

Summary
Spinal muscular atrophy (SMA) is a neurodegenerative disease caused by reduced survival motor neuron (SMN) protein. Recently, SMN dysfunction has been linked to individual aspects of motor circuit pathology in a severe SMA mouse model. To determine whether these disease mechanisms are conserved, we directly compared the motor circuit pathology of three SMA mouse models. The severe SMNΔ7 model exhibits vast motor circuit defects, including degeneration of motor neurons, spinal excitatory synapses, and neuromuscular junctions (NMJs). In contrast, the Taiwanese model shows very mild motor neuron pathology, but early central synaptic loss. In the intermediate Smn2B/- model, strong pathology of central excitatory synapses and NMJs precedes the late onset of p53-dependent motor neuron death. These pathological events correlate with SMN-dependent splicing dysregulation of specific mRNAs. Our study provides a knowledge base for properly tailoring future studies and identifies central excitatory synaptopathy as a key feature of motor circuit pathology in SMA.
Subject areas: Molecular biology, Neuroscience, Molecular neuroscience
Graphical abstract
Highlights
-
•
Comparison of detailed motor circuit pathology across three SMA mouse models
-
•
Motor circuit pathology correlates with dysregulation of specific mRNAs
-
•
Motor neuron death in severe and intermediate SMA models is p53-dependent
-
•
Central excitatory synaptopathy is the most conserved feature of SMA pathology
Molecular biology; Neuroscience; Molecular neuroscience
Introduction
Spinal muscular atrophy (SMA) is the second most common autosomal recessive disorder and most frequent genetic cause of infant mortality. The clinical phenotype of patients with SMA is determined by the disruption of motor circuits causing a proximo-distal progressing muscle atrophy, paralysis, and eventually death in severe cases (Dubowitz, 2009; Tisdale and Pellizzoni, 2015; Wirth, 2021). These impairments of the motor system are caused by homozygous deletion or mutation of the survival motor neuron 1 (SMN1) gene with the retention of the hypomorphic SMN2 gene leading to the ubiquitous deficiency of SMN protein. The copy number of SMN2 and the resulting decrease of functional SMN protein define the onset and severity of SMA in patients (Burghes and Beattie, 2009; Tisdale and Pellizzoni, 2015; Wirth, 2021).
Mouse models of SMA have been a key asset for our understanding of SMN-dependent disease mechanisms. Studies of SMA mouse models revealed that reduced SMN levels disrupt the assembly of spliceosomal small nuclear ribonucleoproteins (snRNPs) and U7 snRNP, which function in pre-mRNA splicing and 3′-end processing of histone mRNAs, respectively (Li et al., 2014; Tisdale and Pellizzoni, 2015; Zhang et al., 2008). SMN deficiency has also been reported to affect axonal transport of mRNAs, endocytosis, and translation among other cellular processes through less understood mechanisms (Donlin-Asp et al., 2016; Hosseinibarkooie et al., 2016; Lauria et al., 2020; Tisdale and Pellizzoni, 2015; Wirth, 2021). Dysregulated RNA splicing of specific genes has been shown to contribute to synaptic loss and motor neuron death in a severe mouse model of SMA (Lotti et al., 2012; Osman et al., 2020; Simon et al., 2017, 2019; Van Alstyne et al., 2018). However, whether these mechanisms, that directly link SMN dysfunction with individual aspects of motor circuit pathology, are conserved in other severe and intermediate forms of SMA mice or patients is unknown.
The most used SMA mouse models are the SMNΔ7, Taiwanese, and Smn2B/- lines. Based on their lifespan, the SMNΔ7 and Taiwanese lines are generally considered as severe models with 2 weeks of survival, while the intermediate Smn2B/- model survives up to four weeks (Ackermann et al., 2013; Bowerman et al., 2012; Hammond et al., 2010; Hsieh-Li et al., 2000; Le et al., 2005; Riessland et al., 2010). In contrast to patients with SMA, the overall lifespan of SMA mice is strongly impacted by the dysfunction of peripheral organs (Hamilton and Gillingwater, 2013; Iascone et al., 2015) and might not necessarily reflect the severity of motor circuit pathology, which include selective motor neuron death, denervation of neuromuscular junctions (NMJs), and loss of central proprioceptive synapses (Groen et al., 2018; Iascone et al., 2015; Shorrock et al., 2019; Tisdale and Pellizzoni, 2015; Wirth, 2021). Additionally, the reported degree of motor neuron loss varies from modest (∼15%) (Eshraghi et al., 2016; Le et al., 2005; Powis and Gillingwater, 2016) to severe (up to 80%) (Cervero et al., 2018; Mattis et al., 2009; Tsai et al., 2008) within each of the three SMA mouse models. These varying results may originate from different methodical approaches and the selective vulnerability of proximal muscle-innervating motor neuron pools, which to date has been described only in SMNΔ7 mice (Fletcher et al., 2017; Mentis et al., 2011; Simon et al., 2017). In accordance, the SMNΔ7 mouse model exhibits pronounced NMJ pathology predominantly in proximal muscles (Kong et al., 2021; Ling et al., 2012; Ruiz et al., 2010) that reliably reflects the proximo-distal disease progression in patients with SMA (Dubowitz, 2009). In contrast, the Taiwanese and Smn2B/- models exhibit only modest NMJ denervation (Bowerman et al., 2012; Courtney et al., 2019b; Lin et al., 2016). Moreover, while the degeneration and dysfunction of proprioceptive synapses onto motor neurons have been shown as one of the earliest pathological events in SMNΔ7 mice (Fletcher et al., 2017; Mentis et al., 2011; Simon et al., 2019; Vukojicic et al., 2019), they have only been investigated at end-stage in the other two models (Ackermann et al., 2013; Cervero et al., 2018; Hosseinibarkooie et al., 2016; Shorrock et al., 2018). Thus, a direct comparison of motor circuit pathology between mouse models with different severity forms of SMA has been missing.
We sought to address this issue by directly investigating the phenotypical, morphological, functional, and molecular alterations that occur in spinal motor circuits of the three SMA mouse models in order to identify shared and distinct features of SMA pathology. We identify variable degrees of selective motor neuron death and NMJ denervation across models with SMNΔ7 mice being most severely affected and Taiwanese generally spared. Intermediate Smn2B/- mice display strong NMJ pathology but minimal death of motor neurons that are limited to very late stages of disease. Nevertheless, we show that p53 is a key driver of SMA motor neuron death in severe and intermediate SMA mouse models. In contrast to the variable degree of motor neuron pathology, selective central excitatory synaptic degeneration, and dysfunction precedes motor neuron death consistently among all mouse models, identifying central synaptopathy as one of the earliest, most well conserved key components in the pathology across mild and severe forms of SMA.
Results
SMNΔ7 mice exhibit more severe motor dysfunction than Taiwanese and Smn2B/- mice
We sought to compare motor phenotype, functional, morphological, and molecular changes in spinal motor circuits of the three most used SMA mouse models: the severe SMNΔ7 (FVB background), Taiwanese (FVB and C57/Bl6 background), and intermediate Smn2B/- (C57/Bl6 background). First, we confirmed previously reported lifespan and body weight analysis (Ackermann et al., 2013; Bowerman et al., 2012; Le et al., 2005; Mentis et al., 2011): SMNΔ7 and Taiwanese (C57/Bl6) mutants die around postnatal day 11 (P11), whereas the Smn2B/- mice had an average life span of 24 days (Figure 1Ai). SMA mutants from all three mouse lines exhibited a strongly reduced body weight than control littermates detectable within the first (SMNΔ7, Taiwanese) and second postnatal week (Smn2B/-) (Figure 1Aii), suggesting vast muscle atrophy. To quantify motor phenotype defects in postnatal mice, we made daily measurements of the righting reflex that is mediated by vestibular pathways, spinal interneurons, proprioceptive afferents, and motor neurons (Bose et al., 1998; Mentis et al., 2011). We observed severe motor dysfunction in the SMNΔ7 mice (Figure 1Aiii), as has been previously reported (Le et al., 2005; Mentis et al., 2011). In contrast, the Taiwanese and Smn2B/- exhibited milder motor impairment, including a slightly increased righting time (Figure 1Aiii) and an unsteady gait at end-stage in agreement with other studies (Ackermann et al., 2013; Bowerman et al., 2012). To test whether the genetic background affects the SMA phenotype, we compared Taiwanese mutants on a C57/Bl6 and FVB background. Interestingly, unaffected control littermates of Taiwanese and Smn2B/- line on a C57/Bl6 background exhibited a delayed development of the righting reflex than the control littermates of FVB SMNΔ7 mice (Figure 1Aiii). A direct comparison of the C57/Bl6 and FVB Taiwanese controls demonstrated that animals on an FVB background show a faster acquisition of the righting reflex than on a C57/Bl6 background, while body weight was not affected (Figure S1B). The FVB background shortens the life span of Taiwanese mutants by 25% than C57/Bl6 mutants (Figure S1Ai) as reported before (Ackermann et al., 2013), but had no specific effect on body weight loss or motor phenotype of SMA mice (Figures S1Aii and S1Aiii). In summary, this phenotypic analysis reveals that all SMA mice show strong bodyweight reduction, but only the SMNΔ7 mouse model displays severe motor dysfunction.
Figure 1.

SMNΔ7 mouse model exhibits the most severe SMA motor phenotype
(A) Survival (i), body weight (ii) and righting time (iii) of SMNΔ7, Taiwanese, Smn2B/- mutants with control littermates. Statistics: Mantel-Cox test for survival (i), multiple t-test with Holm-Sidak method for body weight (ii) and righting time (iii); animal numbers (n) = SMNΔ7: control = 25, SMA = 14; Taiwanese: control = 19, SMA = 18; Smn2B/-: control = 27, SMA = 27.
(B) Western blot analysis of SMN (38 kDa) and tubulin (50 kDa, loading control) protein levels of P10 spinal cords from SMNΔ7, Taiwanese, Smn2B/- mutants.
(C) Normalized quantification (SMN/tubulin) of Western blot intensities from the same groups as in (B). Statistics: one-way ANOVA with Tukey’s correction. n values = 3 per genotype. Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We next determined the disease-causing levels of the SMN protein in spinal cords of all three SMA mouse models at P10 by Western blot analysis. As expected, all SMA mouse models exhibited a strong reduction of SMN compared with their control littermates (Figure S1C). Interestingly, the SMN levels in SMNΔ7 mutants were significantly lower in comparison to Taiwanese and Smn2B/- mice (Figures 1B and 1C), demonstrating that severe SMNΔ7 mice produce smaller amounts of SMN protein compared with the Taiwanese and intermediate Smn2B/- SMA mouse models. Western blot analysis of FVB and C57/Bl6 Taiwanese mutant mice revealed no differences in SMN levels (Figure S1C), excluding the possibility that the genetic background influences SMN expression. Taken together, these results show that the SMNΔ7 mouse model exhibits the most severe motor dysfunction that correlates with the strongest reduction of SMN levels than the Taiwanese and intermediate Smn2B/- mice.
Prominent motor neuron death is a unique feature of the severe SMNΔ7 mouse model
Degeneration of specific spinal motor neurons is the major hallmark of SMA. To evaluate the degree of motor neuron death in different spinal cord regions across mouse models, we quantified the number of choline acetyltransferase (ChAT)+ motor neurons in the whole thoracic T9, lumbar L1, and lumbar L5 spinal cord segments at the respective disease end-stage for all three SMA models (P11 for Taiwanese and severe SMNΔ7; P26 for intermediate Smn2B/-). T9, L1, and L5 medial motor neurons (MMNs) innervate axial and proximal muscles, which are severely affected in patients with SMA (Dubowitz, 2009; Iascone et al., 2015). In contrast, L5 lateral motor neurons (LMNs) innervate predominantly distal hindlimb muscles, which are less vulnerable in human SMA pathology (Dubowitz, 2009; Iascone et al., 2015). SMNΔ7 mice showed consistent loss of axial muscle innervating ChAT + T9, L1, and L5 MMNs with the highest death in L1, but no death of distal muscle innervating L5 LMNs at P11 (Figure 2) in agreement with previous reports (Mentis et al., 2011; Simon et al., 2017, 2019). End-stage P26 Smn2B/- SMA mice showed selective death of L1 motor neurons only (Figure 2). Notably, the soma size of these L1 motor neurons and the length of the L1 spinal segment was selectively reduced in Smn2B/- SMA mice at disease end-stage compared with control littermates (Figures S2A and S2B), suggesting a local pathology within the spinal cord. In contrast, we did not find any significant loss of motor neurons in the spinal cord of either C57/Bl6 or FVB Taiwanese mice at P11 (Figure 2; Figures S2C and S2D). Collectively, our analysis of four distinct spinal motor neuron pools reveals significant differences across the three SMA mouse models, with only the severe SMNΔ7 mouse model exhibiting robust, selective death of motor neurons innervating axial and proximal muscles.
Figure 2.

Prominent motor neuron death in proximal muscle-innervating spinal cord segments of the severe SMNΔ7 mouse model
(A) Immunostaining of T9 (upper panel), L1 (middle panel), and L5 (lower panel) ChAT+ motor neurons (green) of spinal cord sections from control, SMNΔ7, Taiwanese, and Smn2B/- mice at end-stage. Upper and middle panel scale bar = 100μm. Lower panel scale bar = 200μm.
(B-E) Quantification of (B) T9, (C) L1, (D) L5 lateral (LMN), and (E) L5 medial (MMN) motor neurons from the same groups as in (A). Statistics: two-tailed t-test. n values for T9 = SMNΔ7: control = 3, SMA = 3; Taiwanese: control = 3, SMA = 4; Smn2B/-: control = 3, SMA = 4. n values for L1 = SMNΔ7: control = 4, SMA = 3; Taiwanese: control = 7, SMA = 9; Smn2B/-: control = 3, SMA = 4. n values for L5 LMNs = 3 per group; n values for L5 MMNs = SMNΔ7: control = 5, SMA = 3; Taiwanese: control = 3, SMA = 3; Smn2B/-: control = 3, SMA = 3.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Analysis of splicing-dependent mechanisms of p53 activation in severe SMNΔ7 and intermediate Smn2B/- SMA mouse models
Several independent studies reported a strong activation of the p53 pathway in the spinal cord of severe SMNΔ7 and intermediate Smn2B/- mouse models (Baumer et al., 2009; Courtney et al., 2019b; Jangi et al., 2017; Murray et al., 2015; Reedich et al., 2021; Simon et al., 2017, 2019; Van Alstyne et al., 2018). We previously identified nuclear accumulation and phosphorylation of p53 as two distinct mechanisms that converge to trigger selective death of vulnerable motor neurons in the severe SMNΔ7 mouse model (Simon et al., 2017, 2019; Van Alstyne et al., 2018). To determine whether this converging pathway activation of p53 is restricted to the severe form or is commonly activated in SMA, we studied p53 protein expression in different spinal cord segments from all SMA models by immunohistochemistry. We found nuclear accumulation of p53 in ∼25% of the vulnerable L1 motor neurons but not in resistant L5 LMNs of SMNΔ7 mice at P1 (Figures 3A–3C). Perinatal Smn2B/- mutants already exhibited nuclear p53 expression in ∼20% of L1 motor neurons and ∼35% of L5 LMNs (Figures 3A–3C). This p53 nuclear expression increased during disease progression to ∼60% in L1 and T9 motor neurons, and throughout other spinal cord cells in both severe SMNΔ7 and intermediate Smn2B/- SMA models at end-stage, but not in their control littermates (Figures 3A–3C; Figures S3 and S4). In contrast, nuclear p53 accumulation was essentially absent throughout the spinal cord of Taiwanese mice on both backgrounds (Figures 3A–3C; Figures S2C, S3, and S4B). Consistent with the nuclear accumulation and activation of p53, the mRNA levels of p53-regulated genes induced by SMN deficiency (Van Alstyne et al., 2018) were upregulated in the spinal cord of the Smn2B/- and SMNΔ7 but not Taiwanese mice (Figures 3D–3F), while other established SMN-dependent targets such as Chodl mRNA expression and histone H1c 3ʹ end misprocessing events (Van Alstyne et al., 2018) were significantly altered in the spinal cords across SMA mouse models (Figures 3G and 3H). This indicates selective SMN-dependent p53 pathway activation in the intermediate Smn2B/- and severe SMNΔ7 mouse models which is consistent with the observed motor neuron loss.
Figure 3.

p53 nuclear accumulation correlates with snRNP dysfunction in severe and intermediate SMA mouse models
(A) Immunostaining of L1 (upper panel) and L5 (lower panel) ChAT+ motor neurons (green) and p53 (magenta) from control, SMNΔ7, Taiwanese, and Smn2B/- mice at end-stage. Insets show L5 MMNs in higher magnification. Scale bar: upper panel = 100μm, lower panel = 200μm, insets = 20μm.
(B-C) Percentage of p53+ (B) L1 and (C) L5 LMNs of SMNΔ7 at P1, P4, and P11, Taiwanese at P6, P11, and Smn2B/- mice at P1, P10, P22, and P26 with age-matched control littermates. Statistics: two-way ANOVA with Tukey’s correction. n values for p53+ L1 = SMNΔ7: all 3, but SMA P4: 5; Taiwanese: P6 control = 3, SMA = 5; P11 control = 4, SMA = 4; Smn2B/-: P1 control = 3, SMA = 3; P10 control = 3, SMA = 7; P22 control = 4, SMA = 5, P26 control = 4, SMA = 5. n values for p53+ L5 LMNs = SMNΔ7: all 3, but SMA P4: 5; Taiwanese: P11 control = 4, SMA = 3; Smn2B/-: 3 for each age and genotype.
(D-F) RT-qPCR analysis of p53 transcriptional targets (D) Perp, (E) Cdkn1a, and (F) Gtse1 of P10 spinal cords from SMNΔ7, Taiwanese, and Smn2B/- and pooled control mice at P10 and P26 spinal cords of control and mutant Smn2B/- mice.
(G) RT-qPCR analysis of Chodl, (H) histone H1c pre, and (I) p53 mRNA levels of P10 and P26 spinal cords from SMNΔ7, Taiwanese, Smn2B/- mutants with pooled control littermates. n values for mRNA levels P10 control = 6; SMNΔ7 = 4, except for Perp and Gtse1 = 3; Taiwanese = 4; Smn2B/- = 4. n values for mRNA levels P26 Smn2B/- for all n = 3, but Cdkn1a control = 4, SMA = 5.
Statistics: one-way ANOVA with Tukey’s correction. Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we were interested in the mechanisms of p53 induction. Consistent with previous results (Simon et al., 2017; Van Alstyne et al., 2018), the observed increase of p53 protein during disease progression occurs in the absence of changes in p53 mRNA expression at end-stage spinal cords of any SMA mouse model (Figure 3I). A previous study revealed that p53 upregulation in SMNΔ7 mice is caused by snRNP reduction and subsequent mis-splicing of Mdm2 and Mdm4 (two negative regulators of p53) induced by SMN deficiency (Van Alstyne et al., 2018). To explore whether snRNP reduction correlates with p53 upregulation in all mouse models, we quantified the nuclear intensity of SmB – an snRNP core protein whose reduction in the nucleus of SMA motor neurons is an in vivo readout of SMN-dependent disruption of snRNP assembly (Ruggiu et al., 2012; Tisdale and Pellizzoni, 2015) in vulnerable p53+ and p53- L1 motor neurons of SMA mice and controls at P11. Confocal analysis revealed that p53+ motor neurons of SMNΔ7 and Smn2B/- mutants had significantly lower nuclear levels of SmB than controls or SMA motor neurons lacking p53 induction (Figures 4A and 4B). In contrast, we found no SmB reduction in Taiwanese SMA motor neurons that also lack p53 upregulation (Figures 4A and 4B). Thus, these results correlate reduced snRNP levels with p53 activation in all three SMA mouse models.
Figure 4.

Mis-splicing events of Mdm2 and Mdm4 in SMNΔ7 and Smn2B/- mice
(A) Immunostaining of ChAT+ motor neurons (gray), SmB (magenta), and p53 (green) of L1 spinal segments from control, SMNΔ7, Taiwanese, and Smn2B/- mice at P11. Scale bar = 20μm.
(B) Normalized SmB fluorescence intensity of controls, SMNΔ7 p53+, SMNΔ7 p53-, Taiwanese, and Smn2B/- p53+ and Smn2B/- p53- L1 motor neurons at P11. Each point represents SmB fluorescent intensity of a single motor neuron. Data were collected from at least three mice per genotype with pooled controls. Statistics: one-way ANOVA with Tukey’s correction. n values for motor neurons with SmB: control = 131; SMNΔ7 p53- = 28; SMNΔ7 p53+ = 33, Taiwanese = 39; Smn2B/- p53- = 23; Smn2B/- p53+ = 22.
(C) RT-PCR of Mdm2 exon 3 splicing in P10 spinal cord from the same groups as in (A).
(D) Percentage of Mdm2 exon 3 inclusions of the same groups as in (A). Statistics: one-way ANOVA with Tukey’s correction.
(E) RT-PCR of Mdm2 exon 3 splicing in P26 spinal cord from controls and Smn2B/- mice.
(F) Percentage of Mdm2 exon 3 inclusions of the same groups as in (E). Statistics: two-tailed t-test.
(G) RT-PCR of Mdm4 exon 7 splicing of spinal cords at P10 from the same groups as in (A).
(H) Percentage of Mdm4 exon 7 inclusions of P10 spinal cords from the same groups as in (A). Statistics: one-way ANOVA with Tukey’s correction.
(I) RT-PCR analysis of Mdm4 exon 7 splicing of spinal cords from P26 control and mutant Smn2B/- mice.
(J) Percentage of Mdm4 exon 7 inclusions of P26 spinal cords from the same groups as in (I). Statistics: two-tailed t-test. n values for mRNA levels for E3 and E7 inclusions: P10 control = 6; SMNΔ7 = 3; Taiwanese = 4; Smn2B/- = 4. n values for mRNA levels P26 Smn2B/- for all n = 3.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we investigated changes in the levels of alternatively spliced forms of Mdm2 mRNA lacking exon 3 (Mdm2 Δ3) and Mdm4 mRNA lacking exon 7 (Mdm4 Δ7), which have been linked to p53 activation in SMNΔ7 mice (Van Alstyne et al., 2018). RT-PCR analysis of spinal cord tissue revealed an accumulation of Mdm2 Δ3 (Figures 4C–4F) and Mdm4 Δ7 (Figures 4G–4J) mRNAs in end-stage P10 severe SMNΔ7 and P26 intermediate Smn2B/- animals than controls, but not in presymptomatic Smn2B/- mice at P10 (Figures 4C, 4D, 4G, and 4H). A previous study of SMNΔ7 demonstrates that mis-splicing of Mdm2 and Mdm4 occurs earlier and to a much greater extent in motor neurons than in whole spinal cords (Van Alstyne et al., 2018). Consistent with our results (Figures S4A and S4B), increased levels of Mdm2 Δ3 and Mdm4 Δ7 can only be detected at the whole tissue level when p53 expression peaks throughout the spinal cord of SMA mice (Van Alstyne et al., 2018). Accordingly, we found no accumulation of Mdm2 Δ3 and Mdm4 Δ7 transcripts in end-stage Taiwanese mutants (Figures 4C, 4D, 4G, and 4H), which is consistent with the absence of both SmB reduction and p53 nuclear accumulation (Figures 3 and 4B; Figure S4). Taken together, our results suggest conservation of a pathomechanism by which SMN-dependent snRNP dysfunction causes Mdm2 and Mdm4 mis-splicing, leading to p53 nuclear accumulation in severe and intermediate SMA mouse models.
Phosphorylation of p53S18 marks degenerating motor neurons and p53 inhibition prevents their death in severe and intermediate SMA mouse models
We have previously demonstrated that p53 nuclear accumulation is necessary, but not sufficient to execute motor neuron death in the SMNΔ7 mouse model (Simon et al., 2017). Phosphorylation of specific p53’s amino-terminal residues is additionally required for this neurodegenerative process to occur. Importantly, phosphorylation of serine 18 (p-p53S18) of p53 selectively marks degenerating motor neurons in SMNΔ7 mice (Simon et al., 2017). To determine whether p-p53S18 correlated with degenerating motor neurons across SMA mice, we quantified p-p53S18 positive vulnerable L1 and resistant L5 LMNs in all three mouse models during the time course of disease. We confirmed that p-p53S18 accumulation precedes early loss of L1 motor neurons in SMNΔ7 mice at P4 (Figures 5A–5C). Similarly, the onset of p-p53S18 accumulation preceded the very late occurrence of motor neuron death in Smn2B/- mutant mice at P26, when p-p53S18 expression peaked (Figures 5A–5C). Interestingly, p-p53S18 was limited to vulnerable motor neurons and absent in other spinal cord cells and resistant L5 LMNs of both SMA models (Figures S5A–S5C). Furthermore, p-p53S18 accumulation was completely absent in the spinal cord tissue of the Taiwanese model during disease progression (Figures 5A–5C; Figures S5A–S5C), which is consistent with the lack of motor neuron death in this model.
Figure 5.

Phosphorylation of p53S18 consistently marks degenerating motor neurons and inhibition of p53 prevents their death
(A) Immunostaining of ChAT+ motor neurons (green) and p-p53S18 (magenta) of L1 hemicords (upper panel) and higher magnification of L1 motor neuron pools (lower panel) from control, SMNΔ7, Taiwanese, and Smn2B/- mice at end-stage. Scale bar = 100μm.
(B) Number of L1 motor neurons of SMNΔ7 at P1, P4, P11, Taiwanese at P6, P11, and Smn2B/- mice at P10, P22, and P26 with age-matched control littermates. Statistics: two-way ANOVA with Tukey’s correction. n values for L1 MNs = SMNΔ7: all 3, but SMA P4: 5; Taiwanese: P6 control = 3, SMA = 5; P11 control = 7, SMA = 9; Smn2B/-: P10 control = 5, SMA = 6; P22 control = 4, SMA = 5; P26 control = 3, SMA = 4.
(C) Percentage of p-p53S18+ L1 motor neurons of the same groups and ages as in (B). Statistics: two-way ANOVA with Tukey’s correction. n values for p-p53S18+ L1 MNs = SMNΔ7: all 3, but SMA P1: 6 and P4: 4; Taiwanese: P6 control = 3, SMA = 4; P11 control = 3, SMA = 3; Smn2B/-: P10 control = 3, SMA = 3; P22 control = 4, SMA = 3; P26 control = 4, SMA = 3.
(D) RT-qPCR analysis of Stasimon aberrant of P10 spinal cords from the same groups as in (A) and P26 spinal cords of control and mutant Smn2B/- mice. Statistics: one-way ANOVA with Tukey’s correction. n values for mRNA levels P10 control = 6; SMNΔ7 = 3; Taiwanese = 4; Smn2B/- = 4. n values for mRNA P26 = 3 for all.
(E) Immunostaining of ChAT+ L1 motor neurons (green) of control, SMA + DMSO, SMA + PFT from SMNΔ7 and Smn2B/- mice at end-stage. Scale bar = 100μm.
(F, G) Quantification of L1 motor neurons from (F) SMNΔ7 and (G) Smn2B/- line at end-stage. Statistics: one-way ANOVA with Tukey’s correction. n values for L1 MNs = SMNΔ7: control = 4; SMA+DMSO = 3; SMA+PFT = 4; Smn2B/-: control = 3; SMA+DMSO = 3; SMA+PFT = 4.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we performed RT-qPCR analysis for the aberrant splicing of the Stasimon gene (Lotti et al., 2012), a downstream target of SMN deficiency whose dysfunction has been linked to deafferentation as well as p53 phosphorylation and motor neuron death in SMNΔ7 SMA mice (Simon et al., 2019). Consistent with previous studies (Lotti et al., 2012; Osman et al., 2020), we found increased levels of aberrantly spliced Stasimon mRNA in SMNΔ7 and Smn2B/- spinal cord tissue at end-stage (Figure 5D). However, Stasimon mis-splicing was not found in presymptomatic Smn2B/- or end-stage Taiwanese mice at P10 (Figure 5D). Thus, we highlight a timely correlation of Stasimon dysregulation with the observed phosphorylation of p53S18 and motor neuron death. Together, these findings indicate that nuclear accumulation of p53 followed by its phosphorylation is restricted to vulnerable motor neurons and correlates with motor neuron death in both severe and intermediate SMA mouse models, suggesting p-p53S18 as a reliable marker for degenerating motor neurons in SMA.
It has been recently shown that p53 drives motor neuron death in the severe SMNΔ7 mice (Simon et al., 2017, 2019; Van Alstyne et al., 2018). Therefore, we tested whether motor neuron death in the intermediate Smn2B/- mouse model is also p53-dependent. To do so, we performed daily intra-peritoneal (i.p) administration of 2.2mg/kg Pifithrin-α (PFT)—a known chemical inhibitor of p53 transcriptional activity in vivo (Murphy et al., 2004) which has been recently proven to inhibit p53 downstream effectors in SMA mice (Simon et al., 2017, 2021)—to severe SMNΔ7 and intermediate Smn2B/- mice starting presymptomatically at P1 and P10, respectively. Chronic PFT treatment completely rescued the death of the vulnerable L1 motor neurons of end-stage severe SMNΔ7 mice in agreement with previous reports (Simon et al., 2017, 2021) (Figures 5E and 5F). In contrast to two recent studies (Courtney et al., 2019b; Reedich et al., 2021), p53 inhibition by PFT treatment also completely prevented L1 motor neuron loss in the intermediate Smn2B/− model (Figures 5E and 5G). In summary, our results demonstrate that postnatal inhibition of the p53 pathway prevents motor neuron death in both severe and intermediate SMA mice.
NMJ pathology and decreased nerve conduction velocity precede motor neuron death in Smn2B/- and SMNΔ7 mice
Another hallmark of SMA besides motor neuron death is NMJ denervation and dysfunction. Previous studies have investigated distinct subsets of muscles in individual SMA mouse models (Lin et al., 2016; Ling et al., 2012; Murray et al., 2008, 2015). However, a direct comparison of the denervation progression within the same muscles across different models has not been performed yet to unravel conserved features of NMJ pathology in SMA. Therefore, we evaluated the time course and degree of NMJ pathology in all three SMA models. We first analyzed the degree of NMJ denervation in the axial quadratus lumborum (QL) muscle and the distal tibialis anterior (TA) muscle, which are mostly innervated by vulnerable L1/L2 and resistant L4/L5 motor neurons, respectively (Fletcher et al., 2017; Kong et al., 2021; Simon et al., 2017, 2019). To do so, we applied antibodies against the axonal and presynaptic proteins neurofilament and synaptophysin, and α-bungarotoxin as a postsynaptic marker. Although no control QL muscle at any investigated time point showed signs of denervation in all mouse models, we found 10% fully denervated NMJs in the QL muscle of SMNΔ7 mice at P1, which progressively increased to ∼50% at P11 in end-stage mutants (Figures 6A and 6B). Smn2B/- mice exhibited no denervation at birth, underlining the intermediate SMA phenotype, but developed over 30% of fully denervated NMJ in the QL at presymptomatic stage (P10), which slightly increased until end-stage (Figures 6A and 6B). Previous studies reported a very mild beneficial effect of p53 inhibition on NMJ denervation (Courtney et al., 2019b; Simon et al., 2017). Interestingly, we found here that p53 inhibition by PFT treatment did not significantly improve NMJ innervation in SMNΔ7 and Smn2B/- mutants (Figures S5D and S5E), suggesting that the process of NMJ denervation is p53-independent. In contrast to the strong denervation found in SMNΔ7 and Smn2B/- mice, the QL muscle of Taiwanese mutants exhibited only a modest and variable degree of denervated NMJs at end-stage that did not reach statistical significance on either genetic background (Figures 6A and 6B; Figures S2E and S2F). Unlike the observed degenerative events in the QL, the distal TA muscle showed almost no signs of denervation in Taiwanese and SMNΔ7 mice, but ∼25% fully denervated NMJs in end-stage Smn2B/- mice (Figure 6C; Figure S6A). Collectively, these results identify the axial QL muscle as a vulnerable and the TA as a more resistant muscle across models, in agreement with their respective innervation of vulnerable L1 and resistant L5 motor neuron pools, and proximo-distal disease progression in patients with SMA (Dubowitz, 2009).
Figure 6.

Smn2B/- and SMNΔ7 mice exhibit vast NMJ denervation and dysfunction
(A) NMJ staining with bungarotoxin (BTX, red), synaptophysin (Syn, gray), and neurofilament (NF, green) in quadratus lumborum (QL) muscles from control, SMNΔ7, Taiwanese mice at P11, and Smn2B/- mice at P22. Scale bar = 50μm.
(B-C) Percentage of denervation of QL in (B) and tibialis anterior (TA) in (C) of SMNΔ7 at P1, P4, and P11, Taiwanese at P6, P11, and Smn2B/- mice at P1, P10, and P22. Statistics: two-way ANOVA with Tukey’s correction. n values for QL denervation = SMNΔ7: P1 control = 4, SMA = 5; P4 control = 3, SMA = 5; P11 control = 3, SMA = 4; Taiwanese: P6 control = 3, SMA = 3; P11 control = 3, SMA = 6; Smn2B/- = 3 for all groups and ages.
(D) Experiment setup of ex vivo extracellular compound motor action potential (CMAP) recording. L1 ventral root is sucked into the stimulation electrode and the recording electrode is inserted ∼2mm away into the QL.
(E) CMAP recordings from the QL muscle following L1 ventral root (VR) stimulation from the same groups as in (A) at P11. Black triangles indicate stimulation artifact and blue arrows indicate the time from stimulation to response (latency). Scale bars = 1mV, 2ms.
(F, G) Quantification of (F) CMAP amplitude and conduction velocity (CV) recorded from the QL muscle in the same groups as in (A) at P11. Statistics: two-tailed t-test. n values for CMAP and CV = SMNΔ7: control = 9 and SMA = 9; Taiwanese: control = 9, SMA = 8; Smn2B/-: control = 10, SMA = 7.
(H) Example trains of CMAP recordings at 50Hz stimulation from P11 QL muscles of the same groups as in (A). Blue boxes indicate first and tenth muscle response of each train. Scale bars = 0.5mV, 50ms.
(I) Percentage of CMAP amplitude change at 50Hz stimulation from P11 QL muscles of the same groups as in (A) at P11. Statistics: multiple t-test with Holm–Sidak method. Control group is pooled from controls of each individual line. n values for 50 Hz stimulation: control = 24; SMNΔ7 = 5; Taiwanese = 8; Smn2B/- = 7.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we assessed the functional motor output at vulnerable QL muscles at P10 by measuring the compound action potential (CMAP) and axonal conduction velocity (CV) following L1 ventral root stimulation (Figure 6D). CV can be approximated from the latency to the initiation of the CMAP and the distance between stimulating and recording electrodes because synaptic delay is unaltered in SMA mice as we recently reported (Kong et al., 2021). The CMAP response and CV in SMNΔ7 and Smn2B/- mutants showed a great reduction of CMAP amplitude (∼90%) and CV (∼50%) compared with control littermates (Figures 6E–6G). In agreement with our morphological findings, end-stage Taiwanese mutants did not display any reduction in NMJ function or CV (Figures 6E–6G). To test whether the decreased CMAPs are solely owing to NMJ denervation or also impaired transmission, we challenged the QL by applying a train of stimuli at the two frequencies of 10Hz or 50Hz. Control animals of all strains showed reliable responses with minor depression of CMAP during train stimulations (Figures 6H and 6I; Figures S6B and S6C). In contrast, SMNΔ7 and Smn2B/- mutant mice exhibited a ∼40% decline by the end of the stimulus train at both frequencies (Figures 6H and 6I; Figures S6B and S6C), revealing severe impairment of NMJ function in addition to denervation in both models. On the contrary, Taiwanese mutants produced increasing CMAP amplitudes following train stimulation of 50Hz, but not 10 Hz (Figures 6H and 6I; Figures S6B and S6C), suggesting frequency-dependent facilitation induced by altered presynaptic mechanisms that boost neurotransmitter release (Jackman and Regehr, 2017). Together, these data reveal a lack of NMJ pathology in Taiwanese mice, but indicate a correlation between prominent NMJ denervation, synaptic dysfunction, and CV reduction in SMNΔ7 and Smn2B/- mice. Notably, these observed severe NMJ dysfunction and denervation occur in presymptomatic P10 Smn2B/- mice already 2 weeks prior to the first signs of motor neuron death (Figure 5B), confirming NMJ pathology as one of the earliest events in SMA pathology.
Loss of excitatory central synapses precedes motor neuron death in all SMA mouse models
Recent studies identified a consistent reduction of proprioceptive synaptic numbers onto vulnerable motor neurons in all three SMA mouse models at end-stage (Ackermann et al., 2013; Cervero et al., 2018; Hosseinibarkooie et al., 2016; Mentis et al., 2011; Shorrock et al., 2018), suggesting an important role in SMA pathology. To determine when proprioceptive synapses degenerate during the disease progression, we investigated the timing and extent of vesicular glutamate transporter 1 (VGlut1) positive proprioceptive synaptic loss onto vulnerable L1 and resistant L5 LMNs in all three models. We found a continuous increase of proprioceptive synapses onto the soma and dendrites of L1 motor neurons in control animals of all lines during development, which was absent in SMNΔ7 mice as previously reported (Mentis et al., 2011), resulting in a significant reduction of ∼50% at P4 and ∼70% at end-stage (Figures 7A and 7B; and Figure S7A), which suggests either a developmental arrest or active elimination of SMA proprioceptive synapses (Vukojicic et al., 2019). Presymptomatic P6 Taiwanese and P10 Smn2B/- mutants exhibited an already mild reduced number of L1 proprioceptive synapses, which decreased further to ∼50% at end-stage (Figures 7A and 7B; Figure S7A), demonstrating an active elimination of proprioceptive synapses prior to motor neuron death. The genetic background did not influence the synaptic inputs (Figures S2G and S2H). In agreement, proprioceptive synapses on resistant L5 LMNs are moderately, but significantly, reduced at end-stage in all models (Figure 7C; Figure S7B), suggesting initial synaptic pathology of the sensory-motor circuit in resistant spinal segments. Importantly, chronic p53 inhibition did not prevent proprioceptive synaptic loss (Figures S5F and S5G), separating motor neuron death from central synaptic degeneration in SMA motor circuits.
Figure 7.

Degeneration of central excitatory synapses precedes motor neuron death in all SMA mouse models
(A) Immunostaining of VGluT1+ synapses (magenta) and ChAT+ motor neurons (green) of control, SMNΔ7, Taiwanese at P11 and Smn2B/- mice at P22. Scale bar = 20μm.
(B) Number of VGluT1+ synapses on L1 motor neuron somata of SMNΔ7 P1, P4, and P11, Taiwanese at P6, P10, and Smn2B/- mice at P1, P10, and P22. Statistics: two-way ANOVA with Tukey’s correction. n values for L1 VGluT1+ = 3 for all lines and ages.
(C) Number of VGluT1+ synapses on L5 lateral motor neuron somata of SMNΔ7 and Taiwanese at P11, and Smn2B/- mice at P22. Statistics: two-tailed t-test. n values for L5 LMN VGluT1+ = 3 for all lines and ages, except Taiwanese P11 control = 4.
(D) Immunostaining of SmB+ (magenta) and parvalbumin+ (Parv, green) proprioceptive neurons of the L1 dorsal root ganglia (DRG) of the same groups as in (A) at P11. Scale bar = 20μm.
(E) Normalized SmB fluorescence intensity of the same groups as in (A) at P11. Each point represents SmB fluorescent intensity of a single proprioceptive neuron. Data were collected from three mice of each genotype and pooled controls. Statistics: one-way ANOVA with Tukey’s correction. n values for proprioceptive neurons with SmB: control = 93; SMNΔ7 = 50; Taiwanese = 29; Smn2B/- = 25.
(F) Quantification of RT-qPCR of Stasimon aberrant in DRGs of end-stage SMNΔ7, Taiwanese and Smn2B/- mice. Statistics: two-tailed t-test. n values for Stasimon mRNA = 3 per genotype.
(G) Immunostaining of ChAT+ motor neurons (gray) and C-Boutons (white) of L1 spinal cord segments from control, SMNΔ7, Taiwanese at P11 and Smn2B/- mice at P22. Scale bar = 20μm.
(H-I) Number of C-Boutons on (H) L1 motor neuron and L5 lateral motor neuron somata (I) of the same groups at end-stage as in (G). Statistics: two-tailed t-test. n values for C-bouton of L1 and L5 LMN = 3 per genotype.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Previous studies in SMNΔ7 mice implicated Stasimon dysfunction induced by SMN deficiency through the dysregulation of U12 splicing in the loss of afferent synapses that occurs cell autonomously in proprioceptive neurons (Fletcher et al., 2017; Lotti et al., 2012; Osman et al., 2020; Simon et al., 2019). To further evaluate this pathway as a general pathomechanism in SMA, we analyzed the effect of SMN deficiency on the accumulation of spliceosomal snRNPs in the nucleus of proprioceptive neurons of L1 dorsal root ganglions (DRGs) using antibodies against SmB and Parvalbumin, respectively. Importantly, we found strongly reduced snRNP levels monitored by SmB immunostaining in proprioceptive neurons as well as increased amounts of aberrantly spliced Stasimon mRNA in DRGs from all three SMA mouse lines than control littermates (Figures 7D–7F). These results are consistent with splicing-dependent Stasimon dysfunction induced by SMN deficiency as a common pathomechanism that contributes to the degeneration of proprioceptive synapses in SMA.
To decipher whether other types of central synapses are affected in SMA, we quantified excitatory C-boutons onto motor neurons, which contribute to motor function and derive from cholinergic Pitx2 cells located next to the spinal central canal (Zagoraiou et al., 2009). We found that ChAT + C-Boutons were consistently reduced on resistant and vulnerable motor neurons of SMA mutants from all models (Figures 7G–7I), while the number of cholinergic Pitx2 neurons are unaltered (Figures S7C and S7D). Next, we asked whether inhibitory synapses are equally affected. Therefore, we investigated the number of glycinergic and GABAergic synapses onto vulnerable L1 motor neurons by applying antibodies against vesicular GABA transporter (VGAT), a marker for both types of inhibitory synapses (Chaudhry et al., 1998). The number of inhibitory synapses onto motor neurons was unaltered in all SMA mouse models than control littermates at end-stage (Figures S7E and S7F). Taking into account that the number of proprioceptive neurons (Mentis et al., 2011), and Pitx2+ cells as sources of proprioceptive synapses and that C-boutons are resistant to death (Figures S7C and S7D), these results demonstrate a selective excitatory synaptopathy in SMA motor circuits independent of neuronal loss.
Impaired central synaptic neurotransmission affects motor neuron function in all SMA models
To examine the function of the sensory-motor circuit, we measured the monosynaptic ventral root response ex vivo in all models at P11 following the stimulation of the homonymous L1 dorsal root (Figure 8A). This type of ex vivo recordings cannot be performed beyond the second postnatal week owing to insufficient oxygenation of the ex vivo spinal cord. Therefore, this approach was used to investigate the sensory-motor pathology of SMNΔ7 and Taiwanese at end-stage, and that of Smn2B/- mice at presymptomatic stages. As previously described (Mentis et al., 2011; Simon et al., 2017, 2019), the amplitude of the ventral root response which is regulated by the sum of the number and function of proprioceptive synapses and motor neurons participating in the reflex response, was reduced in SMNΔ7 mice by ∼90% compared with control littermates (Figures 8B and 8C), reflecting the strong sensory-motor circuit pathology of this severe SMA mouse model. In contrast, P11 Taiwanese and Smn2B/- mutants exhibited normal ventral root amplitudes (Figures 8B and 8C; black traces of first response), suggesting normal sensory-motor function in both models following a single supramaximal stimulus. However, proprioceptive information in vivo is encoded by repetitive firing. Therefore, we tested proprioceptive neurotransmission under physiological conditions by repetitive stimulation at different frequencies. Although the percentage change in the ventral amplitude following five consecutive stimuli of 0.1Hz remained constant in all models (Figures S8A and S8B), 10Hz stimulation decreased the amplitude in control proprioceptive synapses by ∼50% (Figure 8B, red traces of fifth response and 8D). P11 SMNΔ7 and Taiwanese proprioceptive synapses showed a further significant depression of more than 70% and 80%, respectively (Figures 8B and 8D). Presymptomatic Smn2B/- mutants exhibited a non-significant tendency of synaptic depression at P11 (Figures 8B and 8D), indicating the onset of synaptic dysfunction. In summary, these results indicate that dysfunction accompanies the loss of proprioceptive synapses and both precede motor neuron death in SMA.
Figure 8.

Impaired proprioceptive synaptic function in all SMA models
(A) Experimental setup for ex vivo spinal cord preparation. Extracellular stimulation of L1 dorsal root (DR) and extracellular recording from L1 ventral root (VR).
(B) Representative traces of the first (black) and fifth (red) VR responses recorded at P11 following stimulation of the homonymous L1 DR at 10 Hz from control, SMNΔ7, Taiwanese at P11, and Smn2B/- mice at P11. Scale bars = 0.5mV and 3ms.
(C) Quantification of VR response in mV following stimulation of the homonymous L1 DR at 10Hz from the same groups as in (B) at P11. Statistics: two-tailed t-test. n values for VR response: SMNΔ7: control = 6, SMA = 7; Taiwanese: control = 4, SMA = 4; Smn2B/-: control = 3, SMA = 4.
(D) Quantification of amplitude changes in percent of the monosynaptic ventral root response following 10Hz stimulation at P11 from the same groups as in (B). Control groups were pooled from individual lines. n values for 10Hz stimulation: control = 12; SMNΔ7 = 7; Taiwanese = 5; Smn2B/- = 4. Statistics: multiple t-test with Holm–Sidak method.
(E) Single optical plane confocal images of L1 ChAT+ motor neurons (blue) expressing Kv2.1 channels (gray) from the same groups as in (B) at P11. Scale bar = 20μm.
(F) Percentage of somatic coverage of Kv2.1 expression in L1 motor neurons for the same groups as in (B) at P11. Statistics: two-tailed t-test. n values for Kv2.1 analysis = 3 for each genotype.
Data are presented as mean ± SEM. Asterisks on top of bars without horizontal line indicate significance compared with the control group.
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Finally, we asked whether the reduced excitatory drive affects motor neuron function. We have previously shown that excitatory synaptic drive controls the incorporation of the potassium channel Kv2.1 into the membrane of motor neurons, thereby shaping their ability to fire repetitively (Fletcher et al., 2017). The expression of Kv2.1 in motor neurons was reduced by ∼50% compared with littermate control in all P11 SMA mouse models, indicating shared mechanisms of impaired motor neuron function through reduced excitatory synaptic drive (Figures 8E and 8F), independent of the genetic background (Figures 2I and 2J). Interestingly, while the life span of FVB Taiwanese mice is shortened by 25%, compared with a C57/Bl6 background (Figures S1A and S1B), the motor circuit pathology is not worsened (Figures S2C–S2J), indicating that the life span of SMA mice is limited by the pathology of peripheral organs (Hamilton and Gillingwater, 2013; Iascone et al., 2015). Taken together, early selective degeneration of excitatory synapses prior to motor neuron death is the most constant pathological feature among all SMA mouse models, suggesting central excitatory synaptopathy as a key component for motor circuit pathology in SMA.
Discussion
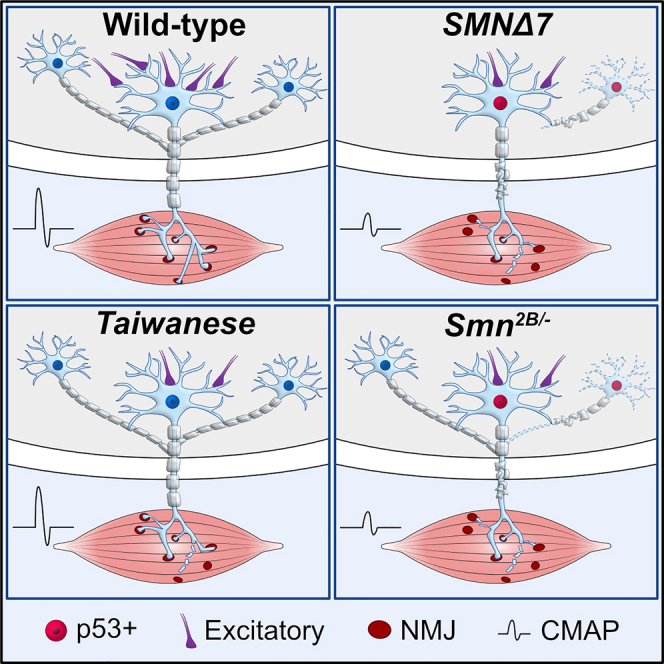
This study describes the first direct comparison of molecular, morphological, and functional changes that occur in severe and intermediate mouse models of SMA to identify conserved mechanisms and temporal series of events that underlie motor circuit pathology (graphical abstract). We identified a variable degree of NMJ pathology and p53-dependent motor neuron death across SMA mouse models. In contrast, early central excitatory synaptic degeneration occurs consistently prior to motor neuron death among all mouse models. Importantly, the progression of motor neuron degeneration and proprioceptive synaptic loss correlates with a reduction in snRNP levels and splicing dysregulation of specific mRNAs that have been previously shown to contribute to these motor circuit defects in SMNΔ7 mice (Simon et al., 2017, 2019; Van Alstyne et al., 2018). Taken together, our study reveals shared and distinct features of motor circuit pathology across different SMA mouse models which will be helpful for designing future studies. Furthermore, it reveals a general mechanism of central excitatory synaptopathy preceding p53-dependent motor neuron death in severe and intermediate forms of SMA.
One major hallmark of severe SMA is the death of motor neurons (Kong et al., 2021). In SMNΔ7 mice, we found significant motor neuron death shortly after birth that increased up to 50% in proximal muscle-innervating spinal segments at end-stage, while distal muscle innervating-motor neuron pools exhibited no loss, consistent with previous studies (Butchbach et al., 2010; Fletcher et al., 2017; Kong et al., 2021; Le et al., 2005; Martinez et al., 2012; Mentis et al., 2011; Rose et al., 2009; Simon et al., 2019, 2021; Van Alstyne et al., 2018). In contrast, we found no motor neuron loss in the Taiwanese model and only minor death of a restricted group of motor neurons of the intermediate Smn2B/- model occurring past the median survival, suggesting that motor neuron death is not a prerequisite to developing an SMA phenotype. These results identify SMNΔ7 as the only mouse model with a robust and selective profile of motor neuron death that resembles the vulnerability observed in patients with SMA. Mechanistically, we previously identified p53 activation as the main driver of motor neuron death in the severe SMNΔ7 model (Simon et al., 2017; Simon et al., 2019; Van Alstyne et al., 2018). In agreement, we detect strong and selective nuclear accumulation of p53 perinatally in vulnerable motor neurons, which progressively extends to all motor neuron pools and other spinal cells in Smn2B/- and SMNΔ7 mice. In contrast, p53 induction is absent in Taiwanese and control mice. We have previously demonstrated that p53 nuclear accumulation is necessary, but requires amino-terminal phosphorylation to execute motor neuron death in the SMNΔ7 mouse model (Simon et al., 2017, 2019). One of these phosphorylation sites, p-p53S18, exclusively marks vulnerable motor neurons at time point of death in Smn2B/- and SMNΔ7 mice, while completely absent in resistant motor neuron pools, corroborating the requirement of p53 phosphorylation as a common mechanism for motor neuron death in SMA. We demonstrate here that SMN-dependent snRNP assembly is significantly reduced in motor neurons of Smn2B/- similar to SMNΔ7 mice (Ruggiu et al., 2012; Van Alstyne et al., 2018), but not in Taiwanese mice. This suggests that although overall SMN levels in the spinal cords of the Taiwanese model are significantly reduced, motor neurons might express sufficient SMN for correctly performing snRNP assembly and alternative splicing to prevent motor neuron death. The short life span of the Taiwanese mice might be explained by SMN-dependent defects in peripheral organs including lung, intestine, and heart (Hamilton and Gillingwater, 2013; Hosseinibarkooie et al., 2016; Schreml et al., 2013). In contrast, we correlate SMN-dependent splicing dysregulation of Mdm2, Mdm4, and Stasimon mRNA with the accumulation and phosphorylation of p53 in both Smn2B/- and SMNΔ7 mice, pointing to conserved mechanisms of motor neuron death in intermediate and severe forms of SMA (Lotti et al., 2012; Simon et al., 2017, 2019; Van Alstyne et al., 2018).
While our results that the pharmacological inhibition of p53 suppresses motor neuron death in both severe SMNΔ7 and intermediate Smn2B/- models of SMA confirm and extend previous studies pointing to the involvement of this pathway in the neurodegenerative process (Simon et al., 2017, 2019, 2021; Van Alstyne et al., 2018), they contrast with the conclusions of recent studies in the Smn2B/- model (Courtney et al., 2019b; Reedich et al., 2021). One of the latter studies reported no rescue effect on motor neuron death by introducing p53 knockout alleles in Smn2B/- mice (Reedich et al., 2021). However, motor neurons located in lower lumbar L4-L6 spinal segments that are resistant to degeneration were investigated. Furthermore, the analysis was performed at a time point (P16) when we did not detect motor neuron loss in any spinal level of C57/Bl6 Smn2B/- mice, a finding confirmed in end-stage Smn2B/- mice on an FVB background (personal communication with Dr. Livio Pellizzoni). The other study used an Smn2B/- model with tamoxifen-inducible postnatal p53 removal in which p53 mRNA levels were only mildly decreased and induction of p53 target genes was not prevented (Courtney, 2019a, 2019b), revealing ineffective inhibition of the p53 pathway. Together, our findings suggest that the dysregulation of alternative splicing induces p53 pathway activation to execute motor neuron death in SMA (Jangi et al., 2017; Nichterwitz et al., 2020; Simic et al., 2000) and possibly other motor neuron diseases (Maor-Nof et al., 2021; Vogt et al., 2018).
In contrast to the variable degree of motor neuron death and NMJ denervation, we found consistent loss of proprioceptive synapses in end-stage SMA mouse models as previously reported (Ackermann et al., 2013; Cervero et al., 2018; Mentis et al., 2011; Shorrock et al., 2018), suggesting that proprioceptive loss is one of the earliest events in SMA pathology. Additionally, we found proprioceptive synaptic dysfunction in all mouse models as reported for SMNΔ7 mice (Fletcher et al., 2017; Mentis et al., 2011; Simon et al., 2017, 2021). However, it has not been clear whether the reduction of synapses is owing to developmental arrest (Tarabal et al., 2014) or active elimination (Mentis et al., 2011; Vukojicic et al., 2019). We demonstrate here that proprioceptive synapses onto motor neurons develop normally, but actively degenerate first in proximal and later in lateral motor circuits before motor neuron death in SMNΔ7 and Smn2B/- SMA models. Motor neuron rescue does not prevent synaptic loss in SMNΔ7 and Smn2B/- mice, confirming that central premotor synapses degenerate in a cell-autonomous manner independent of motor neuron pathology (Fletcher et al., 2017; Imlach et al., 2012; Simon et al., 2016, 2017; Van Alstyne et al., 2018). As previously reported (Cervero et al., 2018; Ling et al., 2010), other excitatory inputs such as cholinergic C-boutons are also consistently reduced, but strikingly inhibitory synapses are not affected throughout mouse lines in our study, demonstrating selective impaired excitatory drive onto motor neurons across mild and severe forms of SMA. Excitatory synaptic drive shapes motor neuron function by potassium channel Kv2.1 incorporation in the membrane (Fletcher et al., 2017). In agreement, we correlate here impaired synaptic drive with reduced Kv2.1 expression in motor neurons in all models. Logically, pharmacological increase of neuronal activity improves motor circuit connectivity and function in SMA mice (Simon et al., 2021). We show here that Pitx2 ChAT+ neurons as sources of cholinergic C-boutons are commonly resistant to death, as previously shown for SMNΔ7 mice (Powis and Gillingwater, 2016). Together with the resistance of proprioceptive neurons to degeneration (Ling et al., 2010; Mentis et al., 2011), this demonstrates a clear central excitatory synaptopathy independent of neuronal death. The mechanisms of synaptic degeneration are not completely understood, but recent studies highlighted the involvement of the UBA1/GARS pathway (Shorrock et al., 2018), Plastin (Ackermann et al., 2013), and Stasimon dysfunction in proprioceptive neurons (Lotti et al., 2012; Osman et al., 2020; Simon et al., 2019), all of which might converge in a yet-to-be-defined pathomechanism that also involves the activation of the classical complement system (Vukojicic et al., 2019). We show here a strong correlation between dysregulated Stasimon splicing and reduced snRNP levels in proprioceptive neurons in DRGs of all mouse models, corroborating the link between snRNP dysfunction and Stasimon-mediated excitatory synaptopathy (Lotti et al., 2012; Osman et al., 2020; Simon et al., 2019). Our results suggest that central synapses are one of the most vulnerable components of SMN-dependent motor circuit pathology across mouse models, suggesting a conserved feature in patients with SMA with different forms of severity.
Besides central synaptopathy, NMJ pathology is one of the earliest events in SMA. We describe here early progressive denervation of proximal muscles prior to motor neuron death that extends later to distal muscles of SMNΔ7 and Smn2B/- mice which reflect the proximo-distal progression of SMA found in humans (Dubowitz, 2009; Iascone et al., 2015; Ling et al., 2012). The Taiwanese model exhibit very mild NMJ denervation in agreement with previous studies, which additionally report reduced NMJ area size and immaturity (Hosseinibarkooie et al., 2016; Lin et al., 2016; Riessland et al., 2017). As NMJ denervation precedes motor neuron death and both underlie motor neuron-autonomous mechanisms (Fletcher et al., 2017; Martinez et al., 2012; Simon et al., 2016), a postulated “dying-back” hypothesis suggests that the lack of retrograde muscle signals following denervation initiates motor neuron degeneration (Dadon-Nachum et al., 2011). Interestingly, we show here that denervation in SMNΔ7 mice is already present at birth and in Smn2B/- mice at P10, but first signs of motor neuron death take place approximately one and two weeks later, respectively in these models. Previous work reported very minor (∼10%) improvement of NMJ denervation following p53 inhibition/ablation (Courtney et al., 2019b; Simon et al., 2017). We found here that p53 has no significant effect on NMJ denervation in agreement with a recent study (Van Alstyne et al., 2018). This shows that p53 plays a very minor role at best in NMJ pathology, in contrast to its strong contribution to motor neuron death, suggesting two independent pathological events. In agreement, replenishment of mis-spliced agrin—a critical organizer of the NMJ—improves denervation, but not motor neuron death (Kim et al., 2017; Zhang et al., 2013). In addition, other vulnerable excitatory neurons, such as cholinergic Pitx2 neurons and proprioceptive neurons exhibit strong synaptic loss without cell death. All these findings argue that motor neuron-intrinsic yet mechanistically independent events drive NMJ denervation and motor neuron death. Our study clearly correlates the cell autonomous events of denervation with NMJ dysfunction and reduced conduction velocity owing to defective axon ensheathment and myelination in SMA mice and patients (Fletcher et al., 2017; Kong et al., 2021; Martinez et al., 2012; Ruiz et al., 2010).
In conclusion, our study reveals common and distinct features of motor circuit pathology across mouse lines, underlining the strength and limitations of each mouse model (graphical abstract). The SMNΔ7 model exhibits the most severe motor circuit pathology. In contrast, the Taiwanese model shows the mildest pathology of motor circuits with minimal motor neuron pathology, but early central synaptopathy, despite a similar survival rate as the SMNΔ7 mice, strengthening the relevance of spinal circuits in SMA. The intermediate Smn2B/- model displays very restricted late occurring p53-dependent motor neuron death with preceding NMJ pathology and central synaptopathy. Our study identifies central excitatory synaptopathy as the most vulnerable component of motor circuit pathology preceding p53-dependent motor neuron death across different severity forms of SMA.
Limitations of the study
It has been demonstrated that Stasimon induces p53-dependent motor neuron death and proprioceptive synaptic loss in the SMNΔ7 mouse model. Although we show here a strong timely correlation of aberrant splicing of Stasimon and both pathological events across all mouse models, we did not demonstrate a causal effect of Stasimon in the Taiwanese and Smn2B/- model because it would have exceeded the scope of this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SMN clone 8 | BD Transd Lab | 610646, RRID:AB_397973 |
| Tubulin DM1A | Sigma | T9026, RRID:AB_1965960 |
| p53 | Leica Novocastra | NCL-p53-CM5p, RRID:AB_563933 |
| p-p53S15 | Cell Signaling | 9284 (Lots: 12,15), RRID:AB_331464 |
| VGluT1 | Synaptic Systems | 135 304, RRID:AB_887878 |
| Synaptophysin | Synaptic Systems | 101-004, RRID:AB_1210382 |
| Neurofilament | Millipore | AB1987, RRID:AB_91201 |
| ChAT | Millipore | AB144P, RRID:AB_2079751 |
| Bungarotoxin | Invitrogen | B35451, RRID:AB_2617152 |
| Kv2.1 | Neuromab | K89/34 concent., RRID:AB_2877280 |
| Parvalbumin | Synaptic Systems | 195 004, RRID:AB_2156476 |
| SmB | Santa Cruz | Sc-130670, RRID:AB_2193856 |
| VGAT | Synaptic Systems | 131004, RRID:AB_887873 |
| 488 Anti-rabbit | Jackson | 711-545-152, RRID:AB_2313584 |
| 488 Anti-goat | Jackson | 705-545-147, RRID:AB_2336933 |
| 488 Anti-chicken | Jackson | 703-545-155, RRID:AB_2340375 |
| Cy3 Anti-rabbit | Jackson | 711-165-152, RRID:AB_2307443 |
| Cy3 Anti-mouse | Jackson | 715-165-150, RRID:AB_2340813 |
| Cy3 Anti-goat | Jackson | 705-165-147, RRID:AB_2307351 |
| Cy5 Anti-goat | Jackson | 705-175-147, RRID:AB_2340415 |
| Cy5 Anti-guinea pig | Jackson | 706-175-148, RRID:AB_2340462 |
| HRP Anti-mouse | Jackson | 115-035-044, RRID:AB_2338503 |
| HRP Anti-rabbit | Jackson | 111-035-003, RRID:AB_2313567 |
| Chemicals, peptides, and recombinant proteins | ||
| Pifithrin-α | Merck | P4359 |
| Experimental models: Organisms/strains | ||
| SMNΔ7 | Jackson | #005025 |
| Taiwanese | Jackson | #005058 |
| Smn2B/- | Bowerman et al. (2012) | Provided by Dr. Kothari |
| Oligonucleotides | ||
| RT-qPCR p53 | Simon et al. (2017) | N/A |
| RT-qPCR Cdkn1a | Ruggiu et al. (2012) | N/A |
| RT-qPCR Perp1 | Van Alstyne et al. (2018) | N/A |
| RT-qPCR Gtse1 | Van Alstyne et al. (2018) | N/A |
| RT-qPCR Stas Aber | Lotti et al. (2012) | N/A |
| RT-qPCR H1c pre | Van Alstyne et al., 2018 | N/A |
| RT-qPCR Chodl | Van Alstyne et al. (2018) | N/A |
| RT-qPCR Gapdh | Ruggiu et al. (2012) | N/A |
| RT-PCR Mdm2 E2-5 | Van Alstyne et al. (2018) | N/A |
| RT-PCR Mdm4 E5-8 | Van Alstyne et al. (2018) | N/A |
| PCR primers for genotyping SMNΔ7 | Simon et al. (2017) | N/A |
| PCR primers for genotyping Taiwanese | Riessland et al. (2010) | N/A |
| PCR primers for genotyping Smn2B/- | Bowerman et al. (2012) | N/A |
| Software and algorithms | ||
| LAS X | Leica | N/A |
| Prism 9 | GraphPad | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Christian M. Simon (Christian.Simon@medizin.uni-leipzig.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Breeding and experiments were performed in the animal facilities of the Faculty of Medicine, University of Leipzig and University of Cologne according to European (Council Directive 86/609/EEC) and German (Tierschutzgesetz) guidelines for the welfare of experimental animals and the regional directorate (Landesdirektion) of Leipzig. Mice were housed in a 12h/12h light/dark cycle with access to food and water ad libitum. The original breeding pairs for SMNΔ7 (Smn+/-; SMN2+/+; SMNΔ7+/+) mice (stock #005025) on FVB background were obtained from Jackson Laboratory. Taiwanese SMA mouse model were originally purchased from the Jackson Laboratory on pure FVB background (FVB.Cg-Tg(SMN2)2Hung Smn1tm1Hung/J, stock #005058). Additionally, these mice were congenitally backcrossed for seven generations to C57Bl/6N background (Ackermann et al., 2013) and bred accordingly to give 50% SMA: 50% heterozygous offspring in each litter (Riessland et al., 2010). The Smn2B/- mice on a C57BL6 background were generated by the Kothary lab (Bowerman et al., 2012) and provided by Dr. Kothary and Dr. Raoul following the breeding scheme (Bowerman et al., 2012). Primers for genotyping: SMNΔ7: forward sequence (5′ to 3′) = GATGATTCTGACATTTGGGATG, reverse sequences (5′ to 3′) = TGGCTTATCTGGAGTTTCACAA and GAGTAACAACCCGTCGGATTC (wild-type band: 325bp, mSmn ko: 411bp). Taiwanese: forward sequences (5′ to 3′) = ATAACACCACCACTCTTACTC and GTAGCCGTGATGCCATTGTCA, reverse sequences (5′ to 3′) = AGCCTGAAGAACGAGATCAGC (wild-type band: 1050bp, mSmn ko: 950bp). Smn2B/-: forward sequence (5′ to 3′) = TTTGGCAGACTTTAGCAGGGC, reverse sequence (5′ to 3′) = AACTCCGGGTCCTCCTTCCT (wild-type band: 500bp, mutant: 700bp). Control animals were littermates of mutants from each individual SMA mouse line. The controls were for Taiwanese = Smn+/-;SMN2tg/+ (Riessland et al., 2010), for Smn2B/- line = Smn2B/+ (Bowerman et al., 2012) and SMNΔ7 = Smn+/+; SMN2+/+; SMNΔ7+/+ (Le et al., 2005). Mice from all experimental groups were monitored daily, body weight measurements and the righting reflex were timed and averaged as established previously (Mentis et al., 2011). Mice with a 25% reduction of body weight and an inability to right were euthanized to comply with German guidelines for the welfare of experimental animals. Righting time was defined as the time for the pup to turn over on all its four limbs after being placed on its back. The cut-off test time for the righting reflex was 60s to comply with German guidelines for the welfare of experimental animals. Approximately equal proportions of mice of both sexes were used and aggregated data are presented since gender-specific differences were not found nor have they been previously reported.
Method details
Injections
Pifithrin-α (PFT) was dissolved in DMSO and delivered daily at a concentration of 2.2mg/kg by IP injection starting at P0.
Electrophysiology
To record the dorsal root to ventral root monosynaptic response, we conducted the experiment as previously established (Mentis et al., 2011; Simon et al., 2017): The animals were decapitated, the spinal cords dissected and removed under cold (∼12°C) artificial cerebrospinal fluid (aCSF) containing 128.35mM NaCl, 4mM KCl, 0.58mM NaH2PO4.H20, 21mM NaHCO3, 30mM D-Glucose, 1.5mM CaCl2.H20, and 1mM MgSO4.7H20. The spinal cord was then transferred to a customized recording chamber. The intact ex vivo spinal cord preparation was perfused continuously with oxygenated (95%O2/5%CO2) aCSF (∼13 mL/min). The dorsal root and ventral root of the L1 spinal segment were placed into suction electrodes for stimulation or recording respectively. The extracellular recorded potentials were recorded (DC – 3kHz, Cyberamp, Molecular Devices) in response to a brief (0.2ms) orthodromic stimulation (A365, current stimulus isolator, WPI, Sarasota, FL) of the L1 dorsal root. The stimulus threshold was defined as the current at which the minimal evoked response was recorded in 3 out of 5 trials. Recordings were fed to an A/D interface HEKA EPC10/2 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) and acquired with HEKA Patchmaster (HEKA Electronics) amplifier at a sampling rate of 10 kHz. Data were analyzed offline using HEKA Patchmaster (HEKA Electronics). The temperature of the physiological solution ranged between 21 and 23°C. Synaptic depression experiments were performed and analyzed for all experimental groups at P11. The dorsal root was stimulated at 0.1Hz or 10Hz for five stimuli and the resulting monosynaptic component of the amplitude recorded and analyzed offline. The amplitude of the second to fifth stimuli was expressed as a percentage of the amplitude to the first stimulus.
To assess functionally NMJs of the QL muscle at P11, the motor neuron axons in the ventral root L1 supplying the QL muscle were stimulated by drawing the ventral root into a suction electrode, having removed the spinal cord, and recorded the compound muscle action potential (CMAP) from the muscle using a concentric bipolar electrode. L1 motor neuron axons were stimulated with five stimuli at 1Hz for peak-to-peak measurements of the maximum CMAP amplitude from five averages and 10Hz or 50Hz stimulation for synaptic depression analysis. The control littermates of each individual mouse line did not differ among each other in functional analysis and could be therefore pooled into a control group for CMAP and monosynaptic response recordings.
Immunohistochemistry
For immunostainings, the spinal cords were either natively dissected with 12°C oxygenated aSCF or perfused with 1x PBS and 4% PFA following 4% PFA postfixation overnight at 4°C. On the following day, the spinal cords were taken out and the specific segments of interest were identified by the ventral roots, briefly washed with PBS and the segment’s length measured to ensure consistency between preparations. Subsequently single segments were embedded in warm 5% agar and serial transverse sections (75 μm) were cut at the Vibratome. The sections were blocked with 5% normal donkey serum in 0.01 M PBS with 0.3% Triton X-100 (PBS-T; pH 7.4) for 90 min and incubated overnight at room temperature in different combinations of the primary antibodies (Table S1: List of antibodies). VGluT1 antibodies were used as a marker for proprioceptive synapses, ChAT antibodies were used to identify C-boutons. VGAT antibodies labeled inhibitory synapses (Table S1: List of antibodies). The following day, after 6 times of 10 min PBS washes, secondary antibody incubations were performed for 3 h with the appropriate species-specific antiserum coupled to Alexa488, Cy3 or Alexa647 (Jackson labs) diluted at 1:1000 in PBS-T. After secondary antibody incubations, the sections were washed 6 times for 10 min in PBS and mounted on slides and cover-slipped with an anti-fading solution made of Glycerol:PBS (3:7) (Simon et al., 2017).
For immunostaining of NMJs, mice were sacrificed or perfused and the muscle was dissected and immediately fixed with 4% PFA overnight. After fixation, single muscle fibers were teased and washed 3 times in PBS for 10 min each followed by staining of the postsynaptic part of the NMJ with α-bungarotoxin (BTX) Alexa Fluor 555 in PBS for 20 min. Subsequently, the muscle fibers were washed 5 times in PBS for 10 min and blocked with 5% donkey serum in 0.01 M PBS with 0.3% Triton X-100 (PBS-T) for 1 h. Rabbit anti-Neurofilament-M (NF) and anti-Synaptophysin-1 (SYP) antibodies to immunolabel the presynaptic aspect of the NMJ were applied in blocking solution overnight at 4°C (Table S1: List of antibodies). The muscle fibers were then washed 3 times for 10 min in PBS. Secondary antibodies were applied for 1 h in PBS-T at room temperature. Finally, the muscle fibers were washed 3 times in PBS for 10 min and mounted on slides covered with Glycerol:PBS (3:7) (Simon et al., 2010).
Confocal microscopy and analysis
Spinal cord sections were imaged using SP8 Leica confocal microscopes. Sections were scanned using a 20x, 40x or 63x objective. Motor neurons were counted offline from z-stack images (collected at 4μm intervals in the z-axis) from an entire spinal cord segment. Control and mutant spinal cord segments were measured from one ventral root to the other with a 10x light microscope to ensure consistency. All control and mutant segments at the same age were of equal length, except for the P26 Smn2B/- mutant L1 segment, which was shorter than control age-matched littermates. Only ChAT+ motor neurons located within the ventral horn containing the nucleus were counted to avoid double counting from adjoining sections. Pitx2 neurons identified as cholinergic neurons next to the central canal cells were quantified from a L1 hemicord.
Images for SmB intensity analysis were acquired using a 63x objective at identical settings for control and SMA samples, analysis was performed using Leica LAS X software by measuring the mean intensity per unit area of the nuclear region of motor neurons and proprioceptive neurons normalized to either ChAT (for motor neurons) or Parvalbumin (proprioceptive neurons) mean intensities of the cytoplasm. The control group was pooled from controls of all three mouse lines to reduce animal numbers. The control littermates of each individual mouse line did not differ among each other, both morphologically and functionally as well as in SmB levels and could be therefore pooled into a control group for SmB analysis.
Quantitative analysis of VGluT1 (proprioceptive synapses), VGAT (inhibitory synapses) and C-boutons (highlighted intense ChAT+ synapses at the motor neuron cell membrane) on motor neurons was performed on image stacks of optical sections scanned using a 40x oil or 63x glycerol objective throughout the whole section thickness at 0.4μm z-steps to include the whole cell body and dendrites of ChAT+ motor neurons. The number of VGluT1+ and ChAT+ synapses were counted over the entire surface of the motor neuron soma as well as on primary dendrites for VGluT1+ synapses up to a distance of 50 μm from the soma using Leica LAS X software as previously established (Mentis et al., 2011). VGAT synapses were quantified as synapses per perimeter of the motor neuron soma. We scanned three different optical planes of each motor neuron to ensure consistency. First, we scanned the optical plane in the center of the motor neuron defined by the largest area of the motor neuron soma and a visible nucleus. From that point, we scanned two additional optical planes with a distance of 6μm above and below from the first plane. Then we quantified the number of VGAT+ synapses onto ChAT+ motor neurons and the perimeter of the soma on each plane by Leica LAS X software. Division of synaptic number by perimeter gave us the synaptic density of each panel. We averaged synaptic density of all three panels for each individual motor neuron. At least 10 motor neurons per mouse were quantified.
For the analysis of muscle innervation, a minimum of 200 randomly selected NMJs per muscle sample were quantified for each biological replicate. Only BTX+ endplates that lack pre-synaptic coverage by both SYP and NF were scored as fully denervated. Kv2.1 coverage was quantified as previously described (Fletcher et al., 2017).
RNA and protein analysis
Whole spinal cords were prepared in 1x LDS buffer (Invitrogen) and resolved using the NuPAGE® precast gel system (Invitrogen) by SDS-PAGE. Extracts (20 μg) were run on Novex® Bis-Tris 12% gels and transferred onto an iBlot2 transfer stack nitrocellulose membrane (Invitrogen) using the iBlot2 Dry Blotting system unit (Invitrogen). After protein transfer, the membranes were blocked for 1 h in 5% non-fat dry milk prepared in 1 x PBS with 0.1% Tween-20. The membranes were then incubated with the corresponding antibodies overnight. Thereafter, the membranes were incubated with IRDye 680RD or 800CW secondary antibodies (Li-cor) followed by visualization using a near-infrared imager (Odyssey; Li-cor) (Blanco-Redondo et al., 2019). Band intensities were analyzed by Fiji ImageJ.
RNA analysis was performed as previously established (Van Alstyne et al., 2018). For RNA analysis, purification of total RNA from mouse spinal cords was carried out using TRIzol reagent (Invitrogen) as per manufacturer’s instructions followed by treatment with RNase-free DNaseI (Ambion). cDNA was generated using RevertAid RT reverse transcription kit (ThermoFisher) with oligo dT primers. RT–PCRs was performed within GoTaq® DNA Polymerase (Promega). RT-qPCR analysis was conducted using SYBR Green (Applied Biosystems) in technical triplicates. The primers used for RT–PCR and RT-qPCR experiments are listed in Table S2: List of Primers. The control group was pooled from controls of all three mouse lines to reduce animal numbers. The control littermates of each individual mouse line did not differ among each other in any morphological, functional or RNA levels and could be therefore pooled into a control group for RNA analysis.
Quantification and statistical analysis
Results are expressed as mean ± standard error of the mean (SEM) from at least three independent experiments using three or more animals per experimental group. Differences between two groups were analyzed by a two-tailed Student's t-test, Mantel-Cox test or multiple t-test with Holm-Sidak method, whereas differences among three or more groups were analyzed by one-way, two-way ANOVA followed by Tukey’s correction or multiple t-test for multiple comparisons as applicable. Each figure legend contains information about which test was used and number of animals. n values indicate number of animals, except labeled differently. GraphPad Prism 9 was used for all statistical analyses and p values are indicated as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Acknowledgments
We are grateful for the helpful comments from Dr. George Mentis, Dr. Livio Pellizzoni, and Dr. Meaghan Van Alstyne. We would like to thank Dr. Rashmi Kothary and Dr. Cédric Raoul for providing the Smn2B/- model. We also like to thank Dr. Stefan Hallermann and Dr. Johannes Hirrlinger for providing reagents and facility access. This work was supported by the German Research Foundation grants SI-1969/2-1, SI-1969/3-1 and SMA-Europe to CMS, and the German Research Foundation [Wi 945/17-1, CRC1451 (project-ID 431549029 – A01), GRK1960 (project ID 233886668 and FOR 2722 (project ID 407176282)], the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 956185 (SMABEYOND) and Center for Molecular Medicine Cologne (project No C18) to BW.
Author contributions
CMS designed and supervised the study. J.M.B., J.K.S.L., F.G., K.S.A., B.B.R., L.S., and E.J. performed the experiments and analyzed the data. E.J. and B.W. provided tissues from the Taiwanese mice and together with T.L. valuable input to the interpretation of the results. C.M.S. and J.M.B. wrote the paper with input from all authors. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103376.
Supplemental information
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ackermann B., Krober S., Torres-Benito L., Borgmann A., Peters M., Hosseini Barkooie S.M., Tejero R., Jakubik M., Schreml J., Milbradt J., et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2013;22:1328–1347. doi: 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E., Talbot K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Redondo B., Nuwal N., Kneitz S., Nuwal T., Halder P., Liu Y., Ehmann N., Scholz N., Mayer A., Kleber J., et al. Implications of the Sap47 null mutation for synapsin phosphorylation, longevity, climbing proficiency and behavioural plasticity in adult Drosophila. J. Exp. Biol. 2019;222:1–15. doi: 10.1242/jeb.203505. [DOI] [PubMed] [Google Scholar]
- Bose P., Fielding R., Ameis K.M., Vacca-Galloway L.L. A novel behavioral method to detect motoneuron disease in Wobbler mice aged three to seven days old. Brain Res. 1998;813:334–342. doi: 10.1016/s0006-8993(98)01033-6. [DOI] [PubMed] [Google Scholar]
- Bowerman M., Murray L.M., Beauvais A., Pinheiro B., Kothary R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 2012;22:263–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Burghes A.H., Beattie C.E. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach M.E., Singh J., Thorsteinsdottir M., Saieva L., Slominski E., Thurmond J., Andresson T., Zhang J., Edwards J.D., Simard L.R., et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervero C., Blasco A., Tarabal O., Casanovas A., Piedrafita L., Navarro X., Esquerda J.E., Caldero J. Glial activation and central synapse loss, but not motoneuron degeneration, are prevented by the Sigma-1 Receptor Agonist PRE-084 in the Smn2B/- mouse model of spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 2018;77:577–597. doi: 10.1093/jnen/nly033. [DOI] [PubMed] [Google Scholar]
- Chaudhry F.A., Reimer R.J., Bellocchio E.E., Danbolt N.C., Osen K.K., Edwards R.H., Storm-Mathisen J. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci. 1998;18:9733–9750. doi: 10.1523/JNEUROSCI.18-23-09733.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney N.L. Dissertation of Courtney; 2019. Motor Neuron Degeneration and Compensatory Sprouting in Mouse Models of Spinal Muscular Atrophy.http://hdlhandlenet/1842/35844 [Google Scholar]
- Courtney N.L., Mole A.J., Thomson A.K., Murray L.M. Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death Dis. 2019;10:515. doi: 10.1038/s41419-019-1727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon-Nachum M., Melamed E., Offen D. The "dying-back" phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Donlin-Asp P.G., Bassell G.J., Rossoll W. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol. 2016;39:53–61. doi: 10.1016/j.conb.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Dubowitz V. Ramblings in the history of spinal muscular atrophy. Neuromuscul. Disord. 2009;19:69–73. doi: 10.1016/j.nmd.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Eshraghi M., McFall E., Gibeault S., Kothary R. Effect of genetic background on the phenotype of the Smn2B/- mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2016;25:4494–4506. doi: 10.1093/hmg/ddw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher E.V., Simon C.M., Pagiazitis J.G., Chalif J.I., Vukojicic A., Drobac E., Wang X., Mentis G.Z. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci. 2017;20:905–916. doi: 10.1038/nn.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen E.J.N., Talbot K., Gillingwater T.H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 2018;14:214–224. doi: 10.1038/nrneurol.2018.4. [DOI] [PubMed] [Google Scholar]
- Hamilton G., Gillingwater T.H. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 2013;19:40–50. doi: 10.1016/j.molmed.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Hammond S.M., Gogliotti R.G., Rao V., Beauvais A., Kothary R., DiDonato C.J. Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PLoS One. 2010;5:e15887. doi: 10.1371/journal.pone.0015887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinibarkooie S., Peters M., Torres-Benito L., Rastetter R.H., Hupperich K., Hoffmann A., Mendoza-Ferreira N., Kaczmarek A., Janzen E., Milbradt J., et al. The Power of human protective Modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 2016;99:647–665. doi: 10.1016/j.ajhg.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Iascone D.M., Henderson C.E., Lee J.C. Spinal muscular atrophy: from tissue specificity to therapeutic strategies. F1000Prime Rep. 2015;7:04. doi: 10.12703/P7-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach W.L., Beck E.S., Choi B.J., Lotti F., Pellizzoni L., McCabe B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman S.L., Regehr W.G. The mechanisms and functions of synaptic facilitation. Neuron. 2017;94:447–464. doi: 10.1016/j.neuron.2017.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M., Fleet C., Cullen P., Gupta S.V., Mekhoubad S., Chiao E., Allaire N., Bennett C.F., Rigo F., Krainer A.R., et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U S A. 2017;114:E2347–E2356. doi: 10.1073/pnas.1613181114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Caine C., Awano T., Herbst R., Monani U.R. Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice. Hum. Mol. Genet. 2017;26:2377–2385. doi: 10.1093/hmg/ddx124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Valdivia D.O., Simon C.M., Hassinan C.W., Delestree N., Ramos D.M., Park J.H., Pilato C.M., Xu X., Crowder M., et al. Impaired prenatal motor axon development necessitates early therapeutic intervention in severe SMA. Sci. Transl Med. 2021;13:1–15. doi: 10.1126/scitranslmed.abb6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F., Bernabo P., Tebaldi T., Groen E.J.N., Perenthaler E., Maniscalco F., Rossi A., Donzel D., Clamer M., Marchioretto M., et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat. Cell Biol. 2020;22:1239–1251. doi: 10.1038/s41556-020-00577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Li D.K., Tisdale S., Lotti F., Pellizzoni L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.L., Chen T.H., Hsu Y.Y., Cheng Y.H., Juang B.T., Jong Y.J. Selective neuromuscular denervation in Taiwanese severe SMA mouse can Be reversed by Morpholino Antisense Oligonucleotides. PLoS One. 2016;11:e0154723. doi: 10.1371/journal.pone.0154723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K.K., Gibbs R.M., Feng Z., Ko C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K.K., Lin M.Y., Zingg B., Feng Z., Ko C.P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D., et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor-Nof M., Shipony Z., Lopez-Gonzalez R., Nakayama L., Zhang Y.J., Couthouis J., Blum J.A., Castruita P.A., Linares G.R., Ruan K., et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR) Cell. 2021;184:689–708 e620. doi: 10.1016/j.cell.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez T.L., Kong L., Wang X., Osborne M.A., Crowder M.E., Van Meerbeke J.P., Xu X., Davis C., Wooley J., Goldhamer D.J., et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 2012;32:8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis V.B., Ebert A.D., Fosso M.Y., Chang C.W., Lorson C.L. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum. Mol. Genet. 2009;18:3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis G.Z., Blivis D., Liu W., Drobac E., Crowder M.E., Kong L., Alvarez F.J., Sumner C.J., O'Donovan M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P.J., Galigniana M.D., Morishima Y., Harrell J.M., Kwok R.P., Ljungman M., Pratt W.B. Pifithrin-alpha inhibits p53 signaling after interaction of the tumor suppressor protein with hsp90 and its nuclear translocation. J. Biol. Chem. 2004;279:30195–30201. doi: 10.1074/jbc.M403539200. [DOI] [PubMed] [Google Scholar]
- Murray L.M., Beauvais A., Gibeault S., Courtney N.L., Kothary R. Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun. 2015;3:55. doi: 10.1186/s40478-015-0231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Nichterwitz S., Nijssen J., Storvall H., Schweingruber C., Comley L.H., Allodi I., Lee M.V., Deng Q., Sandberg R., Hedlund E. LCM-seq reveals unique transcriptional adaptation mechanisms of resistant neurons and identifies protective pathways in spinal muscular atrophy. Genome Res. 2020;30:1083–1096. doi: 10.1101/gr.265017.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman E.Y., Van Alstyne M., Yen P.F., Lotti F., Feng Z., Ling K.K., Ko C.P., Pellizzoni L., Lorson C.L. Minor snRNA gene delivery improves the loss of proprioceptive synapses on SMA motor neurons. JCI Insight. 2020;5:1–16. doi: 10.1172/jci.insight.130574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis R.A., Gillingwater T.H. Selective loss of alpha motor neurons with sparing of gamma motor neurons and spinal cord cholinergic neurons in a mouse model of spinal muscular atrophy. J. Anat. 2016;228:443–451. doi: 10.1111/joa.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedich E.J., Kalski M., Armijo N., Cox G.A., DiDonato C.J. Spinal motor neuron loss occurs through a p53-and-p21-independent mechanism in the Smn(2B/-) mouse model of spinal muscular atrophy. Exp. Neurol. 2021;337:113587. doi: 10.1016/j.expneurol.2020.113587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riessland M., Ackermann B., Forster A., Jakubik M., Hauke J., Garbes L., Fritzsche I., Mende Y., Blumcke I., Hahnen E., et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- Riessland M., Kaczmarek A., Schneider S., Swoboda K.J., Lohr H., Bradler C., Grysko V., Dimitriadi M., Hosseinibarkooie S., Torres-Benito L., et al. Neurocalcin Delta suppression Protects against spinal muscular atrophy in humans and across species by Restoring impaired endocytosis. Am. J. Hum. Genet. 2017;100:297–315. doi: 10.1016/j.ajhg.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose F.F., Jr., Mattis V.B., Rindt H., Lorson C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiu M., McGovern V.L., Lotti F., Saieva L., Li D.K., Kariya S., Monani U.R., Burghes A.H., Pellizzoni L. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol. Cell Biol. 2012;32:126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R., Casanas J.J., Torres-Benito L., Cano R., Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreml J., Riessland M., Paterno M., Garbes L., Rossbach K., Ackermann B., Kramer J., Somers E., Parson S.H., Heller R., et al. Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 2013;21:643–652. doi: 10.1038/ejhg.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock H.K., Gillingwater T.H., Groen E.J.N. Molecular mechanisms Underlying sensory-motor circuit dysfunction in SMA. Front Mol. Neurosci. 2019;12:59. doi: 10.3389/fnmol.2019.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock H.K., van der Hoorn D., Boyd P.J., Llavero Hurtado M., Lamont D.J., Wirth B., Sleigh J.N., Schiavo G., Wishart T.M., Groen E.J.N., et al. UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain. 2018;141:2878–2894. doi: 10.1093/brain/awy237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic G., Seso-Simic D., Lucassen P.J., Islam A., Krsnik Z., Cviko A., Jelasic D., Barisic N., Winblad B., Kostovic I., et al. Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig-Hoffmann disease. J. Neuropathol. Exp. Neurol. 2000;59:398–407. doi: 10.1093/jnen/59.5.398. [DOI] [PubMed] [Google Scholar]
- Simon C.M., Blanco-Redondo B., Buettner J.M., Pagiazitis J.G., Fletcher E.V., Sime Longang J.K., Mentis G.Z. Chronic pharmacological increase of neuronal activity improves sensory-motor dysfunction in spinal muscular atrophy mice. J. Neurosci. 2021;41:376–389. doi: 10.1523/JNEUROSCI.2142-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C.M., Dai Y., Van Alstyne M., Koutsioumpa C., Pagiazitis J.G., Chalif J.I., Wang X., Rabinowitz J.E., Henderson C.E., Pellizzoni L., et al. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017;21:3767–3780. doi: 10.1016/j.celrep.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C.M., Jablonka S., Ruiz R., Tabares L., Sendtner M. Ciliary neurotrophic factor-induced sprouting preserves motor function in a mouse model of mild spinal muscular atrophy. Hum. Mol. Genet. 2010;19:973–986. doi: 10.1093/hmg/ddp562. [DOI] [PubMed] [Google Scholar]
- Simon C.M., Janas A.M., Lotti F., Tapia J.C., Pellizzoni L., Mentis G.Z. A Stem cell model of the motor circuit Uncouples motor neuron death from Hyperexcitability induced by SMN deficiency. Cell Rep. 2016;16:1416–1430. doi: 10.1016/j.celrep.2016.06.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C.M., Van Alstyne M., Lotti F., Bianchetti E., Tisdale S., Watterson D.M., Mentis G.Z., Pellizzoni L. Stasimon contributes to the loss of sensory synapses and motor neuron death in a mouse model of spinal muscular atrophy. Cell Rep. 2019;29:3885–3901.e5. doi: 10.1016/j.celrep.2019.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabal O., Caraballo-Miralles V., Cardona-Rossinyol A., Correa F.J., Olmos G., Llado J., Esquerda J.E., Caldero J. Mechanisms involved in spinal cord central synapse loss in a mouse model of spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 2014;73:519–535. doi: 10.1097/NEN.0000000000000074. [DOI] [PubMed] [Google Scholar]
- Tisdale S., Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015;35:8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai L.K., Tsai M.S., Ting C.H., Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J. Mol. Med. (Berl) 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- Van Alstyne M., Simon C.M., Sardi S.P., Shihabuddin L.S., Mentis G.Z., Pellizzoni L. Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 2018;32:1045–1059. doi: 10.1101/gad.316059.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt M.A., Ehsaei Z., Knuckles P., Higginbottom A., Helmbrecht M.S., Kunath T., Eggan K., Williams L.A., Shaw P.J., Wurst W., et al. TDP-43 induces p53-mediated cell death of cortical progenitors and immature neurons. Sci. Rep. 2018;8:8097. doi: 10.1038/s41598-018-26397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukojicic A., Delestree N., Fletcher E.V., Pagiazitis J.G., Sankaranarayanan S., Yednock T.A., Barres B.A., Mentis G.Z. The classical complement pathway Mediates Microglia-dependent Remodeling of spinal motor circuits during development and in SMA. Cell Rep. 2019;29:3087–3100.e7. doi: 10.1016/j.celrep.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B. Spinal muscular atrophy: in the Challenge Lies a solution. Trends Neurosci. 2021;44:306–322. doi: 10.1016/j.tins.2020.11.009. [DOI] [PubMed] [Google Scholar]
- Zagoraiou L., Akay T., Martin J.F., Brownstone R.M., Jessell T.M., Miles G.B. A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron. 2009;64:645–662. doi: 10.1016/j.neuron.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Pinto A.M., Wan L., Wang W., Berg M.G., Oliva I., Singh L.N., Dengler C., Wei Z., Dreyfuss G. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. U S A. 2013;110:19348–19353. doi: 10.1073/pnas.1319280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.