

Abstract
Sickle cell disease and β-thalassemia are common monogenic disorders that cause significant morbidity and mortality globally. The only curative treatment currently is allogeneic hematopoietic stem cell transplantation, which is unavailable to many patients due to a lack of matched donors and carries risks including graft-versus-host disease. Genome editing therapies targeting either the BCL11A erythroid enhancer or the HBG promoter are already demonstrating success in reinducing fetal hemoglobin. However, where a single locus is targeted, reliably achieving levels high enough to deliver an effective cure remains a challenge. We investigated the application of a CRISPR/Cas9 multiplex genome editing approach, in which both the BCL11A erythroid enhancer and HBG promoter are disrupted within human hematopoietic stem cells. We demonstrate superior fetal hemoglobin reinduction with this dual-editing approach without compromising engraftment or lineage differentiation potential of edited cells post-xenotransplantation. However, multiplex editing consistently resulted in the generation of chromosomal rearrangement events that persisted in vivo following transplantation into immunodeficient mice. The risk of oncogenic events resulting from such translocations therefore currently prohibits its clinical translation, but it is anticipated that, in the future, alternative editing platforms will help alleviate this risk.
Keywords: BCL11A, CRISPR/Cas9, fetal hemoglobin, genome editing, HBG, multiplex editing, sickle cell disease, thalassemia, translocations
Graphical abstract
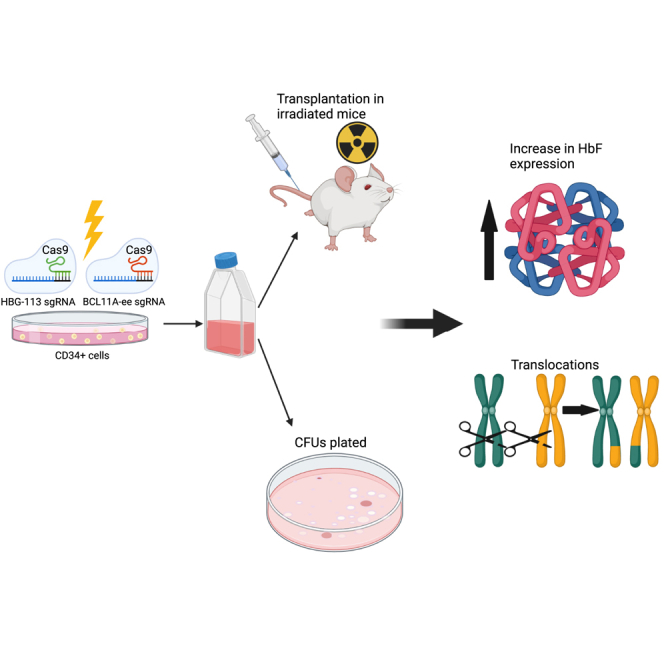
Reinducing fetal hemoglobin benefits patients with sickle cell disease or β-thalassemia. This can be achieved by CRISPR/Cas9 genome editing of BCL11A erythroid enhancer or HBG promoter regions. Targeting both loci for multiplex editing in hematopoietic stem cells results in higher fetal hemoglobin levels but also the development of chromosomal translocations.
Introduction
Sickle cell disease (SCD) and β-thalassemia are among the most common monogenic disorders worldwide and result from genetic alterations in the β-globin gene (HBB).1,2 They cause significant morbidity, reduced quality of life, and early mortality in sufferers.3, 4, 5, 6, 7, 8 The only curative option currently available to patients with β-hemoglobinopathies is allogeneic hematopoietic stem cell transplantation. This option is limited for many patients by a lack of matched donor availability.9 Even when an appropriate donor can be sourced, this treatment option carries significant short- and long-term risks, including graft failure, graft-versus-host disease, infertility, and secondary malignancies.9, 10, 11, 12, 13, 14 An alternative curative approach is offered by genome editing platforms, the most commonly used being Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9).15 One way in which this technology may be applied to the treatment of β-hemoglobinopathies is by the disruption of prespecified areas of the genome in a manner predicted to recapitulate the condition of hereditary persistence of fetal hemoglobin (HPFH). In this naturally occurring, benign variant, individuals retain high levels of fetal hemoglobin (HbF) into adulthood beyond the point at which a physiological switch to adult hemoglobin (HbA) production usually occurs.16 Where HPFH is co-inherited with SCD or β-thalassemia, it ameliorates the disease phenotype and, in some cases, renders individuals entirely asymptomatic.6,17,18
In order to reinduce HbF production, 2 different strategies have been pursued. The first is to target the γ-globin gene (HBG) promoter to recapitulate a natural HPFH 13-nucleotide (13-nt) deletion that disrupts the binding site for the HbF repressor BCL11A at a site located 113 nucleotides upstream of the HBG start codon (HBG-113).19 Hematopoietic stem cells (HSCs) edited at this locus maintained normal long-term engraftment and differentiation potential in the mouse xenograft and in the nonhuman primate autologous transplantation models, and resulted in substantial HbF production.20, 21, 22, 23 An alternative strategy is to inhibit production of the repressor BCL11A itself. Initial attempts to do this by pan-repression of expression across all hematopoietic lineages resulted in engraftment failure and ineffective erythropoiesis in modified cells.24,25 Later discoveries demonstrated no such impairments where an erythroid-specific enhancer motif was instead targeted within the BCL11A intronic sequence (BCL11A-ee).25,26 Phase I clinical trials are now under way to evaluate this genome editing target as a treatment for SCD and β-thalassemia, and promising early data suggest that this approach may offer an alternative functional treatment for these disorders.27 It is estimated that 20% to 30% of stable HbF production is required for functional cure of these disorders,28 but where editing targets a single locus only, reliably achieving and maintaining high in vivo editing frequency and sufficient HbF reinduction remains a challenge. With the aim of maximizing downregulation of the BCL11A/HBG regulatory axis and thereby optimizing HbF reinduction, we investigated the feasibility and efficacy of targeting both loci within the same HSC population in order to simultaneously reduce expression of the BCL11A repressor exclusively in the erythroid lineage and also prevent binding of any residual BCL11A protein to the HBG promoter. Given the likelihood of multiple DNA double-stranded breaks forming within a single cell, we also investigated the toxicities of this approach, including the risk of chromosomal translocation.
Results
Dual HBG-113/BCL11A-ee CRISPR/Cas9 editing in human CD34+ cells enhances HbF reactivation
Single guide RNAs (sgRNA) were employed to disrupt the binding site for BCL11A within both HBG1 and HBG2 promoter regions (HBG-113) and to target the BCL11A-ee locus within human CD34+ cells (Figures 1A and 1B). The HBG-113 sgRNA used in these experiments was previously validated in human CD34+ cells and nonhuman primates,20 and the BCL11A-ee sgRNA targets the same site as that currently under investigation in clinical studies (NCT03745287, NCT03653247, NCT03432364).26,27 These experiments were set up to investigate the hypothesis that editing at both loci would act synergistically and allow greater HbF reinduction than targeting one locus alone. Any negative effects of this dual-editing strategy on engraftment or lineage differential potential of modified HSCs were also investigated. Furthermore, since multiplex CRISPR/Cas9 editing of T cells has been demonstrated to result in the generation of persistent chromosomal translocations,29 it was sought to determine whether this same phenomenon would be observed in CD34+ cells. It was hypothesized that this potential risk could be reduced by separately editing each locus on consecutive days rather than simultaneously, and comparison was made of these 2 methods of delivering dual editing, in their efficacy and translocation generation, with the aim of defining a strategy with the greatest potential therapeutic benefit and lowest risk (Figure 1C).
Figure 1.

Outline of rationale and methodology of CRISPR/Cas9 dual-editing strategy
(A) The BCL11A/HBG axis under physiological conditions dictates the switch from fetal hemoglobin (HbF) to adult hemoglobin (HbA) production in infancy. BCL11A transcription factor binds to the promoter regions of HBG, repressing HbF production in favor of HbA. Circulating hemoglobin in most individuals is composed almost entirely of HbA with negligible amounts of HbF. (B) Following CRISPR/Cas9 disruption of the BCL11A erythroid enhancer (BCL11A-ee), there is reduced production of the BCL11A transcription factor in erythroid progenitor cells. Disruption of the HBG promoter (HBG-113) interferes with binding of residual BCL11A protein. These changes lead to disruption of the BCL11A/HBG axis, allowing reversal of the hemoglobin switch and increased HbF production. (C) Outline of experimental protocol.
Granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood (PB) mononuclear cells were collected in 5 aliquots from 4 different donors by leukapheresis and underwent CD34+ enrichment prior to genome modification. CD34+ cells underwent mock, single or dual genome editing procedures using CRISPR/Cas9 ribonucleoprotein (RNP) electroporation and were cultured in liquid media designed to encourage erythroid differentiation and also plated to assess colony-forming unit (CFU) potential. For dual editing, cells were treated with both sgRNAs simultaneously or separately on consecutive days (22–24 h apart) as described in Figure 1C. Editing efficiency and HbF reinduction were evaluated in cells grown in liquid differentiation media from each reaction.
Editing efficiency was similar between single-edited arms, averaging 46.8% ± 17.8% in the HBG-113 single-edited reactions and 47.7% ± 7.0% in the BCL11A-ee single-edited reactions (mean ± SD). HBG-113 editing in the sequentially dual-edited arm was comparable to the single-edited arm at 49.0% ± 23.6%, but was significantly lower in the simultaneously dual-edited reactions at 30.9% ± 6.2% (mean ± SD) (n = 5, p = 0.0476). At the BCL11A-ee locus, editing was similar between simultaneous and sequentially dual-edited reactions at 36.7% ± 7.6% and 38.5% ± 12.0%, respectively (mean ± SD), both of which trended toward being lower than in the BCL11A-ee single-edited reaction (n = 5, p > 0.05) (Figure 2A).
Figure 2.

Analysis of bulk CD34+ cells in vitro following single, dual, or mock editing
(A) Total HBG-113 and total BCL11A-ee editing efficiency in each reaction (n = 5) (mean ± SD). (B) Frequency of HBG-113 13 nucleotide deletion (n = 5) (mean ± SD). (C) Frequency of BCL11A-ee 13 and 15 nucleotide deletions (n = 5) (mean ± SD). (D) Colony-forming potential of CD34+ cells from each reaction (n = 5) (mean ± SD). (E) Sample flow cytometric plot demonstrating gating strategy to allow assessment of HbA and of HbF-positive cells within bulk cells grown in differentiation media. (F) Ratio of HbF-positive cells to HbA-positive cells in each reaction by flow cytometry, normalized to mock (n = 5) (mean ± SD). (G) Ratio of HbF percentage to HbA percentage in each reaction by HPLC, normalized to mock (n = 4) (mean ± SD). (H) Examples of HPLC traces from each reaction within a single experiment, as well as the HPLC standard demonstrating elution time of HbF and HbA. BCL11A, BCL11A-ee single-edited reactions; HBG, HBG-113 single-edited reactions; nt, nucleotide; Seq, sequential; Sim: simultaneous. Only significant differences (p < 0.05) are shown.
Detailed examination was made regarding the frequency of those precise indel lengths previously reported to be commonly found at each of these particular editing targets and associated with the microhomology-mediated end-joining (MMEJ) pathway.20,25,26,30 At HBG-113, the 13-nt deletion was found in the single-edited and sequentially dual-edited cells at similar mean frequency: 19.9% ± 20.1% and 21.1% ± 15.4%, respectively, but was less commonly observed in the simultaneously dual-edited reactions at only 8.0% ± 8.1% (mean ± SD) (n = 5, p = 0.0222 in comparison with sequential reactions) (Figure 2B). A similar pattern was observed for both the 13- and 15-nucleotide (15-nt) deletions at the BCL11A-ee locus. Mean frequency for the 13-nt deletion was 1.2% ± 1.1% and 1.5% ± 1.7% in single and sequentially edited reactions, respectively, but only 0.7% ± 1.1% in simultaneously dual-edited reactions (mean ± SD) (n = 5, p > 0.05 for all). The same trend was observed for the 15-nt deletion, with frequencies in the single-edited, sequentially and simultaneously dual-edited reactions of 1.9% ± 1.9%, 1.9% ± 1.9%, and 1.1% ± 0.5%, respectively (mean ± SD) (n = 5, p > 0.05 for all) (Figure 2C). Early cell loss was greatest from D0 to D+2 in sequential dual-edited reactions (32.3%), in comparison with single-edited reactions, in which cell count increased by an average of 11.0%, or simultaneous dual-edited reactions, in which cell count increased by 1.0% (n = 5, mean). This likely reflects a combination of cell loss through increased wash and transfer procedures in addition to the effects of 2 electroporation episodes. However, there were no significant differences in the total CFU potential of CD34+ cells from each reaction and distribution of colony types was also comparable between arms (Figure 2D).
HbF reinduction was examined after CD34+ cells were cultured in erythroid differentiation media, both by flow cytometry and by high-performance liquid chromatography (HPLC). In each case, the ratio of HbF to HbA was calculated, and results were normalized by those obtained for the mock-treated reaction in each experiment. Flow cytometric quantification demonstrated that the ratio of cells positive for HbF to cells positive for HbA was increased in comparison with the mock-treated reaction in all edited reactions. Mean normalized HbF/HbA ratio was highest with sequential dual editing, at 2.3 ± 1.0 in comparison with 1.8 ± 1.0 in HBG-113 single-edited cells, 1.4 ± 0.5 in BCL11A-ee single-edited cells, and 1.8 ± 1.0 in simultaneously dual-edited cells (n = 5, mean ± SD). HbF/HbA ratio was significantly higher in the sequentially dual-edited cells when compared with HBG-113 single-edited cells (n = 5, p = 0.0403) and also in comparison with simultaneously dual-edited cells (n = 5, p = 0.0092). Where single-edited arms were compared, there was a trend toward improved HbF reinduction in HBG-113 edited cells in comparison with BCL11A-ee edited cells, despite similar editing efficiencies between these arms (Figures 2E and 2F).
HbF reinduction was also quantified by HPLC. Increased HbF/HbA ratio was observed in all edited reactions in comparison with mock-treated cells, consistent with results of flow cytometric analysis. Again, the greatest increase was in the sequentially dual-edited reactions. Mean normalized HbF/HbA ratio by HPLC was as high as 4.1 ± 2.0 in sequentially dual-edited cells, in comparison with 3.3 ± 1.7 in HBG-113 single-edited cells, 2.2 ± 1.0 in BCL11A-ee single-edited cells, and 3.0 ± 1.6 in simultaneously dual-edited cells (mean ± SD). This difference was statistically significant where sequentially dual-edited reactions were compared with BCL11A-ee single-edited reactions (n = 4, p = 0.0353) and with simultaneously dual-edited reactions (n = 4, p = 0.0128). Consistent with the trend observed on flow cytometric assessment, where HbF/HbA ratio was compared in single-edited arms by HPLC, HbF reinduction appeared to be more effective with targeting of the HBG-113 rather than the BCL11A-ee, but the difference did not reach statistical significance (Figures 2G and 2H).
In summary, CD34+ cells that underwent dual editing at both the BCL11A-ee and HBG-113 loci in a sequential editing protocol, demonstrated editing efficiencies at each locus comparable to those observed in single-edited reactions and were consistently found to express the greatest proportion of HbF regardless of which analytical strategy was employed. Where dual editing was applied simultaneously, however, HBG-113 editing efficiency was compromised, likely explaining the lack of significant improvement in HbF reinduction above that seen in single-edited cells.
Dual HBG-113/BCL11A-ee CRISPR/Cas9 editing in human CD34+ cells generates chromosomal translocations initiated at the DNA double-stranded breaks
The possibility that translocation events may have resulted from the dual-editing approaches was investigated, given the expected generation of multiple DNA double-stranded breaks in the same cell. The presence of chromosomal translocations initiated at each CRISPR/Cas9 target site was first qualitatively assessed by PCR in cells grown in liquid culture using primer pairs designed on each side of the CRISPR/Cas9 cleaving sites aimed at detecting 2 different translocation events (Figure 3A). Chromosomal translocations were consistently detected in both simultaneous and sequential dual-edited reactions from each experiment, as demonstrated by the presence of an amplicon of the expected size, which was absent in single-edited or mock-treated reactions. This amplicon was further confirmed by Sanger sequencing to indeed represent a translocation event generated at the CRISPR/Cas9-induced breakpoints in chromosomes 2 and 11 with sequences that aligned with sections of both BCL11A-ee and HBG-113 regions (Figures 3A and 3B).
Figure 3.

Analysis of translocation events in edited human CD34+ cells
(A) Example of sequences generated by translocation PCR demonstrating dual alignment with both BCL11A-ee and HBG-113. (B) Example of agarose gel electrophoresis image demonstrating presence of translocation product in dual-edited reactions but not single-edited reactions. (C) Frequency of translocation events #1 (HBG F2 + BCL11A R1 primers) and #2 (BCL11A F1 + HBG R2 primers) in vitro by ddPCR (n = 3) (mean ± SD). BCL11A, BCL11A-ee; HBG, HBG-113; Seq, sequential; Sim, simultaneous.
Following the qualitative demonstration of chromosomal translocations in dual-edited reactions, translocation frequency was quantified by digital droplet PCR (ddPCR). For both translocation events evaluated, frequencies were significantly higher where dual editing had been applied simultaneously rather than sequentially. Mean frequency for translocation event #1 (HBG F2 + BCL11A R1 primers) was 0.97% ± 0.29% in simultaneous reactions and 0.11% ± 0.09% in sequential ones (mean ± SD) (n = 3, p = 0.0420). For translocation #2 (BCL11A F1 + HBG R2 primers), frequency was again greater in simultaneous reactions as compared with sequential at 0.58% ± 0.16% and 0.04% ± 0.01%, respectively (mean ± SD) (n = 3, p = 0.0296). The higher reported prevalence of translocation event #1 over translocation event #2 may be due to a genuine difference in frequency, but a differential in the sensitivity of primers/probes between these assays cannot be excluded (Figure 3C).
Together, these results demonstrate that the dual-editing approach induced chromosomal translocations in CD34+ cells, albeit at low frequencies. Translocation frequency was reduced but not effectively prevented by editing each locus separately on consecutive days.
Clonal analysis demonstrates efficient editing at both targets in the same BFU-E and provides evidence of greatest HbF reinduction in dual-edited clones
The results presented thus far are hinting toward an additive effect of HBG-113 plus BCL11A-ee edits on HbF reactivation in sequentially dual-edited cells. However, this dual-editing approach is expected to be most beneficial when the same cell is successfully edited at both targets. Furthermore, the type of indel generated is also predicted to affect HbF reactivation depending on how effectively it interferes with production or binding of BCL11A transcription factor. To address this causal relationship between indel profile and HbF generation at the single-cell level, analysis was performed on single erythroid colonies obtained from CD34+ differentiation in CFU assays to provide information on editing at each of the 4 HBG-113 sites and 2 BCL11A-ee sites per cell (Figure 4A).
Figure 4.

Single BFU-E clonal analyses
(A) Total editing and indel type by HBG-113 or BCL11A-ee locus and by reaction arm in individual BFU-Es. Defined indels are included where frequency was reported as ≥10%, and the difference between sum of defined indels and total reported editing efficiency is presented as undefined indels. (B) Indel patterns observed in BFU-Es following editing at HBG-113 demonstrating a high frequency of 13-nucleotide and small deletions. (C) Indel patterns observed in BFU-Es following editing at BCL11A-ee demonstrating a high frequency of single nucleotide insertion. (D) Number of BFU-Es from each arm edited at one or both loci. (E) HbF percentage of total hemoglobin plotted against editing efficiency in unedited, single- and dual-edited BFU-Es. Reported editing of <5% taken to represent colonies with no editing. (F) HbF percentage of total hemoglobin for reactions with no editing or single HBG-113 editing, plotted against total HBG-113 editing and 13-nucleotide deletion frequency. Reported editing of <5% taken to represent colonies with no editing. (G) HbF percentage of total hemoglobin in BFU-Es with editing of over half available alleles at both HBG-113 and BCL11A-ee, compared with those with over half available alleles edited only at one locus (mean ± SD). BCL11A, BCL11A-ee; HBG, HBG-113.
Where HBG-113 is edited, the most commonly observed indel is the 13-nt deletion, present in ≥1 HBG-113 allele in 27% of all colonies (39% once unedited colonies are excluded), consistent with the activity of the MMEJ repair pathway, which is known to favor this particular deletion length.21, 22, 23 Other indels that were observed to occur relatively commonly at the HBG-113 locus are small deletions of one or 2 nucleotides, consistent with results reported by other groups targeting this site (Figure 4B).21, 22, 23 The indel pattern is markedly different in the case of BCL11A-ee editing. The most common indel is a single nucleotide insertion, present in 27% of burst-forming units (BFUs) (41% after exclusion of unedited colonies). The 13-nt and 15-nt deletions, previously reported to be associated with MMEJ pathway activity,25,26 are seen at frequencies of 4.5% and 2.4%, respectively (7.0% and 3.8%, respectively, in edited colonies) (Figure 4C). Consistent with analyses on bulk cells in culture, editing patterns in single BFU-erythroids (BFU-Es) differ between reaction types. BFU-Es containing larger indels make up a smaller proportion of all edited colonies following simultaneous dual editing in comparison with single or sequential dual editing. This effect is particularly noted at the HBG-113 locus (Figure S1).
Where dual editing was applied simultaneously (n = 110), 42.7% of BFU-Es had at least one allele edited at both HBG-113 and BCL11A-ee, with 9.1% having all alleles at both loci edited. Where dual editing took place sequentially (n = 32), 31.3% of BFU-Es were edited at both loci, with the full complement of alleles at both loci edited in 9.4% (Figure 4D). Given the relatively low number of sequentially dual-edited colonies analyzed, robust comparison between arms is not possible. Further detail on allele numbers edited at each locus in each reaction is given in Figure S2.
A number of single colonies underwent HPLC quantification of hemoglobin (Hb) fractions following Sanger sequencing, which allowed the relationship between editing pattern and HbF reinduction to be examined in detail. Baseline HbF percentage was high even in clones with no editing, consistent with previous reports.23 In unedited BFU-Es, average HbF percentage was 26.7% ± 18.7% (mean ± SD) (n = 8). In BFU-Es with ≥1 HBG-113 allele edited, HbF was 40.1% ± 25.4% (n = 10), and where ≥1 BCL11A-ee allele was edited, HbF was 33.4% ± 21.5% (mean ± SD) (n = 12) (Figure 4E). A wide range of HbF levels were seen within BFU-Es with similarly reported editing frequencies, suggesting that factors in addition to the total number of alleles edited contribute significantly to the HbF reinduction potential of each erythroid clone. The association between total HBG-113 editing and HbF percentage is closely mirrored by that between the 13-nt deletion at this site and HbF (Figure 4F). This suggests that this particular indel size is particularly effective at disrupting the transcription factor binding site in this area and is responsible for much of the HbF reinduction where this locus is disrupted, with other indels having less effect on the Hb ratio. HbF percentage in dual-edited BFU-Es was higher than in single-edited BFU-Es. In colonies with more than half available alleles edited at both BCL11A-ee and HBG-113, mean HbF was 71.3% ± 22.4% (n = 6), whereas it was only 47.1% ± 28.7% with editing of ≥3/4 HBG-113 alleles alone (n = 5), and was significantly lower at 29.6% ± 34.1% where editing was 2/2 alleles at BCL11A-ee only (n = 5, p = 0.0373) (mean ± SD) (Figure 4G). These data demonstrate that dual-edited cells have the highest HbF levels, consistent with results in cells in liquid culture presented above, which demonstrated the highest HbF proportion in sequentially dual-edited reactions. Together, these data confirm the additive effect of dual editing within single cells, leading to maximal HbF generation where both BCL11A-ee and HBG-113 loci have been effectively disrupted.
Dual HBG-113/BCL11A-ee editing does not compromise HSC engraftment and multilineage differentiation in the mouse xenograft model
We assessed the impact of our dual-editing approach on HSC engraftment and differentiation potential in the mouse transplantation model. Twenty-five adult non-obese diabetic (NOD)-severe combined immunodeficiency (SCID)-common γ chain−/− (NSG) mice were transplanted with human mobilized PB CD34+ cells that had undergone either single CRISPR/Cas9 genome editing targeting the HBG-113 (n = 5) or BCL11A-ee region (n = 5), dual editing at both loci delivered simultaneously (n = 5) or sequentially (n = 6), or treated by mock electroporation (n = 4). Two mice had to be euthanized early (one mouse each from the BCL11A-ee single-edited and simultaneously dual-edited arms); remaining mice survived in good health to the point of necropsy 23 weeks posttransplant.
Total human and lineage-specific engraftment were monitored in the PB from 6 weeks postinfusion of human CD34+ cells by PB draws every 3 weeks. Total human engraftment, as defined by human CD45+ (hCD45+) percentage of total CD45+ white blood cells (mouse and human), was similar between mice transplanted with mock, single- or dual-edited cells from each arm of the experiment. The patterns of separate hematopoietic lineage frequency were also similar between the control and all edited groups (Figures 5A and 5B). At necropsy, the frequency of hCD45+ cells and the proportional contribution of each lineage within the bone marrow (BM) and PB remained comparable between experimental arms (Figures 5C and 5D). The fraction of human hematopoietic stem and progenitor cells in the BM, as defined by CD34+CD38low, and of the CD90+CD45RA− subpopulation enriched for HSCs, were also similar between arms (Figures 5E and 5F).31,32 Mouse BM cells were plated into CFU assays with similar results between arms (Figure 5G). Together, these results indicate that dual-editing treatments, whether delivered simultaneously or sequentially, do not impair engraftment, proliferation, or long-term multilineage differentiation of modified HSCs.
Figure 5.

In vivo and ex vivo assessment of engraftment and lineage proliferation over time and at necropsy
(A) Example of flow cytometric gating strategy utilized to determine total human engraftment and delineate human lineages in the PB and BM of humanized mice. (B) Total human engraftment and lineage-specific proportions over time in the PB of transplanted mice (mean ± SD). (C) Total human engraftment in BM and PB of transplanted mice at time of necropsy (mean ± SD). (D) Human lineage analysis in BM and PB of transplanted mice at time of necropsy (mean ± SD). (E) Example of flow cytometric gating strategy for determination of human hematopoietic stem and progenitor cell proportions within BM of transplanted mice at time of necropsy, including assessment of the hematopoietic stem cell-enriched CD90+CD45RA– subpopulation. (F) Human hematopoietic stem and progenitor cells, and hematopoietic stem cell-enriched CD90+CD45RA– cells within the BM of transplanted mice at time of necropsy (mean ± SD). (G) CFUs grown from mouse BM cells (mean ± SD). BCL11A, BCL11A-ee; HBG, HBG-113; Seq, sequential; Sim, simultaneous.
Stable engraftment of dual-edited cells with evidence of translocation events that persist after engraftment
Total HBG-113 editing efficiency within the infusion product transplanted into the mouse cohort was 50.4% in the single-edited cells, 35.4% in the simultaneously dual-edited cells and 74.8% in the sequentially dual-edited cells. BCL11A-ee editing was 55.5% in the single-edited cells, 41.5% in the simultaneously dual-edited reaction, and 43.3% in the sequentially dual-edited cells (Figures 6A and 6B). Although an initial drop in editing was observed in PB at the 9-week time point relative to that reported in the infused cells, both HBG-113 and BCL11A-ee editing levels were subsequently stable to necropsy at 23 weeks. Highest mean editing frequencies at both loci in the PB were observed in the sequential dual-edited mice at necropsy (Figures 6C and 6D): a difference that was only partially explained by higher editing efficiency within the infusion product in this group (Figures S3A and S3B). A greater frequency of HBG-113 editing was found in the BM of sequentially dual-edited mice in comparison with single-edited (p > 0.05) or simultaneously dual-edited mice (p = 0.0063) (Figure 6C). Where BCL11A-ee editing at necropsy was examined, there was also a trend toward highest editing frequencies in the sequentially dual-edited arm, in the BM and spleen (Figure 6D). Frequencies of the MMEJ-induced HBG-113 13-nt deletion and BCL11A-ee 13- and 15-nt deletions generally followed the same trends (Figures S3C and S3D). BM extracted at necropsy was cultured in erythroid differentiation media, and Hb fractions were assessed by flow cytometry and HPLC. HbF proportion was greatest in cells cultured from the BM of sequentially dual-edited mice, with this difference being most marked where analysis was by HPLC (Figures 6E and 6F), consistent with patterns reported above in in vitro analyses.
Figure 6.

Persistence of genome edited cells in vivo is greatest in mice transplanted with sequentially dual-edited cells but translocations persist in vivo
(A) HBG-113 total editing within human cells in the infusion product and subsequently the PB of transplanted mice over time (mean ± SD). (B) BCL11A-ee editing within human cells in the infusion product and subsequently the PB of transplanted mice over time (mean ± SD). (C) HBG-113 total editing within human cells in the PB, BM, and spleen at necropsy (mean ± SD). (D) BCL11A-ee editing within human cells in the PB, BM, and spleen at necropsy (mean ± SD). (E) Example of flow cytometric gating strategy for the assessment of HbA and HbF positivity within cells cultured from mouse BM. (F) Assessment of HbF reinduction within BM cells extracted from mice at necropsy and cultured in differentiation media, by flow cytometry and by HPLC (mean ± SD). (G) Gel electrophoretic image of the results of qualitative translocation PCR conducted on mouse PB samples at 6 weeks posttransplant using HBG-113 F and BCL11A-ee R primers. (H) Quantitative assessment of the frequency of translocation events within human cells in the BM of transplanted mice at necropsy (23 weeks posttransplant) by ddPCR (mean). (I) Quantitative assessment of the frequency of translocation events within human cells in the PB and splenic tissue of mice transplanted with dual-edited reactions at necropsy (mean). BCL11A, BCL11A-ee; HBG: HBG-113; Sim, simultaneous; seq, sequential. Only significant differences (p < 0.05) are shown.
Having previously documented the occurrence of translocation events in vitro following dual-editing approaches, we then sought to determine if these events could be detected after engraftment. The presence of a t(2; 11) chromosomal translocation was demonstrated by qualitative PCR assay in the PB of 4/5 simultaneously dual-edited mice and 2 of 6 sequentially dual-edited mice 6 weeks posttransplant (Figure 6G). Translocation frequency was further analyzed quantitatively by ddPCR in the infusion product, and in mouse BM, PB, and splenic tissue extracted at necropsy. Translocation event #1 (HBG F2 + BCL11A R1 primers) was detected in the infusion product at a frequency of 1.26% in simultaneously dual-edited cells and 0.05% in sequentially dual-edited cells. This persisted in vivo and was detected in the BM of 1 mouse of 4 simultaneously dual-edited mice at a frequency of 0.07% and 4 of 6 sequentially dual-edited mice, with mean frequency of 0.20% ± 0.34%, respectively (mean ± SD). Translocation event #2 (BCL11A F1 + HBG R2 primers) was present in the infusion product at frequencies of 0.71% and 0.03% in simultaneously and sequentially dual-edited reactions, respectively. It was later confirmed to be persistent in the BM of 3 of 4 simultaneously dual-edited mice at a frequency of 0.05% ± 0.04% (mean ± SD), these mice being those without translocation event #1, meaning that all 4 mice in the simultaneous dual-edit arm had a translocation of one type detected in their BM. Translocation event #2 was present in 4 of 6 sequentially dual-edited mice at a frequency of 0.20% ± 0.35%, with only 1 mouse in this arm testing negative for both translocation events (mean ± SD) (Figure 6H). No translocation was present in analyzed mice transplanted with mock-edited (n = 2) or single-edited (n = 4) cells. Detection of the translocation by ddPCR in the PB or spleen at necropsy was less common. Translocation #1 was not detected in the PB of any simultaneously dual-edited mouse but was observed in 2 of 5 sequentially dual-edited mice at frequencies of 0.51% and 0.16%. This same translocation was not detectable in the spleen of any simultaneously dual-edited mouse but was present in the spleen of 1 mouse of 5 sequentially dual-edited mice at 0.37%. Translocation event #2 was not detected in the PB of any dual-edited mouse or the spleen of any simultaneously dual-edited mouse but was present in the spleen of 1 mouse of 5 sequentially dual-edited mice, at 0.24% (Figure 6I).
These data demonstrate robust and stable engraftment of HSCs following dual-editing treatment, with greatest in vivo editing frequencies found in the mice transplanted with sequentially dual-edited CD34+ cells. However, this treatment also resulted in the persistence of cells harboring chromosomal translocations, primarily in the BM, after both simultaneous and sequential dual editing, up to the point of necropsy almost 6 months posttransplant.
Discussion
We report on the application of multiplex CRISPR/Cas9 genome editing targeting both HBG-113 and BCL11A-ee loci to maximize HbF reinduction, as a therapeutic strategy for β-hemoglobinopathies. Sequential targeting of both sites resulted in optimal HbF generation but consistent formation of chromosomal translocations that persist in vivo.
Previously published work has already established both HBG-113 and BCL11A-ee as effective targets for disruption by genome editing, resulting in significant HbF reinduction. The safety of both individual targets is supported by the absence of off-target mutations and lack of impairment to engraftment, lineage differentiation, or erythroid maturation.21, 22, 23,26 However, in the case of each locus being targeted individually, resultant HbF levels have not reliably reached levels expected to confer substantial therapeutic benefit.20, 21, 22, 23,25,26,33 To further increase the efficacy of CRISPR/Cas9 genome editing in reinducing HbF, we investigated a dual-editing strategy in which both the BCL11A-ee and HBG-113 regions are targeted. We demonstrated enhanced HbF production when both loci were sequentially edited both in vitro and also in erythroid cells cultured from the BM of transplanted mice. As the humanized mouse model does not support significant amounts of human erythropoiesis, this ex vivo analysis on erythroid progeny of human HSCs persistent in mouse BM almost 6 months posttransplant provided the closest possible surrogate for an in vivo HbF reactivation assay.
This superiority of the sequential dual-editing strategy with regard to HbF reinduction is consistent with an increase in the sum of total editing events where this strategy is applied, in contrast to single-target editing. Interestingly, this improvement in HbF levels was not observed where simultaneous targeting of both loci was employed, despite a modest improvement in the sum of editing efficiencies in those reactions. We hypothesize that this is due to a reduction in the formation of particular indels that are most effective at disrupting BCL11A production and binding sites, and that are reliant on the MMEJ pathway for their formation. Where dual editing is applied simultaneously, the MMEJ machinery is expected to be recruited at all targeted chromosomal sites in the same cell and may thus become limiting, especially given the presence of 2 HBG genes on each chromosome 11.
MMEJ is already known to be active in the formation of the 13-nt deletion at HBG-113, as well as the 13-nt and 15-nt deletions at BCL11A-ee, all demonstrated here to be reduced in the simultaneously dual-edited reactions in comparison with single-edited or sequentially dual-edited reactions. These larger deletions may contribute disproportionately to the disruption of the BCL11A/HBG axis and therefore reinduction of HbF. This hypothesis also explains why total HBG-113 editing levels are higher in sequential than simultaneous dual-edited reactions, whereas total BCL11A-ee editing is more similar between these arms. Since the MMEJ-dependent 13-nt deletion normally comprises a much greater proportion of the total HBG-113 editing than known MMEJ-dependent indels do in the case of BCL11A-ee editing, saturation of this pathway impacts more significantly upon HBG-113 editing than BCL11A-ee editing. Therefore, sequential dual editing provides the benefits of disrupting both loci within the same HSPC population, without compromising the formation of those large, MMEJ-associated indels, which most effectively disrupt the BCL11A/HBG axis, as was observed with a simultaneous dual-editing strategy.
We found a nonsignificant trend toward higher HbF levels where HBG-113 was targeted for single editing in comparison with BCL11A-ee. This was despite almost identical mean editing efficiencies at these 2 loci. This finding is consistent with recently published data that also found greater gamma-globin mRNA transcription, higher HbF percentages of total Hb, and greater percentages of HbF-positive cells by flow cytometry with editing at the HBG promoter regions in comparison with BCL11A-ee.33 We propose that this finding is due to the particular indel types formed at these loci, which in the case of HBG-113 more effectively disrupt transcription factor binding than indels at BCL11A-ee can manage to disrupt its formation. An alternative reason for this observation may be related to the binding of other transcription factors, such as ZBTB7A, at a similar site within the HBG promoter.34,35 Interfering with transcription factor binding site is thus expected to block regulation by multiple effector proteins, whereas reducing BCL11A production has a more restricted effect on regulation.
The detailed analysis of single erythroid colonies provides granular data on the frequency of a wide spectrum of indel types following CRISPR/Cas9 editing at the 2 targeted loci, and on the associations between single- or dual-editing levels and HbF reinduction. The indel pattern observed following HBG-113 editing is similar to that reported in previous studies in which, in addition to the well-recognized 13-nt deletion, other longer deletion lengths of 11 and 18 nucleotides occur at higher frequency than would be expected due to nonhomologous end-joining alone.21, 22, 23 Given the consistency of these observations, it is highly likely that the formation of these 2 particular nucleotide deletion lengths is also related to activity of the MMEJ pathway. The indel patterns observed at BCL11A-ee are also similar to previous reports in which 13-nt and 15-nt deletions were particularly prevalent, again consistent with contribution by MMEJ pathway activity.25,26 Indel patterns differed between reaction types, with BFU-Es containing larger indels comprising a smaller proportion of edited colonies following simultaneous dual editing than other reactions, particularly at the HBG-113 site. This, again, helps to explain the greater efficacy of sequential over simultaneous dual editing, since the formation of larger indels is preserved with the sequential approach, resulting in more effective disruption of target editing sites.
We acknowledge that there have been reports of significantly higher editing efficiencies achieved with the use of chemically modified sgRNAs.23 However, our results of single erythroid colony analysis demonstrate that even where there has been maximal editing of all available alleles at a single locus (BCL11A-ee), there is still an additional benefit to the disruption of a second, co-operating, genomic target (HBG-113). The additive effect on HbF reinduction offered by HBG-113 disruption within erythroid colonies in which biallelic BCL11A-ee editing was already present is likely due to a combination of incomplete knockdown of BCL11A production even with editing at both BCL11A-ee alleles, alongside the existence of additional gamma-globin-repressor transcription factors that continue to bind to HBG-113 unless this site is also disturbed. Within colonies edited at 1 locus only, HbF percentage remained highly variable between cells with a similar level of editing. This is consistent with our hypothesis that the efficacy of HBG/BCL11A axis disruption is highly dependent on the particular type of indel formed, supported by a previous report demonstrating that a single nucleotide insertion at HBG-113 may allow the core CCAAT box to be preserved and therefore was not associated with HbF reinduction.22 HbF levels in erythroid colonies with biallelic editing of BCL11A-ee were similar to those previously reported by Wu et al.26 and trended toward being lower than those in colonies with editing of all HBG-113 alleles. The higher mean HbF values reported with quadrallelic editing at HBG-113 were also consistent with those previously reported.23
The dual-editing approach, whether applied simultaneously or sequentially, did not impair engraftment, lineage differentiation, or proliferation following transplantation into immunodeficient mice. Editing frequencies were generally highest in the PB and hematopoietic tissues of mice transplanted with sequentially dual-edited cells, and lowest in the simultaneously dual-edited arm, by the end of the experiment. This pattern was not entirely explained by differences in editing frequency within the infusion product and may therefore suggest optimized editing of medium- or long-term repopulating HSCs with the sequential dual-editing approach. However, dual-editing strategies cannot be recommended for clinical translation due to our finding that they consistently resulted in the generation of chromosomal translocation events that persisted in vivo. In vitro, translocations were most frequent where both loci had been targeted simultaneously, but sequential targeting of the 2 loci did not entirely abrogate this risk. Six months following xenotransplantation, translocations were still detectable in both the sequential and simultaneous dual-editing arms, being, in fact, most frequent in the former. The particular t(2; 11) translocations demonstrated in these experiments is only one of a number of potential rearrangement events that may have resulted from on- and off-target DNA breaks. Inversions, large deletions, and other translocations are all possible, and it is considered likely that at least a number of additional rearrangements would be identified, if sought. However, the demonstration that t(2; 11) translocations were consistently formed and persisted in vivo was enough to preclude the dual-editing protocols described here from being recommended for further preclinical or clinical testing, and therefore a search for additional rearrangement events was not justified.
Previously published studies have reported the application of multiplex genome editing to hematopoietic cells in a multitude of scenarios. This approach is already being applied in a number of clinical trials and is being considered for many more. These include the production of chimeric antigen receptor (CAR)-T cells with optimized potency for the treatment of refractory malignancy and to overcome the obstacles of immune incompatibility in hematopoietic lineages.29,36, 37, 38 However, where chromosomal translocations have been sought following multiplex editing in these studies, their presence has usually been confirmed. It is well recognized that chromosomal translocation can serve as a precursor event to malignant, clonal transformation where it occurs in vivo. This property of multiplex genome editing has even been harnessed specifically for the generation of cancer-relevant translocations such as those characteristic of mixed-lineage leukemia (MLL)-rearranged leukemia, anaplastic large cell lymphoma, or Ewing sarcoma, providing evidence that the risk of malignant transformation with multiplex editing approaches is more than just hypothetical.39,40 In some scenarios, the risk of possible future oncogenesis resulting from chromosomal rearrangements may be outweighed by the urgent need to control a more immediately life-threatening malignancy, for example, where CAR-T cells are directed toward aggressive tumors refractory to all other available therapies.29,36,38 However, where multiplex genome editing is considered for the treatment of nonmalignant conditions such as hemoglobinopathies, an inherent future risk of iatrogenic oncogenesis is more likely to outweigh the benefit.
It should be noted that the generation of chromosomal rearrangements is not exclusive to multiplex editing. Multiple DNA double-stranded breaks may be formed in a cell after single-target genome editing, due to simultaneous formation of on- and off-target breaks, although this risk may be reduced by the use of higher-fidelity editing systems.41,42 Even on-target breaks may allow for the formation of chromosomal rearrangement events where more than one target gene is present per chromosome. This is the case with single-target editing of HBG-113 in which large deletions, inversions, and other rearrangements are known to occur due to simultaneous double-stranded DNA breaks forming at the promoter regions of both HBG1 and HBG2.21,23 However, the frequency of rearrangement events, particularly translocations, is predicted to be highest with multiplex editing where multiple chromosomes are specifically targeted with the aim of inducing double-stranded breaks. This is one particular example of a number of well-recognized challenges in the development of safe and effective genome editing therapies for the clinic. Others include off-target mutagenesis; suboptimal reproducibility, and predictability of both on- and off-target effects; and significant social and ethical considerations, including those associated with the high associated cost and limited access to these technologies.43, 44, 45, 46
This report describes the application of multiplex genome editing to augment HbF reinduction in the erythroid progeny of engineered HSCs. We conclude that despite the optimization of HbF reinduction offered by sequentially targeting both the BCL11A-ee and HBG-113 loci, safety concerns raised by the generation of chromosomal translocations that persist in vivo currently preclude the application of this dual-editing strategy in the clinic. Novel platforms such as base editor technologies may, in the future, allow multiple loci to be targeted for editing without the frequent formation of double-stranded DNA breaks, thereby mitigating the risk of chromosomal rearrangement events. Interestingly, base editors have already been validated in the context of HbF reinduction at both the BCL11A47 and HBG sites,48 and are thus ideally suited to build upon our dual-editing results. Such a possibility warrants rigorous investigation in order that patients may safely benefit from the therapeutic advantages of multiplex editing strategies.
Materials and methods
Ethics and animal welfare statements
All mouse experiments were granted approval by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center and University of Washington, under protocol #1864. All animal management conforms to recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.49
NSG mice were bred in-house under approved protocols and in pathogen-free housing conditions. Blood draws (by retro-orbital puncture), tail vein and subcutaneous injections were carried out by appropriately trained animal technicians according to center protocols.
CD34+ enrichment of PB cells
PB mononuclear cells (MNCs) were collected by apheresis from adult donors after mobilization with G-CSF, and cryopreserved at −80°C prior to use. MNCs were thawed using pre-prepared buffer: 1X PBS with 2% fetal bovine serum (FBS) (Atlas Biologicals, Fort Collins, CO), 1% DNAse (DNAse I from bovine pancreas; Millipore, Saint Louis, MO), and 0.2% EDTA (EDTA 0.5M; Millipore). Red cell lysis was carried out if required using hemolysis buffer prepared in-house: 150 mM ammonium chloride, 12 mM sodium bicarbonate, and 0.1 mM EDTA mixed into distilled deionized H2O and sterile filtered (0.22 μM filter units used). Cells were resuspended in Miltenyi buffer that was prepared in advance: 1X PBS with 0.2% of 10% BSA (Sigma Aldrich, Saint Louis, MO) and 2 mM EDTA. CD34+ enrichment was carried out using the CD34 UltraPure Human CD34 + MicroBead enrichment kit (Miltenyi, San Jose, CA) according to the manufacturer's instructions. The following in vitro experiments were undertaken 5 times using 5 separate cryopreserved apheresis products from 4 different donors.
CRISPR/Cas9 editing protocol
The CRISPR/Cas9 editing protocol commenced 1 day following thaw and CD34+-enrichment of PB cells. Cas9 nuclease (Invitrogen Truecut Cas9 Protein V2, 5 μg/μL; ThermoFisher Scientific, Waltham, MA) and sgRNA (Synthego, Menlo Park, CA) were mixed in a 1:5 ratio for ribonucleoprotein (RNP) conjugation, with approximately 60 pmol Cas9 and 300 pmol sgRNA per 1 × 106 CD34+-enriched cells for each editing target. RNPs were introduced into cells via electroporation, using VWR electroporation cuvettes, BTXpress buffer (BTX, Holliston, MA), and a BTX 830 electroporator, as per manufacturer's instructions.
For single-edited reactions, either HBG-113 or BCL11A-ee editing took place on day 0 (D0). For simultaneous dual-edited reactions, full concentration of both HBG-113- and BCL11A-ee-directed RNPs were applied by electroporation simultaneously on D0. For sequential dual-edited reactions, BCL11A-ee editing took place on D0 followed by HBG-113 editing on day 1 (D1), 22 to 24 h later, both utilizing full concentration of reagents. Mock-treated reactions underwent electroporation according to exactly the same protocol as edited reactions, with the omission of RNP reagents.
In vitro assessments
Cell culture
Following editing on D0 ± D1, cells were recovered in SFEM II media (Stemspan; Stemcell Technologies, Seattle, WA) with 1% penicillin streptomycin and 0.1% each of thrombopoietin (TPO), stem cell factor (SCF) and fms-like tyrosine kinase 3 (FLT-3), according to previously published protocol.50 The following day after extraction of cells for CFU assessment (described below), cells were resuspended in differentiation media to encourage erythroid differentiation: IMDM media (ThermoFisher Scientific, Waltham, MA) with 10% FBS (tet-system approved FBS; Clontech, Mountain View, CA), 1% penicillin streptomycin, 0.0075% erythropoietin, 0.0050% cyclosporin A, 0.0020% SCF, and 0.0005% interleukin 3 (IL3) according to previously published protocol.20 Cells were cultured at 37°C in a humid atmosphere with 5% CO2.
Editing efficiency and chromosomal translocation incidence determination
Assessment of editing efficiency was carried out by extraction of DNA from aliquots of cells in liquid culture (DNeasy Blood and Tissues kit; Qiagen, Germantown, MD) followed by PCR amplification of relevant regions. Cells were extracted from liquid culture for editing analysis 4 to 9 days following first edit. Sanger sequencing was carried out on the PCR product followed by TIDE (Tracking of Indels by DEcomposition) analysis (TIDE, Netherlands Cancer Institute, Netherlands) to determine editing frequency.
An initial qualitative assessment of the presence of chromosomal translocation events resulting from the 2 expected breakpoints at the HBG-113 and BCL11A-ee regions was made by PCR. Primer pairs used were: HBG-113-F (HBG-F2) with BCL11A-ee-R (BCL11A-R1) (to pick up translocation event #1), and BCL11A-ee-F (BCL11A-F1) with HBG-113-R (HBG-R2) (to pick up translocation event #2). The presence of the relevant translocation was highly suspected by the amplification of a PCR product of the expected length, demonstrated on agarose gel electrophoresis. Confirmation that such gel bands represented chromosomal translocation was confirmed by Sanger sequencing of PCR products, with an assessment of alignment between the generated sequences and relevant regions of the HBG-113 and BCL11A-ee regions.
In order to quantify the incidence of these 2 translocation events, a ddPCR assay was developed. Two sets of primers and probes were selected based on BCL11A-ee, HBG-113, BCL11A-ee_HBG-113 chimera, and HBG-113_BCL11A-ee chimera sequences generated from PCR reactions described above. The combination of these primer and probe sets of HBGFwd1_BCL11ARev2_BCL11AProbe2 (Set A, with amplicon at 119 base pairs [bp]) and BCL11AFwd2_HBGRev1_HBGProbe1 (Set B, with amplicon at 91 bp) would amplify the translocation site located at HBG-113_BCL11A-ee chimera and BCL11A-ee_HBG-113 chimera translocation sites, respectively. Duplex ddPCR is performed with either primer and probe set A or set B and human RPP30 (Table 1).
Table 1.
Primers and probes designed for ddPCR quantification of chromosomal translocation frequency
| Name | Sequences | Length, bp | Amplicon, bp |
|---|---|---|---|
| HBGFwd1 | TGGCCTCACTGGATACTCTAAGACT | 25 | 119 |
| HBGRev1 | AAACGGTCCCTGGCTAAACTC | 21 | |
| HBGProbe1 | FAM-CTGGCCAACCCATG_MGB | 14 | |
| BCL11AFwd2 | CCACCCTAATCAGAGGCCAAA | 21 | 91 |
| BCL11Rev2 | CATAACACACCAGGGTCAATACAAC | 25 | |
| BCL11Probe2 | FAM-TGCACTAGACTAGCTTC_MGB | 17 | |
| RPP30_Fwd | GATTTGGACCTGCGAGCG | 18 | |
| RPP30_Rev | GCGGCTGTCTCCACAAGT | 18 | |
| RPP30_Probe | HEX-TCTGACCTGAAGGCTCTGCGCG–BHQ | 22 |
The DNA ddPCR was used with ddPCR Supermix for probe (Bio-Rad, Hercules, CA) and the PCR conditions were 95°C for 10 min, then 40 cycles at 94°C for 30 s, 60°C for 1 min, followed by 98°C for 10 min. The RNA-ddPCR was used with One-Step RT-ddPCR Advanced kit for Probes (Bio-Rad). The thermal conditions were 50°C for 60 min for reverse transcription, 95°C for 10 min, then 40 cycles at 94°C for 30 s, 60°C for 1 min for the ddPCR followed by 98°C for 10 min. The translocation and RPP30 copy number were analyzed and calculated with QuantaSoft analysis Pro software.
HbF reinduction assessments
HbF reinduction was analyzed by flow cytometry and HPLC on cells extracted from liquid culture 10 to 14 days after D0. For flow cytometric assessment, cells from liquid culture were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA), as per the manufacturer's instructions. They were then incubated separately with anti-HbF (HbF-1-PE, ThermoFisher) and anti-HbA (Hemoglobin β-FITC, clone 37-8; Santa Cruz Biotechnology, Dallas, TX) antibodies followed by analysis on the Canto 2-1 or Symphony (BD Biosciences) flow cytometers. Total number of cells expressing the relevant Hb fraction was analyzed for a qualitative measure of Hb expression.
Cells were washed in PBS, centrifuged 10 min at 300 × g and lysed by hypotonic pressure in HPLC grade water. Hb was analyzed by ion-exchange HPLC. The different forms of Hb were separated based on their charge at pH 6.5 on a PolyCAT A column (PolyLC, Columbia, MD) with a Prominence chromatograph (Shimadzu, Columbia, MD) and the LC Solution software (Shimadzu) used for peak integration. Elution was achieved with a gradient mixture of buffer A (Tris 40 mM, KCN 3 mM; pH adjusted at 6.5 with acetic acid) and buffer B (Tris 40 mM, KCN 3 mM, NaCl 200 mM; pH adjusted at 6.5 with acetic acid). The wavelength chosen for detection was 418 nm and the flow rate was set at 0.3 mL/min.
CFU and single BFU-E analyses
A total of 400 cells from each reaction were plated per 1 mL methocult media (H4435; Stemcell Technologies, Seattle, WA) on day 2 (D2), with assessments carried out in triplicate for each reaction. Data from an additional arm conducted within one experiment are also included here, in which sequential dual editing was applied with a shorter time lag (6 h) between edits 1 and 2. CFUs were differentiated and counted after a further 13 to 14 days of culture at 37°C with 5% CO2. Single erythroid colonies (BFU-Es) were picked into 1X PBS and dissociated by vortexing. Single BFU-Es arising from edited reactions were split into 2 separate aliquots. They first underwent an assessment of editing efficiency at HBG-113 and/or BCL11A-ee (as appropriate) by PCR, sequencing, and TIDE analysis. HPLC assessment for Hb fraction quantitation was undertaken on the second aliquot. Single BFU-Es arising from mock-edited reactions underwent HPLC only.
To convert total reported editing percentage to estimated number of alleles edited in each BFU-E, limits were set to denote the number of alleles assumed to be edited at each reported editing frequency. For HBG-113, reported editing of 10.0% to 34.9% was taken to represent editing at 1 allele; 35.0% to 59.9%, 2 alleles; 60.0% to 84.9%, 3 alleles, and where editing was reported as ≥85.0%, all 4 alleles were assumed to be edited. For BCL11A-ee, there are 2 potential editing sites per cell. Editing efficiency reported at <20.0% was assumed to represent 0 alleles edited; 20.0% to 59.9% was taken to indicate that 1 allele was edited; and where editing was reported to be ≥60.0%, both alleles were taken to be edited. These figures take into account the fact that TIDE generally under-rather than overreports editing frequencies.51,52
Mouse xenotransplantation, in vivo and ex vivo assessments
Transplantation protocol
Following mock, single or dual editing (as described above, on D0 ± D1), cells were recovered in SFEM II media with cytokines. On D2, adult NSG mice were xenotransplanted with cells from each reaction, with each mouse receiving 1×106 cells resuspended in 1X PBS with 1% heparin (APP Pharmaceuticals, East Schaumburg, IL) to a total volume of 200 μL, preceded by sublethal irradiation of 275 cGy.
Longitudinal assessment of lineage proliferation, persistence of edited cells and chromosomal translocations in vivo
PB was sampled 3-weekly by retro-orbital draw from 6 weeks posttransplantation for lineage assessment and determination, and from 9 weeks determination of editing percentage within circulating leukocytes was also carried out. Lineage assessment was by flow cytometry utilizing antibody panel: anti-human CD45 (hCD45)-PerCP (clone 2D1), anti-mouse CD45 (mCD45)-V500 (clone 30-F11), anti-CD3-APC (clone UCHT1), anti-CD4-V450 (clone RPA-T4), anti-CD8-APC Cy7 (clone SK1), anti-CD14-PE Cy7 (clone M5E2) and anti-CD20-FITC (clone 2H7) (all BD Biosciences), run on the BD FACSCanto II Flow Cytometer (BD Biosciences). For editing analysis, DNA was extracted using QIAamp DNA micro kit (Qiagen) then processed by PCR amplification, sequencing and TIDE analysis, as described above. PB DNA extracted at 6 weeks posttransplant also underwent qualitative translocation PCR assay to determine the presence or absence of chromosomal translocation events in vivo.
Lineage, editing, and translocation assessments at necropsy
Mice underwent euthanasia and necropsy 23 weeks posttransplantation, following maximal PB draw. BM and splenic tissues were harvested. Lineage assessment by flow cytometry was carried out on PB, BM ,and thymic tissues using antibody panel and flow cytometer as for longitudinal lineage assessment. In addition, HSC quantitation within the hCD45+ population in extracted BM was undertaken using antibody panel: anti-hCD45-V450 (clone H130, BD Biosciences), anti-mCD45-PECF594 (clone 30-F11, BD Biosciences), anti-CD38-PerCP/Cy 5.5 (clone HIT2; BioLegend, San Diego, CA), anti-CD34-APC (clone 563, BD Biosciences), anti-CD90-PE Cy7 (clone 5E10, BioLegend) and anti-CD45RA-APC Cy7 (clone 5H9, BD Biosciences), run on the Symphony flow cytometer (BD Biosciences).
Assessment of editing percentage within human cells was undertaken on PB, BM, and splenic tissue from necropsy, as per PB analysis described above. ddPCR quantitation of translocation event frequencies was also undertaken on each of these tissues from necropsy as per protocol above.
CFU and HbF assays on mouse BM
A total of 7 × 104/mL cells were plated in ColonyGEL 1402 (ReachBio, Seattle, WA) in triplicate from each mouse BM, with CFUs differentiated and counted after 14 days. Remaining BM cells were cultured in liquid differentiation media and assessments of Hb fractions made by flow cytometry and HPLC, as described above, on aliquots extracted after 12 days in culture.
Statistical analysis
FlowJo V9 was used for compensation and analysis of flow cytometry results. Excel 2016 for Windows and GraphPad Prism 7 for Windows were used for statistical analysis. Paired t test is applied where each value has an appropriate paired result; unpaired t test is applied in every other case. p values of <0.05 are taken to be significant (∗) and p values of <0.001 are taken to be highly significant (∗∗). Standard t test without multiple testing correction is applied. Presentation of graphical data is with GraphPad Prism 7 for Windows.
For assessment of HbF reinduction, 2 separate calculations are employed. For most analyses, HbF/HbA ratio is taken; however, where single BFU-Es were analyzed by HPLC, in some cases the level of 1 Hb fraction was below the threshold of detection, and was therefore reported as “0.” In order to avoid this value being used as a denominator, an alternative calculation was used: HbF/(HbF + HbA) × 100.
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01 HL136135 and R01 HL147324), from the National Institute of Allergy and Infectious Diseases (R01 AI135953–01, U19 AI096111, and UM1 AI126623), NIH, Bethesda, MD, and charitable support to C.S. from the British Society for Haematology, the UK Thalassaemia Society, and Sheffield Hospitals Charity. H.-P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. C.S. thanks Dr Andrew Chantry, Dr Josh Wright, Professor John Snowden, and Dr Michelle Lawson for their support of her research fellowship and PhD. The authors thank Dr Olivier Negre for processing all HPLC assays; Helen Crawford for her assistance in formatting the figures and preparing the manuscript; and Margaret Cui for laboratory management and support.
Author contributions
C.S. and O.H. designed and carried out the genome editing experiments and wrote the manuscript. S.R advised on flow cytometric assays and revised the manuscript. H.Z. and M.-L.W. designed and carried out the ddPCR assays. M.L. performed experiments and revised the figures. E.F. performed experiments, revised the figures, and prepared the graphical abstract. S.C. performed experiments. K.R.J. and H.-P.K. provided oversight and feedback on the manuscript.
Declaration of interests
H.P.K. has received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Stephanus Family Endowed Chair for Cell and Gene Therapy. H.P.K is or was a consultant to and has or had ownership interests with Rocket Pharmaceuticals, Homology Medicines, VOR Biopharma, and Ensoma. H.P.K has also been a consultant to CSL and Magenta. Other authors have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.10.008.
Supplemental information
References
- 1.Weatherall D.J., Clegg J.B. Inherited haemoglobin disorders: an increasing global health problem. Bull. World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- 2.Modell B., Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piel F.B., Steinberg M.H., Rees D.C. Sickle cell disease. N. Engl. J. Med. 2017;376:1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 4.Inusa B.P.D., Colombatti R., Rees D.C., Heeney M.M., Hoppe C.C., Ogutu B., Hassab H.M., Zhou C., Yao S., Brown P.B., et al. Geographic differences in phenotype and treatment of children with sickle cell anemia from the multinational DOVE study. J. Clin. Med. 2019;8, 2009 doi: 10.3390/jcm8112009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubeck D., Agodoa I., Bhakta N., Danese M., Pappu K., Howard R., Gleeson M., Halperin M., Lanzkron S. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw. Open. 2019;2:e1915374. doi: 10.1001/jamanetworkopen.2019.15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platt O.S., Brambilla D.J., Rosse W.F., Milner P.F., Castro O., Steinberg M.H., Klug P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Eng. J. Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 7.Dhanya R., Sedai A., Ankita K., Parmar L., Agarwal R.K., Hegde S., Ramaswami G., Gowda A., Girija S., Gujjal P., et al. Life expectancy and risk factors for early death in patients with severe thalassemia syndromes in South India. Blood Adv. 2020;4:1448–1457. doi: 10.1182/bloodadvances.2019000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mettananda S., Pathiraja H., Peiris R., Bandara D., de Silva U., Mettananda C., Premawardhena A. Health related quality of life among children with transfusion dependent beta-thalassaemia major and haemoglobin E beta-thalassaemia in Sri Lanka: a case control study. Health Qual. Life Outcomes. 2019;17:137. doi: 10.1186/s12955-019-1207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pidala J., Kim J., Schell M., Lee S.J., Hillgruber R., Nye V., Ayala E., Alsina M., Betts B., Bookout R., et al. Race/ethnicity affects the probability of finding an HLA-A, -B, -C and -DRB1 allele-matched unrelated donor and likelihood of subsequent transplant utilization. Bone Marrow Transpl. 2013;48:346–350. doi: 10.1038/bmt.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson T.M., Fuchs E.J. Allogeneic stem cell transplantation for sickle cell disease. Curr. Opin. Hematol. 2016;23:524–529. doi: 10.1097/MOH.0000000000000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angelucci E., Matthes-Martin S., Baronciani D., Bernaudin F., Bonanomi S., Cappellini M.D., Dalle J.H., Di Bartolomeo P., de Heredia C.D., Dickerhoff R., et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99:811–820. doi: 10.3324/haematol.2013.099747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walters M.C., Storb R., Patience M., Leisenring W., Taylor T., Sanders J.E., Buchanan G.E., Rogers Z.R., Dinndorf P., Davies S.C., et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Blood. 2000;95:1918–1924. [PubMed] [Google Scholar]
- 13.Gluckman E., Cappelli B., Bernaudin F., Labopin M., Volt F., Carreras J., Pinto Simoes B., Ferster A., Dupont S., de la Fuente J., et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129:1548–1556. doi: 10.1182/blood-2016-10-745711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cappelli B., Volt F., Tozatto-Maio K., Scigliuolo G.M., Ferster A., Dupont S., Simoes B.P., Al-Seraihy A., Aljurf M.D., Almohareb F., et al. Risk factors and outcomes according to age at transplantation with an HLA-identical sibling for sickle cell disease. Haematologica. 2019;104:e543–e546. doi: 10.3324/haematol.2019.216788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humbert O., Samuelson C., Kiem H.P. CRISPR/Cas9 for the treatment of haematological diseases: a journey from bacteria to the bedside. Br. J. Haematol. 2021;192:33–49. doi: 10.1111/bjh.16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thein S.L., Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br. J. Haematol. 2009;145:455–467. doi: 10.1111/j.1365-2141.2009.07650.x. [DOI] [PubMed] [Google Scholar]
- 17.Chaouch L., Sellami H., Kalai M., Darragi I., Boudrigua I., Chaouachi D., Abbes S., Mnif S. New deletion at promoter of HBG1 gene in sickle cell disease patients with high HbF level. J. Pediatr. Hematol. Oncol. 2020;42:20–22. doi: 10.1097/MPH.0000000000001626. [DOI] [PubMed] [Google Scholar]
- 18.Weatherall D.J., Clegg J.B., Wood W.G. A model for the persistence or reactivation of fetal haemoglobin production. Lancet. 1976;2:660–663. doi: 10.1016/s0140-6736(76)92469-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer D.E., Orkin S.H. Hemoglobin switching's surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015;33:62–70. doi: 10.1016/j.gde.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O., et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl Med. 2019;11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lux C.T., Pattabhi S., Berger M., Nourigat C., Flowers D.A., Negre O., Humbert O., Yang J.G., Lee C., Jacoby K., et al. TALEN-mediated gene editing of HBG in human hematopoietic stem cells leads to therapeutic fetal hemoglobin induction. Mol. Ther. Methods Clin. Dev. 2019;12:175–183. doi: 10.1016/j.omtm.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Traxler E.A., Yao Y., Wang Y.D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metais J.Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K., et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.J., Heeney M.M., Klatt D., Fogel J., Milsom M.D., et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang K.H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F., Sun C., et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangoul H., Bobruff Y., Cappellini M.D., Corbacioglu S., Fernandez C.M., de la Fuente J., Grupp S.A., Handgretinger R., Ho T.W., Imren S., et al. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: early results from the Climb THAL-111 and Climb SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells (Abstract) Blood. 2020;136:3–4. [Google Scholar]
- 28.Steinberg M.H., Chui D.H., Dover G.J., Sebastiani P., Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 29.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367, eaba7365 doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A., et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6, eaay9392 doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray L., Chen B., Galy A., Chen S., Tushinski R., Uchida N., Negrin R., Tricot G., Jagannath S., Vesole D., et al. Enrichment of human hematopoietic stem cell activity in the CD34+Thy-1+Lin- subpopulation from mobilized peripheral blood. Blood. 1995;85:368–378. [PubMed] [Google Scholar]
- 32.Radtke S., Adair J.E., Giese M.A., Chan Y.Y., Norgaard Z.K., Enstrom M., Haworth K.G., Schefter L.E., Kiem H.P. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl Med. 2017;9, eaan1145 doi: 10.1126/scitranslmed.aan1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamsfus-Calle A., Daniel-Moreno A., Antony J.S., Epting T., Heumos L., Baskaran P., Admard J., Casadei N., Latifi N., Siegmund D.M., et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34(+) HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci. Rep. 2020;10:10133. doi: 10.1038/s41598-020-66309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinjamur D.S., Bauer D.E., Orkin S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018;180:630–643. doi: 10.1111/bjh.15038. [DOI] [PubMed] [Google Scholar]
- 35.Martyn G.E., Wienert B., Kurita R., Nakamura Y., Quinlan K.G.R., Crossley M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood. 2019;133:852–856. doi: 10.1182/blood-2018-07-863951. [DOI] [PubMed] [Google Scholar]
- 36.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R., et al. Multiplex genome-edited T-cell manufacturing platform for "off-the-shelf" adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 37.Gornalusse G.G., Hirata R.K., Funk S.E., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O., et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017;35:765–772. doi: 10.1038/nbt.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valton J., Guyot V., Marechal A., Filhol J.M., Juillerat A., Duclert A., Duchateau P., Poirot L. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol. Ther. 2015;23:1507–1518. doi: 10.1038/mt.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneidawind C., Jeong J., Schneidawind D., Kim I.S., Duque-Afonso J., Wong S.H.K., Iwasaki M., Breese E.H., Zehnder J.L., Porteus M., et al. MLL leukemia induction by t(9;11) chromosomal translocation in human hematopoietic stem cells using genome editing. Blood Adv. 2018;2:832–845. doi: 10.1182/bloodadvances.2017013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piganeau M., Ghezraoui H., De Cian A., Guittat L., Tomishima M., Perrouault L., Rene O., Katibah G.E., Zhang L., Holmes M.C., et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013;23:1182–1193. doi: 10.1101/gr.147314.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corrigan-Curay J., O'Reilly M., Kohn D.B., Cannon P.M., Bao G., Bushman F.D., Carroll D., Cathomen T., Joung J.K., Roth D., et al. Genome editing technologies: defining a path to clinic. Mol. Ther. 2015;23:796–806. doi: 10.1038/mt.2015.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teboul L., Herault Y., Wells S., Qasim W., Pavlovic G. Variability in genome editing outcomes: challenges for research reproducibility and clinical safety. Mol. Ther. 2020;28:1422–1431. doi: 10.1016/j.ymthe.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ormond K.E., Bombard Y., Bonham V.L., Hoffman-Andrews L., Howard H., Isasi R., Musunuru K., Riggan K.A., Michie M., Allyse M. The clinical application of gene editing: ethical and social issues. Per Med. 2019;16:337–350. doi: 10.2217/pme-2018-0155. [DOI] [PubMed] [Google Scholar]
- 45.Abou-El-Enein M., Cathomen T., Ivics Z., June C.H., Renner M., Schneider C.K., Bauer G. Human genome editing in the clinic: New challenges in regulatory benefit-risk assessment. Cell Stem Cell. 2017;21:427–430. doi: 10.1016/j.stem.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Frati G., Miccio A. Genome editing for beta-hemoglobinopathies: Advances and challenges. J. Clin. Med. 2021;10, 482 doi: 10.3390/jcm10030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng J., Wu Y., Ren C., Bonanno J., Shen A.H., Shea D., Gehrke J.M., Clement K., Luk K., Yao Q., et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020;26:535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C., Georgakopoulou A., Mishra A., Gil S., Hawkins R.D., Yannaki E., Lieber A. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal gamma-globin in beta-YAC mice. Blood Adv. 2021;5:1122–1135. doi: 10.1182/bloodadvances.2020003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Research Council, Division on Earth and Life Studies, Institute for Laboratory Animal Research, and Committee for the Update of the Guide for the Care and Use of Laboratory Animals . National Academies Press; 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 50.Humbert O., Peterson C.W., Norgaard Z.K., Radtke S., Kiem H.P. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta-hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018;8:75–86. doi: 10.1016/j.omtm.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sentmanat M.F., Peters S.T., Florian C.P., Connelly J.P., Pruett-Miller S.M. A survey of validation strategies for CRISPR-Cas9 editing. Sci. Rep. 2018;8:888. doi: 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: a method to quantify base editing from Sanger sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.