

Graphical abstract
Keywords: Molecular dynamics, Pharmacophore screening, G-protein coupled receptor GPR120
Highlights
-
•
Hypothesis of the important residues in conserving the GPR120S “ionic-lock”.
-
•
Computational model targeting W277 and N313 for virtual screening of GPR120S ligands.
-
•
Cpd 9 emerged as a potential GPR120S antagonist and anti-cancer treatment.
Abstract
The G-protein coupled receptor, GPR120, has ubiquitous expression and multifaceted roles in modulating metabolic and anti-inflammatory processes. Recent implications of its role in cancer progression have presented GPR120 as an attractive oncogenic drug target. GPR120 gene knockdown in breast cancer studies revealed a role of GPR120-induced chemoresistance in epirubicin and cisplatin-induced DNA damage in tumour cells. Higher expression and activation levels of GPR120 is also reported to promote tumour angiogenesis and cell migration in colorectal cancer. Some agonists targeting GPR120 have been reported, such as TUG891 and Compound39, but to date development of small-molecule inhibitors of GPR120 is limited.
Herein, following homology modelling of the receptor a pharmacophore hypothesis was derived from 300 ns all-atomic molecular dynamics (MD) simulations on apo, TUG891-bound and Compound39-bound GPR120S (short isoform) receptor models embedded in a water solvated lipid bilayer system. We performed comparative MD analysis on protein–ligand interactions between the two agonist and apo simulations on the stability of the “ionic lock” – a Class A GPCRs characteristic of receptor activation and inactivation. The detailed analysis predicted that ligand interactions with W277 and N313 are critical to conserve the “ionic-lock” conformation (R136 of Helix 3) and prevent GPR120S receptor activation. The results led to generation of a W277 and N313 focused pharmacophore hypothesis and the screening of the ZINC15 database using ZINCPharmer through the structure-based pharmacophore.
100 ns all-atomic molecular dynamics (MD) simulations were performed on 9 small molecules identified and Cpd 9, (2-hydroxy-N-{4-[(6-hydroxy-2-methylpyrimidin-4-yl) amino] phenyl} benzamide) was predicted to be a small-molecule GPR120S antagonist. The conformational results from the collective all-atomic MD analysis provided structural information for further identification and optimisation of novel druggable inhibitors of GPR120S using this rational design approach, which could have future potential for anti-cancer drug development studies.
1. Introduction
G protein coupled receptors (GPCRs) are potential drug targets, with more than 40% of approved drugs modulating the action of one or more GPCRs [1]. Through their interaction with a wide number of bioactive molecules, including ions, lipids, amino acids, peptides, proteins, and small organic molecules these receptors play a crucial role in many physiological processes. These processes include the transmission of light and of odorant signals, mediation of neurotransmission and hormonal action, cell growth and the immune defence [1], [2], [3]. Among GPCRs, GPR120, a member of the rhodopsin sub-family was deorphanized in 2005 [4] as a Free fatty acid receptor (FFAR), which has strong affinity for long-chain fatty acids (LCFAs). GPR120 (or free fatty acid 4 receptor, FFAR4) has recently been explored for its role in cancer cell proliferation and tumour angiogenesis through the binding of Omega-3 polyunsaturated fatty acids (ω-3 PUFAs), mainly docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) [5], [6].
In humans, GPR120 is present in two isoforms: a short isoform at 361 amino acids – (GPR120S) and a long isoform at 377 amino acids (GPR120L). GPR120L contains an additional 16 amino acids in the third intracellular loop (ICL3) and is not present in other mammalian species [4], [7], [8], [9], [10]. In humans, GPR120S couples effectively to both Gq/11 and arrestin pathways whereas GPR120L is an arrestin-biased receptor [7], [10], [11]. The functional significance of the two isoforms in humans remains unclear as there is no tissue reported in which GPR120L is expressed selectively and both isoforms have identical endogenous substrate binding sites known as orthosteric binding pockets [8]. Both the short and long isoforms of GPR120 share the same endogenous substrates which bind at the orthosteric pocket and activate the downstream signalling through second messengers [8], [9]. Besides the orthosteric pocket, GPCRs also contain remote binding sites on the receptor surface known as allosteric sites. These allosteric sites can indirectly modulate the receptor signalling by changing the conformation of orthosteric binding pocket upon ligand binding at allosteric sites [9]. When GPR120S is activated by ω-3 PUFAs (orthosteric endogenous ligands), it couples with Gq/11 and induces mobilisation of intracellular calcium [12] while activation by LCFAs and PUFAs also results in β-arrestin2 recruitment [13]. The basal phosphorylation of GPR120S is two-fold higher than that of GPR120L in the absence of endogenous ligands [4], [14].
GPR120 is a potent lipid mediator in metabolic diseases such as obesity and type-2 diabetes mellitus (T2DM) [15]. The anti-inflammatory actions of GPR120 are induced through β-arrestin2/TAB1 interactions, independent of G protein – Gq/G11-coupled pathway [15]. Recent studies have developed potent and selective GPR120 agonists such as GW9508 - a partial agonist of GPR120 [6], TUG-891 - a selective potent agonist of GPR120 [12] and compound 39 (a benzofuran propanoic acid analogue) from Merck & Co., Inc [16], but no orthosteric GPR120 antagonists are available to date (Fig. S1). However, AH7614 (Fig. S1) has been shown to act as a negative allosteric modulator of GPR120. [17], [18]. GPR120 has also been shown to regulate tumour growth and migration of various cancer types, including melanoma and prostate cancers [5]. GPR120 activation results in a signalling cascade that releases lysophosphatidic acid species via activation of cytosolic phospholipase A2 to induce resistance to cisplatin-induced DNA damage in tumour cells. Such induced-chemoresistance was limited in GPR120 ‘knockout’ studies [19]. A similar study in breast cancer cells reported high expression of GPR120 in breast cancer cells upregulated the ABC transporters expressions which induce resistance to epirubicin-induced cell death. [20] In contrast, GPR120 knockdown in breast cancer cells reduced the epirubicin resistance induced by GPR120 agonist treatment.[20]
The Human Protein Atlas (HPA) analysed RNA-seq data from 64 human cell lines and 37 human tissues samples to estimate protein expression of GPR120 [20], with the highest TPM (transcripts per million) of GPR120 being in rectum and colon tissues [5], [6], [21]. Wu and co-workers screened a panel of malignant and non-malignant human colorectal cell lines and reported higher expression of GPR120 in all studied colorectal cancer (CRC) cell lines compared to non-CRC cell lines [6]. GW9508 tested in CRC cell lines demonstrated activation of PI3K/Akt and nuclear factor kB [6]. GW9508 has been reported to be mitogenic for CRC cells [5], [6], but not for prostate cancer cells [22]. Another report by Liu et al [22] showed that the GPR120 agonist EPA activates ribosomal protein S6 kinase β-1 (p70S6K1) in the CRC CaCo2 cell line. GW9508 (at 10 µM dose) stimulates migration of CRC cells whereas TUG-891 (a GPR120 selective agonist) inhibits migration of prostate cancer cells [6], [22]. These studies suggested that GPR120 activation (rather than antagonism as suggested by Wu et al 2013 [6]) might be effective in anticancer therapy. These two results (on CRC cells and prostate cancer cells) stand in contrast and highlight the importance of exploring the mechanisms underlying the opposing effects of GPR120 agonists on proliferation in different types of cancer cells.
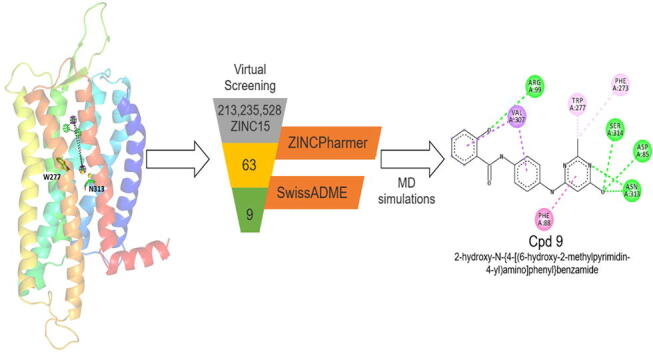
The expansive expression pattern and diverse physiological roles of GPR120 has made it a potential target of multiple therapeutic interventions. For effective structure-based drug design better understanding of the role of residues in the orthosteric binding pocket of the receptor and of agonist-induced subtle conformational changes from ligand-protein interactions is needed. In the absence of an X-ray crystal structure of human GPR120, a 3D homology model of human GPR120S [23] was used to perform structure-based pharmacophore screening (SBPS) through ZINCPharmer with the ZINC database [24] as summarised in Fig. 1. All-atomistic molecular dynamics (MD) simulations of 100 ns were performed on the GPR120S receptor models with SBPS selected hits docked at the orthosteric pocket. The MD studies explored the protein–ligand binding modes and their effect on receptor activation to design potential GPR120 antagonists.
Fig. 1.

General schematic of the methodology applied.
2. Materials and methods
The Irish Centre for High-End Computing (ICHEC) Kay HPC cluster of 336 nodes (13,440 cores) 2.4 GHz Intel Xeon Gold 6148 (Skylake) processors (www.ichec.ie) was used to perform molecular docking and MD simulations. ICHEC provided 60,000 CPU core units for computational processing and 1000 gigabits (GB) of data storage space. The visual analysis and homology model building were carried out on an in-house 8 node (Intel® Core™ i7-4790 CPU @ 3.60 GHz × 8) Linux cluster. For visualization and image rendering PyMol Open-source version 2.1.0 [25] and Biovia DS Client visualizer 2019 [26] were used.
2.1. Molecular docking
GPR120S homology model previously generated using template combination of Orexin OX2 receptor (PDB: 4S0V) and Opioid delta-like receptor (4N6H) (see supplementary appendix A) was used for docking the reference ligands. The generated homology model of GPR120S has previously been used to screen chemical libraries of small molecules in combination of with in vitro screening and structure-active relationship profiling [23]. The successful identification of GPR120-targeting small molecules using the generated GPR120S validated the use of GPR120S homology model in the present computer-aided drug discovery.
TUG-891 [12] and compound 39 [16] were used as reference molecules for predicting binding poses of ligands in the orthosteric pocket by molecular docking. Discovery Studio Pipeline Pilot version 9.1 [26] was used to generate 3D conformations of TUG-891 and compound 39 [12], [16] with stereoisomers and tautomers, which were then energy minimized in the Avogadro suite with the MMFX96 force field [27]. The energy minimised homology model of GPR120S and ligands were pre-processed in AutoDock4 tools (www.autodock.scripps.edu) [28] to assign torsion angles and add polar hydrogens and charge. AutoDock SMINA [29] was used for rigid-flexible docking. AutoDock4 tools’ Grid setting feature was employed to define the binding site grid box, based on the site-specific mutation study of the orthosteric binding pocket residues by Hudson et al 2014 [30] to include residues – R99(TM2), W104 (ECL1), F115 (TM3), W207, F211 (TM5), W277 (TM6) and F304 (TM7), essential for ligand binding. The grid size dimensions used were 40 × 60 × 60 units with spacing of 0.375 Å per unit, and the centre point of the binding pocket set at the xyz coordinates of: x = 61.822, y = 59.75, z = 46.597.
2.2. Protein embedding in lipid bilayer and solvation
The coordinate files of a hydrated, equilibrated 128 molecule POPC lipid bilayer (1-palmitoyl-2oleoyl-sn-glycerophosphocholine) along with lipid parameters for the GROMOS 54a7 force field were obtained from the Automated Topology Force Field Builder (ATB) repository [31].
The coordinate file was then resized using Inflategro2 [32] compatible with GROMACS 5.1 [33] to produce a fully hydrated, 512 molecule lipid bilayer. InflateGro2 incorporated LAMBADA [32] to determine the protein’s longitudinal configuration using a recursive optimization to test different protein orientations thus aligning the membrane and protein. The membrane protein was then automatically embedded into the lipid bilayer patches and clashing lipids removed. The model was stretched in the plane of the lipid bilayer and energy minimized again on contraction allowing optimization of the lipid/protein interface. The entire lipid bilayer was inflated and then slowly compressed around the protein. Each compression step was followed by a round of steepest descent energy minimization to relax the lipid molecules, keeping the protein restrained. The entire system was then solvated with a single-point charge (SPC) water model and neutralized with addition of counter-ions.
2.3. Molecular dynamics simulations of GPR120S-ligand complexes in a phospholipid membrane
MD simulations were performed using the GROMACS version 5.1 package with the GROMOS ffG54a7 force field, extended to improve the lipid components of the force field. The topology and other force field parameters for all ligands were obtained from the ATB repository server [31] and were examined manually for any discrepancies with charges. The final lipid bilayer generated consisted of a pre-equilibrated layer of 512 molecules of POPC molecules.
The systems were then energy minimized for 10,000 steps using the steepest descent algorithm in the GROMACS package. A 500 ps position-restraining simulation was carried out to restrain the protein by 1,000 kJ mol-1 harmonic restraints to relieve the close contacts with POPC and water under NVT (constant Number of particles, Volume, and Temperature) ensemble conditions, with a Vrescale (modified Berendsen) temperature coupler [33]. This was followed by another 5 ns equilibration run under NPT (constant Number of particles, Pressure, and Temperature) ensemble conditions, before a final production run. The MD systems were run at 310 K, i.e., above the phase transition temperature of pure POPC, to ensure that the lipids maintained their proper density, and 1 bar pressure under isothermal-isobaric ensemble [32], [34]. Nosé-Hoover (which is used widely for membrane NPT simulations [32]) temperature and Parrinello-Rahman pressure couplers were used to maintain the temperature and pressure values with the protein, ligands, lipids and water (plus ions) molecules coupled separately with a coupling constant of τt = 0.1 ps. Semi-isotropic pressure coupling was set with τp = 2 ps, allowing the bilayer to deform in the x–y plane independently of the z-axis. A time-step of 2 fs was used throughout, with periodic boundary conditions. LINCS constraint algorithm was used to maintain the geometry of the molecules [35]. Long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method. Van der Waal’s interactions and Coulomb interactions were cut off at 12 Å with updates every five steps. Checkpoint files on production runs were saved every 50 ps.
The trajectory analysis of protein systems was performed by in-built GROMACS tools and visualized in PyMol [25] and XMGRACE (http://plasma-gate.weizmann.ac.il/Grace/). The overall stability of the simulated systems was also checked with respect to temperature, pressure, and potential energy of the systems to check thermodynamic equilibrium during the production simulation runs, confirming the convergence of individual trajectories. The WADDAICA webserver was used to compute the protein–ligand binding free energies of protein–ligand snapshots extracted from the MD trajectory every 5 ns using the Binding Affinity by AI module of WADDAICA [36]. The protein–ligand interaction plots from the MD trajectory were generated using Molecular-dynamics-Interaction-plot (available at https://github.com/tavolivos/Molecular-dynamics-Interaction-plot). Multiple linear regression (MLR) was used to correlate the physicochemical descriptors (obtained from SwissADME [37]) with the binding affinity predictions from WADDAICA [36]. The ChemMine tools webserver was used to develop a structural similarity clustering scatterplot [38]. ChemMine clustering tool converts the Tanimoto similarity matrix into distance matrix to cluster the molecules.
2.4. Structure-based pharmacophore screening
A six-feature pharmacophore model was generated by manual curation of docked reference structures (TUG-891 and compound 39) using the ZINCPharmer web interface [24], [39], which was then screened against the ZINC15 database containing ∼ 230 million purchasable lead-like 3D molecules [24]. The physicochemical and drug-likeness profiling of the screened hits was performed using the online webserver SwissADME [37]. The pharmacophore screening results from ZINCPharmer were downloaded as structure data files (sdf format). The 3D molecules were then prepared and docked against the GPR120S receptor model following the set protocol described in section 2.1. The selected compounds were prepared for 100 ns MD simulations following the protocol described in section 2.2 and 2.3 (see Fig. 1)
3. Results and discussion
3.1. Molecular docking of known GPR120S ligands
In silico investigation of receptor activation commenced with docking of a known agonist to the receptor model in the inactive state (homology model). Previous molecular docking and site-specific mutations studies of GPR120S have revealed that a single arginine residue in TM2 (R99) has a critical interaction between the receptor and the –COOH (carboxylate) of its ligands [7], [30]. Six other residues from the orthosteric binding pocket were defined essential for TUG-891 binding and interaction with GPR120S were: W104 (ECL1), F115 (TM3), W207, F211 (TM5), W277 (TM6) and F304 (TM7) following site-specific mutation studies performed by Hudson et al [30] (Fig. 2a, b). These seven residues were selected as the main criterion for defining the orthosteric binding pocket as well as protein–ligand interactions for binding pose prediction of GPR120S agonists. Over the years, some GPR120 agonists have been developed but to date no orthosteric antagonist of GPR120 is available. For chemical scaffold diversification, besides TUG-891 (EC50 43 nM) [12], [13], [30], another known agonist Compound39 – benzofuran propanoic acid analogue (EC50 97 nM) [16] was used in the molecular docking experiment (Fig. 2c, 2d).
Fig. 2.

(a) Graphical representation of GPR120S energy minimized 3D model embedded in lipid bilayer; (b) Selected seven residues (shown as sticks in orange) in the orthosteric pocket of GPR120S essential for ligand binding. 2D interaction map of TUG-891 (c) and Compound 39 (d). The 3D images were visualized and rendered in PyMol v2.1.0 [25]. The 2D interaction maps were generated in BIOVIA DS Client visualizer 2019 [26]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The best docked pose for TUG-891 yielded a binding score −9.87591 kCal/mol (free energy of binding calculated by AutoDock SMINA). The carboxylate of TUG-891 forms a salt bridge with R99 and a strong T-type (perpendicular) pi-stacking interaction between F115 and the cyclic aromatic core structure of TUG-891 stabilize the ligand into the pocket formed between TM3, TM6 and TM7. Other equitable hydrophobic interactions were also observed to stabilize the docked TUG-891 in the orthosteric binding pocket (Table S1). A molecular docking study conducted by Hudson [12], [13], [30] used the HM of human GPR120S (based on beta-2 adrenoreceptor template; PDB id: 3P0G) to dock TUG-891. An extensive overlap is observed between the binding pocket residues interacting with the agonist in both studies. Similarly, the best docked pose of Compound39 (binding score −9.82688 kCal/mol) also showed favorable hydrophobic interactions and displayed hydrogen bonds with (R99, W277) two out of the seven specific residues as well as hydrogen bonds (T125, N313) with other residues in the orthosteric binding pocket (Table S2).
The results from literature and the present methodologies provide valuable information concerning the optimal GPR120S model generation. To design new GPR120S selective ligands whether agonists or antagonists, optimal interactions with the non-conserved residues involved in the binding pocket need to be considered. These residues are specific to GPR120S and share proximity to the bound ligands.
3.2. MD simulations of GPR120S-ligand complexes
GPCR activation is initiated by binding of an agonist into the orthosteric binding pocket causing reorganization of interhelical interactions. These structural changes propagate towards the intracellular domain via activation switches as they relay the changes from the inactive to the active state [40]. Here the conformational investigation began with an energy minimized and pressure–volume equilibrated systems of human GPR120S (inactive state) embedded in POPC lipid bilayer and solvated SPC water molecules (Fig. 2a). The 300 ns MD simulations of three different systems - an apo system and two agonist-bound systems, TUG-891 and Compound39 were performed to gain further insights on the topology of the binding site and agonist orientations over a time period. The protein backbone of starting structures in all the three systems reached stability with no significant changes in RMSD values after 200 ns timestamp (Figure S2 a). The cysteine bridge between TM2 and ECL2 is highly conserved in most of the Class A GPCRs and is used as an anchor point in modelling the individual backbone of hydrophilic loops as well as forming a ligand trap [40], [34]. The continuous disulfide linkage between the two cysteine residues of GPR120S model (C111 and C194) in TM2 and ECL2 was observed throughout the 300 ns MD production run of all three protein systems (Figure S2 b) supporting the structural stability of the generated GPR120S model.
The RMSF (Root Mean Square Fluctuation) analysis of the protein backbone (Fig. 3) illustrates stable helix domains and high rate of fluctuations in the loop regions only, especially the ECL2 (177–204) and ICL3 (236–252) domains of the ligand-bound protein models compared to the apo protein system. The Compound39-bound system recorded a marked RMSF difference in the ICL1 (65–71) domain compared to TUG-891-bound system suggesting that both the agonists might be inducing conformational changes by two different mechanisms. The low range of fluctuations in the apo protein system suggest that the generated protein model was in a stable inactive state and remained in the inactive state as it might be in global minima due to absence of ligand induced conformational changes during MD simulation run.
Fig. 3.

RMSF (Å) of protein residues recorded during 300 ns MD simulation run of GPR120S in bound (Compound 39 or TUG-891) and unbound forms (apo protein). The intracellular loop (ICL) and extracellular loop (ECL) regions are highlighted as shaded regions. The transmembrane regions in the 3D structures are - TM1: residues 36–65; TM2: 73–101; TM3: 107–141; TM4: 152–175; TM5: 204–233; TM6: 252–289; TM7: 296–324.
The comparative analysis of the putatively active state models (agonist-bound) with the inactive (apo-model) state recorded conformational changes in the residues involved in the ionic lock - R136 of TM3 and D259 of TM6 over the MD production run of 300 ns. The inactive state model retained the closed conformation of ionic lock as both the residues stayed within the range of 2–4 Å – an appropriate distance to form and sustain the hydrogen bond or the salt-bridge, making the inactive state of the model stable. As expected, the active state model of TUG-891-bound human GPR120S recorded the two residues drifting apart over the MD trajectory of 300 ns with the ionic-lock broken within the first 10 ns of MD simulation. At the end of the production run the two residues were ∼ 9–10 Å apart disrupting the salt-bridge. (Fig. 4a). The Compound39-bound protein model demonstrated unexpected behaviour of staying closer to the inactive conformation. The average distance between the two residues (R136 and D259) remained ∼ 4 Å throughout the production run (Fig. 4b). Although the residues were not close enough for salt-bridge formation but with the residues being this close, the intracellular cavity of the receptor might not be able to accommodate the heteromeric G protein.
Fig. 4.

a) Graphical picture of ionic lock closed at simulation time T0 and open conformation at Tavg; representing the average conformation from the 300 ns MD run of the TUG-891 bound protein model; b) Distance between centre of mass of R136(TM3) and D259(TM6) recorded during 300 ns MD production run predicting the effect of bound ligands at the structural conformation of ionic lock residues; c) Distance between centre of mass of R136(TM3) and D259(TM6) recorded during 300 ns MD production re-run. Running average of distance every 1 ns is shown for clarity.
Two possible inferences can be proposed from the Compound39-bound protein simulation: 1) The protein model might be in a local minimum of the energy landscape at that specific conformation [41]; 2) The interaction network pattern of Compound39 carboxylate tail with T125, W277 and N313 (Fig. 4d, 5a), not observed in TUG-891 docking interactions, led the MD trajectory to different conformational changes at the intracellular domain especially at TM3 – ER136M motif (D/ERY) involved in the “ionic lock” formation.
A 300 ns MD re-run from the same starting conformation of Compound39-bound protein system with random initial velocities verified that the variance was reproducible. The comparative analysis of distance between the residues (R136 and D259 – involved in formation of “ionic lock”) from the 300 ns MD rerun reported an increase (average ∼ 5 Å) with a higher range of fluctuations compared to the first 300 ns MD run (average ∼ 3 Å) but were significantly less than those of the TUG-891-bound system (average ∼ 9 Å). The average distance was recorded to be greater than 4 Å limiting the hydrogen bond interactions for the salt bridge / “ionic lock” formation (Fig. 4c).
A recently published study investigated protein–ligand stability from a 200 ns MD simulation of SR13 (a chromane propionic acid analogue – derived from the same Merck & Co. patented series as Compound39) docked to homology model of GPR120S receptor (Uniprot ID: Q5NUL3-2) [42], [43]. This study highlighted the conformational changes the SR13 ligand adopts to enter the binding pocket [42]. However, to the best of our knowledge, these are the first simulations of GPR120S complexed with ligands (TUG-891 and Compound39) at a 300 ns timescale.
As the human GPR120S receptor can bind to the flexible FFAs (PUFAs) as well as a diverse set of rigid compounds such as TUG-891 suggested the existence of different binding conformations in the orthosteric binding pocket of the receptor leading to a cascade effect inducing protein activation. The difference in binding pattern of TUG-891 and Compound39 highlighted important residues (T125, W277 and N313) in the orthosteric binding pocket. Residues T125 and N313 were not analyzed in the previous site-specific mutagenic study while the W277A mutation resulted in loss of receptor activity [30]. The hydrogen bonding analysis of Compound39-bound protein simulations showed an average of ∼ 60% H-bond occupancy between Compound39 and the W277 sidechain during the 300 ns production run accompanied with ∼ 35% and ∼ 10% occupancy for the N313 and T125 sidechains respectively. The interaction network of W277 and / or N313 with Compound39 might be affecting the conformational changes as observed in TUG-891-bound protein simulations.
3.3. W277 and N313-based pharmacophore screening
Based on the inferred significance of the W277 and N313 interaction network with the carboxylate chain of Compound39, a single structure-based pharmacophore model was generated by enumerating the 3D conformation of functional features present in the receptor binding pocket. The docked conformations of TUG-891 and Compound39 were superimposed to generate the pharmacophore model using the ZINCPharmer package [39] which resulted in a six-featured hypothesis consisting of two HBA (Hydrogen bond acceptor), two Ar (Aromatic ring systems) and two Hb (hydrophobic) with preferred chemical features (Fig. 5; Table S3. The selected hypothesis was generated to focus screening on ligands interacting with the W277 and N313 (Fig. 5b) residues with scaffold features of TUG-891 and Compound39 being added to attain rigidity and better anchorage in the binding pocket by interacting with essential binding residues [13], [30] such as R99, W104, F115, W207, F211 and F304.
Fig. 5.

a) Superimposed docked poses of TUG-891 (Cyan stick model) and Compound39 (Green stick model) with GPR120S. TM5 is hidden in the image for a clearer view; b) Developed pharmacophore model with its corresponding chemical features used for screening the ZINC database [25], [39]. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The pharmacophore-based virtual screening (VS) resulted in 63 unique chemical hits identified from the ZINC15 commercial database. Further, selection of these 63 compounds was performed by analysis of physicochemical descriptors computed using SwissADME [37] to predict the druglike and / or lead-like nature of the compounds. In combination with analysis of predicted physicochemical descriptors, an in cerebro assessment was applied to select compounds with diverse and synthesizable scaffolds. Such a well-informed manual selection of ligands after virtual screenings have previously been reported to refine the screening performance [44], [45].
The final screening resulted in 9 best-hits (shown in Table 1) based on the SwissADME predictions over compounds’ physicochemical properties, lipophilicity, bioavailability, and drug-likeness (see supplementary material). These hit compounds were then prepared and docked into the receptor binding pocket following the set protocol (section 2.1) to prepare protein–ligand complexes. The protein–ligand interactions of these 9 compounds were analyzed (Fig. 6a) to confirm W277 / N313 H-bond interaction(s) before preparing the protein–ligand complexes for 100 ns MD production runs following the set protocol (section 2.2 and 2.3).
Table 1.
Assigned nomenclature and docked binding scores (kCal/mol) of the selected pharmacophore-based VS hits based on the SwissADME drug-likeness profiling. Log Po/w – oil/water partition co-efficient; Leadlikeness – 250 ≤ MW ≤ 350; XLOGP ≤ 3.5; Num. rotatable bonds ≤ 7 [37]. The full SwissADME parameters are provided in the supplementary material for Cpds 1–9
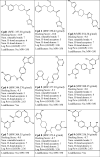 |
Fig. 6.



a) Protein-ligand interaction fingerprint map of the 9 docked compounds with GPR120S; Green – (HB) Hydrogen bond; Yellow – (HP) Hydrophobic interactions; Grey – No interactions; b) Heatmap of protein–ligand binding affinities of snapshots extracted from the 100 ns MD simulation trajectory and scored by webserver WADDAICA [36]; c) Protein-ligand interaction fingerprint map [46] plotting the number of interactions of the residue with the ligands, shows compounds 1, 7 and 9 with conserved W277 and N313 H-bond interactions over the period of 100 ns MD production runs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3.1. MD simulations study of N313-interacting compounds.
Comparative MD simulations for the 100 ns duration of each of the co-complexes of the 9 hit compounds from ZINC database, TUG-891, Compound39 and Apo-model were carried out to evaluate the significance of the N313 interaction in terms of conformational changes to the protein. A MD time scale of 100 ns was chosen based on observations from other studies where100ns MD simulations were shown to be sufficient to illustrate the dynamics of protein–ligand fingerprinting and induced conformational changes in GPCR molecular studies [42], [47], [48]. The protein–ligand binding affinity of the selected 9 compounds over the duration of 100 ns MD simulations predicts the strength of the binding interaction between the receptor and the compounds (Fig. 6b). Cpd 2 and Cpd 8 were predicted to show a continuous decreasing gradient in the binding affinity while Cpd 9 was predicted to show a continuous increase in the binding affinity over time during the MD simulation. Other compounds did not show a consistent pattern of decreasing or increasing binding affinity predictions (Fig. 6b). The binding affinity predictions from the 100 ns MD studies gave insights to the binding stability of docked hits and non-covalent interactions with residues in the binding pocket. Amongst all the compounds, Cpd 1, 7 and 9 were predicted to conserve W277 and N313 H-bond interactions during the 100 ns MD simulations (Fig. 6c). As the binding affinities predicted for this study used only the protein–ligand complex (without solvent) snapshots extracted from the MD simulations, it should be cautioned that solvent (water) molecules also play a key role in non-covalent interactions – forming bridge interactions between ligand and protein [49].
Most of the interactions with Cpd9 (electrostatic or hydrophobic) were observed to be continuous during the simulation. Similarly, the protein–ligand binding affinity prediction over the 100 ns MD simulation suggested a stable binding of Cpd9 in the orthosteric binding pocket of GPR120S (Fig. 6b). In contrast, Cpd 2 showed the lowest binding affinity in the binding pocket compared to the other ligands and Cpd 2 was the only ligand which did not form hydrophobic and /or H-bond interactions with R99 as well as N313 (Fig. 6a and 6b) at the starting conformation. Interestingly, Cpd 7, which had a N313 H-bond interaction (Fig. 6a) at the initial (0 ns) conformation was predicted to have a high binding affinity of −10.8 kCal/mol. However, the binding affinity of Cpd 7 reduced to −10.25 kCal/mol over the MD simulation as the number of N313 H-bond interactions of Cpd 7 reduced (Fig. 6c). The binding affinity predictions (Fig. 6b) of Cpd 7 at 25 ns and 45–55 ns timeframes reported strong binding affinities (-11.48 kCal/mol and −10.85 to −10.91 kCal/mol respectively) compared to the rest of the MD production run. The protein–ligand interaction fingerprinting of the 100 ns MD production run showed that re-formation of H-bonds between Cpd7 and N313 was accompanied by several hydrophobic interactions with other residues in the binding pocket such as F88, F115, W207, I280, I284 and F303. Similar interactions patterns were observed in other ligands (Cpd 1, 4, 5) where an improvement in binding affinities was predicted due to reformation of the N313 H-bond with ligands along with increased hydrophobic interactions. While in-depth analysis of extended MD simulations would further help to understand these changes the present correlative binding affinity predictions and N313 interactions could infer the key role of N313 residue in the ligand binding stability.
As GPR120S was modelled from templates in the inactive form, the binding of ligands with antagonistic activity should keep the receptor in inactive state without causing major conformational changes at the intracellular domain of the receptor. The protein’s structural stability evaluation by RMSF analysis of 100 ns trajectory showed (Fig. 7a) that Cpd 9 and Cpd 1 stabilized the protein backbone as well as reducing the fluctuations of ECL2 (177–203) and ICL3 (236–252) regions observed in Apo, TUG-891 and Compound39 bound protein systems while Cpd 7 bound protein system has recorded the highest range of fluctuations (∼12 Å) in the ICL3 region. Binding of other ligand molecules have an overall similar effect on the protein backbone fluctuations, but higher than that of Cpd 9.
Fig. 7.

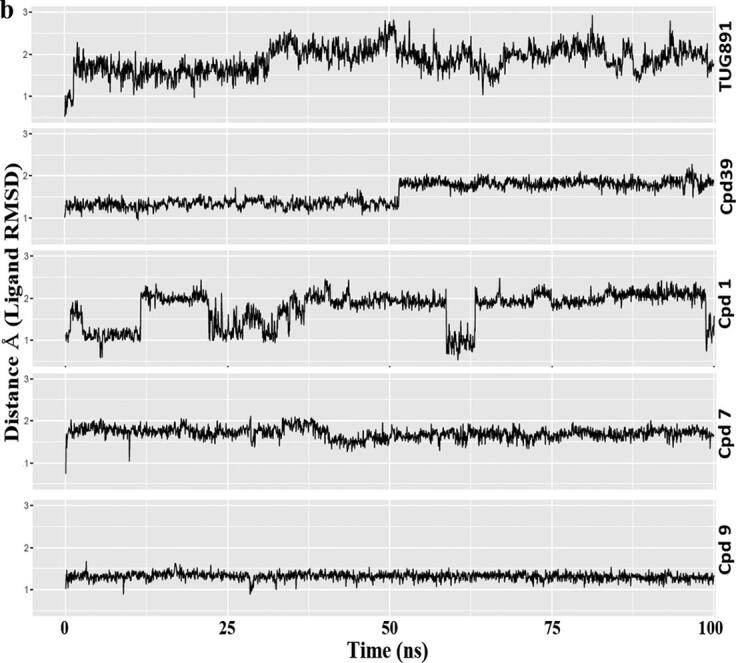
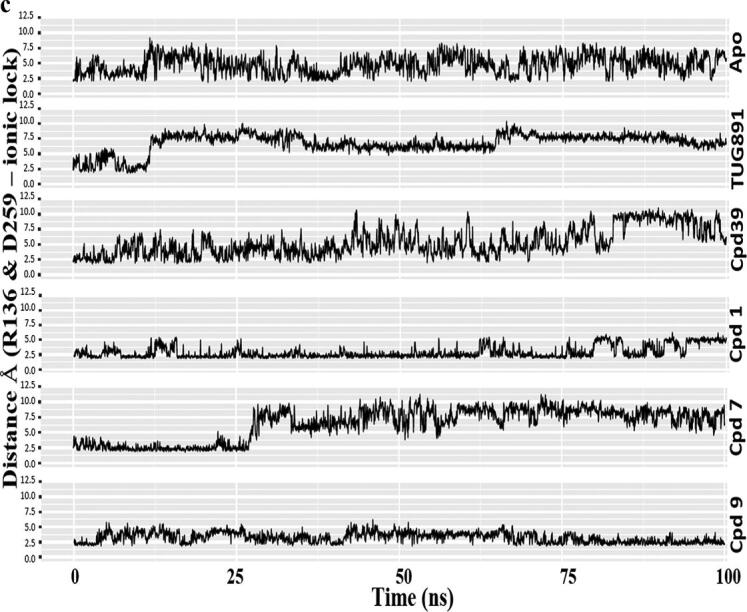
Comparative MD simulation analysis of best hits, compound 1, 7 and 9 with respect to Apo, TUG-891 and Compound39 bound proteins over 100 ns timescale; a) RMSF plot of the protein backbone. The intracellular loop (ICL) and extracellular loop (ECL) regions are highlighted as shaded regions. The transmembrane regions in the 3D structure are - TM1: 36–65; TM2: 73–101; TM3: 107–141; TM4: 152–175; TM5: 204–233; TM6: 252–289; TM7: 296–324; b) RMSD plot of the ligand atoms; c) Distance plot between the center of mass of residues R136(TM3) and D259(TM6) involved in “ionic-lock” conformation.
Focusing on the ligands themselves Cpd 9 and Cpd 7 they were also found to be the most stable in the GPR120S orthosteric binding pocket with RMSD values below ∼ 1.5 Å and 2 Å respectively throughout the 100 ns MD trajectory (Fig. 7b, S3-S7). Such low ligand RMSD values suggest that these two ligands are tightly bound to the orthosteric binding pocket without major changes in their initial docked orientations. It is important to mention that both ligands lead to contrasting effects on the protein-backbone RMSD values – Cpd 9 stabilized the protein-backbone in its initial inactive form whereas Cpd 7 deviated the protein-backbone away from its initial conformation. Cpd 9 stabilized the protein within the timespan of the first 20 ns, keeping the average RMSD of protein model below 4 Å. While the binding of Cpd 7 to GPR120S leads to the highest RMSD values (∼8 Å). Ligand RMSD analysis of Cpd 1 also presented a range of fluctuations with protein backbone RMSD reaching above ∼ 5 Å (Fig. 7b).
Compared with other ligands, the Cpd 9 bound protein model predicted the least movement of the distance between R136 and D259 (involved in the “Ionic lock”) at the intracellular domain of GPR120S (Fig. 7c) that is the site specific for G-protein coupling. The study published by Provasi et al. [50] used inactive and active crystal structures of GPCRs with ligands eliciting different pharmacological actions performing 20 ns MD simulations. The study reported that depending on their pharmacological activity, the ligand bound to the receptor can shift the conformational equilibrium towards active or inactive state of the receptor. A similar conformational shift (Cpd7 – from inactive to active state) as well as stability (Cpd9 – stabilized inactive state) in receptor state was observed in our study. Here, during the 100 ns MD production run of Cpd9 the “ionic lock” remained closed (Fig. 7c, S6), inhibiting the coupling between receptor and G-protein, and hence keeping the receptor in the inactive state.
3.3.2. Multiple linear regression model and structural diversity analysis of the selected compounds
For further analysis, MLR was applied to predict the contribution of physicochemical descriptors (independent variable) such as molecular weight, H-bond donors, H-bond acceptors, logP and topological polar surface area (TPSA) on binding affinity (dependent variable) [51], [52] (Fig. 8). The average binding affinity (obtained from WADDAICA webserver) of the last 20 ns of MD snapshots (from 80 to 100 ns) was used for the MLR model. The performance of the MLR model is expressed in terms of R2, which was found to be 0.799 signifying that ∼ 80% of the data fit the regression model. Cpd 9 with the highest binding affinity value from the WADDAICA server [36], was also predicted to have the highest binding affinity by the MLR model. Indeed, the MLR model agreed with the binding affinity predictions of the WADDAICA webserver but it should be noted that many of the variables used in the MLR model were obtained from other prediction algorithms – such as binding affinity, logP and TPSA.
Fig. 8.
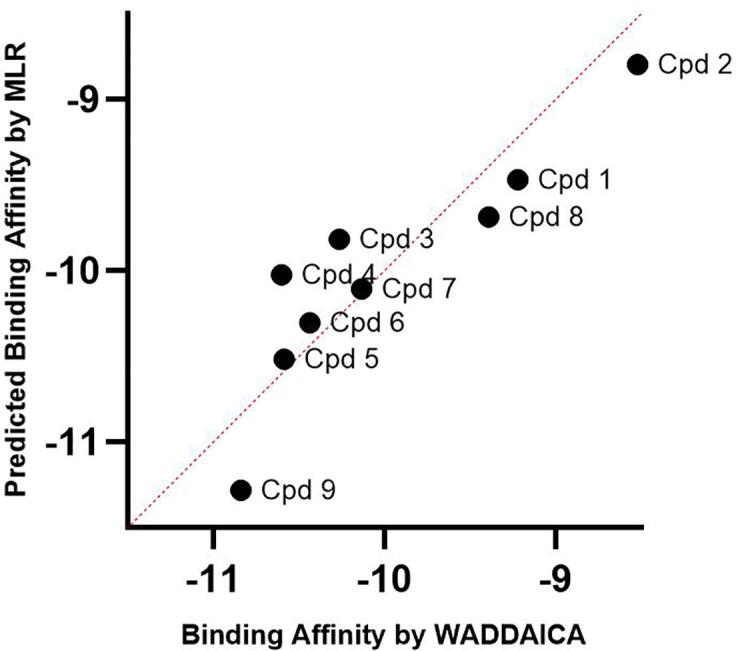
Scatter plot of a multiple linear regression model of the contribution of physicochemical descriptors –molecular weight, number of H-bond acceptors and H-bond donors, TPSA and logP from SwissADME [37] over the WADDAICA [36] binding affinity for the 9 compounds. The full SwissADME parameters are provided in the supplementary material for Cpds 1–9.
The selected compounds Cpd 1–9 were also clustered with the co-crystalized ligands of templates (4N6H-EJ4 and 4S0V-SUV) used for homology model generation and reference ligands used for generation of pharmacophore (TUG-891 and Compound39) to verify the unbiased nature of the screened compounds. The two-dimensional scaling cluster (Fig. 9; Table S4) confirms that screening of Cpd9 and Cpd7 was not biased towards the templates or reference ligands used in the study.
Fig. 9.

Clustering of template ligands (PDBs: 4S0V-SUV and 4N6H-EJ4); agonists (TUG-891 and Compound 39) and selected compounds 1–9 by Two Dimensional similarities with a similarity cutoff of 0.4 [38]. V1 and V2 denotes the variable 1 and 2 for the distance matrix obtained from the Tanimoto similarity matrix.
Finally, with the design and discovery of novel scaffolds by in silico methods, the pharmacokinetic profile and synthesizability of these compounds can be a limiting factor. The proposed antagonist Cpd9 has a promising predicted ADME profile indicating leadlikeness but with a moderate predicted solubility (see supplementary material). Furthermore, a feasible retrosynthesis scheme obtained from CAS SciFinder (https://scifinder.cas.org) confirms the ease to synthesis of this compound (Figure S8).
4. Conclusion
In this study, the MD analysis of 300 ns production runs of agonist bound GPR120S models led to the generation of a pharmacophore hypothesis targeting W277 and N313 residues of GPR120S receptor to discover potential candidates as GPR120S antagonists. The structure-based pharmacophore hypothesis was validated by running MD simulations of pharmacophore identified synthesizable hits over a period of 100 ns suggesting that H-bond interactions of Cpd 9 (2-hydroxy-N-{4-[(6-hydroxy-2-methylpyrimidin-4-yl)amino]phenyl}benzamide) with W277 and N313 stabilize the occupancy of the ligand in the orthosteric pocket and keeps the protein in the inactive form. While the interactions of a phenylimino-phenol analogue - Cpd 7 phases the protein from inactive to active form due to breakage of the ionic lock which can lead to G-protein coupling at the intracellular domain. Further site-specific mutation studies targeting the N313 residue can confirm the importance of N313 interactions in GPR120S antagonist design. Therefore, the insights from the present study can potentially be employed to enhance the selectivity of GPR120S ligands and target interactions with key residues (W277 and N313) to develop novel agonists and antagonists. Further development of a selective and potent GPR120S antagonist could be useful in management of various cancers where GPR120S has been implicated as a tumour-promoting receptor.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors gratefully acknowledge to Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. The authors wish to acknowledge Technological University Dublin (TU Dublin) for providing financial assistance through Fiosraigh Dean of Graduate Research School Award doctoral fellowship to Ajay Pal (Grant No. PB04185).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.11.005.
Contributor Information
Ajay Pal, Email: ajay.pal@tudublin.ie.
James F. Curtin, Email: james.curtin@tudublin.ie.
Gemma K. Kinsella, Email: gemma.kinsella@tudublin.ie.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hauser A.S., Attwood M.M., Rask-Andersen M., Schiöth H.B., Gloriam D.E. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16(12):829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali Daniel Chikere, et al. G protein-coupled receptor, an important target for drug design and screening. EC Pharmacol Toxicol. 2018;6.5:326–333. [Google Scholar]
- 3.Salon J.A., Lodowski D.T., Palczewski K., Perez D.M. The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol Rev. 2011;63(4):901–937. doi: 10.1124/pr.110.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirasawa A., Tsumaya K., Awaji T., Katsuma S., Adachi T., Yamada M., et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11(1):90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 5.Senatorov I., Moniri N. The role of free-fatty acid receptor-4 (FFA4) in human cancers and cancer cell lines. Biochem Pharmacol. 2018;150(2018):170–180. doi: 10.1016/j.bcp.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Q., Wang H., Zhao X., Shi Y., Jin M., Wan B., et al. Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene. 2013;32(49):5541–5550. doi: 10.1038/onc.2013.264. [DOI] [PubMed] [Google Scholar]
- 7.Butcher A., et al. Concomitant Action of Structural Elements and Receptor Phosphorylation Determines Arrestin-3 Interaction with the Free Fatty Acid Receptor FFA4. J Biol Chem. 2014;289(26):18451–18465. doi: 10.1124/mol.113.087783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore K., Zhang Q., Murgolo N., Hosted T., Duffy R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: Comparison with human GPR120 splice variants. Comp Biochem Physiol B: Biochem Mol Biol. 2009;154(4):419–426. doi: 10.1016/j.cbpb.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Chan H., Li Y., Dahoun T., Vogel H., Yuan S. New Binding Sites, New Opportunities for GPCR drug discovery. Trends Biochem Sci. 2019;44(4):312–330. doi: 10.1016/j.tibs.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 10.Watson S.-J., Brown A.J.H., Holliday N.D. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol. 2012;81(5):631–642. doi: 10.1124/mol.111.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davenport, A. et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G Protein-Coupled Receptor List: Recommendations for New Pairings with Cognate Ligands. Pharmacol Rev. 65, 3 (2013), 967-986. DOI: 10.1124/pr.112.007179 [DOI] [PMC free article] [PubMed]
- 12.Hudson B.D., Shimpukade B., Mackenzie A.E., Butcher A.J., Pediani J.D., Christiansen E., et al. The Pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol. 2013;84(5):710–725. doi: 10.1124/mol.113.087783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimpukade B., Hudson B.D., Hovgaard C.K., Milligan G., Ulven T. Discovery of a potent and selective GPR120 agonist. J Med Chem. 2012;55(9):4511–4515. doi: 10.1021/jm300215x. [DOI] [PubMed] [Google Scholar]
- 14.Burns R.N., Moniri N.H. Agonism with the omega-3 fatty acids α-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun. 2010;396(4):1030–1035. doi: 10.1016/j.bbrc.2010.05.057. [DOI] [PubMed] [Google Scholar]
- 15.Moniri N. Free-fatty acid receptor-4 (GPR120): Cellular and molecular function and its role in metabolic disorders. Biochem Pharmacol. 2016;110–111(2016):1–15. doi: 10.1016/j.bcp.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lombardo M., Bender K., London C., Plotkin M.A., Kirkland M., Mane J., et al. Discovery of benzofuran propanoic acid GPR120 agonists: From uHTS hit to mechanism-based pharmacodynamic effects. Bioorg Med Chem Lett. 2016;26(23):5724–5728. doi: 10.1016/j.bmcl.2016.10.054. [DOI] [PubMed] [Google Scholar]
- 17.Watterson K.R., Hansen S.V.F., Hudson B.D., Alvarez-Curto E., Raihan S.Z., Azevedo C.M.G., et al. Probe-Dependent negative allosteric modulators of the long-chain free fatty acid receptor FFA4. Mol Pharmacol. 2017;91(6):630–641. doi: 10.1124/mol.116.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Son S.-E., Kim N.-J., Im D.-S. Development of free fatty acid receptor 4 (FFA4/GPR120) agonists in health science. Biomol. Therap. 2021;29(1):22–30. doi: 10.4062/biomolther.2020.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houthuijzen J.M., Oosterom I., Hudson B.D., Hirasawa A., Daenen L.G.M., McLean C.M., et al. Fatty acid 16:4(n-3) stimulates a GPR120-induced signaling cascade in splenic macrophages to promote chemotherapy resistance. FASEB J. 2017;31(5):2195–2209. doi: 10.1096/fsb2.v31.510.1096/fj.201601248R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X., He S., Gu Y., Wang Q., Chu X., Jin M., et al. Fatty acid receptor GPR120 promotes breast cancer chemoresistance by upregulating ABC transporters expression and fatty acid synthesis. EBioMedicine. 2019;40:251–262. doi: 10.1016/j.ebiom.2018.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uhlen, M. et al. 2017. A pathology atlas of the human cancer transcriptome. Science. 357, 6352 (2017), eaan2507. DOI: 10.1126/science.aan2507 [DOI] [PubMed]
- 22.Liu Z.e., Hopkins M.M., Zhang Z., Quisenberry C.B., Fix L.C., Galvan B.M., et al. Omega-3 fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J Pharmacol Exp Ther. 2015;352(2):380–394. doi: 10.1124/jpet.114.218974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pal A., Curtin J.F., Kinsella G.K. In silico and in vitro screening for potential anticancer candidates targeting GPR120. Bioorg Med Chem Lett. 2021;31:127672. doi: 10.1016/j.bmcl.2020.127672. [DOI] [PubMed] [Google Scholar]
- 24.Sterling T., Irwin J.J. ZINC 15 – ligand discovery for everyone. J Chem Inf Model. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.W.L. DeLano, The PyMol Molecular Graphics System, Schrödinger, LLC. Version 2.1.0. https://github.com/schrodinger/pymol-open-source
- 26.Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017 Dassault Systèmes, San Diego (2016)
- 27.Hanwell M., et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4:1. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.v30:1610.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koes D., et al. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inform Model. 2013;53(8):1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson B.D., Shimpukade B., Milligan G., Ulven T. The Molecular basis of ligand interaction at free fatty acid receptor 4 (FFA4/GPR120) J Biol Chem. 2014;289(29):20345–20358. doi: 10.1074/jbc.M114.561449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koziara K.B., Stroet M., Malde A.K., Mark A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies. J Comput Aided Mol Des. 2014;28(3):221–233. doi: 10.1007/s10822-014-9713-7. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt T., Kandt C. LAMBADA & InflateGRO2: Efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. Biophys J. 2012;102(3):173a. doi: 10.1016/j.bpj.2011.11.938. [DOI] [PubMed] [Google Scholar]
- 33.Bussi G., et al. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1) doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 34.Calmet P., et al. Real time monitoring of membrane GPCR reconstitution by plasmon waveguide resonance: on the role of lipids. Sci Rep. 2016;6:1. doi: 10.1038/srep36181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hess B. P-LINCS: A parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2007;4(1):116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 36.Bai Q., Ma J., Liu S., Xu T., Banegas-Luna A.J., Pérez-Sánchez H., et al. WADDAICA: A webserver for aiding protein drug design by artificial intelligence and classical algorithm. Comput Struct Biotechnol J. 2021;19:3573–3579. doi: 10.1016/j.csbj.2021.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daina A., et al. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Backman T., Cao Y., Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011;39(suppl):W486–W491. doi: 10.1093/nar/gkr320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koes, D. and Camacho, C. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Research. 40, W1 (2012), W409-W414.DOI: 10.1093/nar/gks378 [DOI] [PMC free article] [PubMed]
- 40.Venkatakrishnan A.J., Deupi X., Lebon G., Heydenreich F.M., Flock T., Miljus T., et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature. 2016;536(7617):484–487. doi: 10.1038/nature19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goedecker S. Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems. J Chem Phys. 2004;120(21):9911–9917. doi: 10.1063/1.1724816. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X., Sun H., Wen X., Yuan H. A selectivity study of FFAR4/FFAR1 Agonists by molecular modeling. J Chem Inf Model. 2019;59(10):4467–4474. doi: 10.1021/acs.jcim.9b00735. [DOI] [PubMed] [Google Scholar]
- 43.Adams G.L., Velazquez F., Jayne C., Shah U., Miao S., Ashley E.R., et al. Discovery of chromane propionic acid analogues as selective agonists of GPR120 with in vivo activity in rodents. ACS Med Chem Lett. 2017;8(1):96–101. doi: 10.1021/acsmedchemlett.6b00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voet A., Kumar A., Berenger F., Zhang K. Combining in silico and in cerebro approaches for virtual screening and pose prediction in SAMPL4. J Comput Aided Mol Des. 2014;28(4):363–373. doi: 10.1007/s10822-013-9702-2. [DOI] [PubMed] [Google Scholar]
- 45.Voet A., Callewaert L., Ulens T., Vanderkelen L., Vanherreweghe J.M., Michiels C.W., et al. Structure based discovery of small molecule suppressors targeting bacterial lysozyme inhibitors. Biochem Biophys Res Commun. 2011;405(4):527–532. doi: 10.1016/j.bbrc.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 46.Molecular-dynamics-Interaction-plot. Code Available at: https://github.com/tavolivos/Molecular-dynamics-Interaction-plot
- 47.Chan H., Filipek S., Yuan S. The principles of ligand specificity on beta-2-adrenergic receptor. Sci Rep. 2016;6(1) doi: 10.1038/srep34736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rahman M.M., Saha T., Islam K.J., Suman R.H., Biswas S., Rahat E.U., et al. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J Biomol Struct Dyn. 2021;39(16):6231–6241. doi: 10.1080/07391102.2020.1794974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venkatakrishnan A.J., Ma A.K., Fonseca R., Latorraca N.R., Kelly B., Betz R.M., et al. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc Natl Acad Sci. 2019;116(8):3288–3293. doi: 10.1073/pnas.1809251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Provasi D., Artacho M.C., Negri A., Mobarec J.C., Filizola M., Nussinov R. Ligand-induced modulation of the free-energy landscape of G protein-coupled receptors explored by adaptive biasing techniques. PLoS Comput Biol. 2011;7(10):e1002193. doi: 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Funar-Timofei S., Borota A., Crisan L. Combined molecular docking and QSAR study of fused heterocyclic herbicide inhibitors of D1 protein in photosystem II of plants. Mol Diversity. 2017;21(2):437–454. doi: 10.1007/s11030-017-9735-x. [DOI] [PubMed] [Google Scholar]
- 52.da Silva Costa J., da Silva Lopes Costa K., Cruz J.V., da Silva Ramos R., Silva L.B., Do Socorro Barros Brasil D., et al. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr Pharm Des. 2018;24(5):576–594. doi: 10.2174/1381612823666170711112510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.