

Abstract
Dysregulated alternative splicing (AS) plays critical roles in driving cancer progression, and the underlying mechanisms remain largely unknown. Here, we demonstrated that PHF5A, a component of U2 small nuclear ribonucleoproteins, was frequently upregulated in colorectal cancer (CRC) samples and associated with poor prognosis. PHF5A promoted proliferation and metastasis of CRC cells in vitro and in vivo. Transcriptomic analysis identified PHF5A-regulated AS targets and pathways. Particularly, PHF5A induced TEAD2 exon 2 inclusion to activate YAP signaling, and interference of TEAD2-L partially reversed the PHF5A-mediated tumor progression. Pharmacological inhibition of PHF5A using pladienolide B had potent antitumor activity. Collectively, these data revealed the oncogenic role of PHF5A in CRC through regulating AS and established PHF5A as potential therapeutic target.
Keywords: alternative splicing, PHF5A, colorectal cancer, TEAD2, therapeutic target
Graphical abstract
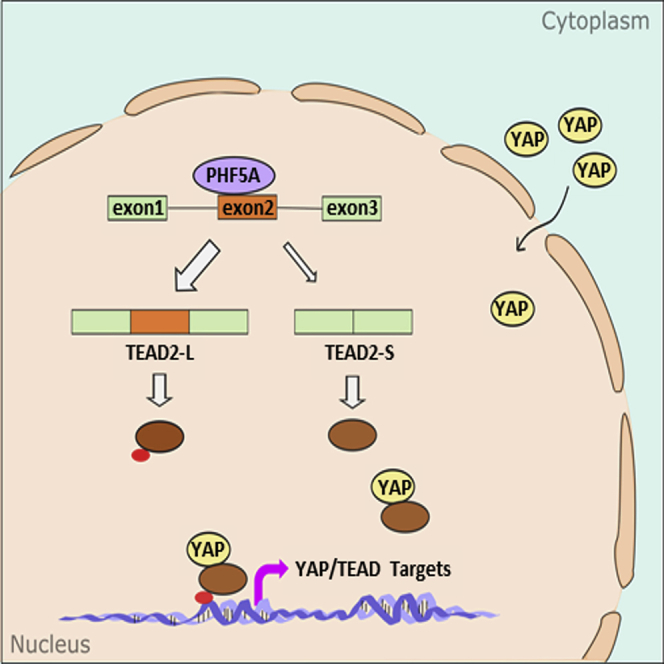
Dysregulated alternative splicing is closely associated with oncological processes. Our study revealed that splicing factor PHF5A was associated with poor prognosis of CRC patients and promoted CRC progression by inducing aberrant splicing of TEAD2 to activate YAP signaling. Splicing modulators targeting PHF5A had potent antitumor activity.
Introduction
Alternative splicing (AS) is one of the prevalent mechanisms of post-transcriptional gene regulation. This process enables the generation of multiple RNA transcripts from a single gene and diversifies the proteome. Aberrant splicing is closely associated with oncological processes, such as proliferation, apoptosis, angiogenesis, and tumor metastasis.1, 2, 3, 4, 5, 6, 7 RNA splicing is catalyzed by two machineries: the major and minor spliceosomes. More than 99.5% of introns in humans are recognized by the major spliceosome, a multi-megadalton protein-RNA complex consisting of five small nuclear ribonucleoproteins (snRNPs; U1, U2, U4, U5, and U6) and more than 200 related proteins. Accumulating evidence has revealed that the main cause of splicing dysregulation is attributed to expression or activity alterations of spliceosome components.8, 9, 10 A recent study even showed that components of spliceosomes could be enriched in apoptotic tumor cell-secreted vesicles to alter RNA splicing in surviving tumor cells and promoted their aggressiveness.11 These findings have raised growing interest in the elucidation of aberrant splicing in cancer development and the manipulation of splicing in cancer treatment.
U2 snRNP, composed of U2 snRNA, SF3a complex, and SF3b complex, is essential for the recognition of branchpoint sequence during early stages of spliceosome assembly.12,13 Several compounds derived from bacterial sources and their analogs have been discovered to target SF3b complex, exerting cytotoxicity to cancer cells.14, 15, 16 Thus, we focused on the splicing factors comprising SF3b complex: SF3B1, SF3B2, SF3B3, SF3B4, SF3B5, SF3B6, and PHD finger protein 5A (PHF5A). Among these genes, PHF5A was most significantly upregulated in colorectal cancer (CRC) specimens in dataset analysis. PHF5A encodes a protein of 110 amino acids with a highly conserved PHD zinc-finger domain.17 As an important component of SF3b complex, PHF5A facilitates interactions between the U2 snRNP and RNA helicases.18 PHF5A could also bind to chromatin through its PHD domain.17,19 Studies have shown that PHF5A plays an important role in regulation of the cell cycle and maintenance of stem cell pluripotency and differentiation.20,21 In certain cancers, PHF5A has been reported to promote tumor progression through regulating the splicing of multiple essential genes.19,20,22,23 However, the role of PHF5A and its regulated AS events in CRC need further exploration.
In this study, we systematically assessed the role of PHF5A in CRC progression and demonstrated the mechanisms by which PHF5A contribute to CRC proliferation and metastasis, focusing on its involvement in regulating the AS of TEAD2 as well as on its clinical relevance.
Results
PHF5A is upregulated in human CRC tissues and correlates with poor prognosis
We first mined Gene Expression Omnibus (GEO) datasets to compare levels of each SF3b component between CRC tissues and normal tissues. Only PHF5A and SF3B3 were upregulated in CRC specimens in both datasets, whereas SF3B4 trended toward downregulation (Figures 1A and S1A and S1B). The upregulation of PHF5A was validated in The Cancer Genome Atlas (TCGA) database (Figure 1A) and paired CRC and adjacent normal tissues at RNA and protein levels (Figures 1B and 1C). Immunohistochemical analysis showed that PHF5A was upregulated in 66% (58/88) of CRC patients (Figure 1D and Table S1). Furthermore, we immunostained 17 pairs of primary CRC and their matched liver metastases, and found that PHF5A expression was elevated in liver metastatic lesions (Figure 1E).
Figure 1.

PHF5A is upregulated in human CRC tissues and activated by H3K27ac
(A) Relative expression of PHF5A mRNA in CRC and normal tissue samples from GEO datasets and TCGA dataset. (B) Relative expression of PHF5A mRNA in paired colorectal cancer and adjacent tissue samples (n = 30). (C) Protein levels of PHF5A in the paired tumor and adjacent normal tissues (n = 9). (D) Representative images of PHF5A IHC staining and statistical analysis of PHF5A score in paired CRC and adjacent tissue samples (n = 88). (E) Representative images of PHF5A IHC staining and statistical analysis of PHF5A expression in human primary CRC samples and corresponding liver metastases (n = 17). (F) Kaplan-Meier analyses for overall survival or disease-free survival of 88 CRC patients with high or low levels of PHF5A protein. (G) ChIP-qPCR assay of the enrichment of H3K27ac on PHF5A promoter relative to immunoglobulin G in normal colon epithelial cells (FHC) and CRC cells (RKO) (n = 3). (H) Genome browser view of H3K27ac ChIP-seq signals at PHF5A promoters in CRC patient samples (GEO: GSE136888). Experiments were repeated at least three times. Data are expressed as mean ± SD.
Next, we evaluated the prognostic value of PHF5A in a cohort of CRC patients. Kaplan-Meier analysis showed that patients with high PHF5A expression had shorter overall survival and disease-free survival compared with low PHF5A expression group (Figure 1F). Elevated PHF5A expression was associated with lymph node metastasis and advanced TNM stage (Table S2). Together, these data indicated that PHF5A was upregulated in CRC and associated with poor clinical outcomes in patients with CRC.
H3K27 acetylation activates PHF5A in CRC
To explore the mechanism of high expression of PHF5A in CRC, we first checked genetic alterations of PHF5A in CRC using TCGA, and found no genomic amplification of PHF5A. Next, we investigated whether epigenetic modification was involved. Histone H3-lysine-4 methylation (H3K4me) and histone H3-lysine-27 acetylation (H3K27ac) are two common histone modifications that positively regulate transcription. Thus, we analyzed the chromatin immunoprecipitation sequencing (ChIP-seq) data of H3K4me and H3K27ac in CRC cells (GEO: GSE71510). Occupancy of H3K27ac but not H3K4me was detected at the promoter region of PHF5A (Figure S2A). Consistently, high enrichment of H3K27ac at the promoter of PHF5A was found by using the UCSC Genome Bioinformatics Site (http://genome.ucsc.edu/) (Figure S2B). A ChIP-qPCR assay revealed the gain of H3K27ac in CRC cells (RKO) compared with normal human colon epithelial cells (FHC) at the PHF5A promoter region (Figures 1G and S2C). Furthermore, ChIP-seq data from CRC patient samples (GEO: GSE136888) confirmed that the H3K27ac peak at the PHF5A promoter region was higher in tumors than that in normal tissues (Figure 1H). Treatment of C646, a histone acetyltransferase (HAT) inhibitor, led to the downregulation of PHF5A expression (Figure S2D). These data suggested that histone acetylation activation of promoter may partly account for the upregulation of PHF5A in CRC.
PHF5A promotes CRC cell proliferation and metastasis
To examine the functional role of PHF5A in CRC progression, we stably overexpressed PHF5A in two CRC cell lines with low basal levels of PHF5A (Figures 2A and S3A). Overexpression of PHF5A promoted the proliferation of CRC cell lines (Figure 2B). In addition, PHF5A enhanced the migration capacity of CRC cells, as judged by wound-healing and transwell migration assays (Figures 2C and 2D). A transwell invasion assay revealed that gain of PHF5A increased the potential of invasiveness in CRC cells (Figure 2E). Conversely, knockdown of PHF5A in two CRC cell lines with high PHF5A basal levels could decrease the proliferation and migration properties of CRC cells (Figures S3B–S3E).
Figure 2.

PHF5A promotes CRC cell proliferation and metastasis
(A) Protein levels of PHF5A in a panel of CRC cell lines and normal colon epithelial cells (FHC). (B) Cell proliferations were measured using CCK-8 assays in HCT-8 or Caco-2 cells stably overexpressing PHF5A. (C) Wound-healing assay and (D) transwell migration assay of CRC cells overexpressing PHF5A or vector control. Scale bar, 20 μm. (E) Transwell invasion assay of CRC cells overexpressing PHF5A or vector control. Scale bar, 20 μm. (F) Xenograft model of nude mice with subcutaneous injection of control and PHF5A overexpressing HCT-8 cells (n = 5 per group). Tumors were removed after 5 weeks, and tumor volume was quantified. (G) Representative images for H&E, PHF5A, and Ki-67 staining in control and PHF5A stably overexpressing xenograft tumors. Scale bar, 20 μm. (H) Representative images of bioluminescent imaging signals in pulmonary metastatic foci by tail-vein injection of control and PHF5A overexpressing HCT-8-luc cells in nude mice. (I) H&E staining (left) in pulmonary metastatic foci, indicated by arrows. The number (right) of metastatic foci in each group (n = 6) were calculated. Scale bar, 20 μm. Experiments were repeated at least three times. Data are expressed as mean ± SD.
We further assessed whether PHF5A promotes CRC growth and metastasis in vivo. HCT8-LV-PHF5A cells and control cells were subcutaneously injected into nude mice. As shown in Figure 2F, tumors of the LV-PHF5A group exhibited increased size and weight compared with control group. Ki-67 levels were also higher in tumors from the PHF5A overexpressing group, suggesting increased cell proliferation in these tissues (Figure 2G). We subsequently developed a metastasis model by injecting luciferase-labeled HCT8-LV-PHF5A cells and control cells into the lateral tail veins of nude mice. Bioluminescent imaging (BLI) showed increased lung metastasis burden in mice injected with LV-PHF5A cells at day 36 (Figure 2H). Histology analysis validated that PHF5A overexpression resulted in larger and more metastatic pulmonary nodules compared with the control group (Figure 2I). Collectively, these findings indicate that PHF5A promotes the growth and metastasis of CRC cells.
Knockdown of PHF5A induces genome-wide alternative splicing events
To gain insights into the mechanism underlying the oncogenic role of PHF5A, we subjected PHF5A-knockdown and control cells to transcriptomic sequencing. The heatmap showed a clear distinction between the two groups (Figure 3A). Gene ontology analysis showed that genes with significant expression changes (>2-fold with adjusted p < 0.05) were associated with cell proliferation, cell migration, and chemotaxis (Figure 3B), supporting that PHF5A is involved in tumor progression. Gene set enrichment analysis indicated that the gene signature reported to be correlated with poor survival was negatively enriched in PHF5A-knockdown cells (Figure 3C).24 These data indicated that depletion of PHF5A implied good survival, consistent with the tumor-promoting role of PHF5A.
Figure 3.

Knockdown of PHF5A induces genome-wide alternative splicing events
(A) Heatmap displaying the deregulated genes by RNA-seq of CRC cells upon PHF5A knockdown. (B) Gene ontology of PHF5A-regulated gene expression events. Fisher’s exact p values were plotted for each category. (C) GSEA of gene signature reported to be associated with poor survival in PHF5A knockdown cells versus control cells. NES, normalized enrichment score. (D) Quantification of the different AS events affected by PHF5A. (E) The relative fraction of each AS event affected positively or negatively by PHF5A. (F) Skipped (green) and included (red) PHF5A-regulated cassette exons plotted by PSI. (G) Gene ontology of PHF5A-regulated AS targets. Fisher’s exact p values were plotted for each enriched functional category.
Next, we analyzed the PHF5A-regulated AS events, and identified 855 AS events with significant change of percentage-spliced-in (PSI) values (PSI ≥ 0.2, Bayes factor ≥ 5). Various types of AS could be regulated by PHF5A, including skipped exon (SE), alternative 3′ splice site (ss) exon (A3SS), alternative 5′ ss exon (A5SS), retained intron (RI), and mutually exclusive exons (MEX) (Figures 3D and 3E). Consistent with the RNA sequencing (RNA-seq) results, a semi-quantitative RT-PCR assay showed that splicing changes of arbitrarily selected target genes were modulated by PHF5A (Figure S4A). SE was the most common AS event regulated by PHF5A (442/855, 51.7%) (Figure 3D), and PHF5A knockdown regulated 153 (153/442, 34.6%) exon skipping and 289 (289/442, 65.4%) exon inclusion changes (Figure 3F), suggesting a dual role for PHF5A as a splicing activator and repressor. Gene ontology analysis of PHF5A-regulated AS events revealed that PHF5A affected genes involved in RNA processing and RNA splicing, which is consistent with the role of PHF5A in RNA splicing (Figure 3G). PHF5A targets were also enriched with cancer-related functions, including cell cycle, cell death, and adherens junction (Figure 3G), again suggesting that PHF5A was involved in cancer cell proliferation and migration.
PHF5A induces TEAD2 exon 2 inclusion to activate YAP signaling
Among the most significant AS events, we noted TEAD2, the transcription factor that mediates Hippo signaling, which was enriched in gene ontology analysis of PHF5A-regulated AS events (Figure 3G). TEAD2 contains an N-terminal domain specifically recognizing the promoter/enhancer of targeted genes through direct binding of DNA and a C-terminal motif that interacts with transcription co-activator YAP to promote transcription of target genes.25 Through the RNA-seq data, we discovered that the exon 2 of full-length TEAD2 (TEAD2-L) could be skipped to generate a short isoform (TEAD2-S) with an alternative coding start site at exon 5 (Figures 4A and S5A). As a result, TEAD2-S protein lacks the N-terminal DNA-binding domain but still contains the C-terminal YAP-binding motif. Since primers flanking exons 1 and 3 generated several non-target bands, we designed specific primers for TEAD2-L and TEAD2-S, respectively (Figure S5A). Consistent with RNA-seq results, overexpression of PHF5A stimulated exon 2 inclusion while knockdown of PHF5A repressed exon 2 inclusion (Figure 4B). Using an antibody against the common amino acid sequence of both TEAD2 isoforms (amino acids 238–249, epitope: EPPDAVDSYQRH), we found that overexpression of PHF5A increased the protein level of TEAD2-L while knockdown of PHF5A suppressed TEAD2-L protein levels in cancer cells (Figure 4C).
Figure 4.

PHF5A induces TEAD2 exon 2 inclusion to activate YAP signaling
(A) The alternative splicing mode of the TEAD2 pre-mRNA. TEAD2-L was produced with exon 2 retention, while TEAD2-S was produced with exon 2 skipping. (B) Inclusion of TEAD2 exon 2 was examined by RT-PCR in HCT8 cells stably overexpressing PHF5A (n = 3) and RKO cells with interference of PHF5A (n = 3). (C) Protein levels of TEAD2-L and TEAD2-S in CRC cells with PHF5A overexpression or knockdown. (D) Binding of TEAD2 pre-mRNAs with PHF5A was detected by RNA immunoprecipitation assay in RKO cells (n = 3). (E) Schematic (left) showing TEAD2 minigene containing exon 1, intron 1, exon 2, intron 2, and exon 3 (TEAD2-wt). The potential PHF5A-binding site is shown in red and deleted in mutant construct (TEAD2-mut). Inclusion of TEAD2 exon 2 was examined by RT-PCR in indicated cells. (F) Real-time PCR analysis of YAP/TEAD target genes (BIRC5, ANKRD1, and CYR61) in indicated cells (n = 3). (G) Co-immunoprecipitation of exogenous TEAD2-L/TEAD2-S and YAP in lysates from HEK293T cells overexpressing HA-TEAD2-L/HA-TEAD2-S and FLAG-YAP. (H) CCK8 assays and (I) transwell assays of HCT-8 cells with expression of YAP, YAP/TEAD2-L, YAP/TEAD2-S, or control (n = 3). Scale bar, 20 μm. Experiments were repeated at least three times. Data are expressed as mean ± SD.
We next elucidated the mechanisms underlying TEAD2 AS. An RNA immunoprecipitation assay showed that PHF5A could directly bind to the endogenous TEAD2 pre-mRNA (Figure 4D). Previous studies reported that PHF5A facilitates recognition of exons with C-rich polypyrimidine splice sites.20 Intriguingly, by scanning the exon 2 and flanking intronic sequences of TEAD2, we found that the 3′ sequence of intron 1 contained C-rich polypyrimidine (Figure S5B). It is also well established that PHF5A recognizes pre-mRNA branchpoint sequences (BPS). By searching BPS (YNYURAY) using the MEME suite, we found three consensus motifs of BPS close to the C-rich polypyrimidine splice sites in the intron 1 of TEAD2 (Figures S5B and S5C).26 Thus, we constructed a TEAD2 minigene plasmid containing genomic DNA fragment of TEAD2 exons 1–3, and a mutant minigene with the putative PHF5A-binding site deletion (Figures 4E and S5B).27 Overexpression of PHF5A promoted exon 2 inclusion in wild-type TEAD2 minigene, but depletion of the predicated PHF5A-binding sites in intron 1 abolished the responsiveness of the TEAD2 minigene to PHF5A (Figures 4E and S5D).
Since TEAD2-S lacks the DNA-binding domain, it may lack transcriptional ability.28 As shown in Figure 4F, expression of YAP alone or co-expression of YAP/TEAD2-L promoted the expression of downstream targets of YAP/TEAD2 (BIRC5, ANKRD1, CYR61), whereas co-expression of YAP/TEAD2-S failed (Figure 4F).29,30 As expected, knockdown of PHF5A inhibited the expression of YAP-activated downstream genes, indicating that PHF5A-induced TEAD2 AS affects YAP signaling (Figure 4F). We further performed a co-immunoprecipitation assay and found that both TEAD2-L and TEAD2-S could bind YAP protein, but TEAD2-L had much stronger binding ability than TEAD2-S (Figure 4G). These data demonstrated that PHF5A promoted TEAD2 exon 2 inclusion to activate YAP signaling. The YAP pathway is known to regulate tumor growth and metastasis.31 The activation of this pathway by TEAD2-L may promote CRC progression. As shown in Figures 4H and 4I, co-expression of YAP with TEAD2-L promoted CRC cell proliferation and migration compared with YAP alone.
Aberrant splicing of TEAD2 is responsible for PHF5A-promoted CRC proliferation and migration
To test whether PHF5A promoted CRC progression through regulating AS, we first examined the effect of the splicing modulator, pladienolide B (PB), which has been reported to target the PHF5A-SF3b complex.32 As shown in Figures 5A–5C, PB reversed the enhanced cell proliferation and migration abilities triggered by PHF5A overexpression. More intriguingly, PB inhibited the PHF5A-induced inclusion of TEAD2 exon 2 (Figure 5D) and the protein levels of TEAD2-L (Figure 5E). In addition, we also examined the effect of another splicing modulator, isoginkgetin, which can prevent the stepwise assembly of spliceosome on pre-mRNAs.33 Similar results were observed upon isoginkgetin exposure (Figures S6A–S6D). These data indicated that PHF5A played its oncogenic role through regulating AS.
Figure 5.

Aberrant splicing of TEAD2 is responsible for PHF5A-promoted CRC proliferation and migration
(A) CCK8 assays of HCT-8 cells stably overexpressing PHF5A or control treated with pladienolide B (PB) (2 nM) (n = 3). (B) Wound-healing and (C) transwell assays of HCT-8 cells stably overexpressing PHF5A or control treated with PB (1 nM) (n = 3). Scale bar, 20 μm. (D) Inclusion of TEAD2 exon 2 was examined by RT-PCR in HCT8 cells stably overexpressing PHF5A upon PB treatment (n = 3). (E) Protein levels of TEAD2-L and TEAD2-S in PHF5A overexpressing CRC cells upon PB treatment. (F) Immunoblotting analysis of TEAD2 protein level in cells transfected with specific siRNAs targeting TEAD2 exon 2 inclusion isoform (TEAD2-L). HCT-8 cells stably overexpressing PHF5A were transfected with siTEAD2-L or siNC, and subjected to (G) CCK8, (H) wound-healing, and (I) transwell assays. Scale bar, 20 μm. Experiments were repeated at least three times. Data are expressed as mean ± SD.
As PHF5A promoted TEAD2 exon 2 inclusion, we next investigated the role of TEAD2-L in PHF5A-promoted tumor progression. To specifically knock down TEAD2-L, we designed two small interfering RNAs (siRNAs) targeting exon 2 of TEAD2 (Figure 5F). As shown in Figures 5G–5I, TEAD2-L depletion abrogated the PHF5A-enhanced proliferation and migration activities of CRC cells. Together, these data demonstrated that aberrant splicing of TEAD2 at least partially contributed to PHF5A-promoted CRC cell proliferation and migration activities.
Correlation between PHF5A expression and TEAD2 splicing in clinical tissues
We further detected the expression of two TEAD2 splicing variants in clinical CRC tissues. Compared with the paired normal tissues, TEAD2-L expression was elevated in the primary CRC specimens alongside increased PHF5A levels. The PSI index of TEAD2 exon 2 was higher in these CRC samples than in matched normal tissues (Figure 6A). Consistently, the protein levels of TEAD2-L were increased in CRC specimens (Figure 6B), implying that TEAD2 AS could play a major role in tumorigenesis. These data supported the role for PHF5A as splicing modulator of TEAD2 in human CRC tissues.
Figure 6.

Correlation between PHF5A expression and TEAD2 splicing in clinical tissues
(A) RT-PCR analysis of TEAD2 isoforms and PHF5A in clinical CRC and paired normal samples (n = 7). PSI of TEAD2 exon 2 in clinical CRC and paired normal tissues are plotted below. (B) TEAD2 protein levels in eight paired CRC tumors and adjacent normal tissues (n = 8). (C) Diagram of PHF5A-regulated TEAD2 splicing and YAP signal activation.
Discussion
Aberrant RNA splicing has been recognized as one of the molecular hallmarks of cancer.34,35 It can contribute to tumorigenesis and progression and can serve as a molecular marker of cancer. Therefore, uncovering the cancer-related splicing factors and AS events would provide new insights into the regulation of cancer. In this study, our data revealed that PHF5A bound to TEAD2 pre-mRNA and promoted the inclusion of TEAD2 exon 2 to produce the TEAD2-L variant. The increased ratio of TEAD2-L/TEAD2-S enhanced the YAP/TEAD2 transcriptional activity, promoting CRC cell proliferation and metastasis (Figure 6C).
Components of the spliceosome machinery have been shown to be dysregulated in cancer.10 Interestingly, we show that the components of SF3b complex were not coordinately altered in CRC, with PHF5A significantly upregulated and SF3B4 downregulated, suggesting that these proteins may have specific functions in the SF3b complex. PHF5A plays critical roles in embryo formation, tissue morphogenesis, and cell pluripotency and differentiation, as well as cancer progression.19,22,36, 37, 38 However, the role of PHF5A in CRC has remained unclear. In this study, we found that PHF5A was upregulated in clinical CRC samples and associated with poor prognosis of patients with CRC. Our biological assays in vitro and in vivo confirmed that PHF5A promoted CRC proliferation and metastasis. We also showed that H3K27ac may partly contribute to the upregulation of PHF5A in CRC. A recent study reported that PHF5A was the target gene of miR-149, which was downregulated in CRC.39 This may be a reason why PHF5A expression remains high upon inhibiting HATs. These findings emphasize the important roles of PHF5A in CRC progression.
AS is regulated by interactions between cis-regulatory pre-mRNA sequences and trans-regulatory splicing factors.40,41 Here we demonstrated that PHF5A promoted the inclusion of TEAD2 exon 2 via binding to its pre-mRNA and recognizing C-rich 3′ ss in intron 1. Inclusion of TEAD2 exon 2 produced a full-length isoform (TEAD2-L), promoting tumor progression and Hippo signaling when co-expressed with YAP. Skipping of exon 2 produced a truncated protein (TEAD2-S), which lacked the N-terminal DNA-binding domain (DBD) but still contained the C-terminal YAP-binding domain. Lack of DBD led to failure of TEAD2-S to promote transcription when co-expressed with YAP. Although TEAD2-S could bind YAP protein, we found that the binding ability was less than that of TEAD2-L, probably due to conformational changes without DBD. We also noticed that the protein levels of two TEAD2 isoforms were different in different cell lines. In CRC cells with a low basal level of PHF5A, TEAD2-S was the main isoform and the protein level of TEAD2-L was quite low (Figures 4C [left] and 5E). Nevertheless, in CRC cells with a high basal level of PHF5A the TEAD2-L level was high, together with the TEAD2-S isoform (Figure 4C, right). These observations supported the role of PHF5A in promoting TEAD2 exon 2 inclusion. Although the splicing of TEAD2 has not been reported previously, Qi et al. showed a similar splicing regulation of TEAD4, suggesting that AS may be a common phenomenon in the TEAD family and the regulation of the Hippo signaling pathway.42
It is possible that PHF5A contributes to cancer progression through other mechanisms as well, as we found that interference of TEAD2 only partially reversed the malignant phenotype triggered by PHF5A overexpression. Through transcriptomic analyses, we found that PHF5A affected many other AS events in cell growth and cell migration. For instance, PHF5A affects splicing of POLDIP3 (Figure S4A), which has been reported to have AS variants with oncogenic roles in cancer.43 Moreover, PHF5A has been reported to regulate gene expression in other ways, such as involving the transcriptional elongation of genes or epigenetic regulation through directly binding histones.19,20 Thus, the other PHF5A-regulated candidates in cancer progression remain to be defined.
For the majority of AS affected genes, the resulting isoforms usually contain in-frame stop codons, causing either degradation by nonsense-mediated mRNA decay or translation of aberrant truncated proteins. Therefore, targeting splicing regulators leads to at least partial loss of function for many essential genes. Some splicing-modulating small molecules or oligonucleotides have already been tested in clinical trials for cancer.44,45 In this study, we revealed that PB, a splicing modulator targeting PHF5A, could inhibit CRC cell proliferation and migration in vitro as well as TEAD2 splicing. These findings indicate PB as a candidate therapeutic agent for inhibiting CRC progression. To date, multiple bacterially derived products (herboxidiene and PB) and their analogs (E7107, spliceostatin A, and sudemycins) have been shown to bind the SF3b components to disrupt the early stages of spliceosome assembly.15,16,46, 47, 48 Further investigation of this family of compounds may be beneficial for CRC as well as other cancers with altered expression of SF3b components.
In conclusion, our study demonstrated the importance of PHF5A in promoting CRC progression through regulating TEAD2 exon2 splicing. We also provided evidence for PB as an antitumor agent through targeting PHF5A. These findings highlight the possibility that manipulation of splicing might provide therapeutic benefit in cancer.
Materials and methods
Cell culture, cell transfection, and treatment
Normal human colonic epithelial cell lines FHC and human colon cancer cell lines HCT8 and HCT116 were maintained in RPMI 1640 medium, and Caco-2, RKO, HT-29, SW480, and SW620 were maintained in Dulbecco's modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin solution (HyClone). Transfection of plasmids was performed using PolyJet Reagent (SignaGen, USA). Transfection of siRNAs was performed using Lipofectamine 3000 (Invitrogen, USA) at a final concentration of 100 nM. Sequences of siRNA (GenePharma, China) against specific targets are listed in Table S3.
The HAT inhibitor C646 was purchased from Medchem Express (USA). The splicing modulator pladienolide B and Isoginkgetin were purchased from R&D Systems (USA).
Animal models
Animal maintenance and experimental procedures were approved by the Institutional Animal Care and Use Committee of Jinling Hospital. Five-week-old female athymic BALB/c nude mice (Cavens, China) were housed and fed in standard pathogen-free conditions. For xenograft assay, 4 × 106 of HCT-8 LV-Control or LV-PHF5A cells were injected subcutaneously into the flanks of nude mice. Three weeks after injection mice were sacrificed, and tumors were dissected and photographed followed by histopathologic examination.
For the metastasis model, 4 × 106 luciferase-labeled HCT-8 LV-Control or LV-PHF5A cells were injected into the tail vein of nude mice. Lung metastasis was monitored via bioluminescence imaging, during which the mice were administered 150 μg of D-luciferin (MCE) intraperitoneally per gram of body weight and anesthetized with isoflurane. Mice were sacrificed 6 weeks after inoculation and consecutive sections of the whole lung were subjected to hematoxylin-eosin staining. The number of metastatic foci on each section was quantified sequentially under microscopy, with repeated metastatic foci not being counted in. After detecting all sections of the whole lung, the number of pulmonary metastatic foci was obtained.
Patients and tissue specimens
All clinical samples were collected with written informed consent from patients, and ethical approval was granted from the Review Board of the Hospital Ethics Committee (Jingling Hospital, Nanjing University). A total of 88 cases of CRC tissues and 17 pairs of CRC tissues and corresponding liver metastasis were collected from the Department of Pathology and Department of Surgery of Jinling Hospital. For the 88 cases of CRC tissues, none of the patients received any preoperative treatment, including chemotherapy and radiotherapy. Tumor stage was defined according to the criteria proposed by the International Union Against Cancer. A tissue microarray block containing 17 pairs of CRC tissues and corresponding liver metastasis was constructed using a tissue microarrayer (Outdo Biotech, Shanghai).
Plasmid construction
Human PHF5A cDNA was PCR amplified using primers PHF5A-5′ and PHF5A-3′, digested by BamHI and XbaI, and ligated into lenti-III vector. The construction work was performed by Genomeditech (Shanghai). Human TEAD2-L (NM_003,598) and TEAD2-S (NM_001,256,662) were synthesized and inserted into pcDNA3.1 vector with HA tagging. Human YAP plasmid was synthesized and inserted into pcDNA3.1 vector with FLAG tagging.
The RG6-TEAD2 minigene was constructed by amplifying genomic sequences spanning exons 1–3 of TEAD2 gene, which were then cloned into the BamHI and AgeI sites of RG6 minigene. Deletion mutant derivatives were made on the basis of the minigene plasmids. The RG6-TEAD2 minigene with mutations was constructed by deleting the predicated PHF5A-binding sites in intron 1. The construction work was performed by Sangon Biotech (Shanghai).
Cell proliferation, cell migration, and invasion assays
For the cell proliferation assay, 2–5 × 103 cells per well were plated into 96-well plates, and cell viabilities were tested every 24 h using Cell Counting Kit 8 (CCK8; KeyGEN BioTECH). For the wound-healing assays, an equal number of cells was plated into 12-well plates with a pipette tip to draw a gap on the plates. Cells that migrated into the cleared section were photographed under a microscope (Nikon) at specific time points. Transwell migration and invasion assays were performed using uncoated and Matrigel-coated Transwell inserts according to the manufacturer’s instructions.
RNA sequencing
Total RNA was extracted from cells using TRIzol reagent (Invitrogen). RNA integrity was verified with the Agilent 2100 Bioanalyzer automated electrophoresis system. Paired-end sequencing was performed on the cDNA library using a HiSeq 2500 instrument (Illumina). The qualified reads were mapped to human reference genome hg38 (GRCh38) using the Hisat2 (version: 2.0.4) split mapping algorithm. The change of splicing isoforms was analyzed using the MISO package, and the results were filtered based on the PSI values. The RNA-seq was assisted by Shanghai Biotechnology.
Quantitative real-time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, USA). cDNA was synthesized by using the PrimeScript RT reagent Kit (TaKaRa, China) according to the manufacturer's instructions. Quantitative real-time PCR was performed using the StepOne Plus Real-Time PCR System (Applied Biosystems, USA) using TB Green (TaKaRa, China). The expression of β-actin was used as an endogenous control. The sequences of primers are listed in Table S4.
Splicing assays with semi-quantitative RT-PCR
Total RNA was isolated and first-strand cDNA generated as described above. PCR was performed with Ex Taq (TaKaRa, China). PCR primers were designed for exons flanking predicted splicing events. The PCR products were separated by agarose gel electrophoresis, detected with Safe-Green (Applied Biological Materials, Canada), and scanned with a Typhoon scanner. The amount of each splicing isoform was quantified using Quantity One software. The primers used for splicing assays are listed in Table S5.
Western blot
Cells were lysed by RIPA buffer containing protease inhibitor cocktail (MedChem Express). Protein concentrations were determined by the BCA Protein Assay Kit (KeyGEN BioTECH). An equal amount of protein was loaded and separated by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad Laboratories). Membranes were incubated with primary antibodies against PHF5A (1:400; Sigma, HPA028885), TEA domain family member 2 (1:500; Abcam, ab92279), HA-Tag (1:1,000; Cell Signaling Technology, 3724s), FLAG Tag (1:1,000; Sigma, F1804), and β-tubulin (1:1,000; Proteintech, 10068-1-AP) overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 h.
Immunohistochemistry
Paraffin-embedded sections were deparaffinized and rehydrated, followed by antigen retrieval. The sections were incubated with primary antibodies against PHF5A (1:50; Sigma, HPA028885) and Ki67 (1:200; Abcam, ab16667), followed by horseradish peroxidase-conjugated secondary antibody. The slides were finally incubated with diaminobenzidine (Dako) and counterstained with hematoxylin. For the 88 cases of CRC tissues, staining strength was scored as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong), and distribution was scored as 0 (0%), 1 (1%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (76%–100%) by positive staining area. The final score of PHF5A in each sample was obtained by multiplying the strength score by the distribution score. For the tissue microarray, the assessment of PHF5A staining was based on the percentage of positively stained cells and staining intensity using Image-scope software (Aperio Technologies).
Immunoprecipitation
Cell pellets were resuspended in lysis buffer (1 L of lysis buffer contains 6 g of Tris-base, 9 g of NaCl, 0.29 g of EDTA, 1% NP-40, and 10% glycerol) for 10 min on ice, followed by centrifugation at 12,000 rpm for 10 min. Anti-HA antibody (5 μg; Cell Signaling, 3724s) was incubated with 20 μL of Dynabeads Protein G (Invitrogen) on the rotating plate for 4 h at 4°C, and washed with wash buffer (1 L of wash buffer contains 6 g of Tris-base, 9 g of NaCl, 0.29 g of EDTA, 1% NP-40, and 10% glycerol) three times. Cell lysates (2 mg) were immunoprecipitated with anti-HA beads on the rotating plate overnight at 4°C, followed by washing four times with wash buffer. Precipitates were purified and analyzed by western blot.
RNA immunoprecipitation
The RNA immunoprecipitation assay was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA) according to the manufacturer's instructions. CRC cells were transfected with PHF5A-expressing plasmids and subjected to RIP experiments using PHF5A antibody (Proteintech, 15554-1-AP). The sequences of primers for RIP assay were CCC AGA CAT TGA GCA GAG C (TEAD2_forward) and CAT ACA TCT TGC CTT CAT CAG A (TEAD2_reverse).
Chromatin immunoprecipitation
The chromatin immunoprecipitation assay was performed using the Magna ChIP HiSens Kit (Millipore) according to the manufacturer's instructions. Chromatin was immunoprecipitated with H3K27ac antibody (Abcam, ab4729) and analyzed using quantitative real-time PCR. The sequences of primers for the ChIP assay were GCG TCA CGG TGA AGG C (PHF5A_forward) and GAT TTG GAT GGC GGA GAT (PHF5A_reverse).
Statistical analysis
All statistical analyses in this study were performed with SPSS 16.0 software (SPSS, USA) or GraphPad Prism (GraphPad Software). Data are presented as mean ± SD. The significance of mean values between two groups was analyzed by two-tailed Student's t test. The differences of quantitative variables among groups were analyzed by ANOVA. Survival curves were plotted using the Kaplan-Meier method and compared by the log-rank test. A p value of less than 0.05 was considered significant.
Data availability
RNA-seq data of this study have been deposited in the Gene Expression Omnibus under the accession number GEO: GSE180042.
Acknowledgments
We thank Yang Yang and Hui Chen for providing technical support. This study was supported by grants from National Natural Science Foundation of China (81972333 to C.C., 81872042 and 82072725 to X.C.) and Foundation of Jiangsu Province (BK20181238 to X.C.).
Author contributions
C.C., X.C., and X.L. designed and supervised the study. Y.C., Y.Z., L.W., and M.W. conducted the experiments and analyzed data. C.H. and M.H. provided computational analysis. Z.L., J.Y., S.H., B.W., Y.C., and C.L. provided technical assistance. H.Y., L.X., J.G., Y.C., T.D., L.R., and Q.W. collected clinical specimens. C.C. and Y.C. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.025.
Contributor Information
Xiaobei Liu, Email: njzylxb@163.com.
Xiaoyuan Chu, Email: chuxiaoyuan000@163.com.
Cheng Chen, Email: chencheng1289@126.com.
Supplemental information
. Clinical characteristics of CRC patients
References
- 1.Bechara E., Sebestyén E., Bernardis I., Eyras E., Valcárcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell. 2013;52:720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Choudhury R., Roy S., Tsai Y., Tripathy A., Graves L., Wang Z. The splicing activator DAZAP1 integrates splicing control into MEK/Erk-regulated cell proliferation and migration. Nat. Commun. 2014;5:3078. doi: 10.1038/ncomms4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y., Chen D., Qian H., Tsai Y., Shao S., Liu Q., Dominguez D., Wang Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26:374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwerk C., Schulze-Osthoff K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell. 2005;19:1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 5.Shehadeh L., Yu K., Wang L., Guevara A., Singer C., Vance J., Papapetropoulos S. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson's disease. PLoS One. 2010;5:e9104. doi: 10.1371/journal.pone.0009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakuma K., Sasaki E., Kimura K., Komori K., Shimizu Y., Yatabe Y., Aoki M. CD44HNRNPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of during epithelial-mesenchymal transition. Gut. 2018;67:1103–1111. doi: 10.1136/gutjnl-2016-312927. [DOI] [PubMed] [Google Scholar]
- 7.Ghigna C., Valacca C., Biamonti G. Alternative splicing and tumor progression. Curr. Genomics. 2008;9:556–570. doi: 10.2174/138920208786847971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sveen A., Kilpinen S., Ruusulehto A., Lothe R., Skotheim R. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35:2413–2427. doi: 10.1038/onc.2015.318. [DOI] [PubMed] [Google Scholar]
- 9.Jensen M., Wilkinson J., Krainer A. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014;21:189–197. doi: 10.1038/nsmb.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anczuków O., Rosenberg A., Akerman M., Das S., Zhan L., Karni R., Muthuswamy S., Krainer A. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlyukov M., Yu H., Bastola S., Minata M., Shender V., Lee Y., Zhang S., Wang J., Komarova S., Wang J., et al. Apoptotic cell-derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell. 2018;34:119–135.e10. doi: 10.1016/j.ccell.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y., Rio D. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015;84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Will C., Lührmann R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011;3:a003707. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 15.Kotake Y., Sagane K., Owa T., Mimori-Kiyosue Y., Shimizu H., Uesugi M., Ishihama Y., Iwata M., Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 16.Hasegawa M., Miura T., Kuzuya K., Inoue A., Won Ki S., Horinouchi S., Yoshida T., Kunoh T., Koseki K., Mino K., et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 2011;6:229–233. doi: 10.1021/cb100248e. [DOI] [PubMed] [Google Scholar]
- 17.Trappe R., Ahmed M., Gläser B., Vogel C., Tascou S., Burfeind P., Engel W. Identification and characterization of a novel murine multigene family containing a PHD-finger-like motif. Biochem. Biophys. Res. Commun. 2002;293:816–826. doi: 10.1016/S0006-291X(02)00277-2. [DOI] [PubMed] [Google Scholar]
- 18.Rzymski T., Grzmil P., Meinhardt A., Wolf S., Burfeind P. PHF5A represents a bridge protein between splicing proteins and ATP-dependent helicases and is differentially expressed during mouse spermatogenesis. Cytogenet. Genome Res. 2008;121:232–244. doi: 10.1159/000138890. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y., Xue M., Shen H., Li X., Ma D., Gong Y., Liu Y., Qiao F., Xie H., Lian B., et al. PHF5A epigenetically inhibits apoptosis to promote breast cancer progression. Cancer Res. 2018;78:3190–3206. doi: 10.1158/0008-5472.CAN-17-3514. [DOI] [PubMed] [Google Scholar]
- 20.Strikoudis A., Lazaris C., Trimarchi T., Galvao Neto A., Yang Y., Ntziachristos P., Rothbart S., Buckley S., Dolgalev I., Stadtfeld M., et al. Regulation of transcriptional elongation in pluripotency and cell differentiation by the PHD-finger protein Phf5a. Nat. Cell Biol. 2016;18:1127–1138. doi: 10.1038/ncb3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hubert C., Bradley R., Ding Y., Toledo C., Herman J., Skutt-Kakaria K., Girard E., Davison J., Berndt J., Corrin P., et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev. 2013;27:1032–1045. doi: 10.1101/gad.212548.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z., Yang X., Liu C., Li X., Zhang B., Wang B., Zhang Y., Song C., Zhang T., Liu M., et al. Acetylation of PHF5A modulates stress responses and colorectal carcinogenesis through alternative splicing-mediated upregulation of KDM3A. Mol. Cell. 2019;74:1250–1263.e6. doi: 10.1016/j.molcel.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Mao S., Li Y., Lu Z., Che Y., Huang J., Lei Y., Wang Y., Liu C., Wang X., Zheng S., et al. PHD finger protein 5A promoted lung adenocarcinoma progression via alternative splicing. Cancer Med. 2019;8:2429–2441. doi: 10.1002/cam4.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen M., Choi T., Nguyen D., Kim J., Jo Y., Shahid M., Akter S., Aryal S., Yoo J., Ahn Y., et al. CRC-113 gene expression signature for predicting prognosis in patients with colorectal cancer. Oncotarget. 2015;6:31674–31692. doi: 10.18632/oncotarget.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian W., Yu J., Tomchick D., Pan D., Luo X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. U S A. 2010;107:7293–7298. doi: 10.1073/pnas.1000293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkinson M., Charenton C., Nagai K. RNA splicing by the spliceosome. Annu. Rev. Biochem. 2020;89:359–388. doi: 10.1146/annurev-biochem-091719-064225. [DOI] [PubMed] [Google Scholar]
- 27.Orengo J., Bundman D., Cooper T. A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res. 2006;34:e148. doi: 10.1093/nar/gkl967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J., Wang C., Chinnaiyan A., et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai D., Ho K., Hao Y., Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71:2728–2738. doi: 10.1158/0008-5472.CAN-10-2711. [DOI] [PubMed] [Google Scholar]
- 31.Zanconato F., Cordenonsi M., Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng T., Tsai J., Puyang X., Seiler M., Peng S., Prajapati S., Aird D., Buonamici S., Caleb B., Chan B., et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017;8:15522. doi: 10.1038/ncomms15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Brien K., Matlin A., Lowell A., Moore M. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 2008;283:33147–33154. doi: 10.1074/jbc.M805556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David C., Manley J. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oltean S., Bates D. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33:5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 36.Trappe R., Schulze E., Rzymski T., Fröde S., Engel W. The Caenorhabditis elegans ortholog of human PHF5a shows a muscle-specific expression domain and is essential for C. elegans morphogenetic development. Biochem. Biophys. Res. Commun. 2002;297:1049–1057. doi: 10.1016/s0006-291x(02)02276-3. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y., Zhu J., Zhang T., Liu J., Li Y., Zhu Y., Xu L., Wang R., Su F., Ou Y., et al. PHD-finger domain protein 5A functions as a novel oncoprotein in lung adenocarcinoma. J. Exp. Clin. Cancer Res. 2018;37:65. doi: 10.1186/s13046-018-0736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Q., Zhang J., Xu S., Jia C., Meng W., Tang H., Zhang X., Zhang Y., Fu B. Knockdown of PHF5A inhibits migration and invasion of HCC cells via downregulating NF-κB signaling. Biomed. Res. Int. 2019;2019:1621854. doi: 10.1155/2019/1621854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Y., Wang Z., Yan Y., Ji L., He J., Xuan B., Shen C., Ma Y., Jiang S., Ma D., et al. Enterotoxigenic Bacteroides fragilis promotes intestinal inflammation and malignancy by inhibiting exosomes-packaged miR-149-3p. Gastroenterology. 2021;161:1552–1566.e12. doi: 10.1053/j.gastro.2021.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Chen M., Manley J. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matlin A., Clark F., Smith C. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 42.Qi Y., Yu J., Han W., Fan X., Qian H., Wei H., Tsai Y., Zhao J., Zhang W., Liu Q., et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016;7 doi: 10.1038/ncomms11840. ncomms11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Yuan J., Wang T., Pan W., Sun S. An alternative POLDIP3 transcript promotes hepatocellular carcinoma progression. Biomed. Pharmacother. 2017;89:276–283. doi: 10.1016/j.biopha.2017.01.139. [DOI] [PubMed] [Google Scholar]
- 44.Eskens F., Ramos F., Burger H., O'Brien J., Piera A., de Jonge M., Mizui Y., Wiemer E., Carreras M., Baselga J., et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 45.Hong D., Kurzrock R., Naing A., Wheler J., Falchook G., Schiffman J., Faulkner N., Pilat M., O'Brien J., LoRusso P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs. 2014;32:436–444. doi: 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- 46.Sakai Y., Yoshida T., Ochiai K., Uosaki Y., Saitoh Y., Tanaka F., Akiyama T., Akinaga S., Mizukami T. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. I. Taxonomy, production, isolation, physicochemical properties and biological activities. J. Antibiot. 2002;55:855–862. doi: 10.7164/antibiotics.55.855. [DOI] [PubMed] [Google Scholar]
- 47.Fan L., Lagisetti C., Edwards C., Webb T., Potter P. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Folco E., Coil K., Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25:440–444. doi: 10.1101/gad.2009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. Clinical characteristics of CRC patients
Data Availability Statement
RNA-seq data of this study have been deposited in the Gene Expression Omnibus under the accession number GEO: GSE180042.