

Abstract
Immune responses to adeno-associated virus (AAV) capsids limit the therapeutic potential of AAV gene therapy. Herein, we model clinical immune responses by generating AAV capsid-specific chimeric antigen receptor (AAV-CAR) T cells. We then modulate immune responses to AAV capsid with AAV-CAR regulatory T cells (Tregs). AAV-CAR Tregs in vitro display phenotypical Treg surface marker expression, and functional suppression of effector T cell proliferation and cytotoxicity. In mouse models, AAV-CAR Tregs mediated continued transgene expression from an immunogenic capsid, despite antibody responses, produced immunosuppressive cytokines, and decreased tissue inflammation. AAV-CAR Tregs are also able to bystander suppress immune responses to immunogenic transgenes similarly mediating continued transgene expression, producing immunosuppressive cytokines, and reducing tissue infiltration. Taken together, AAV-CAR T cells and AAV-CAR Tregs are directed and powerful immunosuppressive tools to model and modulate immune responses to AAV capsids and transgenes in the local environment.
Keywords: CAR Tregs, AAV gene therapy, CAR T regulatory cells, chimeric antigen receptor T-regulatory cells, immunosuppression, immune responses to AAV, immune responses to capsid, immune responses to transgene
Graphical abstract
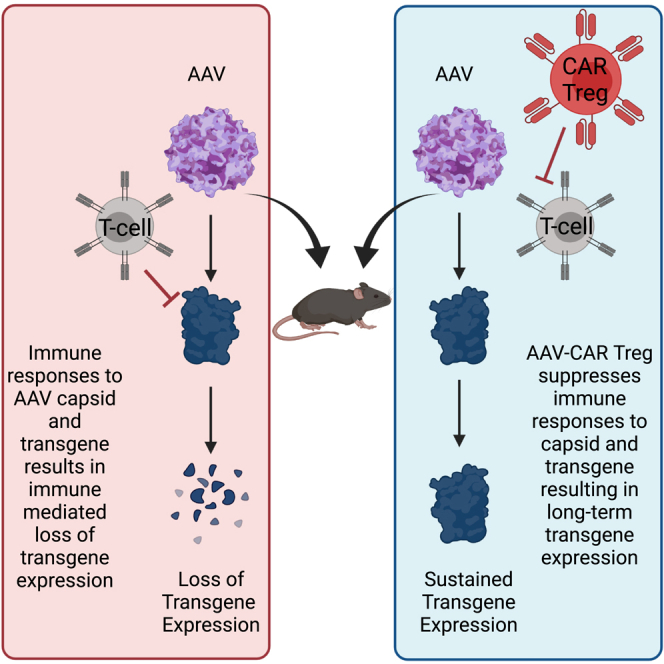
We herein utilized AAV-CAR Tregs to create tolerogenic environments suppressing both capsid and AAV-delivered transgene responses allowing for continued transgene expression from transduced cells. This induced local immune tolerance may ultimately be safer clinically than systemic, broad-spectrum immune suppression.
Introduction
Recombinant adeno-associated virus (AAV) vectors are effective, powerful, and a leading platform for gene delivery. However, significant obstacles to the therapeutic use of AAVs remain, from practical manufacturing limitations to immunological barriers. Capsid-specific T cell responses to AAV-based therapies were not predicted from proof-of-concept studies in animal models but were identified in human studies.1, 2, 3 An early clinical trial for the treatment of hemophilia, via i.v. delivery of recombinant AAV (rAAV), resulted in robust CD8 T cell responses and the loss of transgene expression, which was later shown to be mediated by capsid-specific T cell responses.1, 2, 3, 4, 5 This led to the requirement for immunosuppression to maintain transgene expression in patients treated with AAVs, further complicating the therapeutic impact.6 Despite the observation of immune responses after intravenous (i.v.) administration of AAV, concurrent clinical trials using intramuscular (i.m.) delivery of AAV revealed long-term transgene expression without immune suppression, regardless of significant immune cell infiltration.7, 8, 9, 10 This long-term expression was attributed to the induction of regulatory T cells (Tregs) in the muscles of AAV-injected patients. This infiltration and induction of Tregs suggested an immune mechanism at work in i.m. trials that were not observed in i.v. trials.11,12
Tregs are a subset of immune T cells that express CD4, CD25, and the master transcription factor, forkhead box P3 (FOXP3), although further subsets have been discovered with different properties, such as CD8+ Tregs,13 or type 1 Tregs (Tr1).14 FOXP3 expression was found to be crucial for Treg suppressive activity,15 and studies have shown that transfection of T cells with FoxP3 induces regulatory activity.16,17 Tregs can be classified as natural Tregs, which develop in the thymus, or induced Tregs, which are generated in the periphery.18 Tregs suppress immune responses through secretion of immunosuppressive cytokines, metabolic disruption, modulation of co-stimulation, and direct interaction with effector T cells and other immune cells.19,20 Additionally, they can broadly suppress local environments through bystander suppression as well as induce other suppressive cells by infectious tolerance.21, 22, 23 Thus, Tregs are powerful immunosuppressive cells, and understanding the potential role of infiltrating T cells in liver-directed or muscle-directed AAV gene therapy may be critical for future therapeutic development.
Chimeric antigen receptor (CAR) T therapy has revolutionized immunotherapy with demonstrated clinical efficacy achieved in B cell malignancies.24,25 CAR T cells utilize an extracellular antigen recognition domain from a single chain variable fragment (scFv) of an antibody combined with an intracellular signaling domain. Combining the external scFv with the internal co-stimulatory domains from T cells allows the construct to activate a T cell response in a non-MHC-restricted manner.26 Beyond therapies for cancer, CAR T cells have been designed to target human immunodeficiency virus (HIV), Epstein-Barr virus (EBV), and other viruses.27, 28, 29 Here we generate a CAR against AAV capsid (AAV-CAR T cell) to mimic capsid-specific T cell responses observed in clinical trials.
As the CAR field has grown, it has evolved from mimicking effector T cell responses to creating immunosuppressive CAR T cells by the creation of CAR Tregs. To direct Tregs to a specific antigen, preclinical studies have examined the utility of CAR Tregs for colitis,30, 31, 32 graft-versus-host disease,33,34 hemophilia, and multiple sclerosis.17,35,36 To create these CAR Tregs, most have focused on isolated primary Tregs and transducing them with the CAR construct, except for two studies.17,37 However, the low population of primary Tregs in peripheral blood38 and loss of FOXP3 expression in endogenous Tregs makes this approach intractable.15 Antigen-specific CAR Tregs have great clinical promise for creating specific, directed, and local immunosuppression.
Herein, we generated AAV-CAR Tregs by addition of FoxP3 cDNA downstream of the CAR construct. We show AAV-CAR Tregs suppress capsid-specific immune responses despite anti-capsid antibody responses in vivo. Moreover, AAV-CAR Tregs mitigate immune responses to vector-expressed transgenes by bystander suppression. Altogether, these data suggest that AAV-CAR T cells and AAV-CAR Tregs can be used not only to study anti-vector immune responses but also as therapeutics for localized modulation of the immune responses to AAV capsids and vector-expressed transgenes to enhance the safety and efficacy of clinical applications.
Results
Generation of AAV-CAR T cells and AAV-CAR Tregs
The AAV-CAR construct was developed by cloning an AAV-specific scFv into a third-generation CAR construct (Figure 1A). The AAV-specific monoclonal antibody, D3, has been previously described by the Kleinschmidt group and demonstrated to bind a broad spectrum of AAV capsid variants, including AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV8, and AAV9 and recognizes non-assembled capsid protein as well.39,40 Additionally, they showed that the D3 antibody has no neutralizing ability and recognizes conformational epitopes, including amino acids SRNWLPGPCY, which are highly exposed on the surface of VP3. Recently the Chapman group have studied the structure of the D3 binding site showing the overlap between the D3 antibody epitope and the AAVR binding site.41
Figure 1.

Generation of AAV-CAR Tregs
(A) Schematic representation of transgene construct for AAV-CAR T cell. The AAV-specific scFv is fused to a human IgG1 CH2-CH3 hinge, followed by intracellular CD28 (human) and CD137 (4-1BB) (human) co-stimulatory domains, and CD3ζ (human), under the control of the EF1-alpha promoter. Truncated CD19 (external domain only) were added following the E2A self-cleavage peptide sequences. (B) Schematic representation of the AAV-CAR Treg construct. Murine FoxP3 cDNA, and truncated CD19 (external domain only) were added following the E2A self-cleavage peptide sequences. (C) Schematic diagram of T cell isolation and lentiviral transduction. Pan T cells are isolated from human blood or mouse spleen and activated for 2 days followed by lentiviral-CAR transduction; mixed population of transduced and non-transduced T cells are used in all experiments.
To generate the AAV-CAR Treg construct, Foxp3 cDNA was cloned downstream of the CAR coding sequence followed by an E2A motif providing stable expression of -FOXP3, which is essential for Treg function (Figure 1B). To create AAV-CAR T cells and AAV-CAR Tregs, we transduced CD3+ T cells with lentiviral-CAR vectors or lentiviral-CAR-Foxp3 vectors (Figures 1A–1C). In the latter, we converted CD3+ T cells to the Treg phenotype rather than transducing Tregs with a CAR construct. We utilized CD3+ T cells to maximize cellular input to include CD8+ Tregs as they are a naturally occurring, but rare population that have been shown to be quite immunosuppressive.42 For detection, both CAR constructs included the expression cassette for CD19 truncated protein, containing the extracellular portion but lacking internal signaling domains.
Immunophenotyping of AAV-CAR Tregs
To further examine the induced Treg phenotype in CD3+ cells by expression of FoxP3 from the CAR construct, we characterized transduced cells for relevant Treg markers. Using flow cytometry, we selected cells that expressed FOXP3 and were double positive for CD19, and the T cell markers CD4 or CD8 (Figure S1A). Similar to endogenous Tregs, a majority of the AAV-CAR Tregs were found to be positive for CD25 and cytotoxic T lymphocyte-associated antigen 4 (CTLA4). CD25 signaling is known to regulate the function and stability of Tregs.43 Additionally, Treg expression of CTLA4 is known to disrupt T effector cell function.44 Also, a majority of AAV-CAR Tregs were expressing latency-associated peptide (LAP), with few expressing LAP and glycoprotein A repetitions predominant (GARP), suggesting AAV-CAR Tregs potentially produce immunosuppressive cytokines, interleukin (IL)-10, and transforming growth factor (TGF)-β1, rather than proinflammatory cytokines, such as interferon (IFN)-γ.45 AAV-CAR Tregs were positive for glucocorticoid-induced tumor necrosis factor receptor related (GITR), which inhibits T effector function.46 Consistent with induced Treg phenotype, AAV-CAR Tregs were negative for neuropilin, which distinguishes natural from induced Tregs in vivo.47 Therefore, AAV-CAR Tregs express similar phenotypic markers to endogenous Tregs (Figure 2A).
Figure 2.

AAV-CAR Tregs express Treg phenotype and suppress effector cell proliferation
(A) Flow cytometric analysis of AAV-CAR Tregs. AAV-CAR Treg gating ancestry (Figure S1). AAV-CAR Tregs selected for CD4+and/or CD8+, CD19+, FOXP3-positive cells. Panels show representative fluorescence-activated cell sorting (FACS) profiles from three independent experiments generating AAV-CAR Tregs from three different heathy human donors. (B, C, and D) T cell suppression assay. AAV-CAR T cells were labeled with CellTrace Violet, cocultured with or without AAV-CAR Tregs, and either stimulated with anti-CD3/CD28/CD2 or AAV1-infected HEK293 cells. Flow cytometry was run after 3 and 5 days. (B) Representative FACS profiles. (C) Quantification of MFI of labeled AAV-CAR T cells. (D) Percentage suppression of AAV-CAR T cell proliferation by AAV-CAR Tregs. Data are quantified from three independent experiments using human samples from three healthy donors on the right. Error bars are mean ± SEM; ∗p ≤ 0.05 by paired Student's t test.
AAV-CAR T cells and AAV-CAR Tregs functionality in vitro
A prominent mechanism of action of Tregs is suppression of effector T cell proliferation. Therefore, we tested whether AAV-CAR Tregs could suppress proliferation of AAV-CAR T cells after antigen-specific (AAV1-transduced HEK293 cells) or nonspecific (anti-CD3/CD28/CD2 activator) stimulation. AAV-CAR T cells were labeled with proliferation dye and cocultured with or without stimulated AAV-CAR Tregs. AAV-CAR Tregs suppressed proliferation of AAV-CAR T cells after 3 and 5 days, regardless of stimulation (Figure 2B). Quantification of mean fluorescence intensity (MFI) of labeled AAV-CAR T cells increased when cocultured with AAV-CAR Tregs and nonspecific stimulation, indicating a suppression of fluorescent dye dilution. Similarly, when T cells were cocultured with AAV1-transduced HEK293 cells, the MFI of labeled AAV-CAR T cells increased when cocultured with AAV-CAR Tregs. At both stimulation conditions, this increase was significant at day 5 (p = 0.0318 and 0.0446 for anti-CD3/CD28/CD2 T cell activator and AAV1-HEK293 cells at day 5 respectively) (Figure 2C). Cell counts and viability are shown in Table S1. Further, we quantified the percentage suppression of AAV-CAR T cells by AAV-CAR Tregs. At day 3, AAV-CAR Tregs showed ∼50% suppression of AAV-CAR cells with both AAV1-HEK and anti-CD3/CD28 conditions. The percentage suppression increased at day 5 to ∼80% for both groups (Figure 2D). Together, these data indicate the suppressive activity of AAV-CAR Tregs in response to AAV-transduced cells.
To test the AAV-CAR T cell cytotoxicity and AAV-CAR Treg suppressive activity, as well as broad specificity of capsid recognition, we utilized the previously characterized luciferase-based killing assay (Figures 3A and S2).48,49 Target cells expressing luciferase were transduced with either AAV1, AAV2, AAV3b, AAV5, AAV6, AAV8, AAV9, or rh32.33. Non-transduced cells acted as nonspecific cytotoxicity controls. AAV-CAR T cells were significantly cytotoxic against AAV-transduced target cells compared with non-transduced cells for AAV1, AAV3b, AAV6, AAV8, AAV9, and rh32.33, and trended lower in AAV2 and AAV5. Viability of target cells was reduced in all capsid variants, from ∼17% to 50% depending on the variant, compared with non-transduced cells (Figure 3B). Thus, AAV-CAR T cells can recognize cells transduced with any one of many different AAV capsid variants distributed across several AAV clades. However, when AAV-CAR Tregs were cocultured with the AAV-CAR T cells in the presence of AAV-transduced target cells, the viability of target cells was similar to the cells not transduced with AAV. This indicates the ability of AAV-CAR Tregs to block the cytotoxicity of AAV-CAR T cells. The viability of target cells was significantly increased against all AAV capsid variants ranging from 25% to 40%, compared with culture without AAV-CAR Tregs (Figure 3B), except for AAV2 (NS, ∼14%). There was no impact on the viability of non-transduced target cells when cultured with AAV-CAR T cells or cocultured with AAV-CAR Tregs. Taken together, these results suggest the antigen specificity of the AAV-CAR T cells and AAV-CAR Tregs, but also that the AAV-CAR Tregs can suppress T cell-specific killing. Similar to AAV-CAR T cells, AAV-CAR Tregs recognize numerous AAV capsid variants, suggesting the utility of these immunosuppressive cells. Further, to determine if AAV-CAR Treg suppression is dose dependent, we transduced luciferase-expressing target cells with AAV, followed by coculturing different ratios of AAV-CAR Tregs to AAV-CAR T cells with the transduced target cells. Our data show that AAV-CAR Tregs with 1:1 ratio to AAV-CAR T cells have greater suppressive activity compared with 1:2 and 1:4 ratios, indicating that the suppressive activity of AAV-CAR Treg is dose dependent (Figure S2).
Figure 3.

AAV-CAR Tregs suppress effector cells cytotoxicity against numerous AAV capsid variants
(A) Schematic of a luciferase cytotoxicity assay and suppression of cytotoxicity assay. Luciferase-expressing HEK293 cells (target cells) are either transduced with AAV or non-transduced as a control. Target cells are either cultured with AAV-CAR T cells alone or cocultured with AAV-CAR Tregs. Viability of cells was measured by luminescence. (B) Cytotoxicity of AAV-CAR T cells (blue bars, left) and suppression of cytotoxicity by AAV-CAR Tregs (red bars, left) against AAV1, AAV2, AAV3b, AAV5, AAV6, AAV8, AAV9, rh32.33, or untransduced luciferase-expressing HEK cells as read by cell viability (blue and red bars, right). Percentage viability is determined by luciferase expression measured 24 h after coculture. Cells were cultured at a ratio of 1:10 target cell to AAV-CAR T cell or 1:10:10 of target cell to AAV-CAR T cell to AAV-CAR Treg. Data are the average of three independent experiments using human samples from three healthy donors (within each experiment samples were run in triplicate). Error bars are mean ± SEM; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 by two-way ANOVA with Tukey's multiple comparisons.
Homing of labeled AAV-CAR Tregs to AAV-transduced tissue in vivo
First, to determine whether the AAV-CAR Treg could recognize its epitope in vivo, we stained AAV-transduced tissues with the D3 antibody, from which the CAR Treg was generated, to determine how long the epitope is available for recognition by the antibody. Mice were injected i.m. with AAV8, and muscles were harvested 3 months later and stained with the D3 antibody. We confirmed that, even at 3 months post injection, the D3 elicited positive staining, suggesting that epitopes are available for AAV-CAR Treg recognition at 3 months post injection (Figure S3).
To determine whether AAV-CAR Tregs could traffic to AAV-transduced tissue in vivo, we injected NU/J mice in the right muscle with AAV1 expressing luciferase. One week post AAV1 injection, we i.v. delivered labeled AAV-CAR Tregs or phosphate-buffered saline (PBS). Luminescent imaging revealed localized expression of luciferase in the injected muscles (Figure 4A, left). The luciferase signal was only localized to the injected muscle and was not observed elsewhere or in control animals that did not receive AAV-luciferase vector (Figure 4B). Moreover, labeled AAV-CAR Tregs were observed in the injected muscles of the animals that received AAV1 luciferase (Figure 4A, right). The DiR signal was also significantly higher in the injected muscle compared with the contralateral uninjected muscle or the animals that did not receive AAV1 luciferase (Figure 4C). These data confirm that AAV-CAR Tregs can home to the tissues where antigen is present, specifically to the AAV-injected muscle.
Figure 4.

Characterization of AAV-CAR Tregs homing and AAV-CAR T cells clearance in vivo
(A) Representative images of full-body bioluminescence of AAV1 expressing luciferase (left) and DiR fluorescence of labeled AAV-CAR Tregs (right). (B) Bioluminescence measurement of luciferase in right and left muscles. (C) Fluorescence measurement of DiR in right and left muscles. ∗p ≤ 0.05. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 by two-way ANOVA with Tukey's multiple comparisons. (D) AAT ELISA of animal serum. (E) Anti-capsid ELISA of animal serum. Animals treated with PBS (blue). Animals treated with AAV-CAR T cells (red). Two-way ANOVA repeated measure with Tukey's multiple comparisons was used (n = 6). Error bars are mean ± SEM.
AAV-CAR T cells mimic capsid-specific T cell clearance of transduced cells in vivo
Subsequently, we wanted to test the functionality of the AAV-CAR T cells to clear transgene expression in vivo. To mimic capsid-specific T cell responses observed in clinical trials, we injected C57BL/6 mice via i.m. injection with AAV1 expressing human alpha-1- antitrypsin (hAAT), and, 3 weeks later, we i.v. delivered either AAV-CAR T cells or PBS. Transgene expression was measured in serum over time using a hAAT enzyme-linked immunosorbent assay (ELISA), with both groups having similar alpha-1-antitrypsin (AAT) expression at 3 weeks prior to AAV-CAR T cell delivery. Stable transgene expression was observed in animals treated with PBS, 6 weeks after AAV injection. However, in animals treated with AAV-CAR T cells, transgene expression steadily dropped over 6 weeks (Figure 4D). Anti-AAV1 antibodies increased over time in both groups, indicating anti-capsid antibodies are not responsible for the loss of transgene expression in the AAV-CAR T cell-treated group (Figure 4E). In both groups, anti-AAT levels were below the lower limit of detection as measured by ELISA. At 6 weeks post injection, muscles were collected and immunostained for AAT protein expression. Robust AAT staining was observed in the injected muscle of animals treated with PBS, whereas, in the animals treated with AAV-CAR T cells, reduced staining for AAT was observed, further indicating the ability of AAV-CAR T cells to clear AAT expression (Figure S4). These data suggest that AAV-CAR T cells can clear transduced cells and reduce transgene expression, similar to T cell responses observed in clinical trials.
AAV-CAR Tregs suppress immune responses against AAV capsid in vivo
To test if AAV-CAR Tregs can suppress capsid-specific T cell responses to AAV gene therapy in vivo, we utilized rh32.33 capsid, which has been shown to be highly immunogenic. Although most AAV capsids do not elicit a T cell response in mice, CD8 T cell responses have been observed against the rh32.33 capsid.50 Only in female animals and with i.m. injection did we observe effective clearance of transgene expression (Figures S5A and S5B), hence female C57BL/6 mice were injected with rh32.33-AAT i.m. to induce an immune response against AAV capsid. One week later, we systemically delivered AAV-CAR Tregs or PBS. We added an additional group of polyclonal Tregs, which were isolated and expanded mouse CD4+CD25+ Tregs. In animals treated with PBS control, AAT is initially expressed but then dropped to below the limit of detection after week 7, remaining low for 26 weeks. In both polyclonal Treg and AAV-CAR Treg groups, there was significant and sustained therapeutic transgene expression up to 26 weeks post AAV injection, with greater expression in the AAV-CAR Treg group between weeks 3 and 9 post AAV injection (Figure 5A). Anti-AAT levels were measured by ELISA but in all groups were under the level of detection. Interestingly, anti-capsid antibodies were produced in all three groups. The levels of anti-rh32.33 antibodies were 125 times greater than a less immunogenic capsid AAV1, suggesting greater immunogenicity of rh32.33 over other AAV capsid variants (Figures 5B and 4E). However, despite high levels of anti-rh32.33 antibodies in all groups, loss of transgene expression is only observed in the PBS group, further illuminating the important role of capsid-specific T cell responses in the clearance of transgene expression.
Figure 5.

AAV-CAR Tregs suppress capsid-specific immune response in vivo
(A) Expression of hAAT protein in the serum of animals injected i.m. with rh32.33-AAT followed by AAV-CAR Tregs (red), polyclonal Tregs (orange), or PBS (blue) as measured by ELISA (n = 6 for AAV-CAR Treg and PBS-treated groups, n = 5 for polyclonal Treg-treated group). (B) Anti-rh32.33 antibodies in the serum measured by ELISA. Anti-capsid antibodies increased in all groups over time (n = 6 for AAV-CAR Treg group, n = 7 for PBS group, n = 5 for polyclonal Treg-treated group). (C) Serum level of free active TGF-β1 measured by CBA assay over time. A significant increase in free active TGF-β1 was observed in AAV-CAR Treg group (red) at week 11 post injection compared with the PBS control group (blue). (D) Serum levels of IL-10 measured by CBA assay over time. Two-way ANOVA repeated measure with Tukey's multiple comparisons was used (n = 6 for AAV-CAR Treg and PBS-treated groups, n = 5 for polyclonal Treg treated group for C and D). (E) Quantification of number nuclei in right limb muscles of animals 2 weeks after AAV injection. Each dot represents one animal, which is the average of 10 images per animal (HPF, high-power field). One-way ANOVA, n = 3. Error bars are mean ± SEM; ∗p ≤ 0.05, ∗∗p ≤ 0.01. (F) Representative images of rh32.33-AAT muscles 2 weeks post injection stained for H&E for cellular infiltration. (Upper) scale bar, 527 μm; (lower) scale bar, 131 μm. (G) Representative images of rh32.22-AAT-injected muscles stained by immunohistochemistry for AAT protein (brown), 26 weeks after AAV injection. (Upper) scale bar, 527 μm; (lower) scale bar, 131 μm. ∗AAV-CAR Tregs compared with PBS; #AAV-CAR Tregs compared with polyclonal Tregs; ˆpolyclonal Tregs compared with PBS.
Next, we examined immunosuppressive cytokines in serum. We observed an increase in free active TGF-β1 in the AAV-CAR Treg group compared with the PBS group; this increase was significant at week 11 (Figure 5C). Further, we observed significantly higher IL-10 levels in the AAV-CAR Treg group at weeks 2, 7, and 11 post AAV injection compared with the PBS group (Figures 5D and S6). This suggests AAV-CAR Tregs secrete IL-10 and TFG-β1, which are essential for their suppressive function and agrees with our earlier phenotyping analysis. Two weeks after AAV delivery, muscles were collected and stained with hematoxylin and eosin (H&E). Significant infiltration was noted in the PBS group. Quantification revealed less cellular infiltration, as indicated by fewer nuclei, in the AAV-CAR Treg and polyclonal Treg-treated animals compared with the PBS animals (Figures 5E and 5F). These data suggest AAV-CAR Tregs can reduce cellular infiltration in the injected muscle. Further, at 26 weeks post AAV injection, muscles were immunostained for AAT protein expression. Robust staining of AAT was observed in animals treated with AAV-CAR Tregs or polyclonal Tregs, but no AAT staining was observed in animals treated with PBS (Figure 5G). Subsequently, to validate the loss of transduced cells versus loss of transgene expression, we measured vector genome copies at 26 weeks post injection by quantitative PCR. Greater genome copies were preserved in muscles of the animals treated with AAV-CAR Tregs with less vector genomes evident in the polyclonal Treg and negligible levels in the PBS-treated groups (Figure S7A). Taken together, these data suggest AAV-CAR Tregs can inhibit immune responses to rh32.33 capsid allowing long-term stable levels of expression of the therapeutic AAT transgene.
AAV-CAR Tregs mediated bystander suppression of immune responses to transgene
Although Treg-mediated suppression is stimulated by antigen-specific activation, Tregs can suppress the immune response in a nonspecific manner through bystander suppression. Bystander suppression allows Tregs to create an immunosuppressive microenvironment in addition to specific antigen suppression.21,23 Moreover, a bystander suppressive environment allows for infectious tolerance, mediated by TGF-β, to induce other immunosuppressive cell populations. These induced immunosuppressive cells may be specific for the same or different antigen specificities than the original Tregs.22 Since we observed secretion of IL-10 and TFG-β1 in vivo, we wanted to determine if AAV-CAR Tregs can bystander suppress T cell responses directed to different antigens. To test this, we utilized the in vitro luciferase killing assay. In addition to AAV-CARs, we generated CAR T cells and CAR Tregs against CD20 and utilized Raji cells as targets due to their natural expression of CD20. AAV-CAR T cells alone or as a mixture with CD20-CAR Tregs were incubated with CD20+ target cells, either transduced with AAV6 or non-transduced to test if CD20-CAR Tregs could suppress the cytotoxicity of AAV-CAR T cells. As expected, AAV-CAR T cells reduced the viability of the AAV6-transduced target cells. However, when CD20-CAR Tregs were cocultured, increased cell viability of CD20+ cells was observed, suggesting they suppressed the activity of AAV-CAR T cells. Importantly, no significant cytotoxicity was observed when cells were not transduced with AAV6 by either the AAV-CAR T cells or with the CD20-CAR Tregs (Figure 6A). Subsequently, we tested the ability of AAV-CAR Tregs to suppress the cytotoxicity of the CD20-CAR T cells. AAV-CAR Tregs were able to increase viability of the target cells only when they were transduced with AAV6. When the cells were not transduced with AAV6 or AAV-CAR Tregs were not present, robust cytotoxicity was observed (Figure 6B). These results indicated that the AAV-CAR Tregs can establish a local immunosuppressive environment and suppress T cells with different antigen specificities but also further confirm the specificity of AAV-CAR Tregs’ recognition of antigen as they only suppress, or bystander suppress, when AAV-transduced cells are present.
Figure 6.

AAV-CAR Tregs suppress effector cells cytotoxicity regardless of effector cell antigen specificity in vitro
(A) Cytotoxicity of AAV-CAR T cells and suppression of cytotoxicity of CD20-CAR Tregs against CD20+ Raji cells transduced with AAV6 or non-transduced as a control. (B) Cytotoxicity of CD20-CAR T cells and suppression of cytotoxicity by AAV-CAR Tregs against CD20+ Raji cells. Percentage viability is determined by luciferase expression measured 24 h after coculture. Cells were cultured at a ratio of 1:10 target cell to CAR T cell or 1:10:10 of target cell to CAR T cell to CAR Treg. Data are the average of three independent experiments using human samples from three healthy donors (within each experiment samples were run in triplicate). Error bars are mean ± SEM; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 by two-way ANOVA with Tukey's multiple comparisons.
Next, we examined whether AAV-CAR Tregs, directed toward AAV capsid, can bystander suppress immune responses against vector-delivered transgenes. To test this, we utilized the well-characterized immunogenic transgene, ovalbumin (OVA), expressed by the less immunogenic capsid, AAV1 (Figure 4E). C57BL/6 mice received i.m. delivery of AAV1-OVA followed by either AAV-CAR Tregs, polyclonal Tregs, or PBS. In the PBS group, expression of OVA was initially observed, but was significantly reduced over time, averaging ∼2,100 ng/mL. However, the AAV-CAR Treg group had increasingly high expression of ∼5,500 ng/mL of OVA and the polyclonal Treg group had expression of ∼4,500 ng/mL (Figure 7A). Interestingly, anti-transgene and anti-capsid antibodies are produced by all groups and were not significantly different across groups. Due to the timing of CAR delivery in these experiments, AAV-CAR Tregs are likely introduced too late to block B cell and T helper cell interactions, so they are unable to suppress antibody production. However, only in AAV-CAR Tregs and polyclonal Treg groups was sustained transgene expression observed, suggesting Tregs suppress T cell-mediated clearance of transduced cells (Figures 7B and 7C). We also found that the IL-10 levels in the serum were greater in the polyclonal Treg group than the PBS group, but the IL-10 levels in the AAV-CAR Treg groups were greater than both comparison groups and were significantly greater compared with the PBS-treated group (Figures 7D and S8). Similar to the rh32.33 findings, free active TGF-β1 was elevated in the serum of mice treated with AAV-CAR Tregs compared with the PBS group (Figure 7E).
Figure 7.

AAV-CAR Tregs bystander suppress immune response to AAV-delivered transgene
(A) Expression of OVA protein from animals i.m. injected with AAV1-OVA followed by i.v. AAV-CAR Tregs (red), polyclonal Tregs (orange), or PBS (blue) as measured by ELISA. Serum levels of OVA were significantly greater in AAV-CAR Treg-treated animals compared with PBS. (B) Anti-OVA antibodies detected in the serum by ELISA. (C) Anti-AAV1 antibodies in the serum measured by ELISA. (D) Serum levels of IL-10 measured by CBA assay. (E) Serum level of free active TGF-β1 measured by CBA assay over time. Two-way repeated-measure ANOVA with Tukey's multiple comparisons was used (n = 5 for AAV-CAR Treg and polyclonal Treg-treated groups, n = 3 for the PBS treated group for A, B, C, D, and E). (F) Quantification of number of nuclei in i.m. injected muscles. Each dot represents one animal, which is the average of 10 images per animal. (G) Representative images of H&E-stained limb muscles of mice 16 weeks post i.m. injection with AAV1-OVA. Substantial cellular infiltration (blue) was observed in animals treated with PBS or polyclonal Tregs. (Upper) scale bar, 527 μm; (lower) scale bar, 131 μm. one-way ANOVA, n = 2. Error bars are mean ± SEM; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001. ∗AAV-CAR Tregs compared with PBS.-CAR Tregs compared with PBS.
After 16 weeks, injected muscles were harvested for histology. H&E revealed significant cellular infiltration into the muscle of the PBS-treated animals, with multiple regions containing both focal and diffuse myositis and central nuclei (Figures 7G and S9). Reduced focal and diffuse myositis was observed in animals treated with polyclonal Tregs; however, significantly reduced inflammation was observed in the animals treated with AAV-CAR Tregs compared with both PBS and polyclonal Treg groups (Figures 7G and 7F). Further, the cell death marker caspase-3 staining revealed muscle fibers with central nuclei and caspase-3 staining in the PBS and expanded polyclonal Treg groups, with less staining and central nuclei in the AAV-CAR Treg-treated group (Figure S10). To determine the maintenance of transduced cells, quantitative PCR for vector genome was done at 16 weeks post injection. Corresponding with the transgene expression data, the vector shows greater vector genomes in the animals treated with AAV-CAR Tregs, with less vector genomes evident in polyclonal Treg and PBS-treated groups (Figure S7B). Taken together, AAV-CAR Tregs were able to produce IL-10 and TGF-β1, significantly reduce cellular infiltration and myositis, and allow for strong expression of immunogenic OVA transgene in spite of robust anti-AAV1 and anti-OVA antibody responses, suggesting AAV-CAR Tregs can suppress both capsid and vector-delivered transgene immune responses.
Injection of AAV-CAR Tregs reduced IFN-γ and increased IL-10 production in muscle-derived but not splenic immune cells after ex vivo isolation and restimulation
To further characterize in situ immune responses to AAV-OVA and the induction of tolerance by AAV-CAR Tregs, we isolated immune cells from the injected muscle or spleen and performed an antigen recall experiment. We first injected female C57BL/6 mice with AAV1-OVA. One week later, we injected i.v. either AAV-CAR Tregs, polyclonal Tregs, or PBS. Seven days later, animals were euthanized, and immune cells were isolated from injected muscle and spleen. The isolated immune cells were stimulated with anti-CD3/CD28, AAV1 pool, or SIINFEKL peptide and ovalbumin, or unstimulated (Figure 8A). In animals that received AAV1-OVA i.m. injection but no polyclonal or AAV-CAR Tregs, a significant increase of IFN-γ and IL-4 was observed in the muscle-isolated immune cells, suggesting a robust immune response to OVA was observed after i.m. AAV injection, but blocked by polyclonal or AAV-CAR Tregs (Figures 8B, 8C, S11, S12B, and S12K). IL-10 production was significantly increased in muscle isolated cells treated with AAV1 pool, and, interestingly, induction of IL-10 was significantly greater in animals treated with AAV-CAR Tregs compared with polyclonal Tregs. This suggests that Tregs were specifically induced by AAV capsid and not by the OVA protein (Figures 8D and S12F). Significant increase in free active TGF-β1 was also observed in the group that received AAV-CAR Tregs compared with both polyclonal Treg and PBS groups when cocultured with AAV1 pool, in both muscle- and spleen-isolated immune cells (Figures 8E and S12M). Together, this suggests that animals treated with AAV-CAR Tregs blocked IFN-γ and IL-4 immune responses to OVA transgene-mounted robust immunosuppressive immune responses to AAV capsids locally in the injected muscle. In contrast to muscle-isolated immune cells, immune cells isolated from the spleen showed no significant differences in any of the cytokines tested between stimulated treatment groups, except TGF-β1. These data further indicate that AAV-CAR Tregs home to injected muscle, where they recognize capsid-specific antigens and respond by creating local tolerance to both AAV capsid and delivered transgene.
Figure 8.

Injection of AAV-CAR Tregs reduced IFN-γ and increased IL-10 production in muscular but not splenic immune cells after ex vivo isolation and restimulation
(A) Experimental schematic. (B) Levels of IFN-γ in muscle after ex vivo isolation and restimulation measured by CBA assay. (C) Levels of IL-4 in muscle after ex vivo isolation and restimulation measured by CBA assay. (D) Levels of IL-10 in muscle after ex vivo isolation and restimulation measured by CBA assay. (E) Levels of free active TGF-β1 in muscle after ex vivo isolation and restimulation measured by CBA assay. Two-way ANOVA with Tukey's multiple comparisons was used. Error bars are mean ± SEM; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (n = 3 for AAV-CAR Treg and polyclonal Treg-treated groups, n = 2 for the PBS-treated group for B, C, D and E).
Discussion
Herein we generated AAV-CAR T cells that mimic capsid-specific immune responses observed in AAV clinical trials. Capsid-specific T cell responses were first observed during human liver-directed AAV gene therapy trials and led to loss of transgene expression.4 Such responses were not observed in prior preclinical data or animal models, whereas muscle-directed AAV gene therapy clinical trials have shown long-lived transgene expression and induction of Tregs.7,8,11,51 In contrast, mouse studies have shown liver-directed AAV gene therapy results in tolerogenic responses by the induction of Tregs,52, 53, 54, 55, 56, 57, 58, 59 and muscle-directed gene therapy is more immunogenic.60 Therefore, to mimic in mouse models, the immunogenic immune responses observed in human clinical trials, we chose a muscle-directed approach over a tolerogenic liver-directed approach. Several studies have aimed to mimic the observed human immune response in animals. However, studies in immunodeficient mice,61 canine,62 and non-human primate (NHP)63 models did not mimic human AAV-specific T cell responses. Further studies in murine models were not able to reproduce transgene clearance or specific killing even when an immunodominant ovalbumin SIINFEKL epitope was introduced into the AAV capsid.64 However, we were able to establish an immunocompetent murine model that accurately recapitulates T cell-directed clearance of transduced cells that is not antibody mediated and does not modify the AAV capsid.
In addition, we created AAV-specific Tregs that inhibit immune responses to AAV capsid and vector-delivered transgenes. Steroids are commonly used in AAV trials for broad immunosuppression; however, they do not specifically target capsid-specific T cells,65 and may also deplete Tregs.66 In contrast, AAV-CAR Tregs provide selective immune modulation activated specifically by AAV capsid, creating a local immunosuppressive environment in the presence of transduced cells. Further, a promising model of coadministration of encapsulated rapamycin (ImmTOR) along with AAV vectors results in decreased capsid-specific humoral response, T cell response, and immune cell infiltration into the target organs, with stable transduction and transgene expression.67 The immunomodulatory effect of ImmTOR was inhibited by the depletion of CD25+ T cells, suggesting an important role for Tregs.68,69 Despite the promise of ImmTOR technology, further investigations are necessary to confirm the clinical efficacy and whether immunosuppression by rapamycin will compromise the immune response against infections. Our data show that AAV-CAR Tregs create a suppressive environment activated specifically by AAV capsid. This results in production of inhibitory cytokines and sustained levels of transgene expression regardless of high levels of anti-capsid antibodies. In addition, while still being activated specifically by AAV, AAV-CAR Tregs can suppress both divergent capsid variants and vector-delivered transgenes, allowing for extensive clinical application. The data presented here demonstrate that both polyclonal Tregs and AAV-specific CAR Tregs exert important immune modulatory effects. While the ex vivo specificity of the CAR Tregs is demonstrable, the clinical importance of differences observed between the polyclonal Tregs and AAV-specific CAR Tregs in vivo remains uncertain and requires further study. Future experiments under conditions with improved transduction efficiency of T cells with the CAR Treg lentiviral constructs could clarify this point.
A limiting factor for many clinical applications of AAV gene therapies is the immune response against the vector-delivered transgene. Immune responses to protein replacement therapies and AAV-delivered transgenes have been observed in several diseases, including Duchenne muscular dystrophy (DMD),70,71 Pompe,72,73 and hemophilia B.74,75 To counteract the immune response against AAV-delivered transgenes, multiple approaches have been utilized with varying degrees of efficacy, including even broader multi-agent immunosuppression and more targeted approaches.76, 77, 78 Several studies have focused on testing different promoters, or tissue-specific promoters to reduce immune responses against delivered transgene.79,80 Others have focused on the innate immune response by removing CpG DNA,81 adding antagonists for Toll-like receptor 9 (TLR9)82 or by adding miR binding sites to downregulate transcript expression in antigen-presenting cells.83,84 For curbing immune responses to protein replacement therapy, some studies have focused on the adaptive immune response. One strategy used ex vivo expanded polyclonal Tregs to suppress immune response against coagulation factors 8 and 9 in hemophilia protein replacement therapy. Although transplanted Tregs became undetectable within 2 weeks, suppression persisted for more than 2 months. Additional studies suggested that antigen-specific suppression emerged due to induction of endogenous Tregs or infectious tolerance.76 However, studies show the number of cells needed for infusion of polyclonal Tregs is quite large, and the risk of nonspecific immunosuppression should be considered.85 Further, studies showed that Tregs may be unstable and can convert into T helper-like Treg cells in response to certain immunological environment.15 Although this is a cause for concern in polyclonal expanded Treg studies, or even CAR Tregs studies where CAR Tregs are generated by transduction of CAR constructs into Tregs, our CAR Treg construct is more stable due to FOXP3 expression driven from an exogenous promoter. After development of CAR therapy, studies focused on developing transgene-specific CAR Tregs.35 In one study, factor 8 (FVIII)-specific CAR Tregs were designed to reduce antibody production against factor VIII replacement therapy, but their success in suppression of immune responses to protein replacement suggests potential for suppressing vector-delivered transgene immune response in AAV gene therapy.35 However, in our study, we did not observe suppression of antibody responses to the transgene. Yet, AAV-CAR Tregs can suppress vector-delivered transgene immune responses in addition to capsid-specific immune responses, and, unlike expanded polyclonal cells, they are specifically directed against AAV.
CAR Tregs are extremely powerful tools due to intrinsic properties of Tregs such as bystander suppression and infectious tolerance. The unmatched promise of AAV-CAR Tregs is that a single product can suppress not only many AAV capsid variants but also the vector-delivered transgene without having to create a new CAR construct for every capsid or vector-delivered transgene for the desired therapeutic application. In addition, we designed the AAV-CAR Tregs by driving FoxP3 expression from an exogenous promoter, increasing manufacturing capacity by not requiring Treg selection. This provides a safeguard as all cells expressing CAR should also express FoxP3 as both the CAR and FoxP3 are driven by the same promoter. Therefore AAV-CAR Tregs are not susceptible to conversion to other T cell subsets by downregulation of FoxP3 expression, unlike polyclonal expanded Tregs. Further, AAV-CAR Tregs produce suppressive cytokines, IL-10 and TGF-β1, at greater levels compared with polyclonal Tregs and were able to induce a suppressive environment modulating immune cell infiltration and inflammation, resulting in sustained transgene expression.
Creation of a dual gene and cell therapy is an expensive approach; however, we have designed our system to significantly reduce production and treatment costs, and we believe it has considerable benefits over other approaches. The AAV-CAR recognition allows it to recognize all major capsid variants, allowing for the same AAV-CAR construct to be used in all AAV therapy applications, including different routes, AAV capsid variants, and transgenes. In addition, although the AAV-CAR Treg is specifically activated by AAV capsid, its effect is broad in that it creates a local systemic environment suppressing both capsid and transgene immune responses. This broad yet compartmentalized immune suppression is specific to areas of AAV-transduced cells, unlike other strategies that are broad, nonspecific, and systemic in their suppression. Also, we tried to optimize our product utilizing CD3+ cells as our input cell source and converting them into broadly suppressive Treg cells, allowing us to significantly reduce costs and start with a considerably larger cell population than either CD4+ or low-population Tregs. Further development of this platform technology for both further therapeutic applications as well as significant improvements in suppression, expansion, purification, and manufacturing are currently underway. Lastly, this study has demonstrated the potential therapeutic impact of Tregs in control of immune responses to AAV gene therapy.
Utilizing observations gained from clinical trials, and combining them with emergent CAR technology, we herein have established a unique immunomodulatory therapy for AAV capsid and vector-delivered transgene immune responses. The ability of AAV-CAR Tregs to create tolerogenic environments where transduced cells are sustained while suppressing both capsid and AAV-delivered transgene responses allows their broad clinical application. This form of local peripheral immune tolerance may ultimately be safer clinically than systemic, broad-spectrum immune suppression. Further studies are required to fully elucidate the clinical impact of AAV-CAR Tregs in modulating an immune response to AAV gene therapy.
Materials and methods
AAV-specific CAR-FoxP3 and AAV-specific CAR plasmid design and lentivirus and AAV production
The AAV-specific CAR constructs consist of a previously described scFv of a broadly reactive antibody against the majority of AAV capsid serotypes.39 All constructs were cloned into a third-generation lentiviral plasmid with a CD19 truncated sequence in order to recognize and isolate transduced cells (a kind gift from Dr. Brian Till, Fred Hutchinson Cancer Research Center, Seattle, WA). The scFv is fused to a human IgG1 CH2-CH3 hinge, intracellular CD28 (human), and CD137 (4-1BB) (human) co-stimulatory domains, and CD3ζ (human), under the control of the EF1-alpha promoter (Figure 1A). To create AAV-CAR Tregs, we designed a construct that includes murine FoxP3 cDNA, following the CAR construct expressed from the same promoter using the E2A self-cleavage peptide sequence (Figure 1B). CD20-CAR was a kind gift from Dr. Till. The CD20-CAR Treg construct was made by adding the murine Foxp3 after E2A self-cleavage peptide sequence downstream of a CD20-CAR construct. Third-generation lentiviruses were manufactured using the following plasmids: pHCMV VSVG, pMDLg/pRRE, and pRSV-Rev and AAVs by University of Massachusetts Chan Medical School viral vector core as previously described (Worcester, MA).86, 87, 88
Primary cell culture and cell lines
Cell lines
HEK293 cells were a kind gift from Dr. Gao (University of Massachusetts Chan Medical School, Worcester, MA). Raji cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). HEK293 and Raji cell lines were transduced with lentiviruses that encode firefly luciferase (ffLuc) and blasticidin S (Thermo Fisher Scientific, Waltham, MA) resistance. Cells were placed under blasticidin S selection (10 μg/mL) for multiple generations to produce stable cell lines. HEK293 and Raji cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 (Gibco, Waltham, MA) respectively, supplemented with 10% fetal bovine serum (FBS), 1% sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco).
Primary cells: human
Human T cells were cultured in ImmunoCult-XF T Cell Expansion Medium (StemCell Technologies, Vancouver, Canada) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco), and 50 U/mL IL-2 (BioLegend, San Diego, CA); this formulation will be referred to as human T cell medium. Human AAV-CAR Tregs were cultured in human T cell medium supplemented with 200 U/mL IL-2 and 2 ng/mL TGF-β1(BioLegend).
Primary cells: murine
Murine T cells were cultured in RPMI 1640 medium (Gibco) with 10% FBS (Sigma-Aldrich, Munich, Germany), 1% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma-Aldrich), 0.1 mM minimal essential medium (MEM) non-essential amino acid solution (Sigma-Aldrich), 0.055 mM 2-mercaptoethanol (Gibco), 1% sodium pyruvate, and 200 U/mL IL-2 (BioLegend); this formulation will be referred as murine T cell medium. Murine AAV-CAR Tregs, and polyclonal Tregs, were cultured in murine T cell medium with 2 ng/mL of TGF-β1 (BioLegend) and 1,000 U/mL IL-2.
Human and murine T cell isolation and transduction
Human T cells were isolated from whole blood using RosetteSep Human T Cell Enrichment Cocktail (StemCell Technologies) and stimulated with ImmunoCult Human CD3/CD28/CD2 T Cell Activator (StemCell Technologies) for 48 h. T cells were transduced in RetroNectin (Takara, Kusatsu, S-higa, Japan)-coated plates.89 Briefly, RetroNectin was diluted at a concentration of 30 μg/mL in PBS, and 500 μL of the RetroNectin solution were added to each well of non-tissue-culture-treated 24-well plates (Genesee Scientific, San Diego, CA). The plates were incubated for 2 h at room temperature. Then the RetroNectin solution was aspirated and 500 μL of a blocking solution consisting of Hanks' balanced salt solution (HBSS) (Gibco) plus 2% BSA (Millipore Sigma, St. Louis, MO) were added to each well. The plates were incubated for 30 min at room temperature, then blocking solution was aspirated, and the wells were rinsed with a solution of HBSS containing 2.5% HEPES. Lentiviruses were rapidly thawed and added to each RetroNectin-coated well with multiplicity of infection (MOI) 20 per well in human T cell medium (1 mL per well). The plates were centrifuged for 2 h at 2,000 × g and 32°C, and 1 × 106 stimulated cells/well were plated in RetroNectin-coated plates with 8 μg/well of polybrene (Millipore Sigma) and centrifuged for 10 min at 1,000 × g and 32°C and then were incubated at 37°C overnight. The transduction was repeated the following day. Then the plates were incubated overnight (Figure 1B). Transduced cells were cultured in human T cell medium.
Following euthanasia of naive female C57BL/6J mice, spleens were excised, pooled, and diced in chilled 4°C RPMI 1640 medium containing L-glutamine. Isolated spleens were mechanically dissociated then filtered through a 70-μm cell strainer (Thermo Fisher Scientific). T cells were isolated from splenocytes using an EasySep Mouse T Cell Isolation Kit (StemCell Technologies) and stimulated with 1 μg/mL soluble anti-CD3e NA/LE antibody, and 1 μg/mL anti-CD28 NA/LE antibody (BD Biosciences, San Jose, CA) for 48 h. T cells were transduced in RetroNectin (Takara, )-coated plates as previously described with MOI 200. Murine polyclonal Tregs were isolated with EasySep Mouse CD4+CD25 + Regulatory T Cell Isolation Kit II (StemCell Technologies).
Flow cytometry
Single-cell suspensions were stained as described: intracellular CTLA-4 and intranuclear transcription factor staining for murine FOXP3 was carried out using the FOXP3 staining kit (BD Biosciences) following the manufacturer's recommendations. Dead cells were excluded from all analyses using fixable LIVE/DEAD cell stain (Thermo Fisher Scientific). Data were acquired on a BD LSRII and analyzed using FlowJo software v10.7.1. Antibodies used for phenotyping are listed in Table S2. Experiment was repeated three times with different donor cells for CAR transductions for each repeat.
Luciferase cytotoxicity assay and suppression of cytotoxicity assay
FfLuc+ target cells, either transduced with AAV-GFP (AAV1, AAV2, AAV3b, AAV6, AAV9, rh32.33 with MOI of 1 × 105; AAV5 and AAV8 with MOI of 5 × 106) or not transduced with AAV-GFP, were plated at a concentration of 5 × 103 cells/well in 96-well micro-plates. After 72 h, target cells were centrifuged and supernatant was removed prior to coculturing with either AAV-CAR T cells or AAV-CAR T cells in combination with AAV-CAR Tregs with effector cells at the following ratio: effector:target (E:T) ratio of 10:1 and incubated for 24 h with 5% CO2, at 37°C. D-luciferin (Gold Biotechnology, St. Louis, MO) was added at a concentration of 15 μg/mL. The plate was incubated in the dark for 5–10 min at 37°C, and luminescence flux was measured with Synergy HTX Multi-Mode Microplate Reader (Biotek, Winooski, VT). To achieve minimum luciferase activity, target cells were cultured with 1% sodium dodecyl sulfate (SDS) (Gibco) and untreated target cells were used for maximum luciferase activity. Percentage viability was calculated as previously described.48 The experiment was repeated three times using different donor cells for CAR transductions in each repeat (within each experiment, samples were run in triplicate).
T cell suppression assay
AAV-CAR T cells were stained with CellTrace Violet dye following the manufacturer's recommendations (Invitrogen, Carlsbad, CA) and were cultured alone or cocultured with unstained AAV-CAR Tregs in the presence of either AAV1-transduced HEK293 cells or ImmunoCult Human CD3/CD28/CD2 T Cell Activator (StemCell Technologies). T cells were plated at 5 × 104 cells/well in 96-well plates with human T cell medium for 72–96 h with E:T ratio 10:1. Proliferation of CellTrace Violet-stained cells was measured by flow cytometry. The experiment was repeated three times with different donor cells for CAR transductions for each repeat.
Animals and in vivo models
All procedures involving animals were carried out in accordance with the guidelines of the University of Massachusetts Chan Medical School Animal Care and Use Committee (IACUC). Female (6–12-week-old) inbred C57BL/6J mice (Jackson laboratories, Bar Harbor, ME) were used for modeling anti-AAV capsid immune response. Mice (n = 6) were injected with 5 × 1010 genomic copies (GCs) of AAV1-hAAT in the right muscles. Three weeks later, animals were injected i.v. with either AAV-CAR T cells (5 × 106 cells) or PBS. All groups received 45,000 IU of IL-2 intraperitoneally (i.p.) (BioLegend) followed immediately with i.v. injection, and daily injection of IL-2 for 2 days. Animals were bled by facial vein and euthanized at week 6, and muscle tissues were collected after perfusion.
For suppressing immune response, animals were injected with 5 × 1010 GCs of rh32.33-hAAT (n = 6 for AAV-CAR Treg and PBS-treated groups, n = 5 for polyclonal Treg-treated group) or AAV1-OVA (n = 5 for AAV-CAR Treg and polyclonal Treg-treated groups, n = 3 for PBS-treated group) expressing in the right muscles. Seven days later, the animals were injected i.v. with either AAV-CAR Tregs (2 × 106 cells), polyclonal Tregs (2 × 106 cells), or PBS. All groups received 45,000 IU of IL-2 (i.p.) (BioLegend) immediately after i.v. injection and daily injection of IL-2 for the next 2 days and were bled by facial vein. Animals that received AAT or OVA were sacrificed at week 26 or 16, respectively, and muscle tissues were collected after perfusion.
Tracking AAV-CAR Tregs in vivo
Male NU/J mice (Jackson laboratories) were injected in the right muscle with AAV1 expressing luciferase. Human AAV-CAR Tregs were labeled with DiR (PerkinElmer, Waltham, MA) according to manufacturer's recommendations. One week post AAV injection, animals were injected i.v. with 5 × 106/mouse DiR-labeled AAV-CAR Tregs or PBS. Animals were injected with 100 mg/kg D-luciferin (Gold Biotechnology), anesthetized by isoflurane inhalation, and imaged after 10 min (peak of emission) using IVIS Spectrum CT (PerkinElmer) 24 h after cell injection. The ideal filter imaging conditions were set at 710 nm of excitation and 760 nm of emission for DiR and at 660 nm of emission for luciferase. Signal quantification in specific regions of interest (ROIs) was performed by using fixed-size and fixed-position ROIs throughout the experiments.
ELISA
hAAT ELISA
AAT levels in murine serum samples were quantified by direct ELISA as described.90 Animal serum was diluted 500-fold.
OVA and anti-OVA ELISA
Serum levels of OVA and anti-OVA IgG were determined by ELISA. Briefly, 96-well Immulon 4 HBX (Thermo Scientific) were coated with 2 μg/mL of rabbit anti-OVA polyclonal antibodies (MilliporeSigma) or OVA protein (MilliporeSigma) in 100 μL of coating buffer (Seracare Life Science, Milford, MA) per well. Plates were incubated overnight at 4°C and were washed with wash solution (Seracare Life Science) and incubated with blocking buffer (Seracare Life Science) for 2 h at room temperature. For OVA detection, samples were diluted 100-fold with ELISA diluent (Seracare Life Science), and OVA protein standards (bioWORLD, Dublin, OH) were 2-fold serially diluted with blocking buffer starting from 1,000 ng/mL. Then 100 μL of sample or standard was added to plates and incubated for 1 h at room temperature. After washing two times, peroxidase-conjugated rabbit anti-OVA polyclonal antibody (Rockland Immunochemicals, Limerick, PA) (1:5,000 diluted) was added and incubated for 1 h at room temperature. For anti-OVA IgG1 detection, samples were diluted 1:200, and the mouse anti-OVA IgG1 (Santa Cruz Biotechnology, Dallas, TX) was used as the standard. After a 1-h incubation in OVA-coated plates, wells were washed, horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG1 (Santa Cruz Biotechnology) was added, and plates were incubated for another hour at room temperature. Plates were then washed two times and incubated with 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (Seracare Life Science). Plates were incubated at room temperature in the dark for 30 min, and 50 μL of stop solution (sulfuric acid 2N) was added to each well. Optical density at 450 nm was measured using VersaMax plate reader (Molecular Devices, Sunnyvale, CA). Standard curves for OVA and IgG1 were generated by using the SoftMax Pro software (Molecular Devices).
Anti-AAV ELISA
For anti-capsid mouse IgG1 detection using ELISA, sample wells were coated with 5 × 109 GCs/well of AAV, diluted in coating buffer (Bethyl Laboratories, Montgomery, TX). The standard wells were coated with 100 μL of goat anti-mouse IgG coating antibody (Bethyl Laboratories) and were incubated overnight at 4°C. Next, plates were washed with PBS with 0.1% Tween 20 (PBST) wash solution and blocked with ELISA blocking buffer (Bethyl Laboratories), then incubated for 1 h at 37°C. Samples and standards were added and incubated for 2 h at 37°C. For standards, mouse reference serum (Bethyl Laboratories, RS10-101-6) was used. TMB peroxidase substrate (Seracare Life Science) was added, plates were incubated at room temperature in the dark for 7–10 min, then 50 μL of stop solution (sulfuric acid 2N) was added to each well. Optical density at 450 nm was measured using a VersaMax plate reader (Molecular Devices). Standard curves for IgG1 were generated by using the SoftMax Pro software (Molecular Devices).
Anti-hAAT ELISA
Animals' serum was diluted 200-fold and serum levels of anti-hAAT were determined by ELISA. Briefly, 96-well Immulon 4 HBX (Thermo Scientific) were coated with 9 μg/mL of hAAT (Sigma-Aldrich) in 100 μL of BupH carbonate-bicarbonate buffer (Thermo Scientific) per well. Plates were incubated overnight at 4°C and were washed with PBST wash solution and incubated with 100 μL of 3% BSA in PBST blocking buffer for 1.5 h at 37°C. For anti-hAAT detection, samples were diluted with PBST, and mouse anti-hAAT protein standards (Abcam, Cambridge, UK) were 2-fold serially diluted with blocking buffer starting from 125 ng/mL. Then 100 μL of sample or standard was added to plates and incubated for 1 h at 37°C. After incubation wells were washed, HRP-conjugated goat anti-mouse IgG1 (Santa Cruz Biotechnology) was added, and plates were incubated for another hour at 37°C. The plate was washed five times and TMB peroxidase substrate (Seracare Life Science) was added, plates were incubated at room temperature in the dark for 7–10 min, and 50 μL of stop solution was added to each well. Optical density at 490 nm was measured using a VersaMax plate reader (Molecular Devices). Standard curves for IgG1 were generated by using the SoftMax Pro software (Molecular Devices). In all ELISA experiments, samples were run in triplicate.
Cytokine detection
For the detection of the cytokines, cytometric bead array (CBA) assay was performed using BioLegend LEGENDplex Mouse Th Cytokine Panel (BioLegend, catalog no. 741044) following the manufacturer's recommendations. Serum samples from experimental animals were diluted 1:4. BioLegend's LEGENDplex Mouse/Rat Free Active/Total TGF-β1 assay kit (catalog no. 740490) was used to measure total TGF-β1 in mouse serum. Following the manufacturer's recommendations, samples were treated to release free TGF-β1 from complex before continuing with the assay. Cell culture supernatants were not diluted. Data were acquired on a BD LSRII and analyzed using the LEGENDplex Data Analysis Software Suite.
Immunohistochemistry
Immunohistochemical staining to detect hAAT within myofibers as well as H&E staining was performed after tissues were fixed in 10% neutral buffered formalin for 24 h at room temperature (Fisher Scientific) and embedded in paraffin by University of Massachusetts Chan Medical School Morphology Core (Worcester, MA). Anti-AAT antibody (Fitzgerald, Acton, MA), anti-CD4 antibody (Abcam), and anti-mouse FOXP3 antibody (Abcam) were used as primary antibody. Images were acquired on a Leica DM5500 B microscope (Leica Microsystems, Wetzlar, Germany).
Quantification of nuclei
Images were acquired on a Leica DM5500 B microscope (Leica Microsystems). Ten images were acquired per slide using a ×20 objective. Automated counting of nuclei from the harvested muscles was performed by ImageJ software.
Isolation and stimulation of murine immune cells
Female (6–12-week-old) inbred C57BL/6J mice (Jackson laboratories) were injected with 5 × 1010 GCs of AAV1-OVA in the right muscles. Seven days later, the animals were injected i.v. with either AAV-CAR Tregs (2 × 106 cells), polyclonal Tregs (2 × 106 cells), or PBS. All groups received 45,000 IU of IL-2 i.p. (BioLegend) followed immediately with i.v. injection, and daily injection of IL-2 for 2 days. Animals were euthanized and spleens and injected muscles were collected after PBS perfusion. Immune cells were isolated from spleen as previously described. To isolate immune cells from the injected muscles, tissues were collected in 5 mL of RPMI 1640 medium. Muscles were excised, pooled, diced, and transferred to six-well plate containing RPMI 1640 medium (2 mL/well) supplemented with 0.5 mg/mL DNAse I (Millipore Sigma) and 0.25 mg/mL Liberase TL (Roche, Basel, Switzerland). After 2 h of incubation at 37°C, Liberase was inactivated by adding 8 mL of RPMI 1640 containing 10% FBS. Digested pieces were filtered through a 70-μm cell strainer (Thermo Fisher Scientific). Then cells were washed using RPMI 1640 medium.
The isolated immune cells from muscles and spleen (5 × 104/well) were cocultured in 24-well plate with no stimulation, AAV1 pool (2 μg/mL), SIINFEKL peptide (Genscript, Piscataway, NJ), and ovalbumin (Sigma-Aldrich) (2 μg/mL) or ImmunoCult Human CD3/CD28/CD2 T Cell Activator (following the manufacturer's recommendation) for 72 h. Then the supernatants were collected and used for CBA assay analysis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc, La Jolla, CA) version 8. Results were reported as mean ± SEM with statistically significant differences determined by tests as indicated in figure legends.
Study approval
The present studies in mice were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Chan Medical School.
Data and materials availability
All data associated with this study are present in the paper or the supplementary materials. We received the plasmid encoding CD20-CAR as a gift from Dr. Brian Till at Fred Hutchinson Cancer Research Center under a material transfer agreement (MTA). The plasmid encoding luciferase and blasticidin is available on Addgene via MTA (catalog no. 108542). Correspondence and requests for materials should be addressed to A.M.K. at Allison.keeler@umassmed.edu.
Acknowledgments
Funding was provided by National Heart, Lung, and Blood Institute NHLBI-P01 HL1214 (T.R.F., G.G.) and NHLBI-P01 HL158506 (A.M.K., T.R.F., G.G.)
We are grateful to Dr. Brian Till from Fred Hutchinson Cancer Research Center for providing the lentiviral vector. The authors thank Dr. Abdo Abou-Slaybi and Dr. Mai K. ElMallah for insightful comments. Graphical schematics were created with BioRender.com.
Author contributions
Conceptualization, A.M.K., T.R.F., and A.M.G.; methodology, M.A., A.M.K., and T.R.F.; formal analysis, M.A., A.M.K., and K.S.; investigation, M.A., K.S., M.B., T.N., and Q.T.; resources, A.M.K., T.R.F., G.G., and M.M.; writing – original draft, M.A., A.M.K., and T.R.F.; writing – review & editing, M.A., A.M.K., T.R.F., K.S., M.B., T.N., Q.T., M.M., A.M.G., and G.G.; visualization, M.A. and A.M.K.; project administration, M.A., A.M.K., and T.R.F.; funding acquisition, A.M.K. and T.R.F.
Declaration of interests
M.A., A.M.K., and T.R.F. have submitted a patent (US2020029527). T.R.F. serves as a paid consultant for Ferring Ventures, which is unrelated to the work described here. A.M.K. has an SRA with Kriya Therapeutics, which is unrelated to this work. G.G. is a scientific co-founder of Voyager Therapeutics, Adrenas Therapeutics, and Aspa Therapeutics, and holds equity in the companies. G.G. is inventor on patents related to AAV-based gene therapy, some of which were licensed to commercial entities but are unrelated to this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.10.010.
Supplemental information
References
- 1.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snyder R.O., Miao C., Meuse L., Tubb J., Donahue B.A., Lin H.F., Stafford D.W., Patel S., Thompson A.R., Nichols T., et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat. Med. 1999;5:64–70. doi: 10.1038/4751. [DOI] [PubMed] [Google Scholar]
- 3.Mount J.D., Herzog R.W., Tillson D.M., Goodman S.A., Robinson N., McCleland M.L., Bellinger D., Nichols T.C., Arruda V.R., Lothrop C.D., Jr., High K.A. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99:2670–2676. doi: 10.1182/blood.v99.8.2670. [DOI] [PubMed] [Google Scholar]
- 4.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 5.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E., Ragni M.V., Manno C.S., Sommer J., Jiang H., et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 6.Nathwani A.C., Tuddenham E.G.D., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C., et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brantly M.L., Chulay J.D., Wang L., Mueller C., Humphries M., Spencer L.T., Rouhani F., Conlon T.J., Calcedo R., Betts M.R., et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. U S A. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flotte T.R., Trapnell B.C., Humphries M., Carey B., Calcedo R., Rouhani F., Campbell-Thompson M., Yachnis A.T., Sandhaus R.A., McElvaney N.G., et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α1-antitrypsin: interim results. Hum. Gene Ther. 2011;22:1239–1247. doi: 10.1089/hum.2011.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mingozzi F., Meulenberg J.J., Hui D.J., Basner-Tschakarjan E., Hasbrouck N.C., Edmonson S.A., Hutnick N.A., Betts M.R., Kastelein J.J., Stroes E.S., High K.A. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferreira V., Twisk J., Kwikkers K., Aronica E., Brisson D., Methot J., Petry H., Gaudet D. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPL(S447X)) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum. Gene Ther. 2014;25:180–188. doi: 10.1089/hum.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller C., Chulay J.D., Trapnell B.C., Humphries M., Carey B., Sandhaus R.A., McElvaney N.G., Messina L., Tang Q., Rouhani F.N., et al. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J. Clin. Invest. 2013;123:5310–5318. doi: 10.1172/jci70314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira V., Petry H., Salmon F. Immune responses to AAV-vectors, the Glybera example from bench to bedside. Front. Immunol. 2014;5:82. doi: 10.3389/fimmu.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niederlova V., Tsyklauri O., Chadimova T., Stepanek O. CD8+ Tregs revisited: a heterogeneous population with different phenotypes and properties. Eur. J. Immunol. 2021;51:512–530. doi: 10.1002/eji.202048614. [DOI] [PubMed] [Google Scholar]
- 14.Roncarolo M.G., Gregori S., Bacchetta R., Battaglia M., Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity. 2018;49:1004–1019. doi: 10.1016/j.immuni.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Li Z., Li D., Tsun A., Li B. FOXP3+ regulatory T cells and their functional regulation. Cell Mol. Immunol. 2015;12:558–565. doi: 10.1038/cmi.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (New York, N.Y.) 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 17.Fransson M., Piras E., Burman J., Nilsson B., Essand M., Lu B., Harris R.A., Magnusson P.U., Brittebo E., Loskog A.S. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation. 2012;9:112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilate A.M., Lafaille J.J. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 2012;30:733–758. doi: 10.1146/annurev-immunol-020711-075043. [DOI] [PubMed] [Google Scholar]
- 19.Sojka D.K., Huang Y.H., Fowell D.J. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. 2008;124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vignali D.A.A., Collison L.W., Workman C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thornton A.M., Shevach E.M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 22.Waldmann H., Adams E., Fairchild P., Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunological Rev. 2006;212:301–313. doi: 10.1111/j.0105-2896.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- 23.Karim M., Feng G., Wood K.J., Bushell A.R. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. 2005;105:4871–4877. doi: 10.1182/blood-2004-10-3888. [DOI] [PubMed] [Google Scholar]
- 24.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larson R.C., Maus M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer. 2021;21:145–161. doi: 10.1038/s41568-020-00323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seif M., Einsele H., Löffler J. CAR T cells beyond cancer: hope for immunomodulatory therapy of infectious diseases. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.02711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sautto G.A., Wisskirchen K., Clementi N., Castelli M., Diotti R.A., Graf J., Clementi M., Burioni R., Protzer U., Mancini N. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut. 2016;65:512–523. doi: 10.1136/gutjnl-2014-308316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slabik C., Kalbarczyk M., Danisch S., Zeidler R., Klawonn F., Volk V., Krönke N., Feuerhake F., Ferreira de Figueiredo C., Blasczyk R., et al. CAR-T cells targeting Epstein-Barr virus gp350 validated in a humanized mouse model of EBV infection and lymphoproliferative disease. Mol. Ther. Oncolytics. 2020;18:504–524. doi: 10.1016/j.omto.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elinav E., Adam N., Waks T., Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology. 2009;136:1721–1731. doi: 10.1053/j.gastro.2009.01.049. [DOI] [PubMed] [Google Scholar]
- 31.Elinav E., Waks T., Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. 2008;134:2014–2024. doi: 10.1053/j.gastro.2008.02.060. [DOI] [PubMed] [Google Scholar]
- 32.Blat D., Zigmond E., Alteber Z., Waks T., Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. : J. Am. Soc. Gene Ther. 2014;22:1018–1028. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacDonald K.G., Hoeppli R.E., Huang Q., Gillies J., Luciani D.S., Orban P.C., Broady R., Levings M.K. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koristka S., Kegler A., Bergmann R., Arndt C., Feldmann A., Albert S., Cartellieri M., Ehninger A., Ehninger G., Middeke J.M., et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J. Autoimmun. 2018;90:116–131. doi: 10.1016/j.jaut.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Yoon J., Schmidt A., Zhang A.H., Konigs C., Kim Y.C., Scott D.W. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. 2017;129:238–245. doi: 10.1182/blood-2016-07-727834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mekala D.J., Geiger T.L. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood. 2005;105:2090–2092. doi: 10.1182/blood-2004-09-3579. [DOI] [PubMed] [Google Scholar]
- 37.Smith B.M., Lyle M.J., Chen A.C., Miao C.H. Antigen-specific in vitro expansion of factor VIII-specific regulatory T cells induces tolerance in hemophilia A mice. J. Thromb. Haemost. 2020;18:328–340. doi: 10.1111/jth.14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itoh M., Takahashi T., Sakaguchi N., Kuniyasu Y., Shimizu J., Otsuka F., Sakaguchi S. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 1999;162:5317–5326. [PubMed] [Google Scholar]
- 39.Wobus C.E., Hügle-Dörr B., Girod A., Petersen G., Hallek M., Kleinschmidt J.A. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuck D., Kern A., Kleinschmidt J.A. Development of AAV serotype-specific ELISAs using novel monoclonal antibodies. J. Virol. Methods. 2007;140:17–24. doi: 10.1016/j.jviromet.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Meyer N.L., Hu G., Davulcu O., Xie Q., Noble A.J., Yoshioka C., Gingerich D.S., Trzynka A., David L., Stagg S.M., Chapman M.S. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. eLife. 2019;8:e44707. doi: 10.7554/eLife.44707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y., Ma X., Gong R., Zhu J., Wei L., Yao J. Recent advances in CD8(+) regulatory T cell research. Oncol. Lett. 2018;15:8187–8194. doi: 10.3892/ol.2018.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang K., Gu J., Ni X., Ding Z., Wang Q., Zhou H., Zheng S., Li B., Lu L. CD25 signaling regulates the function and stability of peripheral Foxp3+ regulatory T cells derived from the spleen and lymph nodes of mice. Mol. Immunol. 2016;76:35–40. doi: 10.1016/j.molimm.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Wagner D.L., Peter L., Schmueck-Henneresse M. Cas9-directed immune tolerance in humans—a model to evaluate regulatory T cells in gene therapy? Gene Ther. 2021 doi: 10.1038/s41434-021-00232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elkord E., Abd Al Samid M., Chaudhary B. Helios, and not FoxP3, is the marker of activated Tregs expressing GARP/LAP. Oncotarget. 2015;6:20026–20036. doi: 10.18632/oncotarget.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ronchetti S., Ricci E., Petrillo M.G., Cari L., Migliorati G., Nocentini G., Riccardi C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J. Immunol. Res. 2015;2015:171520. doi: 10.1155/2015/171520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yadav M., Louvet C., Davini D., Gardner J.M., Martinez-Llordella M., Bailey-Bucktrout S., Anthony B.A., Sverdrup F.M., Head R., Kuster D.J., et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 2012;209:1713–1722. doi: 10.1084/jem.20120822. s1711-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown C.E., Wright C.L., Naranjo A., Vishwanath R.P., Chang W.C., Olivares S., Wagner J.R., Bruins L., Raubitschek A., Cooper L.J., Jensen M.C. Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J. Immunol. Methods. 2005;297:39–52. doi: 10.1016/j.jim.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 49.Monjezi R., Miskey C., Gogishvili T., Schleef M., Schmeer M., Einsele H., Ivics Z., Hudecek M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. 2017;31:186–194. doi: 10.1038/leu.2016.180. [DOI] [PubMed] [Google Scholar]
- 50.Mays L.E., Wilson J.M. Identification of the murine AAVrh32.33 capsid-specific CD8+ T cell epitopes. J. Gene Med. 2009;11:1095–1102. doi: 10.1002/jgm.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mueller C., Gernoux G., Gruntman A.M., Borel F., Reeves E.P., Calcedo R., Rouhani F.N., Yachnis A., Humphries M., Campbell-Thompson M., et al. 5 Year expression and neutrophil defect repair after gene therapy in alpha-1 antitrypsin deficiency. Mol. Ther. J. Am. Soc. Gene Ther. 2017;25:1387–1394. doi: 10.1016/j.ymthe.2017.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keeler G.D., Markusic D.M., Hoffman B.E. Liver induced transgene tolerance with AAV vectors. Cell Immunol. 2019;342:103728. doi: 10.1016/j.cellimm.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mingozzi F., Liu Y.L., Dobrzynski E., Kaufhold A., Liu J.H., Wang Y., Arruda V.R., High K.A., Herzog R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobrzynski E., Fitzgerald J.C., Cao O., Mingozzi F., Wang L., Herzog R.W. Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc. Natl. Acad. Sci. U S A. 2006;103:4592–4597. doi: 10.1073/pnas.0508685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Markusic D.M., Hoffman B.E., Perrin G.Q., Nayak S., Wang X., LoDuca P.A., High K.A., Herzog R.W. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol. Med. 2013;5:1698–1709. doi: 10.1002/emmm.201302859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao O., Dobrzynski E., Wang L., Nayak S., Mingle B., Terhorst C., Herzog R.W. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breous E., Somanathan S., Vandenberghe L.H., Wilson J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612–621. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martino A.T., Nayak S., Hoffman B.E., Cooper M., Liao G., Markusic D.M., Byrne B.J., Terhorst C., Herzog R.W. Tolerance induction to cytoplasmic beta-galactosidase by hepatic AAV gene transfer: implications for antigen presentation and immunotoxicity. PLoS One. 2009;4:e6376. doi: 10.1371/journal.pone.0006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobrzynski E., Mingozzi F., Liu Y.L., Bendo E., Cao O., Wang L., Herzog R.W. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104:969–977. doi: 10.1182/blood-2004-03-0847. [DOI] [PubMed] [Google Scholar]
- 60.Herzog R.W., Fields P.A., Arruda V.R., Brubaker J.O., Armstrong E., McClintock D., Bellinger D.A., Couto L.B., Nichols T.C., High K.A. Influence of vector dose on factor IX-specific T and B cell responses in muscle-directed gene therapy. Hum. Gene Ther. 2002;13:1281–1291. doi: 10.1089/104303402760128513. [DOI] [PubMed] [Google Scholar]
- 61.Martino A.T., Basner-Tschakarjan E., Markusic D.M., Finn J.D., Hinderer C., Zhou S., Ostrov D.A., Srivastava A., Ertl H.C., Terhorst C., et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–2233. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun J., Shao W., Chen X., Merricks E.P., Wimsey L., Abajas Y.L., Niemeyer G.P., Lothrop C.D., Monahan P.E., Samulski R.J., et al. An observational study from long-term AAV re-administration in two hemophilia dogs. Molecular therapy. Methods Clin. Dev. 2018;10:257–267. doi: 10.1016/j.omtm.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greig J.A., Limberis M.P., Bell P., Chen S.-J., Calcedo R., Rader D.J., Wilson J.M. Non-clinical study examining AAV8.TBG.hLDLR vector-associated toxicity in chow-fed wild-type and LDLR+/− rhesus macaques. Hum. Gene Ther. Clin. Dev. 2017;28:39–50. doi: 10.1089/humc.2017.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiang Z., Kurupati R.K., Li Y., Kuranda K., Zhou X., Mingozzi F., High K.A., Ertl H.C.J. The effect of CpG sequences on capsid-specific CD8(+) T cell responses to AAV vector gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2020;28:771–783. doi: 10.1016/j.ymthe.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parzych E.M., Li H., Yin X., Liu Q., Wu T.-L., Podsakoff G.M., High K.A., Levine M.H., Ertl H.C.J. Effects of immunosuppression on circulating adeno-associated virus capsid-specific T cells in humans. Hum. Gene Ther. 2013;24:431–442. doi: 10.1089/hum.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mingozzi F., Hasbrouck N.C., Basner-Tschakarjan E., Edmonson S.A., Hui D.J., Sabatino D.E., Zhou S., Wright J.F., Jiang H., Pierce G.F., et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110:2334–2341. doi: 10.1182/blood-2007-03-080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ilyinskii P.O., Michaud A.M., Roy C.J., Rizzo G.L., Elkins S.L., Capela T., Chowdhury A.C., Leung S.S., Kishimoto T.K. Enhancement of liver-directed transgene expression at initial and repeat doses of AAV vectors admixed with ImmTOR nanoparticles. Sci. Adv. 2021;7:eabd0321. doi: 10.1126/sciadv.abd0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kishimoto T.K., Ferrari J.D., LaMothe R.A., Kolte P.N., Griset A.P., O'Neil C., Chan V., Browning E., Chalishazar A., Kuhlman W., et al. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat. Nanotechnol. 2016;11:890–899. doi: 10.1038/nnano.2016.135. [DOI] [PubMed] [Google Scholar]
- 69.Zhang A.-H., Rossi R.J., Yoon J., Wang H., Scott D.W. Tolerogenic nanoparticles to induce immunologic tolerance: prevention and reversal of FVIII inhibitor formation. Cell Immunol. 2016;301:74–81. doi: 10.1016/j.cellimm.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 70.Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G., et al. Dystrophin immunity in Duchenne's muscular dystrophy. New Engl. J. Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flanigan K.M., Campbell K., Viollet L., Wang W., Gomez A.M., Walker C.M., Mendell J.R. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum. Gene Ther. 2013;24:797–806. doi: 10.1089/hum.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corti M., Liberati C., Smith B.K., Lawson L.A., Tuna I.S., Conlon T.J., Coleman K.E., Islam S., Herzog R.W., Fuller D.D., et al. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by Pompe disease. Human gene therapy. Clin. Dev. 2017;28:208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brooks D.A., Kakavanos R., Hopwood J.J. Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol. Med. 2003;9:450–453. doi: 10.1016/j.molmed.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 74.Cao O., Hoffman B.E., Moghimi B., Nayak S., Cooper M., Zhou S., Ertl H.C., High K.A., Herzog R.W. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia B. Molecular therapy. J. Am. Soc. Gene Ther. 2009;17:1733–1742. doi: 10.1038/mt.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel S.R., Lundgren T.S., Spencer H.T., Doering C.B. The immune response to the fVIII gene therapy in preclinical models. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarkar D., Biswas M., Liao G., Seay H.R., Perrin G.Q., Markusic D.M., Hoffman B.E., Brusko T.M., Terhorst C., Herzog R.W. Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol. Ther. Methods Clin. Dev. 2014;1:14030. doi: 10.1038/mtm.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gross D.A., Chappert P., Leboeuf M., Monteilhet V., Van Wittenberghe L., Danos O., Davoust J. Simple conditioning with monospecific CD4+CD25+ regulatory T cells for bone marrow engraftment and tolerance to multiple gene products. Blood. 2006;108:1841–1848. doi: 10.1182/blood-2006-02-011981. [DOI] [PubMed] [Google Scholar]
- 78.Herzog R.W., Mount J.D., Arruda V.R., High K.A., Lothrop C.D., Jr. Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol. Ther. J. Am. Soc. Gene Ther. 2001;4:192–200. doi: 10.1006/mthe.2001.0442. [DOI] [PubMed] [Google Scholar]
- 79.Nathwani A.C., Rosales C., McIntosh J., Rastegarlari G., Nathwani D., Raj D., Nawathe S., Waddington S.N., Bronson R., Jackson S. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun B., Zhang H., Franco L.M., Brown T., Bird A., Schneider A., Koeberl D.D. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol. Ther. 2005;11:889–898. doi: 10.1016/j.ymthe.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 81.Faust S.M., Bell P., Cutler B.J., Ashley S.N., Zhu Y., Rabinowitz J.E., Wilson J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Invest. 2013;123:2994–3001. doi: 10.1172/jci68205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan Y.K., Wang S.K., Chu C.J., Copland D.A., Letizia A.J., Costa Verdera H., Chiang J.J., Sethi M., Wang M.K., Neidermyer W.J., et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021;13:eabd3438. doi: 10.1126/scitranslmed.abd3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Majowicz A., Maczuga P., Kwikkers K.L., van der Marel S., van Logtenstein R., Petry H., van Deventer S.J., Konstantinova P., Ferreira V. Mir-142-3p target sequences reduce transgene-directed immunogenicity following intramuscular adeno-associated virus 1 vector-mediated gene delivery. J. Gene Med. 2013;15:219–232. doi: 10.1002/jgm.2712. [DOI] [PubMed] [Google Scholar]
- 84.Xiao Y., Muhuri M., Li S., Qin W., Xu G., Luo L., Li J., Letizia A.J., Wang S.K., Chan Y.K., et al. Circumventing cellular immunity by miR142-mediated regulation sufficiently supports rAAV-delivered OVA expression without activating humoral immunity. JCI Insight. 2019;4 doi: 10.1172/jci.insight.99052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brunstein C.G., Blazar B.R., Miller J.S., Cao Q., Hippen K.L., McKenna D.H., Curtsinger J., McGlave P.B., Wagner J.E. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol. Blood Marrow Transplant. : J. Am. Soc. Blood Marrow Transplant. 2013;19:1271–1273. doi: 10.1016/j.bbmt.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mueller C., Ratner D., Zhong L., Esteves-Sena M., Gao G. Production and discovery of novel recombinant adeno-associated viral vectors. Curr. Protoc. Microbiol. 2012;26:14D.11.11–14D.11.21. doi: 10.1002/9780471729259.mc14d01s26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sena-Esteves M., Gao G. Production of high-titer retrovirus and lentivirus vectors. Cold Spring Harbor Protoc. 2018;2018 doi: 10.1101/pdb.prot095687. [DOI] [PubMed] [Google Scholar]
- 88.Su Q., Sena-Esteves M., Gao G. Purification of recombinant adeno-associated viruses (rAAVs) by cesium chloride gradient sedimentation. Cold Spring Harbor Protoc. 2020;2020:095604. doi: 10.1101/pdb.prot095604. [DOI] [PubMed] [Google Scholar]
- 89.Kochenderfer J.N., Feldman S.A., Zhao Y., Xu H., Black M.A., Morgan R.A., Wilson W.H., Rosenberg S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cox A., Mueller C. Quantification of murine AAT by direct ELISA. Methods Mol. Biol. (Clifton, N.J.) 2017;1639:217–222. doi: 10.1007/978-1-4939-7163-3_21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.