

Summary
Cell-Specific Mitochondria Affinity Purification (CS-MAP) enables isolation and purification of intact mitochondria from individual cell types of Caenorhabditis elegans. The approach is based on the cell-specific expression of a recombinant hemagglutinin (HA)-tag fused to the TOMM-20 protein that decorates the surface of mitochondria, thereby allowing their immunomagnetic purification. This protocol describes the CS-MAP procedure performed on large populations of animals. The purified mitochondria are suitable for subsequent nucleic acid, protein, and functional analyses.
For complete details on the use and execution of this protocol, please refer to Ahier et al. (2018, 2021).
Subject areas: Cell Biology, Cell separation/fractionation, Metabolism, Model Organisms, Molecular Biology, Protein Biochemistry, Protein expression and purification
Graphical abstract
Highlights
-
•
A quick protocol to isolate cell-specific mitochondria from animals
-
•
Purified mitochondria are functional and can be isolated from multiple cell types
-
•
Mitochondrial DNA and proteins can be analysed on purified mitochondria
-
•
A simple and cost-effective protocol requiring common lab materials
Cell-Specific Mitochondria Affinity Purification (CS-MAP) enables isolation and purification of intact mitochondria from individual cell types of Caenorhabditis elegans. The approach is based on the cell-specific expression of a recombinant hemagglutinin (HA)-tag fused to the TOMM-20 protein that decorates the surface of mitochondria, thereby allowing their immunomagnetic purification. This protocol describes the CS-MAP procedure performed on large populations of animals. The purified mitochondria are suitable for subsequent nucleic acid, protein, and functional analyses.
Before you begin
This protocol requires the use of previously generated C. elegans transgenic lines (see key resources table) expressing the recombinant fusion protein TOMM-20::mKate2::HA under the control of a range of cell-type-specific promoters, which are available through the Caenorhabditis Genetics Center. TOMM-20 (WormBase : WBGene00009092) , the translocase of the outer mitochondrial membrane complex subunit 20, is anchored to the surface of mitochondria and exposes mKate2 and the HA-tag to the cytoplasmic matrix outside the mitochondrion. The mKate2 red fluorophore allows tracking of labelled mitochondria throughout the procedure. The HA-tag (YPYDVPDYA) is used for the immunomagnetic purification of the organelle. This protocol can be used to purify mitochondria from various cell-types in large starting populations of animals. To some extent, mitochondria from a single cell-type can be isolated and enriched when a large population of animals, each with a single labeled cell, are pooled together. However, this limits subsequent analyses to nucleic acids (Ahier et al., 2018). The size of the starting population of animals can vary based on the cell-type used and the desired application. We recommend growing a synchronized population of 5,000 to 10,000 animals on one 100 mm Petri dish containing Nematode Growth Media (NGM) seeded with the Escherichia coli strain OP50 and multiplying the number of dishes to reach the required quantity of animals for the planned application (i.e., nucleic acid, protein, or functional analysis).
Note: OP50 is an Escherichia coli strain conventionally used as the bacterial food source of C. elegans in laboratory contexts. Liquid OP50 culture is spread onto the surface of NGM plates to establish a bacterial lawn.
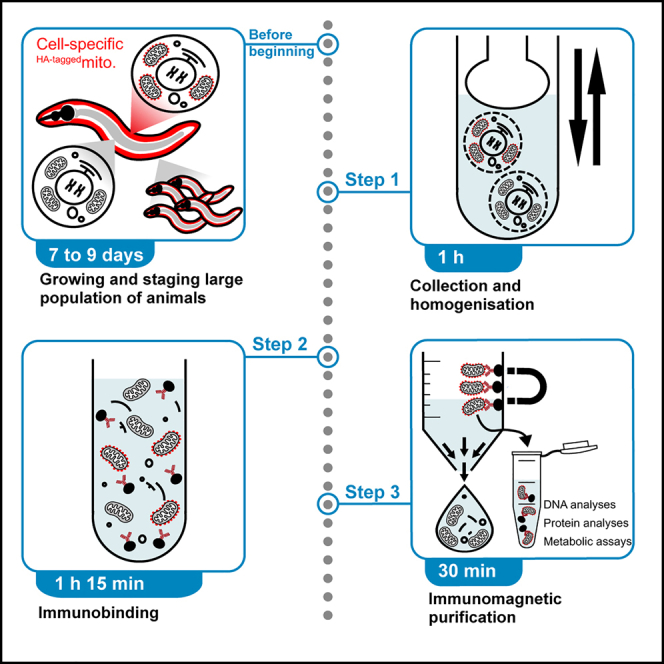
An overview and flowchart of the CS-MAP protocol is presented in Figure 1.
Figure 1.

Overview and flowchart of the CS-MAP protocol
Building the homemade CS-MAP purification device
Timing: 5 min
The CS-MAP homemade purification setup is readily constructed using inexpensive materials common to most molecular biology laboratories (please see materials and equipment and Figures 3 and 4 for additional information regarding the device setup). Once built, the device can be reused multiple times.
-
1.
Use a 4 L sharps waste container (or similar) as a receptacle to collect liquid waste during the CS-MAP procedure.
-
2.
Tape a 96-well PCR tube rack to the top opening of the container.
-
3.
Tape a neodymium cylinder magnet (diameter 15 mm, height 25 mm, weight 60 g, axially magnetized) to the PCR tube rack.
Figure 3.

List of materials required to build the homemade CS-MAP setup
(A–F) (A) A 4 L waste container or similar, (B) a 96-well PCR tube rack holder, (C) a neodymium cylinder magnet (diameter 15 mm, height 25 mm, weight 60 g, axially magnetized), (D) a 1 mL plastic tip, (E) a 25 mL pipette, (F) an electronic pipette (pipettor) or equivalent.
Figure 4.

The homemade CS-MAP setup
(A) Photograph of the assembled device. The magnet and the PCR rack are taped to the top of the container. A 1 mL tip is placed next to the flat surface of the magnet.
(B) Photograph of the device during immunomagnetic immobilization of mitochondria to the tip wall closest to the magnet. Note that the micropipette must remain attached to the tip to prevent the loss of the sample prior to forming a magnetically trapped pellet.
(C) Photograph of the device during the washing step. The 25 mL pipette is placed into the opening of the 1 mL tip and PEB is released by gravity flow by carefully unplugging the pipettor.
In subsequent steps, a 1 mL micropipette tip, a 25 mL pipette, and an electronic pipettor (pipettor) will be required.
Preparing the buffer solutions
-
4.
Prepare a 100 mL stock of Mitochondrial Isolation Buffer (MIB). Add one tablet of protease inhibitor cocktail (S8830, Sigma-Aldrich) to the solution and aliquot into 10 mL volumes before storing at −20°C (see materials and equipment for additional information regarding the recipe). Thaw and keep on ice before use.
-
5.Prepare a 100 mL stock of Phosphate EDTA BSA (PEB) Buffer.
-
a.Add one tablet of protease inhibitor cocktail (S8830, Sigma-Aldrich) to the solution and aliquot into 10 mL volumes before storing at −20°C (see materials and equipment for additional information regarding the recipe).
-
b.Thaw and keep on ice before use.
-
a.
Preparing a large population of staged C. elegans animals
-
6.Transfer 30 to 50 animals of the fourth larval (L4) stage to a 100 mm diameter NGM plate seeded with OP50 bacteria (Stiernagle, 2006).
-
a.Allow the animals to grow in a 20°C incubator for 4–5 days to obtain a mixed-staged animal population. The population’s development must be monitored to ensure that the animals are harvested before they exhaust their food supply of OP50.
-
a.
-
7.
Collect animals by washing the plate with 14 mL of M9 buffer using a pipettor into a 15 mL Falcon conical tube.
-
8.
To release eggs from the animals, centrifuge the tube at 200 × g for 2 min, remove the supernatant and add 1–2 mL of worm bleach solution (see materials and equipment for recipe) to the pelleted animals. Mix gently by inverting the tube 3×.
-
9.
Carefully monitor the bleaching under a dissecting microscope and neutralize the solution when the animals begin to break open (usually after 1–5 min) by adding 12 mL of M9 buffer. Practically, neutralize the solution when at least one of the following are observed: at least 1 to 2 animals adopt a perpendicular shape, several individual animals are seen in fragments, or tissues such as the gonad or intestine are seen extruding from the animal.
CRITICAL: Sodium hypochlorite is a skin and eye irritant that can cause severe skin burns and eye damage following contact or severe respiratory and digestive issues when inhaled or ingested. Wear personal protective equipment including protective gloves, eye shields, and clothing.
-
10.
Rinse the released eggs by pelleting them at 200 × g for 2 min, removing the supernatant, and adding 14 mL of fresh M9 buffer. Repeat this step at least twice to eliminate as many remnants of the worm bleach solution as possible.
-
11.
Resuspend the pellet of eggs in 100 μL of M9, gently swirl the eggs with the pipette tip, and dispatch 3 individual drops of 5 μL onto a glass slide.
-
12.
Count the number of eggs in each drop and estimate the number of eggs per μL by averaging the results of the 3 drops.
-
13.
Pipette a volume of the resuspended egg pellet that contains ∼10,000 eggs onto a 100 mm diameter NGM plate seeded with OP50 bacteria. Repeat as necessary to reach the required quantity of animals for the desired application (see Table 1).
-
14.
Grow animals until the population reaches the L4 larval stage. The L4 larval stage usually occurs around 44–56 h after the bleached eggs have been seeded onto NGM plates (20°C) (Figure 2). At this point the population is ready to be collected for the CS-MAP procedure. We recommend using animals at a mature L4 stage (56 h) to avoid any L3 larval stage contamination. The procedure also works well on young adult animals (56–65 h; Figure 2).
Note: This method of synchronizing the animals does not guarantee a perfectly homogenous population of same-staged animals. Therefore, the growth of the overall population must be monitored. Also, this timeline can vary according to the genetic background of the animals, as certain mutants may be delayed in growth.
Note: The developmental rate of the animals can be increased or decreased by incubation at 25°C or 15°C, respectively.
Table 1.
Size of starting population based on desired application
| Application | Starting population | Quantity of 100 mm NGM plate |
|---|---|---|
| Nucleic acid analyses (Quantitative PCR [qPCR], PCR) as shown previously in Ahier et al. (2018) and (2021). | 10,000 to 20,000 animals | 1 to 4 (according to Escherichia coli OP50 quantity on dishes). |
| Protein analyses (e.g., Western Blot) as shown previously in Ahier et al. (2018). | 50,000 to 100,000 animals | 5 to 20 (according to E. coli OP50 quantity on dishes) |
| Functional analyses (e.g., Seahorse metabolic assay) as shown previously in Ahier et al. (2018). | 50,000 to 100,000 animals | 5 to 20 (according to E. coli OP50 quantity on dishes) |
Figure 2.

Timeline of C. elegans development
The diagram shows the C. elegans developmental timeline at 20°C with approximative hour for each stage transition. Adapted from Byerly et al., 1976.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Anti-HA magnetic beads (100 μg) | Pierce | Cat# 88837; RRID:AB_2861399 |
| Rabbit Anti-HA-Tag Monoclonal Antibody (C29F4; 1:5000) | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| Mouse monoclonal anti-MTCO1 [1D6E1A8] (1:1000) | Abcam | Cat# ab14705; RRID:AB_2084810 |
| Mouse monoclonal anti-ATPB [3D5] (1:1000) | Abcam | Cat# ab14730; RRID:AB_301438 |
| Mouse monoclonal anti-HSP-60 [HSP60] (1:250) | DSHB (Hadwiger et al., 2010) | Cat# HSP60; RRID:AB_10572252 |
| Mouse monoclonal anti-ELT-2 [455-2A4] (1:200) | DSHB (Hadwiger et al., 2010) | DSHB Cat# 455-2A4, RRID:AB_2618114 |
| Mouse monoclonal anti-DLG-1 [DLG1] (1:100) | DSHB (Hadwiger et al., 2010) | DSHB Cat# DLG1, RRID:AB_2314321 |
| IRDye 800CW goat anti-rabbit (1:20000) | LI-COR Biosciences | Cat# 925-32211; RRID:AB_2651127 |
| IRDye 680RD goat anti-mouse (1:20000) | LI-COR Biosciences | Cat# 925-68070; RRID:AB_2651128 |
| Bacterial and virus strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center (CGC) | OP50 |
| Chemicals, peptides, and recombinant proteins | ||
| Proteinase K | Sigma-Aldrich | V3021 |
| SIGMAFAST Protease Inhibitor Cocktail Tablets | Sigma-Aldrich | S8830 |
| Mannitol | Sigma-Aldrich | M4125 |
| Sucrose | Sigma-Aldrich | S7903 |
| BSA | Bovogen Biologicals | BSAS 0.1 |
| PBS 10× | Lonza | 17-517Q |
| KCl | Sigma-Aldrich | P9541 |
| NP40 | Sigma-Aldrich | I8896 |
| Tween 20 | Sigma-Aldrich | P9416 |
| Gelatin from bovine skin | Sigma-Aldrich | G9391 |
| HA peptide | Sigma-Aldrich | I2149 |
| Glycogen | Thermo Fisher Scientific | AM9510 |
| Phenol-chloroform-isoamyl alcohol | Sigma-Aldrich | 77617 |
| 4× Laemmli Sample Buffer | Bio-Rad | 1610747 |
| GeneRuler DNA Ladder Mix | Thermo Fisher Scientific | SM0331 |
| Experimental models: Organisms/strains | ||
| C. elegans: strain N2 (wild-type) L4 stage, hermaphrodite | Caenorhabditis Genetics Center (CGC) | N2 (strain) WormBase: WBStrain00000001 |
| C. elegans: strain SJZ47 (foxSi16 [myo-3p::tomm-20::mKate2::HA::tbb-2 3′ UTR] I) L4 stage, hermaphrodites | Caenorhabditis Genetics Center (CGC) | SJZ47 (Ahier et al., 2018) |
| C. elegans: strain SJZ216 (foxSi44 [rgef-1p::tomm-20::mKate2::HA::tbb-2 3′ UTR] I) L4 stage, hermaphrodites | Caenorhabditis Genetics Center (CGC) | SJZ216 (Ahier et al., 2018) |
| C. elegans: strain SJZ213 (foxSi41 [dpy-7p::tomm-20::mKate2::HA::tbb-2 3′ UTR] I) L4 stage, hermaphrodites | Caenorhabditis Genetics Center (CGC) | SJZ213 (Ahier et al., 2018) |
| C. elegans: strain SJZ204 (foxSi37 [ges-1p::tomm-20::mKate2::HA::tbb-2 3′ UTR] I) L4 stage, hermaphrodites | Caenorhabditis Genetics Center (CGC) | SJZ204 (Ahier et al., 2018) |
| C. elegans: strain SJZ328 (foxSi75 [eft-3p::tomm-20::mKate2::HA::tbb-2 3′ UTR] I) L4 stage, hermaphrodites | Caenorhabditis Genetics Center (CGC) | SJZ328 (Ahier et al., 2018) |
| Oligonucleotides | ||
| CD15 5′-agcgtcatttattgggaagaagac-3′ | (Ahier et al., 2018) | N/A |
| AA113 5′-ccttccaaatactccgtctgc-3′ | (Ahier et al., 2018) | N/A |
| ges-1F1 5′-gctctccgcatgctaatgaatacc-3′ | (Ahier et al., 2018) | N/A |
| ges-1R1 5′-ctaactggtctccattctacatcttc-3′ | (Ahier et al., 2018) | N/A |
| AA178 5′-cataccgaataaacatcagggtaatct-3′ | (Ahier et al., 2021) | N/A |
| AA179 5′-acgggtgttacactatgatgaaga-3′ | (Ahier et al., 2021) | N/A |
| SZ35 5′-aggctaagccggggtaagtt-3′ | (Ahier et al., 2021) | N/A |
| SZ36 5′-gccaaaagcttaaactgcgg-3′ | (Ahier et al., 2021) | N/A |
| Software and algorithms | ||
| Zen2 | Zeiss | version 2.0.0.0 |
| FIJI | (Schindelin et al., 2012) | 1.52p |
| GraphPad Prism v9.1.0 | GraphPad Software | https://www.graphpad.com/ |
| Rotor-Gene Q Pure Detection software v2.3.1 | QIAGEN | https://www.qiagen.com/ |
| Microsoft Office 365 | Microsoft Corporation | https://www.microsoft.com/en-au/microsoft-365 |
| Adobe Illustrator CC 2021 | Adobe | https://www.adobe.com/ |
| Other | ||
| Z2 imager microscope with an Axiocam 506 mono camera | Zeiss | Zeiss |
| Thermo Scientific S1 Pipet Filler | Thermo Fisher Scientific | S1 |
| Serological pipet 25 mL | Falcon | 357525 |
| Sigma Dounce tissue grinder pestle - small clearance (B), working volume 7 mL | Sigma-Aldrich | P1235 |
| Dounce tissue grinder tube working volume 7 mL | Sigma-Aldrich | T0566 |
| NanoPhotometer | Implen | N60 |
| 16-Tube SureBeads™ Magnetic Rack | Bio-Rad | 1614916 |
| Rotor-Gene machine | QIAGEN | NA |
| Odyssey Fc Imager | LI-COR | N/A |
Materials and equipment
A list of materials required to build the homemade CS-MAP setup and a description of it are presented in Figures 3 and 4.
Worm bleach solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Bleach (sodium hypochlorite 8%–14%) | n/a | 4 mL |
| 10 N NaOH | n/a | 5 mL |
| H2O | n/a | 41 mL |
| Total | n/a | 50 mL |
Store at 22°C–25°C , protected from light, for a maximum of 2 weeks. The potency of worm bleach solution decreases over time, partly because of its photosensitive nature.
PEB: Phosphate EDTA BSA Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1× | 20 mL |
| BSA | 1% | 2 g |
| 0.5 M EDTA | 2 mM | 0.8 mL |
| ddH2O | n/a | Up to 200 mL |
| Total | n/a | 200 mL |
Add 1 tablet of protease inhibitor cocktail per 100 mL of buffer prior to use. PEB with protease inhibitor cocktail can be stored at −20°C for a maximum of 6 months. PEB used for washing in the immunomagnetic purification step should be prepared without protease inhibitor cocktail, kept on ice for 1 h, and used within the same day.
MIB: Mitochondrial Isolation Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| KCl | 50 mM | 0.744 g |
| Mannitol | 110 mM | 4 g |
| Sucrose | 70 mM | 4.792 g |
| 0.5 M EDTA | 0.1 mM | 0.04 mL |
| 1 M Tris HCl pH 7.4 | 5 mM | 1 mL |
| ddH2O | n/a | Up to 200 mL |
| Total | n/a | 200 mL |
Add 1 tablet of protease inhibitor cocktail per 100 mL of solution prior to use. MIB with protease inhibitor cocktail can be stored at −20°C for a maximum of 6 months.
M9 buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| KH2PO4 | 3 g | 22 mM |
| Na2HPO4 | 6 g | 42 mM |
| NaCl | 5 g | 86 mM |
| MgSO4 | 0.25 g | 1 mM |
| ddH2O | n/a | Up to 50 mL |
| Total | n/a | 50 mL |
Store at 22°C–25°C for a maximum of 6 months.
MLB: Mitochondrial Lysis Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M KCl | 50 mM | 25 mL |
| 1 M Tris HCl pH 8 | 10 mM | 5 mL |
| 1 M MgCl2 | 2.5 mM | 1.25 mL |
| NP-40 | 0.45% | 2.25 mL |
| Tween-20 | 0.45% | 2.25 mL |
| Gelatin | 0.01% | 50 mg |
| ddH2O | n/a | Up to 500 mL |
| Total | n/a | 500 mL |
Store at 4°C for a maximum of 6 months. Add 0.1 mg·mL−1 proteinase K immediately before use.
RIPA Buffer: Radioimmunoprecipitation Assay Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris HCl pH 8 | 10 mM | 1 mL |
| 0.5 M EDTA | 1 mM | 0.2 mL |
| Triton X-100 | 1% | 1 mL |
| Sodium deoxycholate | 0.1% | 0.1 g |
| SDS | 0.1% | 0.1 g |
| NaCl | 140 mM | 0.8 g |
| ddH2O | n/a | Up to 100 mL |
| Total | n/a | 100 mL |
Store at 4°C for a maximum of 6 months. Add 1 tablet of protease inhibitor cocktail per 100 mL of buffer immediately before use.
TBST: Tris-buffered saline with 0.1% Tween-20 detergent
| Reagent | Final concentration | Amount |
|---|---|---|
| TBS 10× pH 7.5 | 1× | 100 mL |
| Tween-20 | 0.1% | 1 mL |
| ddH2O | n/a | Up to 1 L |
| Total | n/a | 1 L |
Store at 22°C–25°C for a maximum of 6 months.
Step-by-step method details
Animal harvesting and cleaning
Synchronized adult animals are harvested and cleared of E. coli OP50 before being homogenized.
-
1.
Harvest animals (see Experimental models: Organisms/strains of the key resources table for strains to use: myo-3/eft-3/ges-1/rgef-1/dpy-7p::tomm-20::mKate2::HA or N2 [as a control]) by washing them off the plate with two successive volumes of 7.5 mL of M9 buffer into a 15 mL Falcon conical tube using a pipettor. Washing animals off the plate with two successive volumes of M9 buffer ensures that the maximum numbers of animals are collected before beginning the procedure.
Note: If multiple plates are being used per sample, wash the animals off one plate and onto the next with 7.5 mL of M9 buffer to maximise the collection of animals. Continue this process on each successive plate and then collect the animals in a 15 mL Falcon tube at the end. Repeat this sequential collection technique with another 7.5 mL of fresh M9 buffer and add to the Falcon tube.
-
2.
Centrifuge the tube at 200 × g for 2 min and replace the M9 supernatant with 14 mL of fresh M9 buffer.
-
3.
Repeat the previous washing step once more to remove as much OP50 from the sample as possible. See troubleshooting problem 1.
Optional: Rotate the animals in 10 mL of M9 buffer in a 15 mL Falcon tube for 30 min at 30 RPM at 22°C–25°C to clear bacteria from the animals’ intestines. After rotating, pellet the animals by centrifugation at 200 × g for 2 min and wash the pellet once more with 14 mL of M9 buffer.
-
4.
Pellet the animals by centrifugation at 200 × g for 2 min and replace the M9 supernatant with 14 mL of ddH2O to remove traces of the M9 buffer and thus reduce the salinity of the solution.
-
5.
Centrifuge again at 200 × g and remove the ddH2O supernatant. Keep the sample on ice for less than 1 h.
Note: Allow more than 20 min for the processing of multiple samples or multiple plates per sample.
Generating an enriched mitochondrial fraction
The sample is homogenized using a glass Dounce tissue grinder set with a low clearance pestle (usually called B clearance [P1235, Sigma-Aldrich]). A crude animal homogenate is then created by differential centrifugation.
-
6.
Suspend the sample in 1.5 mL of cold Mitochondrial Isolation Buffer (MIB) with protease inhibitor cocktail.
-
7.
Transfer the sample with a 1 mL tip and a micropipette into a cold 7 mL Dounce tissue grinder tube (T0566, Sigma-Aldrich).
Note: The animals tend to adhere to the plastic walls of the pipette tip. To minimize this, animals can be sequentially transferred into the Dounce tissue grinder tube in successive volumes of 300 μL. This reduces the surface area available for animals to adhere to and therefore reduces the animal loss during the transfer.
-
8.Perform 20 to 30 Dounce tissue grinder strokes.
-
a.Dounce the animals by pushing the pestle towards the bottom of the Dounce reservoir in a motion parallel to the tube. Before reaching the bottom of the tube, twist the pestle, keeping the shaft of the pestle parallel to the tube walls, and then pull the pestle out of the buffer.
-
b.Repeat for a total of 20–30 strokes. Strokes must be regularly spaced, relatively fast, and go in and out of the buffer for each individual stroke. A medium to strong resistance against the sample must be experienced during the douncing for the animals to be homogenized.
-
a.
Note: During homogenization, a fraction of animals is broken into multiple fragments and mitochondria are released from cells into the MIB. A fraction of animals in the sample may still be alive but suffering from damage that is severe enough to release some of their mitochondria into the MIB. Over-douncing the sample may potentially fragment mitochondria and generate a large amount of cellular debris that is very difficult to clear during the purification steps described below.
-
9.
Transfer the homogenate into a 1.5 mL microcentrifuge tube.
-
10.After separating 300 μL of the homogenate for use as Input 1, remove intact animals, animal fragments, and large pieces of cellular debris from the remaining 1.2 mL of the animal homogenate from step 9 by a low-speed centrifugation at 200 × g for 5 min.
-
a.Transfer the supernatant to a new 1.5 mL tube.
-
a.
-
11.
Refine the sample further with a second centrifugation at 800 × g for 10 min and transfer the supernatant to a new 1.5 mL tube. The supernatant is now a crude animal homogenate.
Note: A small aliquot of the crude animal homogenate can potentially be saved as Input 2.
-
12.
Pellet the enriched mitochondrial fraction within the crude animal homogenate by centrifugation at 12,000 × g for 10 min.
-
13.
Discard the supernatant and quickly add 20 μL of MIB to the enriched mitochondrial fraction. See troubleshooting problem 2.
-
14.
Resuspend the enriched mitochondrial fraction in MIB by vigorously pipetting the solution up and down for 1 min using a P20 micropipette. As mitochondria can agglutinate, this will help ensure uniform distribution of mitochondria throughout the MIB.
Note: Wild-type (N2) animals generally don’t produce particles that exhibit autofluorescence at a brightness comparable to mKate2-labeled mitochondria. Only some rare red particles can be observed but can easily be discriminated from mKate2-labelled mitochondria by their aberrant shape or brightness.
Figure 6.

Enriched mitochondrial fraction and IPed mitochondria outcomes from the CS-MAP method
The outcomes of wild-type (N2) animals and a strain carrying the transgene myo-3p::TOMM-20::mKate2::HA as a single copy insertion into the universal MosSCI insertion site oxti185 I (SJZ47), which allows the purification of mitochondria specifically from body wall muscle cells (BWM), are presented. One μL of the enriched mitochondrial fraction (upper part of the figure) and 1 μL of the purified mitochondria sample (bottom part of the figure) are shown. The enriched mitochondrial fraction shows uniformly-spread fine debris and some aggregates: (A and B). Note that some large debris is visible (e.g., fragments of pharynx and tips of tails) but is rare and usually removed during the washing step. The enriched mitochondrial fraction of SJZ47 shows numerous mKate2-labelled mitochondria compared to the N2 enriched mitochondrial fraction: (A and B). The IPed mitochondrial fraction shows unbound anti-HA magnetic beads for the N2 control (C) and anti-HA magnetic beads bound to mKate2-labelled mitochondria for SJZ47 (E). Aggregates of mitochondria-magnetic beads are shown in (F). In D), unbound anti-HA magnetic beads are visible. Observations were performed using a fluorescent compound microscope (40× magnification, oil objective, DIC and red channel [excitation wavelength 587 nm, emission wavelength 610 nm]). Scale bar for (A–C). and (E) 100 μm. Scale bar for (D and F) 10 μm.
Binding of anti-HA magnetic beads to HA-taggedmitochondria
The enriched mitochondrial fraction is incubated with anti-HA magnetic beads, which selectively bind tissue-specific HA-taggedmitochondria.
-
15.
Add 400 μL of cold Phosphate EDTA BSA buffer (PEB) with protease inhibitor cocktail to the 20 μL of MIB-suspended enriched mitochondrial fraction and homogenize the sample by vigorously pipetting up and down 10 times using a P200 micropipette set to 200 μL. Transfer the contents into a 0.6 mL round-bottom microtube.
Optional: Consider pipetting up and down more times if clumps are still present in the sample. This step is empirical, as it can be difficult to spot clumps of mitochondria under the microscope once they are diluted in 400 μL of PEB.
Note: Performing the next incubation step in a smaller 0.6 mL round-bottom microtube minimizes the contact of mitochondria with the plastic walls of the tube, thereby reducing their loss.
-
16.Carefully wash 100 μg of anti-HA magnetic beads (88837, Pierce) in a 1.5 mL microcentrifuge tube by adding 1 mL of cold PEB with protease inhibitor cocktail and flicking gently to mix.
-
a.Place the microcentrifuge tube into a standard magnetic rack (SureBeads, Bio-Rad or equivalent) and leave for 1–2 min until the magnetic beads have assembled against the magnet.
-
b.Carefully remove clear PEB before removing the tube from the magnetic rack and repeating the washing process at least 6 times.
-
a.
-
17.
Resuspend the clean anti-HA magnetic beads in 100 μL of PEB with protease inhibitor cocktail.
Note: 100 μg of anti-HA magnetic beads are used per sample. If the experiment involves multiple samples, calculate the μg of beads required in total and wash together. To obtain the greater quantity of mitochondria required for protein extraction, up to 200 μg of anti-HA magnetic beads have been used per sample.
-
18.
Add the washed 100 μg of anti-HA magnetic beads (in 100 μL of PEB with protease inhibitor cocktail) to the 400 μL sample from step 15.
-
19.
Wrap Parafilm around the closed lid of the tube to prevent it from opening and incubate at 4°C under gentle rotation, using an Intelli-Mixer RM-2 (ELMI) on program F1 at 8 RPM for 1 h.
Note: Incubation can be increased to 2 h with similar results.
Immunomagnetic purification of mitochondria
Mitochondria bound by immunomagnetic beads are column-purified on the homemade CS-MAP setup (Figure 4). The procedure is shown step-by-step in Methods video S1: Purification of mitochondria.
-
20.
Carefully pipette up the sample with a 1 mL tip and a P1000 micropipette.
-
21.
Position the tip – still connected to the micropipette – into the magnetic field of the magnet (adjacent to one of the flat surfaces of the magnet) on the PCR rack of the homemade CS-MAP device (see Figure 4B and 5).
-
22.
Once a visible pellet of magnetic beads is visible on the wall of the tip (after 1 min), carefully remove the micropipette from the tip, without disturbing the tip or magnet. When the micropipette is released, the supernatant will drain into the waste bucket.
-
23.
Using a pipettor and a 25 mL plastic serological pipette, aspirate 25 mL of cold PEB.
Note: PEB used for washing does not require the protease inhibitor cocktail.
-
24.
Plug the tip of the 25 mL pipette into the top of the 1 mL tip and carefully unplug the pipette from the pipettor without disrupting the 1 mL tip or magnet. Allow the PEB to flow through the 1 mL tip and into the waste bucket. This step washes the magnetic beads that remain held in place by the magnet.
Note: If an air bubble forms in the 1 mL tip preventing uniform washing, reattach the pipettor to the 25 mL pipette and very briefly re-aspire less than 1 ml of liquid. This causes a slight backflow in liquid that refills the 1 ml tip and can clear the bubble. Once the bubble is removed, unplug the pipette from the pipettor once again.
-
25.Once all the PEB has passed through the column, remove the 25 mL pipette and discard.
-
a.Next, remove the 1 mL tip from the magnetic field, place it into a 1.5 mL microcentrifuge tube, and elute the bead pellet by adding 1 mL of PEB into the tip.
-
b.Reconnect the P1000 micropipette to the tip containing the beads and pipette up and down a few times to resuspend the beads (see Methods video S1).
-
a.
Note: For a low stringency wash, only 1 wash with 25 mL of PEB is sufficient, so directly proceed to step 28. A low stringency wash improves the yield of immunomagnetic-purified mitochondria at the expense of purity. For a higher stringency wash, proceed to step 24. A high stringency wash involves a second wash with 25 mL of PEB and improves the purity of immunomagnetic-purified mitochondria at the expense of yield. A low stringency wash is preferred when protein and metabolic analyses will be performed but a high stringency wash is required for analysis of nucleic acids.
-
26.
Using a standard magnetic rack (SureBeads, Bio-Rad, or equivalent), pellet the magnetic beads and replace the PEB used for the elution with 1 mL of fresh PEB.
-
27.
Vortex the sample 3 times for 2 s each.
-
28.
Place the tube under gentle rotation for 10 min at 4°C.
-
29.
Perform a second wash with 25 mL of cold PEB on the CS-MAP homemade setup (steps 18 to 23).
-
30.
Remove PEB using a standard magnetic rack and resuspend the immunomagnetic-purified (IPed) mitochondria in 20 μL of cold MIB with protease inhibitor cocktail. The sample of immunomagnetic-purified (IPed) mitochondria is ready to use and must be kept on ice. See troubleshooting problem 3 and problem 4 for potential issues.
Figure 5.

Pellet of immunomagnetic mitochondria
Mitochondria attached to magnetic beads aggregate on the wall of the tip closest to the magnet after 1 min. White arrow: magnetic bead pellet.
Step 1) A 1 mL micropipette tip containing the immunomagnetic mitochondria is positioned next to the flat surface of the magnet. Step 2) the immunomagnetic mitochondria are trapped by the magnet to the wall of the 1 mL micropipette tip closest to the magnet (1 min waiting). Step 3) A 25 mL pipette containing PEB is plugged into the top of the 1 mL micropipette tip to create a transitory washing column and the sample is washed using gravity flow. Step 4) Purified immunomagnetic mitochondria are collected with PEB and concentrated into a reduced volume of MIB.
Expected outcomes
Immunomagnetic-purified (IPed) mitochondria are resuspended in 20 μL of cold MIB and kept on ice. The sample is ready to use for subsequent applications.
Enriched mitochondrial fraction and IPed mitochondria outcomes from the CS-MAP method are presented in Figure 6.
The quality of the sample should be evaluated using a fluorescence compound microscope. To do so, 1 μL of the IPed mitochondria sample is placed on a glass slide and covered with a coverslip. Samples are observed using a 40× oil objective (EC Plan-Neofluar 40×/1.30 Oil DIC M27) with DIC or Normarski and the red fluorescence channel (excitation wavelength 587 nm, emission wavelength 610 nm, light source intensity 50%, exposure time 3 s, binning mode 2.2). Differences between control experiments performed using N2 animals and strains of interest are indicative of purified mitochondria from the cell-type of interest. It is normal for multiple fluorescently labelled mitochondria to be bound to a single anti-HA magnetic bead or form aggregates with multiple anti-HA magnetic beads (Figures 6E and 6F). The number of fluorescently labelled mitochondria can vary according to the cell-type from which they were purified. DIC observation allows confirmation of the absence of fine (and possibly coarse) debris which was not visible in the enriched mitochondrial fraction (Figures 6A vs 6C).
Ideally, the IPed mitochondria should be used or processed immediately for any subsequent application even though mitochondria are still visually normal for up to 4 h after purification when kept on ice or at 4°C. We have previously used IPed mitochondria for different applications, including PCR, Western blot, and functional Seahorse assays (Ahier et al., 2018). IPed mitochondria must be prepared accordingly to the target application. If required, the number of purified mitochondria can also be visually quantified.
Nucleic acids analyses (qPCR and PCR)
The nucleotide composition of the mitochondrial genome (mutation types and levels) can vary between tissues types (Hahn and Zuryn, 2019). As such, it is often informative to analyze mtDNA after CS-MAP. For such analyses (e.g., PCR or quantitative PCR), a standard magnetic rack (SureBeads, Bio-Rad or equivalent) is used to pellet the magnetic beads and replace the MIB with 100 μL of mitochondrial lysis buffer (MLB).
Pause point: Sample can be frozen in 100 μL of MLB at −80°C for 1 month for later processing at the same time as Input 1.
Samples are lysed for 1 h at 65°C before inactivating proteinase K at 95°C for 15 min. A standard phenol-chloroform DNA extraction is then performed, followed by an ethanol precipitation of DNA using 10 μg of glycogen as a carrier. The DNA pellet is resuspended in 20 μL of 10 mM Tris pH 8.0, 1 mM EDTA buffer. It can be difficult to accurately quantify mtDNA using a NanoPhotometer (N60, IMPLEN); however, 0.01–1 μL is generally used for PCR applications.
Note: Input 1 can be thawed, resuspended in 200 μL of MLB, and processed in parallel to the IPed mitochondria sample. The sample should be processed similarly to the IPed mitochondria sample except for the lysing step which should be performed for 2–3 h at 65°C, as the sample contains fragments of animals and entire animals. Because of the presence of nuclear DNA (nDNA), mtDNA cannot be quantified in Input 1. Using a NanoPhotometer (N60, IMPLEN), total DNA can be quantified and diluted to 10 ng·μL−1. For PCR applications, 0.01–1 μL is used. If required, Input 2 can be processed as the IPed mitochondria sample.
The enrichment of mtDNA after CS-MAP may reflect the enrichment of the mitochondrial fraction and can be evaluated, as seen in Figure 7. We usually use a semi-quantitative PCR approach where we amplify a sequence of the mtDNA that we normalize to an amplified target of the nDNA of the animal (Figure 7A). This approach provides an overview of the purity of the sample and helps verify the use of this sample for further analyses. Alternatively, if a more accurate quantification is required, a qPCR measuring the ratio of mtDNA to nDNA in Input 1 compared to IPed mitochondria can be performed (Figures 7B and 7C).
Figure 7.

Quantification of mtDNA enrichment after CS-MAP
Two different types of quantification (semi-quantitative and quantitative PCR (qPCR)) were performed on the same samples for comparison.
(A) Semi-quantitative PCR showing the enrichment of mitochondrial DNA (mtDNA) over nuclear DNA (nDNA) after CS-MAP. To evaluate mtDNA enrichment, the primers AA113 and CD15 (Ahier et al., 2018) were used with Phusion DNA Polymerase (Biolabs, M0530) under the following cycle conditions: 98°C for 30 s, followed by 34 cycles of 98°C for 10 s, 65°C for 15 s, and 72°C for 1 min. To evaluate nDNA levels, the primers ges-1F1 and ges-1R1 (Ahier et al., 2018) were used with the aforementioned polymerase under the following cycle conditions: 98°C for 30 s, followed by 34 cycles of 98°C for 10 s, 55°C for 15 s, and 72°C for 1 min. 1 μL of the input diluted at 10 ng·μL-1 and 1 μL of the CS-MAP sample were used per reaction. nDNA has occasionally been detected in the CS-MAP sample. GeneRuler DNA Ladder Mix (SM0331, Thermo Scientific) was used for this gel.
(B and C) The samples used in A) were analyzed by qPCR using a Rotor-Gene Q real-time PCR (QIAGEN) machine. To quantify mtDNA levels, the primers AA178 and AA179 were used, while the primers SZ35 and SZ36 (Ahier et al., 2021) were used to quantify nDNA. qPCR was performed using the following cycle conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 58°C for 40 s using the Sensifast SYBR No-ROX Kit (BIO-98005, Bioline). 1 μL of the input diluted at 10 ng·μL-1 and 1 μL of the undiluted CS-MAP sample were used per technical triplicate. The efficiency of enrichment can vary due to experimental variability and/or cell-type used. We usually observe a 20- to 50-fold enrichment with some rare cases where nuclear DNA was not detected within 40 qPCR cycles. (B) Table showing the obtained Ct values and the calculation steps of the applied comparative Ct method. ΔCt corresponds to nDNA average Ct value - mtDNA average Ct value and relative enrichment (RE) corresponds to 2ΔCt. (C) Quantification of mtDNA enrichment relative to nDNA. An 18.4-fold enrichment was observed in this experiment. IP: ImmunoPurified mitochondria; Input: Corresponding input 1.
Protein analyses (western blot)
For standard Western blot assays, the total or ½ of the total sample of immunopurified (IPed) mitochondria are loaded into a 10-well gel (1.5 mm, 1653365, Bio Rad) Mini-PROTEAN (1658030, Bio-Rad) electrophoresis system. As we have never been able to efficiently elute HA- taggedmitochondria from the anti-HA magnetic beads using a HA peptide (I2149, Sigma-Aldrich), total protein concentration cannot be quantified using a standard BCA Protein Assay. See troubleshooting problem 5 for further information. Protein is extracted from IPed mitochondria by lysing the sample in 15 μL of Radioimmunoprecipitation Assay (RIPA) buffer for 20 min on ice. Then, using a standard magnetic rack (SureBeads, Bio-Rad or equivalent), the magnetic beads (still coated with their anti-HA antibodies) are pelleted and the lysate, with mitochondrial proteins, is collected. This step greatly reduces the presence of mouse HA-antibodies in the protein sample. 5 μL of 4× Laemmli Sample Buffer (1610747, Bio-Rad) is added to the sample which is then denatured at 100°C for 5 min, centrifuged at 12,000 × g for 10 min, and loaded into one well of an electrophoresis gel. Alternatively, the IPed mitochondria can be resuspended in 20 μL of standard 1× Laemmli Sample Buffer and denatured by boiling at 100°C for 5 min, centrifuged at 12,000 × g for 10 min, and loaded in one well of an electrophoresis gel.
Note: Input 1 can be thawed, resuspended in 200 μL of RIPA buffer with protease inhibitor cocktail, and lysed for 30 min on ice. Animal tissues can be dounced (50 strokes) with a 2 mL Dounce tissue grinder (D8938, Sigma-Aldrich) and potentially sonicated with a probe sonication system on ice (5 × 30 s, amplitude: 10%–40%; Sonics VibraCell VCX130), as shown previously (Cummins et al., 2019). The sample should be centrifuged for 30 min at 18,710 × g at 4°C, with the supernatant kept as the protein lysate. 5 μL of 4× Laemmli Sample Buffer (1610747, Bio-Rad) should be added to 15 μL of the protein lysate and the sample denatured at 100°C for 5 min, centrifuged at 12,000 × g for 10 min, and entirely loaded in one well of an electrophoresis gel. Alternatively, Input 1 can be denatured by boiling at 100°C for 5 min in 100 μL of standard 1× Laemmli Sample Buffer with 5–20 μL loaded into the well of an electrophoresis gel system.
Protein extracts obtained from Input 1 and IPed mitochondria can be used to assess the enrichment of mitochondrial proteins over proteins from different cellular compartments by Western blot, thereby reflecting the enrichment of the mitochondrial fraction. Protein samples are loaded on a 10% polyacrylamide gel and blotted on an Immobilon-FL PVDF membrane (IPFL00010) previously activated with ethanol. The membrane is blocked with 5% bovine serum albumin (BSA) in TBST buffer before incubation with primary and secondary antibodies (the latter coupled to either IRDye 680RD or 800RD) and imaged using an Odyssey Fc Imager (Li-COR). We usually verify the enrichment of mitochondrial proteins compared to the nuclear Erythroid-Like-Transcription (ELT-2) factor monoclonal antibody [455-2A4] from the Developmental Studies Hybridoma Bank (DSHB, Iowa). Alternatively, the cell adhesion protein Drosophila Discs LarGe homolog-1 (DLG-1) monoclonal antibody ([DLG1], DHSB, Iowa). ELT-2 and DLG-1 are abundantly detected by Western blot in Input 1 but are absent from the IPed mitochondrial fraction. To observe an example of ELT-2, see Figure 3D of previously published work (Ahier et al., 2018). For DLG-1, data is not shown. To confirm the presence of mitochondrial proteins in Input 1 and the IPed mitochondrial fraction, a non-exhaustive list of antibodies can be successfully used (Table 2). Antibodies against MTCO1 (1D6E1A8), ATPB (3D5), HSP-60 (HSP60), and HA-tag (C29F4, for TOMM-20::mKate2::HA bait detection) work efficiently for Western blot applications, as seen in Figures 3B and 3D in previous publications (Ahier et al., 2018).
Note: PVDF membrane is incubated with primary antibodies for 12–16 h at 4°C under gentle rocking in 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween 20 detergent (TBST).
Note: PVDF membrane is incubated with secondary antibodies (Goat anti-mouse - IRDye 680RD or Goat anti-rabbit- IRDye 800CW, both diluted at 1:20000) in 5% BSA in TBST for 1 h at 22°C–25°C under gentle rocking. The membrane is washed 3 × 10 min in TBST between primary and secondary incubations and before image acquisition.
Table 2.
Experimental details about the antibodies used for Western blot
| Target | Antibody | Host | Subcellular localisation | Working dilution |
|---|---|---|---|---|
| ELT-2 | [455-2A4] | Mouse | Nucleus | 1:200 |
| DLG-1 | [DLG1] | Mouse | Cellular membrane | 1:100 |
| MTCOI/CTC-1 | 1D6E1A8a | Mouse | Mitochondrion | 1:1000 |
| ATPB/ATP-2 | 3D5 | Mouse | Mitochondrion | 1:1000 |
| HSP60/HSP-60 | HSP60 | Mouse | Mitochondrion | 1:250 |
| TOMM-20::mKate2::HA | C29F4 | Rabbit | Mitochondrion | 1:5000 |
Do not boil the sample before loading on gel.
Functional assays
For functional assays, cell-specific mitochondria must be purified in the morning and assayed the same day (within 4 h of purification). As their elution from the anti-HA magnetic beads using a HA peptide is inefficient, mitochondria are used attached to anti-HA magnetic beads. The whole sample, or potentially ½ of it, can be used to measure oxygen consumption rate as in the Seahorse metabolic assay using the Seahorse XFe96 FluxPak kit (ref. SEA102601100, Agilent) and an Agilent Seahorse XFe Analyzer (Ahier et al., 2018).
Note: Input 1, which was kept on ice during the CS-MAP procedure, can also be used in the assay.
Quantification and statistical analysis
The number of purified mitochondria can be quantified by imaging a glass slide without a coverslip of 2 μL of the sample from step 29 (immunomagnetic purification of mitochondria), as shown in Figure 8. As the mitochondria are still bound to the anti-HA magnetic beads, a magnet can be used to carefully attract IPed mitochondria toward the border of the liquid drop to concentrate them into a small crescent shape (Figures 8A and 8B). The concentrated IPed mitochondria are then imaged at 20× magnification (objective: EC plan-Neofluar 20×/0.50 M27, without immersion) under a fluorescence compound microscope (light source intensity 53%, exposure time 11 s, excitation wavelength 587 nm, emission wavelength 610 nm, binning mode 2.2) to create a montage of the whole crescent. Imaging must be performed quickly before the sample dries out. The total number of mKate2-labelled mitochondria can be quantified with a tally counter using the Grid plugin (Wayne Rasband) in FIJI (Schindelin et al., 2012).
Figure 8.

Quantification of mKate2-labelled IPed mitochondria obtained from SJZ47 myo-3p::TOMM-20::mKate2::HA
IPed mitochondria were attracted to the edge of the sample using a magnet.
(A and B) An approximative and manual assembling of a crescent shape formed by the beads and mKate2-labelled HA-taggedmitochondria is shown. The white dotted line indicates the front of the crescent.
(C–F) A portion of the crescent shape structure formed by the beads and mitochondria is shown. (C and D) mKate2-labelled HA-taggedmitochondria bound to anti-HA magnetic beads. (E and F) Anti-HA magnetic beads saturated with HA-peptide block the binding of the HA-taggedmitochondria (negative control). Mitochondria were quantified using FIJI and the Grid plugin (shown as a yellow grid). Observations were performed with a fluorescence compound microscope using DIC and red channel (excitation wavelength 587 nm, emission wavelength 610 nm), at 20× magnification, non-immersed. Scale bar: 100 μm.
Limitations
This protocol is not efficient on young larval stages (i.e., L1 to L3) due their small size and physical resistance to Dounce-based homogenization. Performing this protocol on a mixed staged population can lead to the presence of intact young larval stage animals in the final purified mitochondria sample, which may compromise downstream applications.
The expression pattern of the TOMM-20::mKate2::HA transgene can influence the efficiency of the protocol. Even though we have shown that this protocol can be applied to purify mitochondria from a single cell in a large population of animals (Ahier et al., 2018), final yields of purified mitochondria can be low. Furthermore, some cells can be refractory to physical homogenization (e.g., pharyngeal muscle cells).
We have never been able to efficiently elute HA-tagged mitochondria from anti-HA magnetic beads using a HA peptide. Similar issues have been reported for 3×HA-tagged mitochondria (Kuhnert et al., 2020). We believe that this is due to the strong interaction between the HA-tag and the HA antibody. The presence of HA antibodies in the sample can potentially preclude further mass spectrometric analyses.
We have established the CS-MAP method exclusively using a HA tag as our efforts to develop the method using a FLAG (DYKDDDDK) tag were unsuccessful. Even though the reason for this failure remains unclear, similar results have been observed previously (Chen et al., 2016).
Troubleshooting
Problem 1
Washing the animals in M9 buffer does not appear to clear them of bacteria as the sample still appears cloudy after several washes (animal harvesting and cleaning step 2).
Potential solution
Although E. coli OP50 bacteria are relatively easy to clear with M9 solution, extra washes can be performed if the sample still appears cloudy. Other strains of bacteria (e.g., E. coli HB101) can be more difficult to clear from the animal sample. Unidentified bacterial contamination can elude removal by washing as it can remain stuck to the animal. This contamination can later adhere to the anti-HA magnetic beads. Therefore, contamination can preclude the normal procedure and should be removed by additional washes where necessary.
Problem 2
The enriched mitochondrial fraction contains low amounts of mKate2-labeled mitochondria (generating an enriched mitochondrial fraction step 13).
Potential solution
1) The expression of the TOMM-20::mKate2::HA transgene in a limited number of cells can explain the subsequent low yield of extracted mKate2-labelled mitochondria. In this case, starting from a larger population of animals may be beneficial. However, the douncing step is the most common source of variability in the quality of the enriched mitochondrial fraction. Indeed, the Dounce tissue grinder set erodes after multiple uses and becomes less efficient. When douncing efficiency is lost, less mitochondria are released. Attempting to counteract this by over-douncing can create more debris and generate fragmented mitochondria. Instead, a new Dounce should be used. Additionally, we observed inconsistent outcomes when douncing samples suspended in volumes less than 1.5 mL. Optimizing the number of douncing strokes has been tried with variable outcomes. 2) Perform a proper calibration of the red channel of the microscope (with an N2 control condition). Even though settings are dependent on the microscope, several seconds of exposure time may be required to observe mKate2-labelled mitochondria. Saved microscope settings can be reused in the future.
Problem 3
Low yields of mKate2-labeled IPed mitochondria are obtained at the end of the CS-MAP procedure (immunomagnetic purification of mitochondria step 30).
Potential solution
1) First of all, assess the quality of the enriched mitochondrial fraction. If too few mitochondria are present, do not proceed to the next step. Note that the enriched mitochondrial fraction shown in Figure 6B is especially good, but approximately half as many mKate2-labelled mitochondria as the quantity shown here is still suitable for proceeding to the next step. 2) Ensure that the magnetic beads have been carefully washed before use. Any traces of detergent or NaN3, in which beads are stored, will affect or destroy mitochondria. 3) Avoid bacterial contamination when growing animals. Some bacteria can stick to the magnetic beads and decrease their efficiency. 4) Excessive large debris and aggregates in the enriched mitochondrial fraction can affect the subsequent immunopurification step. Try to disrupt aggregates by pipetting the sample up and down (as described in steps 13 and 14 of generating an enriched mitochondrial fraction).
Problem 4
High levels of debris, nuclear proteins, or nuclear DNA are contaminating the final IPed mitochondria sample (immunomagnetic purification of mitochondria step 30).
Potential solution
1) Normally, the stringent washing procedure presented in this protocol prevents this issue. Nevertheless, experimental variation can occur and the presence of obvious debris among mKate2-labelled mitochondria can be assessed using a fluorescent compound microscope. If required, an extra wash can be performed. Also, the washing efficiency can be monitored using anti-HA magnetic beads blocked with HA peptide (I2149, Sigma-Aldrich). Ten μg·mL−1 of HA- peptide can be used to block 100–200 μg of anti-HA magnetic beads. As anti-HA magnetic beads blocked with HA peptide cannot bind HA-labeled mitochondria, a properly washed sample should be free of mKate2-labelled mitochondria. Ultimately, the presence of nuclear protein contamination can be assessed by Western Blot (see expected outcomes – proteins analyses) while potential nuclear DNA contamination can be assessed by semi-quantitative PCR or qPCR, as described in expected outcomes – nucleic acid analyses, Figure 7, and previous publications (Ahier et al., 2018, 2021). 2) Monitor the quality of PEB used for washing. The level of BSA in this buffer makes it highly prone to contamination; therefore, it is advised to prepare it immediately prior to use or keep it frozen at −20°C.
Problem 5
Western blot analyses of the IPed mitochondria sample are compromised because of the detection of the mouse anti-HA antibody (used for CS-MAP) due to cross reaction with an anti-mouse secondary antibody to detect other proteins of interest (expected outcomes – protein analyses).
Potential solution
HA-taggedmitochondria cannot be eluted efficiently using a HA peptide (I2149, Sigma-Aldrich). Therefore, when HA-taggedmitochondria bound to HA-beads are processed for a Western blot, as described in expected outcomes – protein analyses, mitochondrial proteins and mouse anti-HA antibodies are detected on the same membrane. This precludes detection of most proteins between 50 and 25 kDa using a primary antibody raised in mice, as the relevant secondary antibody will strongly cross-detect both the light and heavy chains of the anti-HA IgG antibody, masking any other signal. To avoid this, we lyse the HA-taggedmitochondria bound to HA-beads with RIPA buffer on ice before using a suitable magnet or magnetic rack to transfer only the supernatant to a new tube. As the inner and outer mitochondrial membranes are disrupted through this process, this method facilitates the extraction of most mitochondrial proteins with the exception of the TOMM-20::mKate2::HA fusion protein, which will partially remain attached to the HA-beads.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Steven Zuryn, E-mail: s.zuryn@uq.edu.au.
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by NHMRC project grants GNT1128381 (to S.Z.) and GNT1162553 (to S.Z.), ARC discovery grants DP200101630 (to S.Z.), a Clem Jones Centre for Ageing Dementia Research flagship grant (to S.Z.), and a Stafford Fox senior research fellowship (to S.Z.). Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). Some antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB), created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Author contributions
A.A. carried out experiments. S.Z. contributed to the interpretation of experiments. A.A. and S.Z. designed and interpreted experiments. A.A., T.O., and S.Z. wrote the paper. All authors approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100952.
Contributor Information
Arnaud Ahier, Email: a.ahier@uq.edu.au.
Steven Zuryn, Email: s.zuryn@uq.edu.au.
Data and code availability
This study did not generate/analyse datasets and codes.
References
- Ahier A., Dai C.Y., Kirmes I., Cummins N., Hung G.C.C., Gotz J., Zuryn S. PINK1 and parkin shape the organism-wide distribution of a deleterious mitochondrial genome. Cell Rep. 2021;35:109203. doi: 10.1016/j.celrep.2021.109203. [DOI] [PubMed] [Google Scholar]
- Ahier A., Dai C.Y., Tweedie A., Bezawork-Geleta A., Kirmes I., Zuryn S. Affinity purification of cell-specific mitochondria from whole animals resolves patterns of genetic mosaicism. Nat. Cell Biol. 2018;20:352–360. doi: 10.1038/s41556-017-0023-x. [DOI] [PubMed] [Google Scholar]
- Byerly L., Cassada R.C., Russell R.L. The life cycle of the nematode Caenorhabditis elegans. I. Wild-type growth and reproduction. Dev. Biol. 1976;51:23–33. doi: 10.1016/0012-1606(76)90119-6. [DOI] [PubMed] [Google Scholar]
- Chen W.W., Freinkman E., Wang T., Birsoy K., Sabatini D.M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell. 2016;166:1324–1337 e11. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins N., Tweedie A., Zuryn S., Bertran-Gonzalez J., Gotz J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019;38:e99360. doi: 10.15252/embj.201899360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtubay J.I., Goni F.M., Gomez-Fernandez J.C., Otamendi J.J., Macarulla J.M. Triton X-100 solubilization of mitochondrial inner and outer membranes. J. Bioenerg. Biomembr. 1980;12:47–70. doi: 10.1007/BF00745012. [DOI] [PubMed] [Google Scholar]
- Hadwiger G., Dour S., Arur S., Fox P., Nonet M.L. A monoclonal antibody toolkit for C. elegans. PLoS One. 2010;5:e10161. doi: 10.1371/journal.pone.0010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A., Zuryn S. The cellular mitochondrial genome landscape in disease. Trends Cell. Biol. 2019;29:227–240. doi: 10.1016/j.tcb.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Kuhnert F., Stefanski A., Overbeck N., Drews L., Reichert A.S., Stuhler K., Weber A.P.M. Rapid single-step affinity purification of HA-tagged plant mitochondria. Plant Physiol. 2020;182:692–706. doi: 10.1104/pp.19.00732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. WormBook; 2006. Maintenance of C. elegans; pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Step 1) A 1 mL micropipette tip containing the immunomagnetic mitochondria is positioned next to the flat surface of the magnet. Step 2) the immunomagnetic mitochondria are trapped by the magnet to the wall of the 1 mL micropipette tip closest to the magnet (1 min waiting). Step 3) A 25 mL pipette containing PEB is plugged into the top of the 1 mL micropipette tip to create a transitory washing column and the sample is washed using gravity flow. Step 4) Purified immunomagnetic mitochondria are collected with PEB and concentrated into a reduced volume of MIB.
Data Availability Statement
This study did not generate/analyse datasets and codes.