete DMEM media and transfer to a T-150 tissue culture flask. Culture flasks at 37°C and 5% CO2.
Note: Always start lentivirus prep with freshly thawed HEK293T cells to ensure higher viral yield. Around 2–3 80% confluent T-150 flasks are needed for one batch of lentiviral prep.
-
3.
24 h prior to transfection, harvest HEK293T cells by washing flasks with 10 mL warm PBS and adding 3 mL warm 1× Trypsin-EDTA(0.05%). Place flasks in the incubator for 2–5 min and neutralize trypsin with 3× the volume of warm complete DMEM media.
Note: Cells should be ∼80% confluent three days post-thaw. HEK293T cells are very loosely adherent thus do not require a long incubation time with Trypsin. Use Trypsin to harvest cells to prevent cell clumping.
-
4.
Transfer cell suspension to a 50 mL falcon tube and centrifuge at 300 × g for 5 min. Resuspend cell pellet with warm complete DMEM media and count cells using Trypan Blue.
Note: Use antibiotic-free complete DMEM media to plate cells prior to transfection as it may interfere with Lipofectamine 2000 reagent.
-
5.Plate 9 million HEK293T cells in 15 mL of antibiotic-free complete DMEM media per 15 cm NUNC tissue-culture treated plate. Incubate plates at 37°C and 5% CO2.
-
a.Re-seed a T-150 flask with 1.5–2 million cells in 20 mL of complete DMEM media for lentiviral titration.
-
a.
Note: Typically, three 15 cm plates are required to obtain sufficient viral particles per prep, scale up as needed. Carefully place plates into incubator ensuring that plates are even to make sure media covers each plate evenly.
-
6.
Transfection can begin 24 h after plating HEK293T cells, at this point cells should be ∼70%–80% confluent.
Note: Measure concentration of all plasmids used prior to transfection using a spectrophotometer that measures the concentration and purity of nucleic acids.
-
7.
For each plate, prepare two 15 mL falcon tubes with 4 mL of Opti-MEM media labeled Tube A and Tube B.
-
8.
Prepare transfection mixture by following schematic in Figure 2.
-
9.
Slowly add the DNA and Lipofectamine mixture to each plate drop wise making sure to cover the entire plate area. Swirl plate carefully and slowly to evenly distribute the mixture.
-
10.
Incubate plates at 37°C and 5% CO2.
Alternatives: Lipofecatamine (lipid based transfection agent) can be substituted with chemical methods such as calcium phosphate for the transfection. However, the amount of total DNA added should be optimized using this method. Refer to Brown et al. (2020) and Follenzi and Naldini (2002) for protocols on LV production using calcium phosphate.
-
11.
12–16 h after transfection, carefully aspirate media and replace with 15 mL warm antibiotic-containing complete DMEM media supplemented with 1 mM of sodium butyrate.
Note: Sodium butyrate is added as it has been shown to enhance HIV-1 derived vector production (Olsen and Sechelski, 1995).
-
12.
Incubate plates at 37°C and 5% CO2.
Note: From this point forward, you will be working with virus samples. Ensure you are following proper BSL-2 guidelines when handling lentivirus.
-
13.
36–48 h after transfection, lentivirus released in the supernatant can be harvested for viral concentration.
Note: Pre-cool the SW-32 Ti ultracentrifuge rotor and buckets at 4°C before starting the harvest.
-
14.
Aspirate viral containing supernatant from the 15 cm plates and transfer to 50 mL falcon tubes.
-
15.
Centrifuge the 50 mL falcon tubes at 2000 × g for 10 min at 4°C to sediment any collected cells.
-
16.
Filter supernatant using a 150 mL 0.45 μM PES filter to remove cell debris.
Alternatives: A filter with a pore size of 0.22 μM can also be used to improve purity. However, a size of 0.45 μM is used in this protocol to avoid any loss of viral particles during the filtration.
-
17.
Add filtrate into 38.5 mL ultracentrifuge tubes and use cold PBS to bring tube within 0.5 cm of the edge to prevent centrifuge tube from collapsing.
-
18.
Carefully balance the centrifuge tubes before placing into the ultracentrifuge buckets.
-
19.
Centrifuge buckets at 130,000 × g for 1 h 40 min at 4°C with minimal deceleration.
-
20.
Carefully remove ultracentrifuge tube from ultracentrifuge bucket and pour the supernatant into a 20% bleach bucket.
-
21.
Keep the ultracentrifuge tube inverted and blot the excess supernatant onto paper towels for up to 1 min to prevent viral pellet from drying out.
-
22.
Immediately place the ultracentrifuge tube on ice and let it sit for 10 min to begin viral resuspension.
Note: From this point forward only use micropipette filter tips when handling the virus.
-
23.
Add between 50–100 μL of cold PBS and start to slowly scrape the bottom of the ultracentrifuge tube using a micropipette filter tip.
Note: Set the micropipette to a low volume (∼40 μL) and do not go to the second stop while resuspending to avoid formation of bubbles.
Note: The volume of PBS added to resuspend the viral pellet can be adjusted depending on your experience with the particular LV. For example, you can add a higher volume of PBS to lentivirus preps that have higher yields. If you consistently obtain a low viral yield see Troubleshooting 3.
-
24.
Using the same micropipette filter tip, slowing resuspend the pellet by gently pipetting up and down and wash the sides of the ultracentrifuge tube.
-
25.Once viral pellet is fully resuspended aliquot virus into 1.5 mL microcentrifuge tubes and immediately place into −80°C freezer.
-
a.Prepare one aliquot of 13 μL to use for the lentiviral titration.
-
a.
-
26.
Decontaminate anything that has come into contact with the virus using 70% ethanol.
Note: Minimize the production of aerosols when working in BSL2 conditions.
Figure 2.

Flowchart depicting how to prepare transfection mixture
(1) Add required amount of plasmid DNA to Tube B and mix gently by inverting tube 3 times (3×). (2) Add 120 uL of Lipofectamine 2000 to Tube A and mix gently by inverting tube 3 times. (3) Incubate Tube A for 5 min at 20°C–22°C. (4) Add Tube B to Tube A, mix gently by inverting tube 3 times. (5) Incubate for 20 min at 20°C–22°C. (6) Add transfection mixture to each plate.
Lentivirus titration on HEK293T cells
The lentivirus stock should be titrated using HEK293T cells prior to NK cell transduction. To determine the titer, HEK293T cells are first transduced with serial dilutions of the lentivirus stock and they are then stained to measure the expression of the transduction marker by flow cytometry. The titer is calculated using the following formula: Titer (TU/mL)= ((# of HEK293T cells transduced × % Transgene-positive cells × Dilution factor)/100). The expected titer for the lentivirus containing the HER2 CAR construct used in this study is between 3 × 107–2 × 108 TU/mL.
-
27.
Harvest HEK293T cells from a T-150 flask and count using Trypan Blue.
Note: Perform two separate counts and take the average to ensure the cell count is accurate.
-
28.Plate 30,000 HEK293T cells per well in a 24-well tissue culture plate.
-
a.Resuspend cells to 6 × 104 cells/mL using complete DMEM media.
-
b.Up to 6-wells will be needed for each lentivirus batch, using dilutions 10−2 to 10−6 and one control well with no virus.
-
c.Resuspend the cell suspension 3 times and add 500 μL per well.
-
a.
-
29.
Incubate plate at 37°C and 5% CO2 for at least 3 h to ensure HEK293T cells have adhered.
-
30.Prepare lentivirus serial dilutions following Figure 3.
-
a.Prepare dilutions using 1.5 mL microcentrifuge tubes in complete DMEM media.
-
b.Mix each dilution thoroughly by pipetting or vortexing.
-
a.
-
31.
Thoroughly mix and add 500 μL of each dilution per well.
-
32.
Add 500 μL of complete DMEM media to the no virus control well.
-
33.
Incubate plate at 37°C and 5% CO2 for 72 h.
Alternatives: We find that assessing transgene expression after 72 h post HEK293T cell transduction is adequate to obtain a determination of LV titer. However, if a highly accurate titer is needed waiting 5 days post transduction could minimize the proportion of HEK293T cells that express the transgene but has not been successfully integrated.
-
34.Carefully harvest the transduced HEK293T cells using 1000 μL micropipette filter tips.
-
a.Slowly pipette up and down first in the centre of each well and then around the edges.
-
a.
-
35.
Transfer cells to 5 mL polysterene tubes with caps.
-
36.
Centrifuge at 300 × g for 5 min and decant supernatant.
-
37.
Resuspend cell pellet, add 2 mL of cold PBS and invert capped tubes to mix.
-
38.Centrifuge at 300 × g for 5 min and decant supernatant.
-
a.Prepare a 1:1000 dilution of viability dye using eBioscience Fixable Viability Dye eFluor 780 in PBS.
-
a.
-
39.
Resuspend cell pellet and add 100 μL of viability dye mixture to each tube.
-
40.Incubate at 4°C for 30 min in the dark.
-
a.Prepare antibody mix during the incubation by adding 1 μL of VioBright FITC Mouse Anti-Human CD271 (LNGFR) antibody into 49 μL of FACS buffer (1:50 dilution) per test.
-
a.
-
41.
Add 2 mL of FACS buffer and invert capped tubes to mix.
-
42.
Centrifuge at 300 × g for 5 min and decant supernatant.
-
43.
Resuspend cell pellet and add 50 μL of antibody mix to each tube.
-
44.
Incubate at 4°C for 30 min in the dark.
-
45.
Add 2 mL of FACS buffer and invert capped tubes to mix.
-
46.
Centrifuge at 300 × g for 5 min and decant supernatant.
-
47.
Resuspend cell pellet at add 100 μL of 2% PFA mix to each tube.
-
48.
Incubate at 4°C for 1 h in the dark.
-
49.
Add 2 mL of FACS buffer and invert capped tubes to mix.
-
50.
Centrifuge at 300 × g for 5 min and decant supernatant.
-
51.
Resuspend cell pellet and acquire samples by flow cytometry.
-
52.
Decontaminate anything that has come into contact with the virus using 70% ethanol.
Note: Minimize the production of aerosols when working in BSL2 conditions.
Figure 3.

Flowchart of serial dilutions for stock lentivirus titration on HEK293T cells.
Transduction of expanded NK cells with HER2 CAR lentivirus construct
This step describes the process of transducing expanded NK cells with 3rd generation LVs. Prior to NK cell transduction, determine the number of NK cells you need to transduce to perform downstream functional assays. In addition, you need to determine the amount of lentivirus needed to transduce desired number of wells at the desired MOI. Typically an MOI of 5 results in sufficient transduction efficiency. This is based on the LV titers typically obtained with our construct after titration on HEK293T cells. However, the MOIs used should be optimized depending on the size of your CAR lentiviral construct.
Note: Primary NK cells should be expanded by co-cultured with irradiated K562 mb-IL21 feeder cells at a 2:1 ratio for at least 2 weeks before transduction.
-
53.A day before transduction, replenish expanded NK cells with irradiated K562 mb-IL21 cells at a 1:1 ratio.
-
a.Seven days after the last replenishment, count NK cells with Trypan Blue.
-
b.Resuspend the co-culture to a final concentration of 0.5 × 106 cells/mL in complete RPMI media supplemented with 100 IU/mL of IL-2.
-
a.
-
54.
Plate 200 μL of the co-culture into a 96-well U bottom plate for a total of 1 × 105 cells per well.
-
55.
Incubate plate for 12–18 h at 37°C and 5% CO2.
-
56.18–24 h after plating, centrifuge 96-well U bottom plate at 300 × g for 10 min.
-
a.During the centrifugation, prepare the NK cell transduction media.
-
a.
-
57.
Carefully remove the supernatant from each well, and resuspend cells with 90 μL of NK cell transduction media using a multichannel pipette.
-
58.
Place plate in the incubator (37°C and 5% CO2) while you prepare the viral suspension.
-
59.
Prepare viral suspension by diluting the stock with the appropriate amount of cold PBS to add 10 μL of viral suspension per well at an MOI of 5.
Note: Thaw your lentivirus containing the CAR construct and the control vector on ice. If the lentivirus titre is <5 × 107 TU/mL do not dilute in PBS.
Example: To calculate the amount of lentivirus needed to transduced 5 wells at an MOI of 5 use the following calculation (use virus titer obtained from the HEK293T titration):
| ((1 × 105 × MOI × # of wells)/virus titer(TU/mL)) × 1000 = Total amount of virus needed (μL) |
-
60.
Once the virus suspension is prepared, obtain 96-well plate from the incubator and add 10 μL of virus suspension per well.
-
61.
Add 10 μL of PBS to the non-transduced control wells.
-
62.
Immediately centrifuge the plate at 1000 × g for 45 min at 32°C as the spinoculation step.
Note: In our experience, we find that while spinoculation is not necessary for lentiviral transduction of human T cells, it does enhance the transduction efficiency of expanded NK cells. However, this step may be skipped depending on the size and titer of your lentiviral construct but should be optimized.
-
63.
Gently resuspend the wells using a micropipette and incubate plate for 12–18 h at 37°C and 5% CO2.
-
64.
The next day, centrifuge the plate at 300 × g for 10 min.
-
65.
Carefully remove the supernatant and resuspend cells to a final volume of 100 μL using complete RMPI media supplemented with 100 IU/mL of IL-2.
-
66.
Three to four days after transduction, HER2 CAR-NK cells can be stained to assess HER2 CAR transgene expression.
Assessing HER2 CAR expression by flow cytometry
This procedure describes how to assess expression of the HER2 CAR transgene after NK cell lentiviral transduction. The HER2 CAR transgene used in this study is under the EF1a promoter and the transduction marker is under the CMV promoter, the construct is described in Hammill et al. (2015).
-
67.
Pool wells containing transduced HER2 CAR- and control NK cells (non-transduced and control vector transduced) into a 5 mL polystyrene tube.
-
68.
Wash cells with 2 mL of complete RPMI media and centrifuge at 300 × g for 5 min.
-
69.
Carefully discard the supernatant and count cells with Trypan Blue.
-
70.
Transfer 100,000 cells of the HER2 CAR-, control vector- and non-transduced NK cells to a 96-well U bottom plate for staining with the surface stain cocktail.
Surface stain master mix
| Reagent | Dilution | Amount per test (μL) |
|---|---|---|
| Anti-CD56 BV421 | 1:20 | 2.5 |
| Anti-CD3 APC-H7 | 1:20 | 2.5 |
| Anti-IgG Fc PE | 1:20 | 2.5 |
| Anti-NGFR APC | 1:20 | 2.5 |
| FACS Buffer | N/A | 40 |
| Total | 50 |
-
71.Prepare additional control wells using 100,000 cells per well.
-
a.Plate one well with the HER2 CAR-NK cells HER2 Fc chimera protein control.
-
b.Plate four wells with the non-transduced NK cells for the unstained, viability dye compensation and fluorescence minus one (FMO) control wells.
-
a.
Control well set up
| HER2 Fc chimera | CD56 BV421 | CD3 APC-H7 | IgG Fc PE | NGFR APC | |
|---|---|---|---|---|---|
| Unstained | – | – | – | – | – |
| HER2 Fc chimera protein control | – | X | X | X | X |
| CD56 BV421 FMO | X | – | X | X | X |
| CD3 APC-H7 FMO | X | X | – | X | X |
Note: FMOs for the IgG Fc PE and NGFR APC can be included but the transgene positive gates should be set based on the fully stained non transduced NK cell control sample.
-
72.Resuspend the remainder of the cells to a final concentration of 1 × 106 cells/mL in complete RMPI media supplemented with 100 IU/mL IL-2.
-
a.Incubate at 37°C and 5% CO2 until functional assays.
-
a.
Note: Functional assays should be performed within 1–2 weeks after initial transduction as we have found transgene expression is downregulated over time and CAR-NK cells cannot be further expanded.
-
73.
Spin the plate at 300 × g for 5 min to pellet the cells.
-
74.
Remove supernatant and wash cells with 200 μL of PBS.
-
75.Spin the plate 300 × g for 5 min and discard PBS.
-
a.Prepare a 1:1000 dilution of viability dye (Fixable Viability Stain 510) in PBS.
-
a.
-
76.Add 100 μL of the viability dye and incubate at 4°C for 30 min in the dark.
-
a.Prepare the HER2 Fc chimera protein mix during the incubation by adding 2.5 μL of recombinant ErbB2/HER2 Fc chimeric protein to 25 μL of FACS buffer per test.
-
a.
Note: You can also perform viability staining with Fixable Viability Stain 510 at 37°C for 7 min.
-
77.
Spin the plate at 300 × g for 5 min and discard supernatant.
-
78.
Wash cells with 200 μL of FACS buffer, spin the plate at 300 × g for 5 min and discard supernatant.
-
79.Add 25 μL of the HER2 Fc chimera protein dilution and incubate in the dark for 30 min at 20°C–22°C.
-
a.Do not add HER2 Fc chimera protein to the extra HER2 CAR-NK cell control well.
-
a.
-
80.
Spin the plate at 300 × g for 5 min and discard supernatant.
-
81.
Wash cells with 200 μL of FACS buffer, spin the plate at 300 × g for 5 min and discard supernatant.
-
82.
Add 50 μL of the surface stain master mix and incubate 4°C for 30 min in the dark.
Surface stain master mix
| Reagent | Dilution | Amount per test (μL) |
|---|---|---|
| Anti-CD56 BV421 | 1:20 | 2.5 |
| Anti-CD3 APC-H7 | 1:20 | 2.5 |
| Anti-IgG Fc PE | 1:20 | 2.5 |
| Anti-NGFR APC | 1:20 | 2.5 |
| FACS Buffer | N/A | 40 |
| Total | 50 |
Alternatives: Anti-CD3 APC-H7 can be swapped out of this panel with Anti-CD3 PerCp-Cy5.5 and used at the same dilution, in that case you can use the fixable viability dye eBioscience Fixable Viability Dye eFluor 780 instead of Fixable Viability Stain 510.
-
83.
Spin the plate at 300 × g for 5 min and discard supernatant.
-
84.
Wash cells with 200 μL of FACS buffer, spin the plate at 300 × g for 5 min and discard supernatant.
-
85.
Add 100 μL of 2% PFA mixture and incubate at 4°C for 30 min in the dark.
-
86.
Spin the plate at 300 × g for 5 min and discard supernatant.
-
87.
Wash cells with 200 μL of FACS buffer, spin the plate at 300 × g for 5 min and discard supernatant.
-
88.
Resuspend cells to 200 μL of FACS buffer.
Note: Prepare single color compensation controls using compensation beads prior to sample acquisition by flow cytometry.
-
89.
Transfer cells to appropriate tubes for flow cytometry and perform sample acquisition.
Note: Ensure that HER2 CAR-NK cells have a minimum of 15% CAR transgene expression before functional assays (Troubleshooting 5).
Flow cytometry-based HER2 CAR-NK cell cytotoxicity assay
This step describes how to prepare target cells for the flow cytometry-based cytotoxicity assay and how to plate the effector and target cells. We recommend using 100,000–200,000 target cells but this number can be reduced to 50,000 cells. We recommend to include at least three Effector:Target (E:T) ratios in the cytotoxicity assay to obtain a killing curve.
-
90.
Label a 96-well plate layout with all of your conditions, follow Figure 4 as an example.
Note: Due to the high susceptibility of SKBR3 breast cancer cells to expanded NK cell killing we chose the 1:1, 0.5:1 and 0.25:1 E:T ratios to assess in vitro anti-tumor function. The following protocol uses 100,000 target cells per well.
-
91.Prepare effector NK cells for plating.
-
a.Transfer cells to a 15 mL falcon tube and centrifuge at 300 × g for 5 min.
-
b.Carefully remove supernatant and count cells with Trypan Blue.
-
c.Resuspend NK cells with complete RPMI media to a final concentration needed to add 100 μL of effector cells at the highest E:T ratio.
-
a.
Figure 4.

Example of a cytotoxicity assay plate layout comparing the in vitro anti-tumor function of non-transduced, control vector-, and HER2 CAR-transduced NK cells.
Example: If the highest E:T ratio is a 1:1, resuspend the effector NK cells to a final concentration of 1 × 106 cells/mL in order to add 100,000 NK cells in 100 μL.
-
92.
Set effector NK cells aside while you prepare the target cells.
Optional: One day before the cytotoxicity assay, split SKBR3 target cells 1:2 to ensure high tumor cell viability on the day of the assay.
-
93.
Harvest SKBR3 target cells by washing with 10 mL warm PBS and adding 5 mL of 1× Trypsin-EDTA (0.05%).
-
94.
Neutralize trypsin by adding 3 times the volume of complete DMEM media.
-
95.
Transfer cells to a 50 mL falcon tube and centrifuge at 300 × g for 5 min.
-
96.
Discard supernatant and wash cells with 20 mL of PBS.
-
97.
Centrifuge at 300 × g for 5 min.
-
98.Discard supernatant and count cells with Trypan Blue.
-
a.Resuspend cells with a low volume for counting since they will need to be highly concentrated for CFSE-labeling.
-
a.
Alternatives: Other fluorescent cell labelling reagents can be used such as Cell Trace Violet (Thermo Fisher).
-
99.
Resuspend total amount of SKBR3 cells needed for the cytotoxicity assay at a concentration of 10 × 106 cells/mL in PBS in a 50 mL falcon tube.
-
100.Prepare 5 μM CFSE solution.
-
a.Prepare a series of three 15 mL falcon tubes labeled A, B, C. Add 990 μL DMSO to Tube A; Add 2.70 mL PBS to Tube B; Add 9 mL of PBS to Tube C.
-
b.Add 10 μL of CFSE (50 mM) stock into Tube A and vortex.
-
c.Take 300 μL from Tube A and add it to Tube B. Vortex.
-
d.Take 1 mL from Tube B and add it to Tube C. Vortex.
-
a.
Note: Working concentration of CFSE can be prepared earlier but should be stored at 4°C protected from light before use.
-
101.
Add an equal volume of CFSE mixture from Tube C to the resuspended target cells.
Example: If a total of 10 million target cells are resuspended in 1 mL of PBS, add 1 mL of CFSE mixture from Tube C.
-
102.
Immediately vortex at a low-med speed, making sure cells are properly mixed.
-
103.
Incubate the CFSE-labeled cells at 37°C for 15 min.
Note: Protect the CFSE-labeled cells from light using tinfoil.
-
104.
After incubation, quench the unbound CFSE by topping up the 50 mL falcon tube with 10% FBS-PBS for 10 min at 20°C–22°C in the dark.
-
105.
Centrifuge at 300 × g for 5 min.
Note: The CFSE-labeled cell pellet can look slightly green at this point.
-
106.
Wash cells with 20 mL of PBS and centrifuge at 300 × g for 5 min.
-
107.
Discard supernatant and resuspend cells in a low amount of complete RMPI media.
-
108.Count cells using Trypan Blue and resuspend to 1 × 106 cells/mL with complete RPMI media.
-
a.The target cells are now ready to be plated.
-
a.
-
109.Plate the effector NK cells according to each E:T ratio and top up wells to a total volume of 100 μL with complete RPMI media.
-
a.1:1 E:T; Add 100 μL of NK cells.
-
b.0.5:1 E:T; Add 50 μL of NK cells and 50 μL of complete RPMI media.
-
c.0.25:1 E:T; Add 25 μL of NK cells and 75 μL of complete RPMI media.
-
a.
-
110.
Prepare the tumor only control wells by adding 100 μL of complete RPMI media.
-
111.Use a multichannel pipette to add 100 μL of CFSE-labeled target cells to wells containing the effector NK cells and the tumor only wells.
-
a.Plate one extra well with the target cells to use as the CFSE-only control well to adjust voltages during sample acquisition.
-
a.
-
112.
Incubate cytotoxicity assay 96-well plate at 37°C and 5% CO2 for 5 h.
-
113.
Spin plate at 300 × g for 5 min and discard supernatant.
Optional: Carefully collect ∼150 μL of the cell-free supernatants using a multichannel pipette and transfer to a new 96-well U or V bottom plate. Place plate in −20°C freezer for cytokine analysis.
-
114.Wash wells using 200 μL of PBS, spin plate at 300 × g for 5 min and discard supernatant.
-
a.Prepare a 1:1000 dilution of viability dye using eBioscience Fixable Viability Dye eFluor 780 in PBS.
-
a.
-
115.
Add 100 μL of fixable viability dye and incubate at 4°C for 30 min in the dark.
-
116.
Spin plate at 300 × g for 5 min and discard supernatant.
-
117.
Wash wells with 200 μL of FACS buffer, spin plate at 300 × g for 5 min and discard supernatant.
-
118.
Add 100 μL of 1% PFA mixture and incubate at 4°C for 1 h in the dark.
-
119.
Spin plate at 300 × g for 5 min and discard supernatant.
-
120.
Wash wells with 200 μL of FACS buffer, spin plate at 300 × g for 5 min and discard supernatant.
-
121.
Resuspend plate with 200 μL of FACS buffer.
-
122.
Transfer samples to appropriate tubes for flow cytometry and perform sample acquisition.
Expected outcomes
The expected purity of the expanded CD3-CD56superbright NK cells is demonstrated in Figure 5.
Figure 5.

Purity of CD3-CD56superbright expanded NK cells after 3 week expansion
The CD3-negative and CD56-positive gates were set using the corresponding FMOs. Samples were acquired using the LSRFortessa Flow Cytometer and analyzed with FlowJo Software.
The expected titer for the lentivirus containing the HER2 CAR construct in this study is between 3 × 107 and 2 × 108 TU/mL, use Figure 6 as a guide to set the transgene-positive gates after the lentiviral titration.
Figure 6.

Example flow plots of transgene expression on HEK293T cells 72h after lentiviral titration
To determine the lentivirus titer use the formula: Titer (TU/mL)= ((30,000 × % Transgene-positive cells X Dilution factor)/100). Set the transgene-positive gate on the sample that was stained but did not obtain any lentivirus and use the dilution factor which results in >10% transgene-positive cells. Samples were acquired using the LSRFortessa Flow Cytometer and analyzed with FlowJo Software.
The transduction efficiency of the CAR construct can vary between donors, generally we obtain at least 15% transduction efficiency, see Figure 7 for an example.
Figure 7.

Example flow plots of transgene expression on HER2 CAR- and control vector-transduced expanded NK cells
(A) Percent transgene expression in the non transduced, HER2 CAR-, and control vector transduced NK cells.
(B) Extent of transgene expression in the transduced NK cells compared to the non transduced control. Samples were acquired using the LSRFortessa Flow Cytometer and analyzed with FlowJo Software.
To determine the percent-specific lysis of the control and HER2 CAR-expressing effector NK cells first determine the percentage of dead target cells using the gating strategy defined in Figure 8. Calculate percent-specific lysis using the following formula:
| % specific lysis = ((% tumor cell death – % basal tumor cell death)/(100 – % basal tumor cell death)) × 100 |
Figure 8.

Example flow plots of in vitro cytotoxicity assay
Singlets are discriminated (FSC-H v. FSC-A) and CFSE-positive tumor cells are gated. Create a dead cell gate (APC-Cy7 positive) based on the samples that only contain tumor cells and no effector cells (basal). Apply dead cell gate to the non-transduced, control vector, and HER2 CAR-transduced expanded NK cell samples. Samples were acquired using the LSRFortessa Flow Cytometer and analyzed with FlowJo Software.
Limitations
The following protocol can be used to generate human CAR-NK cells with transduction efficiencies ranging between 15 and 80%. However, a main limitation is that transgene expression is downregulated over time thus functional assays should be performed within one to 2 weeks of CAR-NK cell generation. Protocols that use retrovirus instead of lentivirus or include sequences that prevent promoter silencing in their CAR plasmid may yield CAR-NK cells with stable transgene expression (Colamartino et al., 2019; Liu et al., 2020). Additionally, other promoters such as SFFV may lead to greater transduction efficiencies however we only used the EF1alpha and CMV promoters in our studies.
Another limitation is that the transduction protocol has been optimized for use with human NK cells that have been expanded using genetically engineered K562 feeder cells. This protocol may require further optimization for the transduction of cytokine activated NK cells which are typically more difficult to transduce. However, we have also used the protocol to successfully generate CAR-NK cells from the NK-92 human NK cell line as described in Portillo et al. (2021).
Troubleshooting
Problem 1
Low viability and slow expansion of NK cells from starting PBMCs (Before you begin steps 14–19).
Potential solution
Using fresh PBMCs as opposed to cryopreserved PBMCs to expand the NK cells usually yields a higher viability and expansion rate. In addition, changing the media and adding IL-2 every 2 days is crucial to maintain a good viability of the rapidly proliferating NK cells in culture. Do not use IL-2 aliquots that have been thawed and stored at 4°C for longer than 2–3 days.
Problem 2
A non-specific CD3+ T cell population has expanded in the expanded NK cell culture (Before you begin step 19).
Potential solution
Perform a CD3+ positive selection using commercial isolation kits to remove the contaminating CD3+ T cells and continue replenishing the culture. Additionally, CD3+ cells can also first be depleted from the starting PBMCs using an immunomagnetic CD3+ isolation kit. If the CD3+ T cell population continues to expand, use PBMCs from a different donor.
Problem 3
Low lentiviral yield after ultracentrifugation (step 25).
Potential solution
The transfer vector containing your desired transgene might be too large to efficiently generate 3rd generation lentivirus using this protocol. We find that lentiviral titer and LV production efficiency decreases as the size of the transgene packaged in the LV increases. In this case, the 2nd generation lentivirus packaging system could be used since all the packaging genes are encoded within only one vector as opposed to two like in the 3rd generation system. Using the 2nd generation system has shown to increased viral yields 50-fold (Cribbs et al., 2013). However, 3rd generation LVs are preferred for clinical applications due to the added biosafety aspect.
Problem 4
Low lentiviral titer (<1 × 107 TU/mL) is obtained after lentiviral production (Titer will be determined after step 51).
Potential solution
If the LV titer is lower than 1 × 107 TU/mL after ultracentrifugation there was an issue during LV production. Ensure that HEK293T cells have a viability between 90 and 95% at every plating step. We recommend that HEK293T cells are no more than 80% confluent during the transfection step to prevent cells lifting off the plate during the media change the next day as this can highly impact viral titer. It is also critical that your starting plasmids have high purity for successful HEK293T transfection. Using commercially available plasmid isolation kits such as the PureLink™ HiPure Plasmid Maxiprep Kit (Thermo Fisher) is recommended to obtain high purity. You may also need to optimize the amount of plasmids used for the transfection.
Problem 5
A low transduction efficiency (<15%) is obtained after NK cell transduction (Transduction efficiency will be determined after step 89).
Potential solution
In our experience we find that lentivirus preps with a titer >3 × 107 TU/mL are necessary to yield CAR-NK cells with high transgene expression. In addition, we have found that using expanded NK cell cultures that have not been expanded beyond 8 weeks typically yield higher transduction efficiencies. The MOIs needed to transduce NK cells may also need to be optimized. The CAR lentiviral construct used in this study is ∼10 Kb, and we find MOIs 3–8 are sufficient to yield >15% transduction efficiencies. However, higher MOIs may be needed for larger lentiviral constructs. We also find that transgene efficiency can also be donor dependent.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Ali A. Ashkar (ashkara@mcmaster.ca).
Materials availability
The study did not generate new unique reagents.
Acknowledgments
This work was funded by a Canadian Institute of Health Research (CIHR) grant (20009360) awarded to A.A.A. A.A.A is a holder of a Tier 1 Canada Research Chair in Natural Immunity and NK Cell Function. The graphical abstract and all other figures were created using bio-render.com.
Author contributions
A.L.P wrote the original draft. A.L.P and R.H contributed to the methodology. A.L.P prepared the figures. A.L.P and A.A.A revised the draft. A.A.A supervised the work and acquired funding.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Ana L. Portillo, Email: portilal@mcmaster.ca.
Ali A. Ashkar, Email: ashkara@mcmaster.ca.
Data and code availability
No data or code was generated or analyzed in this protocol.
References
- Brown L.Y., Dong W., Kantor B. An improved protocol for the production of lentiviral vectors. STAR Protoc. 2020;1:100152. doi: 10.1016/j.xpro.2020.100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colamartino A.B.L., Lemieux W., Bifsha P., Nicoletti S., Chakravarti N., Sanz J., Roméro H., Selleri S., Béland K., Guiot M., et al. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front. Immunol. 2019;10:2873. doi: 10.3389/fimmu.2019.02873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs A.P., Kennedy A., Gregory B., Brennan F.M. Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnol. 2013;131:1–8. doi: 10.1186/1472-6750-13-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman C.J., Senyukov V.V., Somanchi S.S., Phatarpekar P.V., Kopp L.M., Johnson J.L., Singh H., Hurton L., Maiti S.N., Huls M.H., et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7:e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A., Naldini L. In: Gene Therapy Protocols. Morgan J.R., editor. Humana Press; 2002. HIV-based vectors; pp. 259–274. [Google Scholar]
- Hammill J.A., VanSeggelen H., Helsen C.W., Denisova G.F., Evelegh C., Tantalo D.G.M., Bassett J.D., Bramson J.L. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J. Immunother. Cancer. 2015;3:1–11. doi: 10.1186/s40425-015-0099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammill J.A., Afsahi A., Bramson J.L., Helsen C.W. In: Methods in Molecular Biology. Ursini-Siegel, editor. Humana Press Inc.; 2016. Viral engineering of chimeric antigen receptor expression on murine and human T lymphocytes; pp. 137–157. [DOI] [PubMed] [Google Scholar]
- Liu E., Marin D., Banerjee P., MacApinlac H.A., Thompson P., Basar R., Kerbauy L.N., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J.C., Sechelski J. Use of Sodium Butyrate to Enhance Production of Retroviral Vectors Expressing CFTR cDNA. Hum. Gene Ther. 1995;6:1195–1202. doi: 10.1089/hum.1995.6.9-1195. [DOI] [PubMed] [Google Scholar]
- Portillo A.L., Hogg R., Poznanski S.M., Rojas E.A., Cashell N.J., Hammill J.A., Chew M.V., Shenouda M.M., Ritchie T.M., Cao Q.T., et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. IScience. 2021;24:102619. doi: 10.1016/j.isci.2021.102619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H., Figliola M., Dawson M., Huls H., Olivares S., Switzer K., Mi T., Maiti S., Kebriaei P., Lee D., et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71:3516–3527. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data or code was generated or analyzed in this protocol.