

Abstract
Dementia with Lewy bodies (DLB) is epitomized by the pathognomonic manifestation of α-synuclein-laden Lewy bodies within selectively vulnerable neurons in the brain. By virtue of prion-like inheritance, the α-synuclein protein inexorably undergoes extensive conformational metamorphoses and culminate in the form of fibrillar polymorphs, instigating calamitous damage to the brain’s neuropsychological networks. This epiphenomenon is nebulous, however, by lingering uncertainty over the quasi “pathogenic” behavior of α-synuclein conformers in DLB pathobiology. Despite numerous attempts, a monolithic “α-synuclein” paradigm that is able to untangle the enigma enshrouding the clinicopathological spectrum of DLB has failed to emanate. In this article, we review conceptual frameworks of α-synuclein dependent cell-autonomous and non-autonomous mechanisms that are likely to facilitate the transneuronal spread of degeneration through the neuraxis. In particular, we describe how the progressive demise of susceptible neurons may evolve from cellular derangements perpetrated by α-synuclein misfolding and aggregation. Where pertinent, we show how these bona fide mechanisms may mutually accentuate α-synuclein-mediated neurodegeneration in the DLB brain.
Keywords: Alzheimer’s disease, Parkinson’s disease, Braak hypothesis, Oligomers, Fibrils
Background
The emergence of “Dementia with Lewy bodies” (DLB), as a notion, was entirely based on geographically-defined schools of thought and revolutionary pathological analyses at a time when neurogenetics was beginning to contribute to our understanding of this understudied yet devastating condition [1, 2]. Once reputed to be a clinical syndrome depicted in isolated case reports, DLB has been purported to be the second most prevalent neuropathologic substrate of dementia [3–5] characterized by visual hallucinations, parkinsonism, and a fluctuating clinical course in older individuals [6, 7]. Contingent upon the nature of the population probed and the diagnostic criteria adapted, estimates from community-based clinical cohorts have been as high as one-quarter of all demented cases over the age of 65 [5, 8], parallel to approximations from post-mortem successions which have spanned between 15 and 25% [3, 9]. Reminiscing over 50 years of DLB research, the first inroads of this remarkable evolution commenced with James Parkinson’s intricate narrative of a Lewy body-like disorder vaguely identified as Shaking Palsy [10], which is what we now call Parkinson’s disease (PD) [11]. Despite the comprehensive portrayal of psychomotor manifestations, the unprecedented monograph made no allusion to the pathological and cognitive aspects of the disease, ultimately concluding that the intellect was ‘unharmed’ [10]. It was not until 1912 that Friedrich Heinrich Lewy began to explicate Parkinson’s call for pathological clarification [12] when he showcased the first depiction of eosinophilic intracytoplasmic and intraneuritic inclusions in the substantia innominata and dorsal vagal nuclei of autopsied PD brains [1, 13]. In less than a decade later, the term “Lewy body” (corps de Lewy) would go on to be enunciated for the first time by Konstantin Nikolaevich Tretiakoff [14], who documented the occurrence of analogous proteinaceous inclusions in the post-mortem substantia nigra of parkinsonian subjects [1, 14]. After a hiatus lasting more than 50 years, Kenji Kosaka went on to propose a new disease entity which he collectively termed “diffuse Lewy body disease” following his discovery of the first autopsy case with ‘Lewy-like-bodies’ pathology [15, 16]. The clinicopathological manifestations of individuals with diffuse cortical Lewy body disease were elucidated by Gibb and colleagues 3 years later [17, 18], and in 1992, the first operational consensus of senile dementia of Lewy body type (SDLT) was defined [19]. In spite of these early circumstantial nomenclatures and metaphors, the currently accepted conceptual framework formulated for early and prodromal diagnosis of DLB was first published in 1996 by the internationally renowned Consortium on DLB [6, 20].
Hitherto, delineating the nosological horizon of DLB diagnoses with respect to both Alzheimer’s disease (AD) and Parkinson’s disease dementia (PDD) has been an obstinate dilemma that reflects the fundamental pathobiology of the disease. Although DLB was first recognized as a rare pathological malady [21], it was gradually comprehended that α-synuclein-immunoreactive Lewy bodies [22] were recurrent ramifications in dementia cases with plaque pathology [23–26] and in PDD, specifically in cases where the latter had advanced to encompass cognitive deficits [27, 28]. Be that as it may, whether these juxta-nuclear inclusions represent an adaptive manifestation of a cytoprotective response or a defective self-preservation mechanism at the cellular level has yet to be singled out. Over the years, semi-quantitative assessments of autopsy-proven cases evaluating the neuroanatomical vulnerability between Lewy body biogenesis and DLB clinical phenotypes have reported contradictory repercussions. In a fraction of DLB cases, the approximated amount of cortical Lewy bodies in relation to total neuronal count correlates faintly with the severity and duration of cognitive and neuropsychiatric symptoms [29–32]. On the other hand, several lines of evidence have disparately verified a pronounced correlation between neocortical Lewy body densities and the clinical indices of dementia severity accompanying long-standing DLB [33, 34]. This dichotomy of regional pathology was also ascertained in neurochemical profiles of DLB patients [35, 36], where paradoxical alterations in neurochemical levels did not always correlate with cognitive domains [37]. Although these traits persist at the core of DLB, our conceptualization of the disease is evolving, with mounting evidence that α-synuclein-mediated synaptic aberrances discerned at predilection sites of neuronal depletion and Lewy body morphogenesis [38, 39] precede the clinical onset of DLB [40–44]. In post-mortem DLB brain samples, the subcellular localization of hyper-phosphorylated [45–47], detergent-insoluble [48, 49], and proteinase K-resistant [40, 50] α-synuclein aggregates in presynaptic-enriched fractions was affiliated with profound synaptic degeneration [51, 52]. These provocative findings are reminiscent of the observation that Lewy bodies are formed by microtubule-dependent cytoprotective aggresomes that sequester and compartmentalize synaptosomal α-synuclein aggregates from the neuronal periphery [53, 54]. Mechanistically, the former phenomenon is somewhat anecdotal and has been disputed, yet it would be thoroughly congruous with the supposition that DLB is a primary synaptopathy.
Emerging parallels posit that the intermolecular dynamics underlying the transformation of α-synuclein from its benign soluble state into fibrillized pathogenic inclusions entails a complex series of modifications that extend beyond the process of inclusion formation. According to the most simplistic viewpoint, physicochemical factors that enhance α-synuclein levels, catalyze its misfolding, or incite its translocation from its physiological presynaptic organization could possibly provoke de novo LB pathology [23, 55–62]. Supporting a role in causation, DLB-associated hereditary mutations in α-synuclein were also unambiguously consistent with the high densities of Lewy bodies in the subcortical nuclei, parahippocampus, amygdala, and cortex. The fact that this notation holds true in familial DLB kindreds with α-synuclein locus multiplications shows that α-synuclein expression is integral in determining whether the gradual degeneration of susceptible neurons prevails [63–66]. Yet, despite being implicated across a continuum of clinical phenomenologies [67], the pathogenic behavior of α-synuclein species in the DLB brain remains inadequately axiomatic. Recent reports connote that the insoluble filamentous α-synuclein in LBs may not be the prime neurotoxic culprit underlying neuronal dyshomeostasis and clinical variability. In fact, complementary cellular experiments have broadly featured the noxious insults of soluble α-synuclein oligomers as an adjuvant to the spreading of Lewy pathology [68, 69]. Thus, in this review, we explicatively explore experimental contexts highlighting the seminal contributions of α-synuclein conformers to the pathogenesis of DLB as an underappreciated pathological paradox of the DLB pathocascade. In particular, we assimilate and delineate the molecular prototypes that have led to new insights into understanding the mechanistic role that α-synuclein abnormalities dictate during their subsequent propagation in the DLB brain. Such a deliberation is, of necessity, selective rather than exhaustive, and its perspective may exhilarate the development of a tantalizing α-synuclein-centric therapeutic armamentarium capable of addressing the diverse biological defects underlying DLB pathophysiology.
Dementia with Lewy bodies: a pathonosological dilemma
Notwithstanding the designation it has attained in the classification of nosologists [6], DLB is an enigmatic clinical entity of a nature highly insidious since its clinicopathological overlaps with AD and PDD are clearly arbitrary and arduous to define [70–72]. In recent years, there have been considerable attempts to demarcate the neuropsychological features that distinctively characterize the disorder and distinguish it from AD and PDD [73–76]. These methodical proceedings have been corroborated by retrospective neuropathological analyses that have identified anomalous patterns of brain aberrations in DLB [22, 77–83]. Given the well-defined criteria, patients with DLB have been shown to display subcortical pathological similarities to that of PDD, but with fewer LBs and a milder degree of neuronal cell depletion in the substantia nigra and other brainstem nuclei (e.g., locus coeruleus and dorsal vagal nucleus) [71, 81, 84, 85]. Accordingly, diffusely scattered cortical Lewy bodies were also primarily concentrated in the neocortex, transentorhinal cortex, entorhinal cortex, hippocampus, amygdala, insula, and cingulate [86–88]. Conversely, the pursuit for neuropsychological features that may potentially distinguish DLB from AD was obfuscated by the fact that a substantial majority of DLB patients have concomitant AD pathology (i.e., neurofibrillary tangles, neuritic plaques) that is adequate to meet the neuropathological diagnostic criteria for AD [20, 89, 90]. This pattern of AD pathology was ascertained indistinguishably throughout the DLB brain and was, for the most part, identical to ‘pure’ AD as it plagued cortical areas that were afflicted by Lewy pathology [91]. Adding to the intricacy of diagnostic considerations, both patients with DLB and pure AD typically displayed widespread deficiencies in cortical choline acetyltransferase (ChAT) levels, although the degree of reduction was considerably greater in DLB [92–94]. Unlike their AD counterparts, DLB patients are further characterized by the dopaminergic deafferentation of the striatum due to selective degeneration of pigmented substantia nigra neurons [95, 96]. Given the resemblances in the nature and dissemination of neuropathological alterations in DLB and AD, it is not surprising that both entities are at first classified by the insidious onset of cognitive deterioration with no other conspicuous neurological abnormalities [6, 70, 97–99]. Early-onset neuropsychiatric features such as memory deficits, delusions, and delusional misidentifications frequently predate cognitive impairments, and with time, patients inexorably progress to severe dementia. Indeed, the clinical manifestations are so comparable that DLB patients are habitually misdiagnosed as having possible or probable AD [89, 100]. There are, however, certain clinical features that manifest with a greater incidence in patients with DLB as opposed to those with pure AD [101]. These unequivocal attributes consist of spontaneous motor characteristics of parkinsonism, history of rapid eye movement (REM) sleep behavior disorder (RBD), recurring vivid visual hallucinations, and fluctuating cognition with marked disparities in attention or awareness [6, 19, 20, 89, 100, 102]. Despite this, the prevailing commonalities in dementia syndromes engendered by DLB and AD remain undisputed, which raises the question of whether Lewy body pathology contributes eminently to the syndrome beyond the realms of AD pathology [34, 103]. The relevance of Lewy-related pathology to the pathogenic mechanisms accountable for eliciting the wide phenotypic spectrum of DLB is still controversial. Numerous clinicopathological correlates have failed to associate LB density with the severity of parkinsonism, recurrent falls, cognitive fluctuations, visual hallucinations, delusions, age of onset, and disease duration [31, 88, 104–106].
Perceived by some as the “Holy Grail of DLB,” Lewy bodies (LBs) are intracytoplasmic eosinophilic inclusions predominantly expressed in selectively vulnerable neuronal perikarya of the DLB brain [5, 107, 108]. While the majority of LBs are single and spherical in shape and morphology, a substantial fraction of neurons is almost invariably accompanied by multiple or pleomorphic LBs [17, 91, 109]. From an ultrastructural perspective, classic brainstem LBs are marked by the occurrence of filamentous and amorphous granular material, with an argyrophilic core that lacks discernible detail and a peripheral halo that has radially arranged 10 nm filaments [110–115]. In specific vicinities of the brain, for instance, the dorsal vagal motor nucleus and the basal nucleus of Meynert, distribution of analogous inclusions are detected within neuronal processes and are often denominated as ‘intraneuritic’ LBs [15, 21, 116]. Intraneuritic LBs are presumably identified via immunohistochemical characterization [15] and should be discerned from Lewy neurites (LNs) [117], which are sometimes capricious and not distinguishable through conventional histopathology [118, 119]. The thread-like LNs were first detected in the hippocampus [118] but are also abundant in various brain structures, including the cingulate gyrus, entorhinal cortex, amygdala, and basal ganglia [120, 121]. Remarkably, there is a rapid expansion in the relationship of neuritic pathology to cognitive severity in DLB, especially since the constellation of neurons affected with LBs can be relatively small in a minority of cases [34, 122, 123]. In addition to the well-circumscribed hyaline inclusions, some neuronal populations develop lesions that bear a striking resemblance to the antigenic determinants of LBs [124] but are poorly circumscribed and easily disregarded in routine histological preparations [91]. These pale-staining eosinophilic lesions are typified as pre-Lewy bodies or pale bodies and are substantially more common in pigmented nigral neurons [91, 125]. Contrariwise, similar inclusions within cortical neurons are referred to as cortical LBs [21, 126], which tend to be confined to pyramidal and small non-pyramidal neurons in lower cortical layers (layers V and VI of the neocortex) [17, 127]. At the immunoelectron microscopic level, cortical LBs and LNs appear to be made up of haphazardly arranged 10 nm filaments in the halo but lack a distinctive dense core with a matted network of filaments [118]. Occasionally, however, a spectrum of cortical LBs consume a hyaline appearance similar to brainstem LBs [17, 119, 128, 129].
Revolutionary paradigmatic advances in biochemical and immunohistochemical techniques further illustrated that the primary structural constituent of LB-laden neurons is α-synuclein [23, 25, 130–133]. The α-synuclein deposited within LBs and LNs are conventionally detergent-insoluble [23] and are subjected to a plethora of post-translational modifications, with N- and C-terminal truncations, SUMOylation, nitration, ubiquitination, and Serine 129 phosphorylation being the predominant modifications [47, 55–57, 59, 134]. In fact, the immunostaining of human post-mortem DLB brains with serine-129 phosphorylated α-synuclein antibodies displayed a staggeringly profound accumulation of α-synuclein in LBs and LNs, in contrast to the visualization by phosphorylation-independent antibodies. Although there was some initial skepticism, deposits of Lewy threads and Lewy dots were comparably immune-positive for this post-translationally modified form of α-synuclein [135, 136]. As such, it is enticing to postulate that cell types in specific brain regions may well accumulate discretely modified forms of α-synuclein, which could potentially have insinuations in deciphering the clinico-pathological subtypes of DLB. Coincidently, a detailed molecular dissection of LB biogenesis revealed that ubiquitin, a small heat-shock protein involved in energy-dependent protein degradation, and the ubiquitin-binding protein p62, manifest in most classical and cortical LBs [137, 138]. Thus far, a repertoire of proteins implicated across various cellular programs has been detected within the confines of LBs via immunohistochemical quantification. Initially, an antibody-based profiling approach unveiled proteins that were engaged in lysosomal and proteasomal degradation, cell cycle regulation, and mitochondrial function. Alternatively, an in-depth proteomic investigation of LBs purified from DLB cortices exposed proteins that had relevance for oxidative stress, signal transduction and apoptosis, synaptic transmission and vesicular transport, folding and intracellular trafficking, and the ubiquitin–proteasome system [139]. More recently, with the advent of correlative super-resolution microscopy, dysmorphic organelles, vesicular structures, subtypes of lipid constituents, and membranous fragments surfaced as pivotal components of LBs [140, 141]. Furthermore, the authentication of neuropathological hallmarks and cytoskeletal proteins in LBs [142–145] essentially challenged the unresolved interactability and spatio-temporal dynamics of these heterogeneous inclusions. Tau protein, a microtubule-associated scaffolding element of the AD neurofibrillary tangle, was sporadically associated with a subset of LBs [146]. Likewise, the hyper phosphorylation of tau at Ser396 was discernible in synaptic-enriched fractions of DLB, AD, and PD brains [147], while α-synuclein genetic variability has been shown to modulate neurofibrillary tau pathology [148]. Overall, this posits that compensatory mechanisms may emerge in the face of concomitant α-synuclein and tau burden, which construes the frequent phenotypic overlap between DLB and AD.
The α-synuclein architecture
An inherently disordered protein preponderantly disseminated in the brain [149, 150], the α-synuclein assimilated new-found relevance when its’ aberrant conformations nucleated the intracytoplasmic inclusion bodies of Lewy body diseases [25, 131]. Since a protein’s homology and configurational dynamics are coupled to its function, a concerted effort was subsequently initiated to designate the sequence and biophysical determinants that command α-synuclein’s proteome integrity and aberrant behavior. A highly divergent 14-kDa protein (140 amino acids) [151], the α-synuclein is defined by a lysine-rich membrane binding N-terminal amphipathic domain (residues 1–60), an aggregation indispensable central hydrophobic core (residues 61–95), and a disordered, acidic carboxy-terminal tail (residues 96–140) that is rich in proline, glutamate, and aspartate [152, 153]. The N terminus domain of α-synuclein comprises seven 11 residue imperfect tandem repeats, with a highly conserved central hexamer motif (KTKEGV) that wields an intrinsic tendency to approximately form two amphipathic alpha-helices (amino acids 3–37 and 45–92) in an antiparallel manner, flanking a truncated bridging region [152, 154–156]. By virtue of numerous repeats, the architectural flexibility of this domain enables the polypeptide to precisely potentiate three turns of the helix and establish contact preferentially with high curvature membranes [156–161]. Remarkably, the α-helical conformational conversion adopted upon membrane surface binding is stabilized by acidic phospholipid headgroups, signifying a synergistic interplay between the membrane and lysines positioned at opposing ends of the helix [156]. It is also riveting to note that all known genetic mutations affiliated with synucleinopathies—A53T, A53E, A30P, E46K, G51D, and H50Q are clustered within this domain [64, 162–168]. While these missense mutations, with the exemption of A30P, A53E, and G51D, have shown to escalate the tendency of α-synuclein to concoct morphologically insoluble discrete aggregates [164, 169–176], the bona fide mechanism through which these mutations prompt aggregation has not been convincingly presented. Nonetheless, the nonconservative E46K mutant is perhaps the only hereditary mutation that transpires in a clinical picture reminiscent of DLB since the clinical phenotype of E46K patients centered around severe and rapid disease evolvement alongside early-onset parkinsonism [64]. The crystalized hydrophobic central core of α-synuclein (residues 61–95), otherwise called the non-amyloid-β component of Alzheimer’s disease amyloid (NAC) [131, 151], encompasses two supplementary KTKEGV sequence motifs and a highly amyloidogenic segment that is accountable for the conformational plasticity of the protein [177]. Intriguingly, the deletion or disruption of sizable subdivisions within this domain immensely abrogated the oligomerization and fibrillogenesis of α-synuclein [178], accentuating the salience of NAC domains in the pathogenic cascade of α-synuclein aggregation [179–181] and its’ self-assembly into amyloid fibrils [182–185]. Originally projected to be a requisite for perpetuating the solubility of the protein, the C terminus of human α-synuclein (residues 96–140) contains 15 acidic amino acids (10 Glutamate and 5 Aspartate residues) and 5 Proline residues (P108, P117, P120, P128, and P138) that help to circumvent the spontaneous conglomeration of aggregation-prone conformations by masking the hydrophobic NAC domain [152, 154, 186, 187]. Owing to its higher proportion of charged residues and low hydrophobicity, it is conceivable that this domain undergoes specific phosphorylation at discrete sites [188, 189] and is responsible for the intrinsically unstructured topology of α-synuclein. As radical as it may seem, there is now compelling evidence that the obliteration of C terminus, in tandem with alterations to the domain hydrophobicity, may possibly compromise the membrane-binding machinery and enhance the aggregation propensity of α-synuclein [190–195].
Understanding the biology of α-synuclein: membrane remodeling and synaptic behavior
The presynaptic confinement of α-synuclein has become well entrenched ever since its discovery as a phosphoprotein that is vastly enhanced in synaptic boutons, which sprout from axons of diverse neurochemical phenotypes [154, 196, 197]. Yet, in spite of the initial affiliation with synaptic vesicles, α-synuclein is not consistently existent in all presynaptic terminals and curiously appears to be among the last proteins to approach the evolving synapse during synaptogenesis [198]. The discrepancies of these explications in neurodegenerative disorders have led to the fundamental question of how α-synuclein localizes to the biochemically distinct synaptic termini. Unswerving with its portrayal as a weakly associated peripheral membrane protein, it has been presumed that α-synuclein depends on the N-terminus domain to mediate membrane binding in cells [160, 174, 199, 200]. However, subcellular biochemical fractionation of brain tissue stipulates that an immense majority of synucleins behave almost entirely as soluble monomeric proteins, with an extremely weak affinity towards native synaptic vesicles [201–203]. In an endeavor to visually trace the dynamics and intrinsic mobility of α-synuclein in live intact cultured cells, hippocampal neurons derived from transgenic mice were transfected with GFP-labelled synuclein while individually isolated presynaptic boutons were exposed to photobleaching. Interestingly, the fluorescence recovery following photobleaching was persistently rapid, implying that the protein is exceedingly mobile [201]. On the contrary, when this experimental paradigm was extended to fluorescently-tagged human α-synuclein in individual cortical neurons of transgenic mice, signal recovery was prominently delayed and less exhaustive in synapses with increased synuclein expression, reputedly due to substantial in vivo aggregation [204]. Despite its greatly diminished tethering with cellular membranes, α-synuclein conversely recuperates more sluggishly than GFP after photobleaching [201], raising the plausibility that the N-terminal membrane-binding domain of α-synuclein may be spearheading the interplay. Supporting this notion, the A30P point mutation notably eliminates the synaptic enrichment of α-synuclein, obliterates the interaction of α-synuclein with artificial [205] and native biological membranes, and expedites fluorescence recovery to that of GFP following photobleaching [201, 206]. Amid these studies, counter-intuitive chemistry has alternatively described α-synuclein’s N-terminal region as a membrane curvature sensor, whose apparent “design defect” translates into an extraordinary ability to unambiguously adsorb to the surface of highly curved biological membranes (Fig. 1) [200, 207–209]. Accounting for this marked predilection is the hydrophobic fragment of the N-terminal amphipathic region, which harbors a sequence of threonines within the repeat that compromises the synergism of α-synuclein with membranes scrupulously as a means to attain specificity for high curvatures [210]. In conjunction with the aforementioned repertoire of membrane curvature dynamics, α-synuclein notoriously appears to possess a precise inclination towards the assemblies of lipid rafts, a membrane micro-domain that is negligible in fluidity but fortified with cholesterol and saturated acyl chains [201, 211–214]. Indeed, the A30P mutational derangement of lipid rafts prevented the agglomeration of α-synuclein in presynaptic boutons [201], complementing the relevance of this interaction for neurons. Recapitulating the specificity observed in vitro, biochemical methods have analogously detailed that α-synuclein necessitates a “mish-mash” of phospholipids with oleoyl and polyunsaturated acyl chains, signifying that it may irrevocably identify the phase boundary that ensues between membranes that vary in fluidity [215]. Remarkably, there was an irrefutable prerequisite for the acidic headgroup appertaining to polyunsaturated acyl in preference over the oleoyl chain, albeit the curvature-sensitive membrane tethering [215]. In accordance with this consideration, α-synuclein has been found to influence lipid packing within cholesterol-enriched raft-like domains [216], suggesting that α-synuclein is not only congregated by these structures but also effectuates their structural remodeling. Furthermore, α-synuclein has a notoriety for being implicated as a fatty acid-binding protein [217] that modulates the uptake of polyunsaturated fatty acids to catalyze the formation of highly soluble lipid-dependent oligomers [218–221]. Affirming a role for this phenomenon in α-synuclein deficient mice, astonishing changes were observed in acyl chain composition [222], while fatty acid uptake and metabolism turned out to be exaggerated [223–225]. Nonetheless, subtle alterations in acyl chain orientational order may modulate membrane fluidity and trafficking, and variations in α-synuclein expression level appear to augment clathrin-mediated endocytosis [226].
Fig. 1.
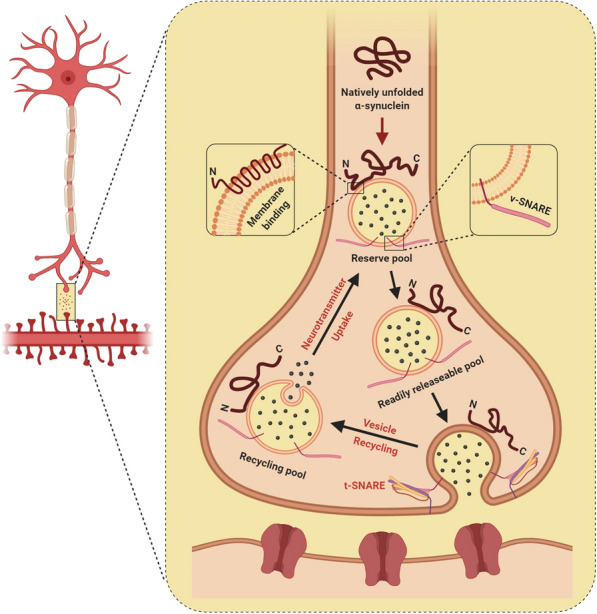
This schematic highlights the regulatory roles of α-synuclein in harmonizing synaptic homeostasis at the presynaptic terminal by coordinating vesicle trafficking, vesicle refilling, and the interactions between vesicle-associated SNARE (v-SNARE), membrane-associated SNARE (t-SNARE), and neurotransmitter release. Figures were created with Biorender.com
Notwithstanding the tremendous diversity in original publications, the dauntingly complex presynaptic enrichment of α-synuclein [197, 198] and its contiguous interaction with synaptic membranes have emphatically implied a regulatory role in the molecular machinery that mediates synaptic neurotransmission (Fig. 1) [193, 227–231]. Electrophysiological analysis of α-synuclein deficient mice has previously demonstrated a strikingly selective deterioration in hippocampal synaptic responses, following sustained high-frequency tetanic stimulations that exhaust the docked and reserve pool of synaptic vesicles [227, 232]. Concurrent with the altered responses, electron microscopic examination of mice lacking α-synuclein further displayed profound deficiencies in the rejuvenation of docked pools from the reserve pool. In a similar fashion, the repression of α-synuclein expression through antisense oligonucleotide pharmacokinetics prompts a significant diminishment in the attainability of reserve synaptic-vesicle pool in cultured hippocampal neurons [228]. On the basis of these observations, α-synuclein unexpectedly appears to orchestra the refilling and mobilization of synaptic vesicles from the reserve pool to the site of synaptic vesicular release. Perhaps not surprising, compelling support for this hypothesis came from transgenic model organisms and viral gene-delivery systems that were employed to over-express human α-synuclein, which elegantly demonstrated considerable deficits in synaptic vesicle exocytosis with diminutions in neurotransmitter release [233–238]. Contemporary ultrastructural paradigms subsequently publicized that the overabundance of α-synuclein precipitates a depletion in “readily releasable” vesicles [236] and physiologically disrupts the reclustering of synaptic vesicles following endocytosis, instigating a decrement in the synaptic vesicle recycling pool density [235]. Under similar circumstances, α-synuclein in overexpressing neurons unequivocally diminished dopamine reuptake in dopaminergic terminals [237] and impeded the intersynaptic vesicular dynamics between presynaptic boutons, resulting in a smaller reserve pool of vesicles [234]. The plausible portrayal of α-synuclein in harmonizing synaptic homeostasis, however, is not assiduously affiliated to its precise connectivity with synaptic vesicles. The multifarious α-synuclein has been shown to interact with synaptic proteins [239] that facilitate exocytic and synaptic vesicle motility, such as the phospholipase D2 and Rab GTPase-activating proteins [240, 241]. In this context, α-synuclein engaged as a tightly coordinated chaperone for the trimeric supramolecular SNARE complex by dramatically orchestrating the dissipation and coordinating the recruitment, safeguarding, and rigorous configuration of this multifaceted machinery [242]. These elucidations raise the possibility that α-synuclein dictates the trafficking of synaptic vesicles, regulation of vesicle exocytosis, and may theoretically mediate a more refined reciprocal phenomenon by domineering synaptic homeostasis-affiliated proteins (Fig. 1). The fact that synuclein deficient synapses are physiologically relevant connotes that synucleins are not integral denominators of the neurotransmitter release machinery but are redundantly essential for the long-term synchronization and maintenance of presynaptic efficacy [243]. Akin to the expunction of synucleins, the transgenic expression of α-synuclein in cysteine-string protein-α (CSPα)-ablated mice was able to abrogate progressive neurodegeneration [244], indicative of a neuroprotective role for α-synuclein in protecting nerve terminals against lethality. These observations appear to be mediated by a downstream mechanism that relies upon phospholipid binding, as the A30P α-synuclein mutant, which is greatly impaired in membrane binding, failed to rescue CSPα knockout mice from the deleterious consequences of CSPα deficiency.
The α-synuclein alchemy: conformational heterogeneity and biological consequences
Over the last decennary, tangible evidence suggests that α-synuclein is able to stochastically fluctuate through conformational space and undergo dramatic internal reorganization from their primitive states to form intermolecular β-sheet-rich entities; ranging from monomers, tetramers, and higher-order soluble oligomers to large insoluble fibrillar polymorphs (Fig. 2a, b) [245–250]. The abstruse heterogeneity and rapid interconversion tendency to assume higher-order aggregates, however, have made it exceedingly laborious to reconcile high-resolution structural details for the native assembly and naturally-occurring multimeric conformations. Years of elegant work have previously demonstrated that recombinant α-synuclein isolated from bacterial expression systems, under native or denaturing environments, subsists arbitrarily as stable unfolded monomers with insubstantial secondary structural features [153, 245, 251–253]. This ostensibly anomalous behavior was further ratified by the analysis of purified α-synuclein, which revealed a compact monomeric state for native α-synuclein that counteracts spontaneous assemblage, arrogates α-helical conformation upon phospholipid tethering, and withstands conformational metamorphoses prior to oligomerization and fibrillogenesis [254, 255]. While these annotations stipulated that native α-synuclein is rudimentarily an ill-defined monomer that adopts multiple forms of compact conformers depending on its cellular localization and membrane interactions, a lingering ambiguity endured concerning the apparent native size of the protein. Consequently, further investigations on the α-synuclein native state were reignited several years later, and this time, suggesting that α-synuclein behaves intrinsically as dynamic α-helical tetramers that are impervious to aggregation and fibril assembly [256–258]. In parallel, complementary biophysical efforts by several groups insinuated that only a small proportion of α-synuclein assembles into α-helical trimers and tetramers while the majority of species perseverated as deranged monomers [257, 259, 260]. Concertedly, these illustrations sparked off an alleged controversy and effectuated a reappraisal of the native state of α-synuclein. Even though synonymous inquisitions in the mouse brain reaffirmed that the predominant native form of α-synuclein is an inherently disordered monomer with minimal secondary structure [246, 261], conformationally diverse α-synuclein multimers were also present in post-mortem brain tissue [251]. This continuum of conformational states, embraced by native α-synuclein, evolved through a series of equilibria that adopted distinct assemblies under precise stress-induced constraints or upon interaction with specific lipids, ligands, proteins, or biological membranes [161, 168, 251, 262–265]. Considering the heterogeneity and diversity of oligomeric variants that were of disease relevance [69, 266–271], numerous propositions have accordingly explored the molecular and structural determinants that influence α-synuclein’s initial facile oligomerization. Compellingly, mildly acidic milieus [272], low fractions of negatively charged lipids [273], and specific post-translational modifications [189, 274–277] regulate α-synuclein oligomerization. Besides, α-synuclein interacts with polyunsaturated fatty acids [220], lipid vesicles [278], transition metals (Fe2+, Cu2+, and Zn2+) [254, 276, 279], and drug-like molecules such as dopamine [280] to promote its’ self-assembly and formation into β-sheet rich soluble aggregates. Upon assembly, α-synuclein oligomers sequentially undergo conformational switching to become proteinase-K-resistant oligomeric precursors that generate significant levels of oxidative stress before metamorphosing into downstream fibrils [281]. These oligomeric states of α-synuclein may chemically permutate in equilibrium, remodeling their mechanical behavior, either engaging as on-pathway intermediates or kinetically trapped assemblies from which fibrillation is no longer conducive [282].
Fig. 2.

Under pathological circumstances, α-synuclein aggregation could potentially take place in affiliation with the cellular membrane or in the cytosol. a Membrane-bound monomeric α-synuclein assumes an α-helical structure, but at elevated levels, the monomer endures conformational change to generate membrane-bound β-sheet structures that self-associate to form oligomers and fibrils. b In the cytoplasm, unfolded monomers fluctuate through conformational space to form unstable dimers, which undergo reorganization to generate oligomers of varying morphologies that eventually transform into fibrils. The erratic accretion of these fibrils leads to the amassing of intracytoplasmic Lewy bodies. c During α-synuclein fibrillogenesis, oligomers and amyloid fibrils are immensely noxious, compromising microtubule dynamics, endoplasmic reticulum–Golgi trafficking, and mitochondrial function. Figures were created with Biorender.com
In spite of the wealth of empirical data, there is no unanimity on whether oligomers or downstream fibrils are the more noxious entity. Fastidious observations of human α-synuclein-expressing mesencephalic neurons suggested that oligomers are more noxious since α-synuclein purified from DLB brain extracts displayed escalated levels of soluble, lipid-dependent α-synuclein oligomers [220]. In a similar vein, the pioneering characterization of oligomeric landscape in post-mortem DLB brain homogenates validated the existence of soluble α-synuclein species that exacerbated phospholipid membrane integrity [69, 271]. The diagnostic utility of cerebrospinal fluid (CSF) α-synuclein species to distinguish DLB patients from AD individuals further reported a unique trend of high-oligomeric-α-synuclein CSF levels, albeit a lack of correlation with cognitive performance [270]. Conversely, while exosomal α-synuclein levels correlated with the severity of cognitive impairment in DLB patients, isolated DLB-patient CSF-derived exosomes governed the oligomerization of soluble α-synuclein in recipient cells [283]. Intriguingly, the inoculation of DLB brain-derived exosomes into healthy rodent brain tissue was sufficient to propagate α-synuclein aggregation, bolstering the hypothesis that pathogenic α-synuclein oligomers may be preferentially sorted into exosomes to facilitate α-synuclein fibril growth [284]. The adaptation of an in vitro amplification technique, designated “real-time quaking-induced conversion (RT-QUIC),” jointly implied that the oligomeric forms of α-synuclein are the seeding species that cause prion-like propagation [285]. Coherent with these findings, α-synuclein-associated mutations (A53T and A30P) accelerated oligomerization, not fibrillization [286], while stereotaxic inoculation of oligomer-forming rather than fibril-promoting α-synuclein variants induced severe in vivo deterioration [287]. Additionally, much evidence has been adduced to suggest that α-synuclein oligomers may dysregulate cell function and culminate in synaptotoxicity [252, 286, 288] by eliciting lysosomal leakage [289], compromising mitochondrial activity [290, 291], dishevelling microtubule dynamics [292], and physically altering the axonal transport of presynaptic proteins [234] (Fig. 2c). While it is conceivable that α-synuclein toxicity involves distinct intermediates on the pathway en route to fibril formation, a series of kinetic signatures insinuate that the process involving the conversion of oligomers to fibrils itself contributes to α-synuclein-mediated neurodegeneration. Expression of α-synuclein mutants designed to promote fibril formation propensity (S129A) was toxic to rat models [293, 294], whereas mutations that impede α-synuclein oligomerization and fibrillogenesis (S87E) catalyze considerably less α-synuclein aggregation [274]. Likewise, complementary expression of artificial mutant variants of α-synuclein (E57K and E35K) resulted in conformationally snared oligomers that were profoundly noxious [288] to dopaminergic neurons in animal models of synucleinopathies [287]. Conjointly, mutations that augment α-synuclein oligomerization unveiled high neurotoxicity, but a sustained progressive loss of dopaminergic neurons was undisputedly reliant on the potential of α-synuclein variants to form fibrils [295]. As such, one may presume that the fibrillary α-synuclein assemblies might be significantly more pathogenic than their precursor on-assembly pathway oligomers [176, 296] since the nigral inoculation of α-synuclein fibrils aggravated motor deficits and induced more pronounced Lewy body/Lewy neurite-like inclusions [297]. In support of this proposition, the Lewy-associated filamentous α-synuclein enriched in detergent-insoluble fractions of DLB patients projected the ability to alter neuritic outgrowth in human iPSC-derived neurons [69]. The revelation of α-synuclein assemblies with distinct strains of pathological fibrils further led to the premise that the fibrillar polymorphs display differential seeding and fibril-templating efficiencies that may account for the heterogeneous neurotoxic phenotypes in DLB [176, 298, 299]. As innocuous as it seems, the structural rationale underlying fibril polymorphism is that the α-synuclein fibrils are made up of two intertwining protofilaments, and the discrepancies in the assembly of these protofilaments may give rise to discrete conformational polymorphs [300, 301]. Nevertheless, it remains obscure if preformed fibrils which maturate in LBs may instigate disease-specific cytopathology. Traditionally, whilst the omnipresence of cortical LB densities are pathognomonic for cognitive dysfunctions [24], there is limited evidence to reinforce a correlative relationship between LB burden and the severity of dementing phenotypes [27, 88, 108, 302]. In the course of addressing the malleability and dynamism of LB composition with respect to the lack of overt phenotype, key pathological events were often reported to be imperiled and overwhelmed prior to LB biogenesis. These cellular insults include synaptic collapse [40], reduced neurofilament mRNA levels [303], the amassing of specialized axonal proteins [304], and the implementation of apoptotic signaling cascades [305]. With these caveats in mind, it is tempting to speculate that the acquisition of deleterious hallmarks is instead galvanized by ill-defined heterogeneous oligomers. Taken together, the mechanistic relationship between oligomers and fibrils remains to be clarified since a well-founded conjecture is complicated by the heterogeneity of diverse experimentally observed states. Despite the fact that oligomers are conceivably implicated in the breakdown of neuronal homeostasis, the stable protuberant nature of fibrillar α-synuclein assemblies appears to be the most competent at propagating itself both in vitro and in vivo. The inconvenient truth is that the “oligomer” and “fibril” nomenclatures lacked the fidelity and mechanistic precision required for the critical appraisal of physiological entities. Rather, numerous conformations of these assemblies exist, which commands their biophysical profile, and may account for unique strains of polymorphic aggregates resulting in distinct pathobiological traits [298, 306].
Beyond α-synuclein propagation: from transmission to DLB pathogenesis
The central tenet of presynaptic α-synuclein fidelity, under physiological circumstances, is that the α-synuclein is an intracellular synaptic protein that coalesces with vesicles [197, 231]. Yet, under distinct pathological milieus, parsimonious explanations theorized that the toxic oligomeric species of α-synuclein could be eliminated from neuronal cells through unorthodox secretory pathways [307–310]. As such, perturbation of the intracytoplasmic degradation pathways, for instance, the autophagic signaling network [311, 312], might jeopardize and catapult the pathological release of α-synuclein in degenerating neurons [313, 314]. In principle, the sequestration of extracellular α-synuclein oligomers entails unconventional exocytosis in clear-core synaptic vesicles [313, 315], exosome-mediated discharge [283, 308, 316–318], and infiltration from the donor membrane [319, 320]. Circumstantially, these pathogenic α-synuclein aggregates are unequivocal in substantiating trans-synaptic and intracellular transmissibility through the neuraxis [308, 313, 321], where they govern intraneuronal aggregation [322] and are ‘primed’ to exacerbate neuroinflammation [323–326]. Exemplifying the molecular basis of this stereotyped spread, various models of Lewy-prone systems have displayed robust induction of α-synuclein inclusion pathology [327] in human neuronal precursor grafts and human fetal grafted neurons [323, 328–332]. Currently, there are several theoretically acclaimed neuropathological grading paradigms to evaluate the topographic trajectory of α-synuclein, entailing a semiquantitative staging of α-synuclein to address the chronological severity in discrete brain regions. The hierarchical caudo-rostral dissemination of Lewy body-related pathology in LBD, systematically proposed by Heiko Braak and Kelly Del Tredici, has been spatiotemporally interpreted to corroborate a cohesive hypothesis of unidirectional α-synuclein distribution through specific pre-established neuroanatomical circuits [120, 333–335]. In an influential series of histopathological evaluations, the provocative “Braak hypothesis” first outlined that the dorsal motor nucleus of the vagus nerve, and to a lesser extent, the olfactory system, serve as peripheral entry points for the α-synucleinopathy (Stage 1). Essentially, these misfolded versions of α-synuclein travel transneuronally in a stereotypic fashion across the medulla and pontine tegmentum (Stage 2), imprinting the retention of α-synuclein in somal Lewy inclusions and provoking the polymerization of neighboring synuclein deposits in the midbrain and amygdala (Stage 3). It then follows that the α-synuclein transcellularly deposits deeper into the temporal cortex (Stage 4) and neocortex (Stages 5 and 6), exposing the cardinal cognitive deficits observed in LBD [106, 120, 333–335]. Suffice it to say here, that in DLB, which at first manifests with clinical dementia and only sporadically with extrapyramidal signs, this topographical α-synuclein-centric expansion map is not favorable. Overall, though, it was consequently reasoned that α-synuclein pathology might initially transpire in neocortical and limbic areas. Marui et al. [41], in their seminal investigation of the neuroanatomic contiguity of DLB pathology, explored the regional focality of Lewy-related α-synuclein dissemination in the brain of cases that fulfilled the clinicopathological diagnostic criteria for DLB. Based essentially on topographic proximity, the brain regions reviewed encompassed the hippocampus (CA2/CA3), the amygdala (cortical and accessory basal nuclei), and the superior frontal, middle temporal, insular, transentorhinal, and entorhinal cortices. Compellingly, the widespread evolution of Lewy pathology defined exclusively by a morphological staging scheme engaged the amygdala first, followed by various limbic regions, most notably the transentorhinal, insular, entorhinal, and CA2/3 cortices, and, ultimately, the neocortex [41]. But even so, one fundamental drawback of this framework was that the olfactory bulb, which is implicated in virtually all of the DLB cases, was not cross-examined [336]. This forestalled the feasibility of drawing firm inferences about how the olfactory bulb proteotype might hypothetically tailor into such a staging template. Nevertheless, these findings are particularly consequential since they disclosed that olfactory and limbic structures are most susceptible and succumb early in the disease process. Among the many amygdaloid subnuclei, the cortical areas are the ones that receive projections directly from the olfactory bulb [337, 338]. These evaluations have led to the assumption that the distribution of Lewy pathology in the amygdala of DLB cases mirrors the input from the olfactory bulb. Yet, the predictive validity of this staging scheme is ambiguous, considering there was a lack of correlation between the α-synuclein stages and the clinical severity of dementia and psychiatric comorbidities. According to these considerations, it is inferable that the pathoarchitectonic pattern of DLB is not just confined to the systematic progression of α-synuclein pathology but also to the subsequent influence of more-deterministic AD-relevant pathologies in anatomically related foci [339]. Consistent with this supposition, Rey et al. [340] unveiled unprecedented insights into the spatiotemporal pattern and transneuronal propagation of pathologic α-synuclein from the olfactory bulb. In a notable series of experiments, the authors inoculated recombinant α-synuclein fibrils into the olfactory bulb of wild-type mice and probed their transmissibility through the neural circuitry. Within 1-month post-inoculation, α-synuclein pathology was preliminarily disseminated in the ipsilateral entorhinal and piriform cortices, the cortical amygdaloid nuclei, and the contralateral and ipsilateral anterior olfactory nuclei. By 3 months, α-synuclein-positive inclusions have progressively spread sequentially to the hippocampus, the basal amygdaloid nuclei, and the insular, ectorhinal, and orbitofrontal cortices. At 6 months, the spatial spreading of α-synuclein aggregates evolved considerably to the nucleus of the lateral olfactory tract, the olfactory tubercle, the central amygdala, the dentate gyrus, the CA2/CA3 fields of the hippocampus, the thalamus, and the hypothalamus. Eventually, by 12 months post-inoculation, the seeded α-synuclein inclusions were able to serially propagate to the associative neocortex, the locus coeruleus, the substantia nigra, and the raphe nuclei [340]. Despite assertions that the pathogenic templating of endogenous α-synuclein amass in a connectome-dependent manner, there remains an unsatisfying disconnect between α-synuclein deposits and its subsequent engagement with additional brain regions 2 years post-inoculation [341]. The shortcoming fueled speculation that transmission of α-synuclein pathology is likely to be perturbed by premature α-synuclein-induced neuronal loss, which compromises the neural circuit integrity, or by proteolytic mechanisms that facilitate α-synuclein degradation. This notion has gained traction since no α-synuclein-positive inclusions were disseminated in mouse brains 21 months post-nasal inoculation of DLB-associated α-synuclein fibrils, further insinuating that α-synuclein aggregates cannot possibly navigate through the nasal mucosa [342]. Tantalizing as it may be, although dense α-synucleinopathy expanded beyond the limbic and associative neocortical connectomes, distinctive inclusion bodies were only ‘occasionally’ concentrated in the substantia nigra [340]. One might connote that in terms of clinical correlates, such template-directed sequential dispersal of Lewy pathology would likely be associated with early-onset dementia and subsequent parkinsonism, mimicking the clinical phenotype of DLB (Fig. 3). In this structure-oriented view, the iatrogenic transmission of α-synuclein pathology from the olfactory bulb is able to extend beyond the olfactory limbic axis and substantially infiltrate the associative neocortex [340]. Of paramount importance, the preponderance of cortical amygdaloid-predominant α-synuclein pathology originated from the olfactory bulb during the earliest stage of spatial dissemination. Similarly, the proposed stereotypical expansion of incidental LB pathology by Marui et al. [41] acknowledged that the cortical nuclei of the amygdala were indeed the primary site in which α-synuclein inclusions are first discerned. Together, these directional predilections buttress the conjecture that, in DLB, the cortical zone of the amygdala may represent a preferential locus for α-synuclein deposition wherein pathologic aggregates are reached via the olfactory bulb. If this corollary is valid, it raises the intriguing possibility that the translation of this discernible gradient in regional hierarchy into a unidirectional chronological sequence may provide the basis for a hypothetical DLB staging construct (Fig. 3). In the same way that a caudorostral trajectory subsists in PD, a predefined nasal route with stereotyped incursions of the α-synuclein pathology across limbic and neocortical networks could also be conceivable in DLB. This spatiotemporal axis, proposed by the aforementioned conceptualization, would presumably result in a clinical picture in which overt cognitive deficits appear early, thus bringing forth a phenotype resembling DLB. Although mechanistically distinguishable, auxiliary determinants such as concomitant AD-type pathology may, in part, exacerbate the corruptive templating and DLB-specific spatiotemporal progression of α-synuclein lesions [339]. These, in turn, may influence the complementarity of molecular surfaces that interact to instigate the seeding cascade of afflicted areas, rendering certain brain regions selectively susceptible to further abnormal protein deposition. Moreover, the revelation of α-synuclein-driven phenotypic diversity in pathology is contingent upon neurons that are already burdened with concomitant Aβ- and tau-mediated pathological insults. In a self-perpetuating process, these subsets of compromised neurons tend to engage as trigger sites that synergize with lesioned assemblies to precipitate synuclein-based deposit accumulation in the neocortex. The concurrent manifestation of hyperphosphorylated tau, Aβ, and α-synuclein inclusions in the broad sense thus revamps the topographical accrual of anomalous proteinaceous entities and thereby influences the nature of the ensuing disease [339].
Fig. 3.

Schematic representation of the hypothetical DLB staging construct with stereotyped incursions of α-synuclein pathology across limbic and neocortical networks that might support the occurrence of early dementia with parkinsonism. Figures were created with Biorender.com
To this end, the perpetual mechanisms ascribed to the imminent spreading of extracellular α-synuclein oligomers have been warranted by endocytosis-mediated access [313, 343, 344], transmembrane penetration [158, 319, 345], trans-synaptic transmission [346, 347], and membrane receptor internalization [313, 344, 348]. Once within the recipient cell, internalized α-synuclein oligomers incapacitate the recipient cell by engaging as a molecular blueprint that further impels the morphogenesis of intracellular Lewy body-like aggregates [179, 324]. In fact, the topology of these induced inclusions argues in favor of a rate-limiting nucleation mechanism that pre-eminently heightens the cross-linking of assembly-competent oligomers, which is redirected by cooperative oligomeric growth and exponential fibril elongation by monomeric addition [272, 278, 349]. At the right stoichiometry, the binary switch between nucleation and elongation necessitates disordered oligomeric intermediates to adopt dynamically ordered configurations that are impervious to devolution and capable of evoking further fibrillation [350]. Such fine-tuning of fibrillation, in turn, can be catalyzed by α-synuclein-harboring familial mutants [351, 352] and posttranslational modifications such as truncation [353], phosphorylation [55], oxidation [354], nitration [355], glycation [356], and acetylation [357]. On this basis, the presence of seeding-competent fibrils can override the initial lag period of the primary nucleation phase [272], resulting in de novo secondary nucleation, which rudimentarily facilitates the generation of new entities on the surface of existing fibrils [358]. In analogy to mammalian prions, the so-called “nucleation-dependent polymerization” is usually invoked to illustrate the perplexing sequelae of intracellular oligomer and fibril propagation; and is thought to encipher the evolving neuroanatomical spread of α-synuclein pathology. This catastrophic assembly has been observed in a cell-based construct in which the inoculation of recombinant α-synuclein fibrils channeled the engagement of endogenous α-synuclein and self-amplification of Lewy body-like inclusions [179, 324]. In a similar fashion, the introduction of brain homogenates comprising α-synuclein protofibrils and fibrils considerably enhanced α-synuclein pathology and propagation in genetically engineered α-synuclein mice [359, 360]. Indeed, the suggestion that α-synuclein may propagate like a prion is entering a whole new realm of clinical relevance, given that it could unravel the connectomic distribution of Lewy pathology and aetiological heterogeneity across DLB. Regardless, the template-mediated prion-like amplification of pathogenic α-synuclein doesn’t appear to be solely defined by the ‘nearest neighbor’ rule or the strength of neuroanatomic connectivity. The engagement of neuronally expressed lymphocyte-activation gene 3 (LAG-3) demonstrated a high binding affinity towards preformed α-synuclein fibrils that initiated endocytosis from the extracellular milieu. What is more, the knockdown of LAG-3 substantially attenuated the cellular uptake of α-synuclein fibrils, diminishing the pathology set in motion [361]. Building on this finding, extracellular α-synuclein oligomers preferentially interact with the prion protein, eliciting a wide repertoire of signaling cascades that culminates with neuronal dysfunction [362]. Hence, according to these interpretations, the network signatures corresponding to the robust development of Lewy-type pathology may be actuated by cell- or region-autonomous mechanisms. Along the same line of thought, a low regional expression of native α-synuclein has been attributed to sequentially vulnerable brain areas that do not develop Lewy body-like α-synuclein inclusions [363] while attenuated cellular expression was prohibitive to intracellular aggregation [364]. In this regard, lower expression levels of physiological α-synuclein within distinct neuronal subpopulations may hinder the consequent amassing of intracellular aggregates by constraining the initiation nucleation phase in pathology-laden brains. Conceptually, these observations may represent a far end of a continuum that begins with extracellular α-synuclein seeds that presumptively choreographs specific patterns of neuronal vulnerability in a prion-like fashion. While transcellular propagation in vivo remains conjecture, such experimental systems necessitate rigorous substantiation, preferably by the observation that α-synuclein spontaneously disseminate aggregation through pre-existing neural networks.
Concluding remarks
A blurring of the traditional distinction between physiologically relevant α-synuclein and its robust cytopathological signature in the DLB brain has heightened unwarranted apprehension over the neuropathological transmissibility of α-synuclein. The unsalutary intersection of disease-linked determinants that accelerate α-synuclein oligomerization has obscured the precise role of α-synuclein oligomers, fibrils, and prion-like α-synuclein propagation in the etiopathogenesis of DLB. Expanding the experimental momentum from genetically engineered systems to the DLB brain's neural circuits continues to be a constraining facet and mandates the exploitation of in vivo culture models derived from α-synuclein-rich brain extracts. The mechanistic commonality geared towards abating α-synuclein synthesis, toxicity, and aggregation are currently being scrutinized in preclinical models and lend credence to the establishment of mechanism-based therapeutics in DLB. We believe that more emphasis on unraveling the molecular mechanisms that unify the self-templating capacity of α-synuclein will substantially broaden our understanding of the pathocascades leading to neurodegenerative dementias such as DLB.
Acknowledgements
All authors appreciate the support from Jeffrey Cheah School of Medicine and Monash University Malaysia.
Disclosure
The authors have nothing to disclose.
Abbreviations
- DLB
Dementia with Lewy bodies
- PD
Parkinson’s disease
- SDLT
Senile dementia of Lewy body type
- AD
Alzheimer’s disease
- PDD
Parkinson’s disease dementia
- LBs
Lewy bodies
- LB
Lewy body
- LNs
Lewy neurites
- NAC
Non-amyloid-β component of Alzheimer’s disease amyloid
- GFP
Green Fluorescent Protein
- SNARE
Soluble NSF Attachment Protein Receptor
- CSPα
Cysteine-string protein-α
- LBD
Lewy body dementias
- LAG-3
Lymphocyte-activation gene 3
Authors’ contributions
CS critically reviewed the literature, wrote the manuscript and conceived the figures. IP, TS and HJO edited the final manuscript. All authors read and approved the final manuscript.
Funding
C.S. is a recipient of the Monash University Malaysia Graduate Research Merit Scholarship.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christopher Simon, Email: christopher.simon@monash.edu.
Tomoko Soga, Email: tomoko.soga@monash.edu.
Hirotaka James Okano, Email: hjokano@jikei.ac.jp.
Ishwar Parhar, Email: ishwar@monash.edu.
References
- 1.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9(1):13. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 2.Kosaka K. Dementia with Lewy bodies. Tokyo: Springer; 2017. History and latest concepts of Lewy body disease and dementia with Lewy bodies; pp. 3–9. [Google Scholar]
- 3.Heidebrink JL. Is dementia with Lewy bodies the second most common cause of dementia? J Geriatr Psychiatry Neurol. 2002;15(4):182–187. doi: 10.1177/2F089198870201500402. [DOI] [PubMed] [Google Scholar]
- 4.Kane JP, Surendranathan A, Bentley A, Barker SA, Taylor J-P, Thomas AJ, Allan LM, McNally RJ, James PW, McKeith IG. Clinical prevalence of Lewy body dementia. Alzheimer’s Res Ther. 2018;10(1):19. doi: 10.1186/s13195-018-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller C, Ballard C, Corbett A, Aarsland D. The prognosis of dementia with Lewy bodies. Lancet Neurol. 2017;16(5):390–398. doi: 10.1016/s1474-4422(17)30074-1. [DOI] [PubMed] [Google Scholar]
- 6.McKeith IG, Galasko D, Kosaka K, Perry E, Dickson DW, Hansen L, Salmon D, Lowe J, Mirra S, Byrne E. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 7.Taylor J-P, McKeith IG, Burn DJ, Boeve BF, Weintraub D, Bamford C, Allan LM, Thomas AJ, T O'Brien J. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020;19(2):157–169. doi: 10.1016/s1474-4422(19)30153-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shergill S, Mullan E, D'ath P, Katona C. What is the clinical prevalence of Lewy body dementia? Int J Geriatr Psychiatry. 1994;9(11):907–912. doi: 10.1002/gps.930091107. [DOI] [Google Scholar]
- 9.Perry RH, Irving D, Tomlinson BE. Lewy body prevalence in the aging brain: relationship to neuropsychiatric disorders, Alzheimer-type pathology and catecholaminergic nuclei. J Neurol Sci. 1990;100(1–2):223–233. doi: 10.1016/0022-510X(90)90037-N. [DOI] [PubMed] [Google Scholar]
- 10.Parkinson J. An essay on the shaking palsy. J Neuropsychiatry Clin Neurosci. 2002;14(2):223–236. doi: 10.1176/jnp.14.2.223. [DOI] [PubMed] [Google Scholar]
- 11.Obeso J, Stamelou M, Goetz C, Poewe W, Lang A, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S. Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov Disord. 2017;32(9):1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holdorff B. Friedrich Heinrich Lewy (1885–1950) and his work. J Hist Neurosci. 2002;11(1):19–28. doi: 10.1076/jhin.11.1.19.9106. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues e Silva AM, Geldsetzer F, Holdorff B, Kielhorn FW, Balzer-Geldsetzer M, Oertel WH, Hurtig H, Dodel R. Who was the man who discovered the “Lewy bodies”? Mov Disord. 2010;25(12):1765–1773. doi: 10.1002/mds.22956. [DOI] [PubMed] [Google Scholar]
- 14.Lees AJ, Selikhova M, Andrade LA, Duyckaerts C. The black stuff and konstantin nikolaevich tretiakoff. Mov Disord. 2008;23(6):777–783. doi: 10.1002/mds.21855. [DOI] [PubMed] [Google Scholar]
- 15.Kosaka K. Lewy bodies in cerebral cortex. Report of three cases. Acta Neuropathol. 1978;42(2):127–134. doi: 10.1007/bf00690978. [DOI] [PubMed] [Google Scholar]
- 16.Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20:73–78. doi: 10.1046/j.1440-1789.2000.00301.x. [DOI] [PubMed] [Google Scholar]
- 17.Gibb W, Esiri M, Lees A. Clinical and pathological features of diffuse cortical Lewy body disease (Lewy body dementia) Brain. 1987;110(5):1131–1153. doi: 10.1093/brain/110.5.1131. [DOI] [PubMed] [Google Scholar]
- 18.Gibb W, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51(6):745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKeith IG, Perry R, Fairbairn A, Jabeen S, Perry E. Operational criteria for senile dementia of Lewy body type (SDLT) Psychol Med. 1992;22(4):911–922. doi: 10.1017/s0033291700038484. [DOI] [PubMed] [Google Scholar]
- 20.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. doi: 10.1212/wnl.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree—a new disease? Clin Neuropathol. 1984;3(5):185. [PubMed] [Google Scholar]
- 22.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci. 1998;95(11):6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba M, Nakajo S, Tu P-H, Tomita T, Nakaya K, Lee V, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152(4):879. [PMC free article] [PubMed] [Google Scholar]
- 24.Hurtig H, Trojanowski J, Galvin J, Ewbank D, Schmidt M, Lee V-Y, Clark C, Glosser G, Stern M, Gollomp S. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology. 2000;54(10):1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 25.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol. 1998;152(2):367. [PMC free article] [PubMed] [Google Scholar]
- 26.Cersosimo MG. Propagation of alpha-synuclein pathology from the olfactory bulb: possible role in the pathogenesis of dementia with Lewy bodies. Cell Tissue Res. 2018;373(1):233–243. doi: 10.1007/s00441-017-2733-6. [DOI] [PubMed] [Google Scholar]
- 27.Mattila P, Rinne J, Helenius H, Dickson DW, Röyttä M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100(3):285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- 28.Lin C-H, Yang S-Y, Horng H-E, Yang C-C, Chieh J-J, Chen H-H, Liu B-H, Chiu M-J. Plasma α-synuclein predicts cognitive decline in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2017;88(10):818–824. doi: 10.1136/jnnp-2016-314857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buldyrev S, Cruz L, Gomez-Isla T, Gomez-Tortosa E, Havlin S, Le R, Stanley H, Urbanc B, Hyman B. Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias. Proc Natl Acad Sci. 2000;97(10):5039–5043. doi: 10.1073/pnas.060009897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colosimo C, Hughes A, Kilford L, Lees A. Lewy body cortical involvement may not always predict dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2003;74(7):852–856. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Isla T, Growdon W, McNamara M, Newell K, Gomez-Tortosa E, Hedley-Whyte E, Hyman B. Clinicopathologic correlates in temporal cortex in dementia with Lewy bodies. Neurology. 1999;53(9):2003–2003. doi: 10.1212/wnl.53.9.2003. [DOI] [PubMed] [Google Scholar]
- 32.de Vos RA, Jansen EN, Stam FC, Ravid R, Swaab DF. ‘Lewy body disease’: clinico-pathological correlations in 18 consecutive cases of Parkinson’s disease with and without dementia. Clin Neurol Neurosurg. 1995;97(1):13–22. doi: 10.1016/0303-8467(94)00060-J. [DOI] [PubMed] [Google Scholar]
- 33.Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 2003;106(1):83–88. doi: 10.1007/s00401-003-0705-2. [DOI] [PubMed] [Google Scholar]
- 34.Samuel W, Alford M, Hofstetter CR, Hansen L. Dementia with Lewy bodies versus pure Alzheimer disease: differences in cognition, neuropathology, cholinergic dysfunction, and synapse density. J Neuropathol Exp Neurol. 1997;56(5):499–508. doi: 10.1097/00005072-199705000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Dalfo E, Albasanz J, Martin M, Ferrer I. Abnormal metabotropic glutamate receptor expression and signaling in the cerebral cortex in diffuse Lewy body disease is associated with irregular α-synuclein/phospholipase C (PLCβ1) interactions. Brain Pathol. 2004;14(4):388–398. doi: 10.1111/j.1750-3639.2004.tb00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry R, McKeith I, Perry E. Dementia in Parkinsonism. New York: Springer; 1997. Lewy body dementia—clinical, pathological and neurochemical interconnections; pp. 95–109. [DOI] [PubMed] [Google Scholar]
- 37.Sabbagh MN, Corey-Bloom J, Tiraboschi P, Thomas R, Masliah E, Thal LJ. Neurochemical markers do not correlate with cognitive decline in the Lewy body variant of Alzheimer disease. Arch Neurol. 1999;56(12):1458–1461. doi: 10.1001/archneur.56.12.1458. [DOI] [PubMed] [Google Scholar]
- 38.Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J Neurosci. 2009;29(11):3365–3373. doi: 10.1523/jneurosci.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: a shared mechanism in neurological disease. Ageing Res Rev. 2016;28:72–84. doi: 10.1016/j.arr.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 40.Kramer ML, Schulz-Schaeffer WJ. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27(6):1405–1410. doi: 10.1523/jneurosci.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marui W, Iseki E, Nakai T, Miura S, Kato M, Uéda K, Kosaka K. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195(2):153–159. doi: 10.1016/s0022-510x(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 42.Calo L, Wegrzynowicz M, Santivañez-Perez J, Grazia Spillantini M. Synaptic failure and α-synuclein. Mov Disord. 2016;31(2):169–177. doi: 10.1002/mds.26479. [DOI] [PubMed] [Google Scholar]
- 43.Whitfield DR, Vallortigara J, Alghamdi A, Howlett D, Hortobágyi T, Johnson M, Attems J, Newhouse S, Ballard C, Thomas AJ. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer’s disease: association with cognitive impairment. Neurobiol Aging. 2014;35(12):2836–2844. doi: 10.1016/j.neurobiolaging.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 44.Bereczki E, Francis PT, Howlett D, Pereira JB, Höglund K, Bogstedt A, Cedazo-Minguez A, Baek J-H, Hortobágyi T, Attems J. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement. 2016;12(11):1149–1158. doi: 10.1016/j.jalz.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Muntané G, Ferrer I, Martinez-Vicente M. α-Synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience. 2012;200:106–119. doi: 10.1016/j.neuroscience.2011.10.042. [DOI] [PubMed] [Google Scholar]
- 46.Walker DG, Lue L-F, Adler CH, Shill HA, Caviness JN, Sabbagh MN, Akiyama H, Serrano GE, Sue LI, Beach TG. Changes in properties of serine 129 phosphorylated α-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol. 2013;240:190–204. doi: 10.1016/j.expneurol.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colom-Cadena M, Pegueroles J, Herrmann AG, Henstridge CM, Muñoz L, Querol-Vilaseca M, Martín-Paniello CS, Luque-Cabecerans J, Clarimon J, Belbin O. Synaptic phosphorylated α-synuclein in dementia with Lewy bodies. Brain. 2017;140(12):3204–3214. doi: 10.1093/brain/awx275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell BC, Li Q-X, Culvenor JG, Jäkälä P, Cappai R, Beyreuther K, Masters CL, McLean CA. Accumulation of insoluble α-synuclein in dementia with Lewy bodies. Neurobiol Dis. 2000;7(3):192–200. doi: 10.1006/nbdi.2000.0286. [DOI] [PubMed] [Google Scholar]
- 49.Klucken J, Ingelsson M, Shin Y, Irizarry MC, Hedley-Whyte E, Frosch MP, Growdon JH, McLean PJ, Hyman BT. Clinical and biochemical correlates of insoluble α-synuclein in dementia with Lewy bodies. Acta Neuropathol. 2006;111(2):101–108. doi: 10.1007/s00401-005-0027-7. [DOI] [PubMed] [Google Scholar]
- 50.Tanji K, Mori F, Mimura J, Itoh K, Kakita A, Takahashi H, Wakabayashi K. Proteinase K-resistant α-synuclein is deposited in presynapses in human Lewy body disease and A53T α-synuclein transgenic mice. Acta Neuropathol. 2010;120(2):145–154. doi: 10.1007/s00401-010-0676-z. [DOI] [PubMed] [Google Scholar]
- 51.Klucken J, McLean PJ, Gomez-Tortosa E, Ingelsson M, Hyman BT. Neuritic alterations and neural system dysfunction in Alzheimer’s disease and dementia with Lewy bodies. Neurochem Res. 2003;28(11):1683–1691. doi: 10.1023/a:1026061021946. [DOI] [PubMed] [Google Scholar]
- 52.Revuelta GJ, Rosso A, Lippa CF. Neuritic pathology as a correlate of synaptic loss in dementia with Lewy bodies. Am J Alzheimer’s Dis Other Dement. 2008;23(1):97–102. doi: 10.1177/1533317507310565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10(12):524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 54.McNaught KSP, Shashidharan P, Perl DP, Jenner P, Olanow CW. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16(11):2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 55.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 56.Anderson JP, Walker DE, Goldstein JM, De Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739–29752. doi: 10.1074/jbc.m600933200. [DOI] [PubMed] [Google Scholar]
- 57.Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM-Y, Trojanowski JQ, Mann D, Iwatsubo T. Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J Biol Chem. 2002;277(50):49071–49076. doi: 10.1074/jbc.m208046200. [DOI] [PubMed] [Google Scholar]
- 58.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 1998;436(3):309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 59.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM-Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 60.Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VMY, Trojanowski JQ. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol. 2000;157(5):1439–1445. doi: 10.1016/s0002-9440(10)64781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahul-Mellier A-L, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW, Lashuel HA. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci. 2020;117(9):4971–4982. doi: 10.1073/pnas.1913904117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miners JS, Renfrew R, Swirski M, Love S. Accumulation of α-synuclein in dementia with Lewy bodies is associated with decline in the α-synuclein-degrading enzymes kallikrein-6 and calpain-1. Acta Neuropathol Commun. 2014;2(1):1–11. doi: 10.1186/s40478-014-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ikeuchi T, Kakita A, Shiga A, Kasuga K, Kaneko H, Tan C-F, Idezuka J, Wakabayashi K, Onodera O, Iwatsubo T. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch Neurol. 2008;65(4):514–519. doi: 10.1001/archneur.65.4.514. [DOI] [PubMed] [Google Scholar]
- 64.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann Neurol Off J Am Neurol Assoc Child Neurol Soc. 2004;55(2):164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 65.Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, Hernandez DG, Nalls MA, Clark LN, Honig LS. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet. 2014;23(23):6139–6146. doi: 10.1093/hmg/ddu334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singleton A, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra AN, Lincoln S. [alpha]-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841–842. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 67.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 68.Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT. Brain-permeable small-molecule inhibitors of Hsp90 prevent α-synuclein oligomer formation and rescue α-synuclein-induced toxicity. J Pharmacol Exp Ther. 2010;332(3):849–857. doi: 10.1124/jpet.109.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanderson JB, De S, Jiang H, Rovere M, Jin M, Zaccagnini L, Hays Watson A, De Boni L, Lagomarsino VN, Young-Pearse TL. Analysis of α-synuclein species enriched from cerebral cortex of humans with sporadic dementia with Lewy bodies. Brain Commun. 2020;2(1):fcaa010. doi: 10.1093/braincomms/fcaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansen LA, Samuel W. Criteria for Alzheimer’s disease and the nosology of dementia with Lewy bodies. Neurology. 1997;48(1):126–132. doi: 10.1212/wnl.48.1.126. [DOI] [PubMed] [Google Scholar]
- 71.Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018;16(1):1–16. doi: 10.1186/s12916-018-1016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levy G, Levin B, Engelhardt E. The nosology of Lewy body disorders from analytic-epidemiologic and statistical vantage points. Mov Disord. 2020 doi: 10.1002/mds.28288. [DOI] [PubMed] [Google Scholar]
- 73.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimer’s Dis. 2006;9(s3):417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 74.Cormack F, Aarsland D, Ballard C, Tovée MJ. Pentagon drawing and neuropsychological performance in Dementia with Lewy bodies, Alzheimer’s disease, Parkinson’s disease and Parkinson’s disease with dementia. Int J Geriatr Psychiatry. 2004;19(4):371–377. doi: 10.1002/gps.1094. [DOI] [PubMed] [Google Scholar]
- 75.Walker M, Ayre G, Cummings J, Wesnes K, McKeith I, O’brien J, Ballard C. Quantifying fluctuation in dementia with Lewy bodies, Alzheimer’s disease, and vascular dementia. Neurology. 2000;54(8):1616–1625. doi: 10.1212/wnl.54.8.1616. [DOI] [PubMed] [Google Scholar]
- 76.Ferman TJ, Smith GE, Boeve BF, Graff-Radford NR, Lucas JA, Knopman DS, Petersen RC, Ivnik RJ, Wszolek Z, Uitti R. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623–636. doi: 10.1080/13854040500376831. [DOI] [PubMed] [Google Scholar]
- 77.Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain. 2004;127(4):791–800. doi: 10.1093/brain/awh088. [DOI] [PubMed] [Google Scholar]
- 78.Minoshima S, Foster NL, Sima AA, Frey KA, Albin RL, Kuhl DE. Alzheimer’s disease versus dementia with Lewy bodies: cerebral metabolic distinction with autopsy confirmation. Ann Neurol Off J Am Neurol Assoc Child Neurol Soc. 2001;50(3):358–365. doi: 10.1002/ana.1133. [DOI] [PubMed] [Google Scholar]
- 79.Noe E, Marder K, Bell KL, Jacobs DM, Manly JJ, Stern Y. Comparison of dementia with Lewy bodies to Alzheimer’s disease and Parkinson’s disease with dementia. Mov Disord. 2004;19(1):60–67. doi: 10.1002/mds.10633. [DOI] [PubMed] [Google Scholar]
- 80.Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P, Perry R, O’brien J. White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer’s disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatry. 1999;67(1):66–72. doi: 10.1136/jnnp.67.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ballard C, Ziabreva I, Perry R, Larsen J, O'brien J, McKeith I, Perry E, Aarsland D. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931–1934. doi: 10.1212/01.wnl.0000249130.63615.cc. [DOI] [PubMed] [Google Scholar]
- 82.Skogseth R, Hortobágyi T, Soennesyn H, Chwiszczuk L, Ffytche D, Rongve A, Ballard C, Aarsland D. Accuracy of clinical diagnosis of dementia with Lewy bodies versus neuropathology. J Alzheimers Dis. 2017;59(4):1139–1152. doi: 10.3233/jad-170274. [DOI] [PubMed] [Google Scholar]
- 83.Walker L, Stefanis L, Attems J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies—current issues and future directions. J Neurochem. 2019;150(5):467–474. doi: 10.1111/jnc.14698. [DOI] [PubMed] [Google Scholar]
- 84.Tsuboi Y, Uchikado H, Dickson DW. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord. 2007;13:S221–S224. doi: 10.1016/s1353-8020(08)70005-1. [DOI] [PubMed] [Google Scholar]
- 85.Jellinger KA. Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm. 2018;125(4):615–650. doi: 10.1007/s00702-017-1821-9. [DOI] [PubMed] [Google Scholar]
- 86.Double K, Halliday G, Krill J, Harasty J, Cullen K, Brooks W, Creasey H, Broe G. Topography of brain atrophy during normal aging and Alzheimer’s disease. Neurobiol Aging. 1996;17(4):513–521. doi: 10.1016/0197-4580(96)00005-x. [DOI] [PubMed] [Google Scholar]
- 87.Kosaka K, Iseki E, Odawara T, Yamamoto T. Cerebral type of Lewy body disease. Neuropathology. 1996;16(1):32–35. doi: 10.1111/j.1440-1789.1996.tb00152.x. [DOI] [Google Scholar]
- 88.Gomez-Tortosa E, Newell K, Irizarry M, Albert M, Growdon J, Hyman BT. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999;53(6):1284–1284. doi: 10.1212/wnl.53.6.1284. [DOI] [PubMed] [Google Scholar]
- 89.Merdes A, Hansen L, Jeste D, Galasko D, Hofstetter C, Ho G, Thal L, Corey-Bloom J. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology. 2003;60(10):1586–1590. doi: 10.1212/01.wnl.0000065889.42856.f2. [DOI] [PubMed] [Google Scholar]
- 90.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42(11):1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 91.Gómez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT. α-Synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000;99(4):352–357. doi: 10.1111/j.1749-6632.2000.tb06899.x. [DOI] [PubMed] [Google Scholar]
- 92.Reid RT, Sabbagh MN, Corey-Bloom J, Tiraboschi P, Thal LJ. Nicotinic receptor losses in dementia with Lewy bodies: comparisons with Alzheimer’s disease. Neurobiol Aging. 2000;21(5):741–746. doi: 10.1016/s0197-4580(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 93.Tiraboschi P, Hansen L, Alford M, Masliah E, Thal L, Corey-Bloom J. The decline in synapses and cholinergic activity is asynchronous in Alzheimer’s disease. Neurology. 2000;55(9):1278–1283. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 94.Tiraboschi P, Hansen LA, Alford M, Merdes A, Masliah E, Thal LJ, Corey-Bloom J. Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Arch Gen Psychiatry. 2002;59(10):946–951. doi: 10.1001/archpsyc.59.10.946. [DOI] [PubMed] [Google Scholar]
- 95.Ince PG, Perry EK, Morris CM. Dementia with Lewy bodies. A distinct non-Alzheimer dementia syndrome? Brain Pathol. 1998;8(2):299–324. doi: 10.1111/j.1750-3639.1998.tb00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perry EK, Marshall E, Perry RH, Irving D, Smith CJ, Blessed G, Fairbairn AF. Cholinergic and dopaminergic activities in senile dementia of Lewy body type. Alzheimer Dis Assoc Disord. 1990;4(2):87–95. doi: 10.1097/00002093-199040200-00003. [DOI] [PubMed] [Google Scholar]
- 97.Heitz C, Noblet V, Phillipps C, Cretin B, Vogt N, Philippi N, Kemp J, de Petigny X, Bilger M, Demuynck C. Cognitive and affective theory of mind in dementia with Lewy bodies and Alzheimer’s disease. Alzheimer’s Res Ther. 2016;8(1):10. doi: 10.1186/s13195-016-0179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Breitve MH, Hynninen MJ, Brønnick K, Chwiszczuk LJ, Auestad BH, Aarsland D, Rongve A. A longitudinal study of anxiety and cognitive decline in dementia with Lewy bodies and Alzheimer’s disease. Alzheimer’s Res Ther. 2016;8(1):1–6. doi: 10.1186/s13195-016-0171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thomas AJ, Mahin-Babaei F, Saidi M, Lett D, Taylor JP, Walker L, Attems J. Improving the identification of dementia with Lewy bodies in the context of an Alzheimer’s-type dementia. Alzheimer’s Res Ther. 2018;10(1):27. doi: 10.1186/s13195-018-0356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, Thal L, Pay M, Hofstetter R, Klauber M. The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology. 1990;40(1):1–1. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- 101.Peavy GM, Edland SD, Toole BM, Hansen LA, Galasko DR, Mayo AM. Phenotypic differences based on staging of Alzheimer’s neuropathology in autopsy-confirmed dementia with Lewy bodies. Parkinsonism Relat Disord. 2016;31:72–78. doi: 10.1016/j.parkreldis.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fritz NE, Kegelmeyer DA, Kloos AD, Linder S, Park A, Kataki M, Adeli A, Agrawal P, Scharre DW, Kostyk SK. Motor performance differentiates individuals with Lewy body dementia, Parkinson’s and Alzheimer’s disease. Gait Posture. 2016;50:1–7. doi: 10.1016/j.gaitpost.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 103.Samuel W, Galasko D, Masliah E, Hansen LA. Neocortical Lewy body counts correlate with dementia in the Lewy body variant of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55(1):44–52. doi: 10.1097/00005072-199601000-00005. [DOI] [PubMed] [Google Scholar]
- 104.Harding A, Broe G, Halliday G. Visual hallucinations in Lewy body disease relate to Lewy bodies in the temporal lobe. Brain. 2002;125(2):391–403. doi: 10.1093/brain/awf033. [DOI] [PubMed] [Google Scholar]
- 105.Gomez-Tortosa E, Irizarry M, Gomez-Isla T, Hyman B. Clinical and neuropathological correlates of dementia with Lewy bodies. Ann N Y Acad Sci. 2000;920(1):9–15. doi: 10.1007/s004010051135. [DOI] [PubMed] [Google Scholar]
- 106.Weisman D, Cho M, Taylor C, Adame A, Thal L, Hansen L. In dementia with Lewy bodies, Braak stage determines phenotype, not Lewy body distribution. Neurology. 2007;69(4):356–359. doi: 10.1212/01.wnl.0000266626.64913.0f. [DOI] [PubMed] [Google Scholar]
- 107.Shults CW. Lewy bodies. Proc Natl Acad Sci. 2006;103(6):1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001;102(4):355–363. doi: 10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- 109.Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, Donaghy P, Morris C, Taylor J-P, Thomas A. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14(1):1–18. doi: 10.1186/s13024-019-0306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256(3):270–279. doi: 10.1007/s00415-009-5243-y. [DOI] [PubMed] [Google Scholar]
- 111.Duffy PE, Tennyson VM. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus caeruleus in Parkinson’s disease. J Neuropathol Exp Neurol. 1965;24(3):398–414. doi: 10.1097/00005072-196507000-00003. [DOI] [Google Scholar]
- 112.Roy S, Wolman L. Ultrastructural observations in Parkinsonism. J Pathol. 1969;99(1):39–44. doi: 10.1002/path.1710990106. [DOI] [PubMed] [Google Scholar]
- 113.Fukuda T, Tanaka J, Watabe K, Numoto RT, Minamitani M. Immunohistochemistry of neuronal inclusions in the cerebral cortex and brain-stem in Lewy body disease. Pathol Int. 1993;43(10):545–551. doi: 10.1111/j.1440-1827.1993.tb03230.x. [DOI] [PubMed] [Google Scholar]
- 114.Galloway P, Mulvihill P, Perry G. Filaments of Lewy bodies contain insoluble cytoskeletal elements. Am J Pathol. 1992;140(4):809. [PMC free article] [PubMed] [Google Scholar]
- 115.Galvin JE, Lee V, Schmidt ML, Tu P-H, Iwatsubo T, Trojanowski JQ. Pathobiology of the Lewy body. Adv Neurol. 1999;80:313. [PubMed] [Google Scholar]
- 116.Dickson DW, Davies P, Mayeux R, Crystal H, Horoupian D, Thompson A, Goldman J. Diffuse Lewy body disease. Acta Neuropathol. 1987;75(1):8–15. doi: 10.1007/bf00686786. [DOI] [PubMed] [Google Scholar]
- 117.Forno LS. Progress in Parkinson research. Boston: Springer; 1988. The neuropathology of Parkinson’s disease; pp. 11–21. [Google Scholar]
- 118.Dickson DW, Ruan D, Crystal H, Mark M, Davies P, Kress Y, Yen S-H. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2–3 neurites specific to DLBD. Neurology. 1991;41(9):1402–1402. doi: 10.1212/wnl.41.9.1402. [DOI] [PubMed] [Google Scholar]
- 119.Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52(3):183–191. doi: 10.1097/00005072-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 120.Braak H, Del Tredici K, Rüb U, De Vos RA, Steur ENJ, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 121.Braak H, Sandmann-Keil D, Gai W, Braak E. Extensive axonal Lewy neurites in Parkinson’s disease: a novel pathological feature revealed by α-synuclein immunocytochemistry. Neurosci Lett. 1999;265(1):67–69. doi: 10.1016/s0304-3940(99)00208-6. [DOI] [PubMed] [Google Scholar]
- 122.Kraybill ML, Larson EB, Tsuang D, Teri L, McCormick W, Bowen J, Kukull W, Leverenz J, Cherrier M. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005;64(12):2069–2073. doi: 10.1212/01.wnl.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Connor DJ, Salmon DP, Sandy TJ, Galasko D, Hansen LA, Thal LJ. Cognitive profiles of autopsy-confirmed Lewy body variant vs pure Alzheimer disease. Arch Neurol. 1998;55(7):994–1000. doi: 10.1001/archneur.55.7.994. [DOI] [PubMed] [Google Scholar]
- 124.Pappolla M, Shank D, Alzofon J, Dudley A. Colloid (hyaline) inclusion bodies in the central nervous system: their presence in the substantia nigra is diagnostic of Parkinson’s disease. Hum Pathol. 1988;19(1):27–31. doi: 10.1016/s0046-8177(88)80312-5. [DOI] [PubMed] [Google Scholar]
- 125.Dale G, Probst A, Luthert P, Martin J, Anderton B, Leigh P. Relationships between Lewy bodies and pale bodies in Parkinson’s disease. Acta Neuropathol. 1992;83(5):525–529. doi: 10.1007/BF00310030. [DOI] [PubMed] [Google Scholar]
- 126.Gibb W, Luthert P, Janota I, Lantos P. Cortical Lewy body dementia: clinical features and classification. J Neurol Neurosurg Psychiatry. 1989;52(2):185–192. doi: 10.1136/jnnp.52.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wakabayashi K, Hansen LA, Masliah E. Cortical Lewy body-containing neurons are pyramidal cells: laser confocal imaging of double-immunolabeled sections with anti-ubiquitin and SMI32. Acta Neuropathol. 1995;89(5):404–408. doi: 10.1007/bf00307643. [DOI] [PubMed] [Google Scholar]
- 128.Okazaki H, Lipkin LE, Aronson SM. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol. 1961;20(2):237–244. doi: 10.1097/00005072-196104000-00007. [DOI] [PubMed] [Google Scholar]
- 129.Kosaka K, Oyanagi S, Matsushita M, Hori A, Iwase S. Presenile dementia with Alzheimer-, Pick-and Lewy-body changes. Acta Neuropathol. 1976;36(3):221–233. doi: 10.1007/bf00685366. [DOI] [PubMed] [Google Scholar]
- 130.Arima K, Ueda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka-Uehara H, Kawai M. Immunoelectron-microscopic demonstration of NACP/α-synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 1998;808(1):93–100. doi: 10.1016/s0006-8993(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 131.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 132.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett. 1997;239(1):45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- 133.Irizarry MC, Growdon W, Gomez-Isla T, Newell K, George JM, Clayton DF, Hyman BT. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J Neuropathol Exp Neurol. 1998;57(4):334–337. doi: 10.1097/00005072-199804000-00005. [DOI] [PubMed] [Google Scholar]
- 134.Rott R, Szargel R, Shani V, Hamza H, Savyon M, Abd Elghani F, Bandopadhyay R, Engelender S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc Natl Acad Sci. 2017;114(50):13176–13181. doi: 10.1073/pnas.1704351114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M. Accumulation of phosphorylated α-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62(6):644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- 136.Obi K, Akiyama H, Kondo H, Shimomura Y, Hasegawa M, Iwatsubo T, Mizuno Y, Mochizuki H. Relationship of phosphorylated α-synuclein and tau accumulation to Aβ deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol. 2008;210(2):409–420. doi: 10.1016/j.expneurol.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 137.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155(1):9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 138.Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of α-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62(12):1241–1253. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- 139.Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, Jin J, Pan C, Shin J, Zhu D. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007;17(2):139–145. doi: 10.1111/j.1750-3639.2007.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gai W, Yuan H, Li X, Power J, Blumbergs P, Jensen P. In situ and in vitro study of colocalization and segregation of α-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol. 2000;166(2):324–333. doi: 10.1006/exnr.2000.7527. [DOI] [PubMed] [Google Scholar]
- 141.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci. 2019;22(7):1099–1109. doi: 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 142.Forno LS, Norville RL. Ultrastructure of Lewy bodies in the stellate ganglion. Acta Neuropathol. 1976;34(3):183–197. doi: 10.1007/bf00688674. [DOI] [PubMed] [Google Scholar]
- 143.Ikeda K, Hori A, Bode G. Progressive dementia with ‘diffuse Lewy-type inclusions’ in cerebral cortex. Arch Psychiatr Nervenkr. 1980;228(3):243–248. doi: 10.1007/bf00342349. [DOI] [PubMed] [Google Scholar]
- 144.Yagishita S, Itoh Y, Amano N, Nakano T. Atypical senile dementia with widespread Lewy type inclusion in the cerebral cortex. Acta Neuropathol. 1980;49(3):187–191. doi: 10.1007/bf00707105. [DOI] [PubMed] [Google Scholar]
- 145.Tomonaga M. Neurofibrillary tangles and Lewy bodies in the locus ceruleus neurons of the aged brain. Acta Neuropathol. 1981;53(2):165–168. doi: 10.1007/bf00689998. [DOI] [PubMed] [Google Scholar]
- 146.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62(4):389–397. doi: 10.1093/jnen/62.4.389. [DOI] [PubMed] [Google Scholar]
- 147.Muntane G, Dalfo E, Martinez A, Ferrer I. Phosphorylation of tau and α-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related α-synucleinopathies. Neuroscience. 2008;152(4):913–923. doi: 10.1016/j.neuroscience.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 148.Peuralinna T, Oinas M, Polvikoski T, Paetau A, Sulkava R, Niinistö L, Kalimo H, Hernandez D, Hardy J, Singleton A. Neurofibrillary tau pathology modulated by genetic variation of α-synuclein. Ann Neurol Off J Am Neurol Assoc Child Neurol Soc. 2008;64(3):348–352. doi: 10.1002/ana.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.George JM. The synucleins. Genome Biol. 2001;3(1):1–6. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lavedan C. The synuclein family. Genome Res. 1998;8(9):871–880. doi: 10.1101/gr.8.9.871. [DOI] [PubMed] [Google Scholar]
- 151.Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero D, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci. 1993;90(23):11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem. 2005;280(10):9595–9603. doi: 10.1074/jbc.m411805200. [DOI] [PubMed] [Google Scholar]
- 153.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307(4):1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 154.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15(2):361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 155.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21(6):249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 156.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 157.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci. 2004;101(22):8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu M, Li J, Fink AL. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278(41):40186–40197. doi: 10.1074/jbc.m305326200. [DOI] [PubMed] [Google Scholar]
- 159.McClain SM, Ojoawo AM, Lin W, Rienstra CM, Murphy CJ. Interaction of Alpha-synuclein and its mutants with rigid lipid vesicle mimics of varying surface curvature. ACS Nano. 2020;14(8):10153–10167. doi: 10.1021/acsnano.0c03420. [DOI] [PubMed] [Google Scholar]
- 160.Iyer A, Roeters SJ, Schilderink N, Hommersom B, Heeren RM, Woutersen S, Claessens MM, Subramaniam V. The impact of N-terminal acetylation of α-synuclein on phospholipid membrane binding and fibril structure. J Biol Chem. 2016;291(40):21110–21122. doi: 10.1074/jbc.m116.726612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.O'Leary EI, Jiang Z, Strub M-P, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-synuclein. J Biol Chem. 2018;293(28):11195–11205. doi: 10.1074/jbc.ra118.002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J. Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28(6):811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 163.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 164.Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R. G51D α-synuclein mutation causes a novel Parkinsonian–pyramidal syndrome. Ann Neurol. 2013;73(4):459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 165.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 166.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel α-synuclein missense mutation in Parkinson disease. Neurology. 2013;80(11):1062–1064. doi: 10.1212/wnl.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35(9):2180.e2181–2180, e2185. 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed]
- 168.Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, Woodhams PJ, Mela I, Lashuel HA, Phillips JJ. Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun. 2020;11(1):1–15. doi: 10.1038/s41467-020-16564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ghosh D, Sahay S, Ranjan P, Salot S, Mohite GM, Singh PK, Dwivedi S, Carvalho E, Banerjee R, Kumar A. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry. 2014;53(41):6419–6421. doi: 10.1021/bi5010365. [DOI] [PubMed] [Google Scholar]
- 170.Giasson BI, Uryu K, Trojanowski JQ, Lee VM-Y. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274(12):7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 171.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in α-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280(9):7800–7807. doi: 10.1074/jbc.m411638200. [DOI] [PubMed] [Google Scholar]
- 172.Mahul-Mellier A, Vercruysse F, Maco B, Ait-Bouziad N, De Roo M, Muller D, Lashuel H. Fibril growth and seeding capacity play key roles in α-synuclein-mediated apoptotic cell death. Cell Death Differ. 2015;22(12):2107–2122. doi: 10.1038/cdd.2015.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P. Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J Biol Chem. 1999;274(14):9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 174.Doherty CP, Ulamec SM, Maya-Martinez R, Good SC, Makepeace J, Khan GN, van Oosten-Hawle P, Radford SE, Brockwell DJ. A short motif in the N-terminal region of α-synuclein is critical for both aggregation and function. Nat Struct Mol Biol. 2020;27(3):249–259. doi: 10.1038/s41594-020-0384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lázaro DF, Dias MC, Carija A, Navarro S, Madaleno CS, Tenreiro S, Ventura S, Outeiro TF. The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol Commun. 2016;4(1):1–15. doi: 10.1186/s40478-016-0402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Boyer DR, Li B, Sun C, Fan W, Zhou K, Hughes MP, Sawaya MR, Jiang L, Eisenberg DS. The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc Natl Acad Sci. 2020;117(7):3592–3602. doi: 10.1073/pnas.1917914117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hashimoto M, Takenouchi T, Mallory M, Masliah E, Takeda A. The role of NAC in amyloidogenesis in Alzheimer’s disease. Am J Pathol. 2000;156(2):734. doi: 10.1016/s0002-9440(10)64777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.El-Agnaf OM, Jakes R, Curran MD, Wallace A. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 1998;440(1–2):67–70. doi: 10.1016/s0014-5793(98)01419-7. [DOI] [PubMed] [Google Scholar]
- 179.Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM-Y. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci. 2009;106(47):20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ryan P, Xu M-M, Davey AK, Kassiou M, Mellick GD, Rudrawar S. O-GlcNAcylation of truncated NAC segment alters peptide-dependent effects on α-synuclein aggregation. Bioorg Chem. 2020;94:103389. doi: 10.1016/j.bioorg.2019.103389. [DOI] [PubMed] [Google Scholar]
- 181.Srivastava T, Raj R, Dubey A, Kumar D, Chaturvedi RK, Sharma SK, Priya S. Fast kinetics of environmentally induced α-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci Rep. 2020;10(1):1–12. doi: 10.1038/s41598-020-75361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Giasson BI, Murray IV, Trojanowski JQ, Lee VM-Y. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. 2001;276(4):2380–2386. doi: 10.1074/jbc.m008919200. [DOI] [PubMed] [Google Scholar]
- 183.Han H, Weinreb PH, Lansbury PT., Jr The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by β-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol. 1995;2(3):163–169. doi: 10.1016/1074-5521(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 184.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525(7570):486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xu L, Nussinov R, Ma B. Coupling of the non-amyloid-component (NAC) domain and the KTK (E/Q) GV repeats stabilize the α-synuclein fibrils. Eur J Med Chem. 2016;121:841–850. doi: 10.1016/j.ejmech.2016.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Levitan K, Chereau D, Cohen SI, Knowles TP, Dobson CM, Fink AL, Anderson JP, Goldstein JM, Millhauser GL. Conserved C-terminal charge exerts a profound influence on the aggregation rate of α-synuclein. J Mol Biol. 2011;411(2):329–333. doi: 10.1016/j.jmb.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Meuvis J, Gerard M, Desender L, Baekelandt V, Engelborghs Y. The conformation and the aggregation kinetics of α-synuclein depend on the proline residues in its C-terminal region. Biochemistry. 2010;49(43):9345–9352. doi: 10.1021/bi1010927. [DOI] [PubMed] [Google Scholar]
- 188.Sato H, Kato T, Arawaka S. The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: a review of in vivo models. Rev Neurosci. 2013;24(2):115–123. doi: 10.1515/revneuro-2012-0071. [DOI] [PubMed] [Google Scholar]
- 189.Oueslati A, Fournier M, Lashuel HA. Progress in brain research. Amsterdam: Elsevier; 2010. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson’s disease pathogenesis and therapies; pp. 115–145. [DOI] [PubMed] [Google Scholar]
- 190.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109–140 on α-synuclein aggregation in vitro. Biochemistry. 2004;43(51):16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- 191.McLean PJ, Hyman BT. An alternatively spliced form of rodent α-synuclein forms intracellular inclusions in vitro: role of the carboxy-terminus in α-synuclein aggregation. Neurosci Lett. 2002;323(3):219–223. doi: 10.1016/s0304-3940(02)00154-4. [DOI] [PubMed] [Google Scholar]
- 192.Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM-Y. Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42(28):8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 193.Lautenschläger J, Stephens AD, Fusco G, Ströhl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat Commun. 2018;9(1):1–13. doi: 10.1038/s41467-018-03111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Afitska K, Fucikova A, Shvadchak VV, Yushchenko DA. Modification of C terminus provides new insights into the mechanism of α-synuclein aggregation. Biophys J. 2017;113(10):2182–2191. doi: 10.1016/j.bpj.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Torpey J, Madine J, Wood A, Lian L-Y. Cyclophilin D binds to the acidic C-terminus region of α-synuclein and affects its aggregation characteristics. Sci Rep. 2020;10(1):1–10. doi: 10.1038/s41598-020-66200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8(8):2804–2815. doi: 10.1523/jneurosci.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, De Silva HR, Kittel A, Saitoh T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14(2):467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 198.Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Dev Brain Res. 1997;99(1):87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 199.Bartels T, Ahlstrom LS, Leftin A, Kamp F, Haass C, Brown MF, Beyer K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys J. 2010;99(7):2116–2124. doi: 10.1016/j.bpj.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Runfola M, De Simone A, Vendruscolo M, Dobson CM, Fusco G. The n-terminal acetylation of α-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci Rep. 2020;10(1):1–10. doi: 10.1038/s41598-019-57023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fortin DL, Troyer MD, Nakamura K, Kubo S-I, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci. 2004;24(30):6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kahle PJ, Neumann M, Ozmen L, Müller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van der Putten H, Probst A. Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20(17):6365–6373. doi: 10.1523/jneurosci.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hannestad JK, Rocha S, Agnarsson B, Zhdanov VP, Wittung-Stafshede P, Höök F. Single-vesicle imaging reveals lipid-selective and stepwise membrane disruption by monomeric α-synuclein. Proc Natl Acad Sci. 2020 doi: 10.1073/pnas.1914670117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT. In vivo imaging of α-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS ONE. 2010;5(5):e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein. J Mol Biol. 2002;315(4):799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- 206.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273(41):26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 207.Jensen MB, Bhatia VK, Jao CC, Rasmussen JE, Pedersen SL, Jensen KJ, Langen R, Stamou D. Membrane curvature sensing by amphipathic helices a single liposome study using α-synuclein and annexin B12. J Biol Chem. 2011;286(49):42603–42614. doi: 10.1074/jbc.m111.271130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys J. 2010;99(7):2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cholak E, Bugge K, Khondker A, Gauger K, Pedraz-Cuesta E, Pedersen ME, Bucciarelli S, Vestergaard B, Pedersen SF, Rheinstädter MC. Avidity within the N-terminal anchor drives α-synuclein membrane interaction and insertion. FASEB J. 2020 doi: 10.1096/fj.202000107r. [DOI] [PubMed] [Google Scholar]
- 210.Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz J-M, Antonny B, Jackson CL. α-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol. 2011;194(1):89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Badawy SMM, Okada T, Kajimoto T, Hirase M, Matovelo SA, Nakamura S, Yoshida D, Ijuin T, Nakamura S-I. Extracellular α-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J Biol Chem. 2018;293(21):8208–8216. doi: 10.1074/jbc.ra118.001986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Emanuele M, Esposito A, Camerini S, Antonucci F, Ferrara S, Seghezza S, Catelani T, Crescenzi M, Marotta R, Canale C. Exogenous alpha-synuclein alters pre-and post-synaptic activity by fragmenting lipid rafts. EBioMedicine. 2016;7:191–204. doi: 10.1016/j.ebiom.2016.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Casalis L, Parisse P, Perissinotto F, Rondelli VM, Scaini D, Legname GA, Stani C. Interaction of alpha-synuclein with raft containing model lipid membranes: morphology and structure. Biophys J. 2020;118(3):391a. doi: 10.1016/j.bpj.2019.11.2225. [DOI] [Google Scholar]
- 214.Casalis L, Perissinotto F, Scaini D. Iron mediated interaction of alpha synuclein with lipid rafts in model membrane systems. Biophys J. 2018;114(3):244a. doi: 10.1016/j.bpj.2017.11.1360. [DOI] [PubMed] [Google Scholar]
- 215.Kubo S-I, Nemani VM, Chalkley RJ, Anthony MD, Hattori N, Mizuno Y, Edwards RH, Fortin DL. A combinatorial code for the interaction of α-synuclein with membranes. J Biol Chem. 2005;280(36):31664–31672. doi: 10.1074/jbc.m504894200. [DOI] [PubMed] [Google Scholar]
- 216.Leftin A, Job C, Beyer K, Brown MF. Solid-state 13C NMR reveals annealing of raft-like membranes containing cholesterol by the intrinsically disordered protein α-synuclein. J Mol Biol. 2013;425(16):2973–2987. doi: 10.1016/j.jmb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. α-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci. 2001;98(16):9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Assayag K, Yakunin E, Loeb V, Selkoe DJ, Sharon R. Polyunsaturated fatty acids induce α-synuclein-related pathogenic changes in neuronal cells. Am J Pathol. 2007;171(6):2000–2011. doi: 10.2353/2Fajpath.2007.070373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Perrin RJ, Woods WS, Clayton DF, George JM. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem. 2001;276(45):41958–41962. doi: 10.1074/jbc.m105022200. [DOI] [PubMed] [Google Scholar]
- 220.Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron. 2003;37(4):583–595. doi: 10.1016/S0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 221.Sharon R, Bar-Joseph I, Mirick GE, Serhan CN, Selkoe DJ. Altered fatty acid composition of dopaminergic neurons expressing α-synuclein and human brains with α-synucleinopathies. J Biol Chem. 2003;278(50):49874–49881. doi: 10.1074/jbc.M309127200. [DOI] [PubMed] [Google Scholar]
- 222.Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barceló-Coblijn GC, Nussbaum RL. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking α-synuclein. Mol Cell Biol. 2005;25(22):10190–10201. doi: 10.1128/mcb.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Golovko MY, Faergeman NJ, Cole NB, Castagnet PI, Nussbaum RL, Murphy EJ. α-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of α-synuclein palmitate binding. Biochemistry. 2005;44(23):8251–8259. doi: 10.1021/bi0502137. [DOI] [PubMed] [Google Scholar]
- 224.Golovko MY, Rosenberger TA, Færgeman NJ, Feddersen S, Cole NB, Pribill I, Berger J, Nussbaum RL, Murphy EJ. Acyl-CoA synthetase activity links wild-type but not mutant α-synuclein to brain arachidonate metabolism. Biochemistry. 2006;45(22):6956–6966. doi: 10.1021/bi0600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Golovko MY, Rosenberger TA, Feddersen S, Færgeman NJ, Murphy EJ. α-Synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. J Neurochem. 2007;101(1):201–211. doi: 10.1111/j.1471-4159.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- 226.Ben Gedalya T, Loeb V, Israeli E, Altschuler Y, Selkoe DJ, Sharon R. α-Synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic. 2009;10(2):218–234. doi: 10.1111/j.1600-0854.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci. 2002;22(20):8797–8807. doi: 10.1523/jneurosci.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM-Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20(9):3214–3220. doi: 10.1523/jneurosci.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yamada K, Iwatsubo T. Extracellular α-synuclein levels are regulated by neuronal activity. Mol Neurodegener. 2018;13(1):1–8. doi: 10.1186/s13024-018-0241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, Roy S. Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci. 2019;116(23):11113–11115. doi: 10.1073/pnas.1903049116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Vargas KJ, Schrod N, Davis T, Fernandez-Busnadiego R, Taguchi YV, Laugks U, Lucic V, Chandra SS. Synucleins have multiple effects on presynaptic architecture. Cell Rep. 2017;18(1):161–173. doi: 10.1016/j.celrep.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho W-H, Castillo PE, Shinsky N, Verdugo JMG, Armanini M, Ryan A. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25(1):239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 233.Yavich L, Tanila H, Vepsäläinen S, Jäkälä P. Role of α-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24(49):11165–11170. doi: 10.1523/jneurosci.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J Neurosci. 2010;30(24):8083–8095. doi: 10.1523/jneurosci.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65(1):66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun J-C, Schneggenburger R, Knott GW. Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123(5):653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- 237.Lundblad M, Decressac M, Mattsson B, Björklund A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc Natl Acad Sci. 2012;109(9):3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26(46):11915–11922. doi: 10.1523/jneurosci.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Atias M, Tevet Y, Sun J, Stavsky A, Tal S, Kahn J, Roy S, Gitler D. Synapsins regulate α-synuclein functions. Proc Natl Acad Sci. 2019;116(23):11116–11118. doi: 10.1073/pnas.1903054116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Payton JE, Perrin RJ, Woods WS, George JM. Structural determinants of PLD2 inhibition by α-synuclein. J Mol Biol. 2004;337(4):1001–1009. doi: 10.1016/j.jmb.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 241.Dalfo E, Ferrer I. α-Synuclein binding to rab3a in multiple system atrophy. Neurosci Lett. 2005;380(1–2):170–175. doi: 10.1016/j.neulet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 242.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chandra S, Fornai F, Kwon H-B, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE. Double-knockout mice for α-and β-synucleins: effect on synaptic functions. Proc Natl Acad Sci. 2004;101(41):14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell. 2005;123(3):383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 245.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 246.Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native brain α-synuclein. Nature. 2013;498(7453):E4–E6. doi: 10.1038/nature12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, Van Rossum M, Goldfarb D. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. 2016;530(7588):45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 248.Fakhree MA, Nolten IS, Blum C, Claessens MM. Different conformational subensembles of the intrinsically disordered protein α-synuclein in cells. J Phys Chem Lett. 2018;9(6):1249–1253. doi: 10.1021/acs.jpclett.8b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rey NL, Bousset L, George S, Madaj Z, Meyerdirk L, Schulz E, Steiner JA, Melki R, Brundin P. α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun. 2019;7(1):1–18. doi: 10.1186/s40478-019-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ilie IM, Nayar D, Den Otter WK, Van Der Vegt NF, Briels WJ. Intrinsic conformational preferences and interactions in α-synuclein fibrils: insights from molecular dynamics simulations. J Chem Theory Comput. 2018;14(6):3298–3310. doi: 10.1021/acs.jctc.8b00183. [DOI] [PubMed] [Google Scholar]
- 251.Gould N, Mor DE, Lightfoot R, Malkus K, Giasson B, Ischiropoulos H. Evidence of native α-synuclein conformers in the human brain. J Biol Chem. 2014;289(11):7929–7934. doi: 10.1074/jbc.c113.538249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4(11):1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 253.Fauvet B, Mbefo MK, Fares M-B, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287(19):15345–15364. doi: 10.1074/jbc.m111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein a possible molecular link between parkinson’s disease and heavy metal exposure. J Biol Chem. 2001;276(47):44284–44296. doi: 10.1074/jbc.m105343200. [DOI] [PubMed] [Google Scholar]
- 255.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in α-synuclein fibril formation. J Biol Chem. 2001;276(14):10737–10744. doi: 10.1074/jbc.m010907200. [DOI] [PubMed] [Google Scholar]
- 256.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci. 2011;108(43):17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Dettmer U, Newman AJ, von Saucken VE, Bartels T, Selkoe D. KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc Natl Acad Sci. 2015;112(31):9596–9601. doi: 10.1073/pnas.1505953112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM. The dynamic structure of α-synuclein multimers. J Am Chem Soc. 2013;135(10):3865–3872. doi: 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- 260.Binolfi A, Theillet F-X, Selenko P. Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature? Biochem Soc Trans. 2012 doi: 10.1042/bst20120096. [DOI] [PubMed] [Google Scholar]
- 261.Fauvet B, Fares M-B, Samuel F, Dikiy I, Tandon A, Eliezer D, Lashuel HA. Characterization of semisynthetic and naturally nα-acetylated α-synuclein in vitro and in intact cells implications for aggregation and cellular properties of α-synuclein. J Biol Chem. 2012;287(34):28243–28262. doi: 10.1074/jbc.m112.383711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ghosh S, Kundu A, Chattopadhyay K. Small molecules attenuate the interplay between conformational fluctuations, early oligomerization and amyloidosis of alpha synuclein. Sci Rep. 2018;8(1):1–16. doi: 10.1038/s41598-018-23718-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Dasari AK, Kayed R, Wi S, Lim KH. Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. 2019;58(25):2814–2821. doi: 10.1021/acs.biochem.9b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kaur U, Lee JC. Unroofing site-specific α-synuclein–lipid interactions at the plasma membrane. Proc Natl Acad Sci. 2020;117(32):18977–18983. doi: 10.1073/pnas.2006291117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97(1):92–107.e110. doi: 10.1016/j.neuron.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163–172. doi: 10.1001/jamaneurol.2016.4547. [DOI] [PubMed] [Google Scholar]
- 267.Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci. 2012;32(10):3301–3305. doi: 10.1523/jneurosci.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wilkaniec A, Lenkiewicz AM, Czapski GA, Jęśko HM, Hilgier W, Brodzik R, Gąssowska-Dobrowolska M, Culmsee C, Adamczyk A. Extracellular alpha-synuclein oligomers induce parkin s-nitrosylation: relevance to sporadic Parkinson’s disease etiopathology. Mol Neurobiol. 2019;56(1):125–140. doi: 10.1007/s12035-018-1082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ. Formation of toxic oligomeric α-synuclein species in living cells. PLoS ONE. 2008;3(4):e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Hansson O, Hall S, Öhrfelt A, Zetterberg H, Blennow K, Minthon L, Nägga K, Londos E, Varghese S, Majbour NK. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimer’s Res Ther. 2014;6(3):1–6. doi: 10.1186/alzrt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Paleologou KE, Kragh CL, Mann DM, Salem SA, Al-Shami R, Allsop D, Hassan AH, Jensen PH, El-Agnaf OM. Detection of elevated levels of soluble α-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain. 2009;132(4):1093–1101. doi: 10.1093/brain/awn349. [DOI] [PubMed] [Google Scholar]
- 272.Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TP, Linse S, Dobson CM. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci. 2014;111(21):7671–7676. doi: 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ferreon ACM, Gambin Y, Lemke EA, Deniz AA. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci. 2009;106(14):5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Oueslati A, Paleologou KE, Schneider BL, Aebischer P, Lashuel HA. Mimicking phosphorylation at serine 87 inhibits the aggregation of human α-synuclein and protects against its toxicity in a rat model of Parkinson’s disease. J Neurosci. 2012;32(5):1536–1544. doi: 10.1523/jneurosci.3784-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Souza JM, Giasson BI, Chen Q, Lee VM-Y, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275(24):18344–18349. doi: 10.1074/jbc.m000206200. [DOI] [PubMed] [Google Scholar]
- 276.Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E. Oxidative stress induces amyloid-like aggregate formation of NACP/α-synuclein in vitro. NeuroReport. 1999;10(4):717–721. doi: 10.1097/00001756-199903170-00011. [DOI] [PubMed] [Google Scholar]
- 277.Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim H-Y, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci. 2010;30(9):3184–3198. doi: 10.1523/jneurosci.5922-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Galvagnion C, Buell AK, Meisl G, Michaels TC, Vendruscolo M, Knowles TP, Dobson CM. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 2015;11(3):229–234. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Moons R, Konijnenberg A, Mensch C, Van Elzen R, Johannessen C, Maudsley S, Lambeir A-M, Sobott F. Metal ions shape α-synuclein. Sci Rep. 2020;10(1):1–13. doi: 10.1038/s41598-020-73207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Conway KA, Rochet J-C, Bieganski RM, Lansbury PT. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294(5545):1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 281.Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Fink AL. The aggregation and fibrillation of α-synuclein. Acc Chem Res. 2006;39(9):628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 283.Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, Mollenhauer B, Schneider A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain. 2016;139(2):481–494. doi: 10.1093/brain/awv346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ngolab J, Trinh I, Rockenstein E, Mante M, Florio J, Trejo M, Masliah D, Adame A, Masliah E, Rissman RA. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun. 2017;5(1):1–10. doi: 10.1186/s40478-017-0445-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, Yoshida M, Murayama S, Mishima K, Nishida N. Prion-like seeding of misfolded α-synuclein in the brains of dementia with Lewy body patients in RT-QUIC. Mol Neurobiol. 2018;55(5):3916–3930. doi: 10.1007/s12035-017-0624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Conway KA, Lee S-J, Rochet J-C, Ding TT, Williamson RE, Lansbury PT. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S. In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci. 2011;108(10):4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, Adame A, Trejo-Morales M, Gerez J, Picotti P. Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain. 2014;137(5):1496–1513. doi: 10.1093/brain/awu057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Hashimoto M, Kawahara K, Bar-On P, Rockenstein E, Crews L, Masliah E. The role of α-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci. 2004;24(3):343–352. doi: 10.1385/jmn:24:3:343. [DOI] [PubMed] [Google Scholar]
- 290.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27(34):9220–9232. doi: 10.1523/jneurosci.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. α-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157(2):401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Alim MA, Ma Q-L, Takeda K, Aizawa T, Matsubara M, Nakamura M, Asada A, Saito T, Xkaji M, Yoshii M. Demonstration of a role for α-synuclein as a functional microtubule-associated protein. J Alzheimer’s Dis. 2004;6(4):435–442. doi: 10.3233/jad-2004-6412. [DOI] [PubMed] [Google Scholar]
- 293.Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N. The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci. 2008;105(2):763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, Sage D, Abbas-Terki T, Iwatsubo T, Unser M, Aebischer P. Phosphorylation does not prompt, nor prevent, the formation of α-synuclein toxic species in a rat model of Parkinson’s disease. Hum Mol Genet. 2009;18(5):872–887. doi: 10.1093/hmg/ddn417. [DOI] [PubMed] [Google Scholar]
- 295.Taschenberger G, Garrido M, Tereshchenko Y, Bähr M, Zweckstetter M, Kügler S. Aggregation of αSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 2012;123(5):671–683. doi: 10.1007/s00401-011-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J. 2012;102(12):2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522(7556):340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 298.Sokratian A, Ziaee J, Kelly K, Chang A, Bryant N, Wang S, Xu E, Li JY, Wang S-H, Ervin J. Heterogeneity in α-synuclein fibril activity correlates to disease phenotypes in Lewy body dementia. Acta Neuropathol. 2021;141(4):547–564. doi: 10.1007/s00401-021-02288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Van der Perren A, Gelders G, Fenyi A, Bousset L, Brito F, Peelaerts W, Van den Haute C, Gentleman S, Melki R, Baekelandt V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020;139(6):977–1000. doi: 10.1007/s00401-020-02157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9(1):1–10. doi: 10.1038/s41467-018-05971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H. Cryo-EM structure of alpha-synuclein fibrils. Elife. 2018;7:e36402. doi: 10.7554/eLife.36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ballard CG, Jacoby R, Del Ser T, Khan MN, Munoz DG, Holmes C, Nagy Z, Perry EK, Joachim C, Jaros E. Neuropathological substrates of psychiatric symptoms in prospectively studied patients with autopsy-confirmed dementia with Lewy bodies. Am J Psychiatry. 2004;161(5):843–849. doi: 10.1176/appi.ajp.161.5.843. [DOI] [PubMed] [Google Scholar]
- 303.Bergeron C, Petrunka C, Weyer L, Pollanen MS. Altered neurofilament expression does not contribute to Lewy body formation. Am J Pathol. 1996;148(1):267. [PMC free article] [PubMed] [Google Scholar]
- 304.Katsuse O, Iseki E, Marui W, Kosaka K. Developmental stages of cortical Lewy bodies and their relation to axonal transport blockage in brains of patients with dementia with Lewy bodies. J Neurol Sci. 2003;211(1–2):29–35. doi: 10.1016/s0022-510x(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 305.Tompkins MM, Hill WD. Contribution of somal Lewy bodies to neuronal death. Brain Res. 1997;775(1–2):24–29. doi: 10.1016/s0006-8993(97)00874-3. [DOI] [PubMed] [Google Scholar]
- 306.Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature. 2018;557(7706):558–563. doi: 10.1038/s41586-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113(5):1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 308.Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011;25(1):326–336. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42(3):360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci. 2013;110(19):E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Ho PW-L, Leung C-T, Liu H, Pang SY-Y, Lam CS-C, Xian J, Li L, Kung MH-W, Ramsden DB, Ho S-L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA) Autophagy. 2020;16(2):347–370. doi: 10.1080/15548627.2019.1603545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, Bergmann A, Rockenstein E, Adame A, Marxreiter F. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14(1):98–119. doi: 10.1080/15548627.2017.1395992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee S-J. Assembly-dependent endocytosis and clearance of extracellular a-synuclein. Int J Biochem Cell Biol. 2008;40(9):1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 314.Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, Rammonhan M, Wong YC, Xie Z, Akamatsu W. Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci. 2019;39(29):5760–5772. doi: 10.1523/jneurosci.3085-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Lee H-J, Patel S, Lee S-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J Neurosci. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30(20):6838–6851. doi: 10.1523/jneurosci.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Guo M, Wang J, Zhao Y, Feng Y, Han S, Dong Q, Cui M, Tieu K. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143(5):1476–1497. doi: 10.1093/brain/awaa090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, Kou L, Yin S, Liu L, Huang J. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019;10(3):1–15. doi: 10.1038/s41419-019-1404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Tsigelny IF, Sharikov Y, Wrasidlo W, Gonzalez T, Desplats PA, Crews L, Spencer B, Masliah E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012;279(6):1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci. 2008;105(50):19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Angot E, Steiner JA, Tomé CML, Ekström P, Mattsson B, Björklund A, Brundin P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE. 2012;7(6):e39465. doi: 10.1371/journal.pone.0039465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Zhang S, Eitan E, Wu T-Y, Mattson MP. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol Aging. 2018;61:52–65. doi: 10.1016/j.neurobiolaging.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee S-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM-Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Lee H-J, Suk J-E, Patrick C, Bae E-J, Cho J-H, Rho S, Hwang D, Masliah E, Lee S-J. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285(12):9262–9272. doi: 10.1074/jbc.m109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH. Temporal evolution of microglia and α-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain. 2019;142(6):1690–1700. doi: 10.1093/brain/awz104. [DOI] [PubMed] [Google Scholar]
- 327.Hoban DB, Shrigley S, Mattsson B, Breger LS, Jarl U, Cardoso T, Wahlestedt JN, Luk KC, Björklund A, Parmar M. Impact of α-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized α-synuclein rat model of PD. Proc Natl Acad Sci. 2020;117(26):15209–15220. doi: 10.1073/pnas.2001305117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C, Winner B, Winkler J, Masliah E. α-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci. 2008;28(16):4250–4260. doi: 10.1523/JNEUROSCI.0066-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Karlsson J, Petersén Å, Gidö G, Wieloch T, Brundin P. Combining neuroprotective treatment of embryonic nigral donor tissue with mild hypothermia of the graft recipient. Cell Transplant. 2005;14(5):301–309. doi: 10.3727/000000005783983089. [DOI] [PubMed] [Google Scholar]
- 330.Karlsson J, Emgård M, Gidö G, Wieloch T, Brundin P. Increased survival of embryonic nigral neurons when grafted to hypothermic rats. NeuroReport. 2000;11(8):1665–1668. doi: 10.1097/00001756-200006050-00014. [DOI] [PubMed] [Google Scholar]
- 331.Frodl EM, Duan W-M, Kupsch A, Brundin P. Human embryonic dopamine neurons xenografted to the rat: effects of cryopreservation and varying regional source of donor cells on transplant survival, morphology and function. Brain Res. 1994;647(2):286–298. doi: 10.1016/0006-8993(94)91328-5. [DOI] [PubMed] [Google Scholar]
- 332.Kordower JH, Freeman TB, Olanow CW. Neuropathology of fetal nigral grafts in patients with Parkinson’s disease. Mov Disord. 1998;13:88–95. doi: 10.1002/mds.870130303. [DOI] [PubMed] [Google Scholar]
- 333.Braak H, Rüb U, Gai W, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110(5):517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 334.Jellinger KA. Is Braak staging valid for all types of Parkinson’s disease? J Neural Transm. 2019;126(4):423–431. doi: 10.1007/s00702-018-1898-9. [DOI] [PubMed] [Google Scholar]
- 335.Adler CH, Beach TG, Zhang N, Shill HA, Driver-Dunckley E, Caviness JN, Mehta SH, Sabbagh MN, Serrano GE, Sue LI. Unified staging system for Lewy body disorders: clinicopathologic correlations and comparison to Braak staging. J Neuropathol Exp Neurol. 2019;78(10):891–899. doi: 10.1093/jnen/nlz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Beach TG, White CL, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J, Bachalakuri J, Henry-Watson J. Olfactory bulb α-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009;117(2):169–174. doi: 10.1007/s00401-008-0450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Price JL. Comparative aspects of amygdala connectivity. Ann N Y Acad Sci. 2003;985(1):50–58. doi: 10.1111/j.1749-6632.2003.tb07070.x. [DOI] [PubMed] [Google Scholar]
- 338.Sah P, Faber EL, Lopez de Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83(3):803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- 339.Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J. Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015;129(5):729–748. doi: 10.1007/s00401-015-1406-3. [DOI] [PubMed] [Google Scholar]
- 340.Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM-Y, Brundin P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016;213(9):1759–1778. doi: 10.1084/jem.20160368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, Lee VMY, Brundin P. Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 2018;135(1):65–83. doi: 10.1007/s00401-017-1792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(4):1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34(28):9364–9376. doi: 10.1523/jneurosci.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Delenclos M, Trendafilova T, Mahesh D, Baine AM, Moussaud S, Yan IK, Patel T, McLean PJ. Investigation of endocytic pathways for the internalization of exosome-associated oligomeric alpha-synuclein. Front Neurosci. 2017;11:172. doi: 10.3389/fnins.2017.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Ludtmann MH, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun. 2018;9(1):1–16. doi: 10.1038/s41467-018-04422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Diógenes MJ, Dias RB, Rombo DM, Miranda HV, Maiolino F, Guerreiro P, Näsström T, Franquelim HG, Oliveira LM, Castanho MA. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci. 2012;32(34):11750–11762. doi: 10.1523/jneurosci.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Pacheco CR, Morales CN, Ramírez AE, Muñoz FJ, Gallegos SS, Caviedes PA, Aguayo LG, Opazo CM. Extracellular α-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J Neurochem. 2015;132(6):731–741. doi: 10.1111/jnc.13060. [DOI] [PubMed] [Google Scholar]
- 348.Bieri G, Gitler AD, Brahic M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol Dis. 2018;109:219–225. doi: 10.1016/j.nbd.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Wood SJ, Wypych J, Steavenson S, Louis J-C, Citron M, Biere AL. α-Synuclein fibrillogenesis is nucleation-dependent Implications for the pathogenesis of Parkinson’s disease. J Biol Chem. 1999;274(28):19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- 350.Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA, Abramov AY, Gandhi S, Wood NW, Cremades N. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci. 2016;113(9):E1206–E1215. doi: 10.1073/pnas.1524128113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Conway K, Lee SJ, Rochet JC, Ding T, Harper J, Williamson R, Lansbury P., Jr Accelerated oligomerization by Parkinson’s disease linked α-synuclein mutants. Ann N Y Acad Sci. 2000;920(1):42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 352.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322(5):1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 353.Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL. Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci. 2005;102(6):2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human α-synuclein in vitro. FEBS Lett. 2002;517(1–3):239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 355.Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human α-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 2003;542(1–3):147–152. doi: 10.1016/s0014-5793(03)00367-3. [DOI] [PubMed] [Google Scholar]
- 356.Vicente Miranda H, Szegő ÉM, Oliveira LM, Breda C, Darendelioglu E, de Oliveira RM, Ferreira DG, Gomes MA, Rott R, Oliveira M. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain. 2017;140(5):1399–1419. doi: 10.1093/brain/awx056. [DOI] [PubMed] [Google Scholar]
- 357.de Oliveira RM, Vicente Miranda H, Francelle L, Pinho R, Szegö ÉM, Martinho R, Munari F, Lázaro DF, Moniot S, Guerreiro P. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 2017;15(3):e2000374. doi: 10.1371/journal.pbio.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TP, Sparr E, Linse S. Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. 2017 doi: 10.1017/S0033583516000172. [DOI] [PubMed] [Google Scholar]
- 359.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012;209(5):975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener. 2018;13(1):1–11. doi: 10.1186/s13024-018-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin J-H. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016 doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Ferreira DG, Temido-Ferreira M, Miranda HV, Batalha VL, Coelho JE, Szegö ÉM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M. α-Synuclein interacts with PrP C to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017;20(11):1569. doi: 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- 363.Erskine D, Patterson L, Alexandris A, Hanson PS, McKeith IG, Attems J, Morris CM. Regional levels of physiological α-synuclein are directly associated with Lewy body pathology. Acta Neuropathol. 2018;135(1):153–154. doi: 10.1007/s00401-017-1787-6. [DOI] [PubMed] [Google Scholar]
- 364.Luna E, Decker SC, Riddle DM, Caputo A, Zhang B, Cole T, Caswell C, Xie SX, Lee VM, Luk KC. Differential α-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018;135(6):855–875. doi: 10.1007/s00401-018-1829-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.