

Abstract
Background and Objectives
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form of human prion disease and typically occurs in middle to late life. sCJD in early adulthood is extremely uncommon. The purpose of this report is to raise awareness of cases of sCJD in young patients that are not associated with a genetic mutation or acquired prion disease risk factors.
Methods
We describe the clinical presentation, diagnostic workup, and postmortem examination of a 22-year-old man with sCJD.
Results
The patient presented with a rapidly progressive neurocognitive disorder consisting of early and prominent psychiatric symptoms. CSF real-time quaking-induced conversion (RT-QuIC) was indeterminate, and brain MRI was suggestive of prion disease. Neuropathologic examination and the absence of a genetic mutation and acquired prion disease risk factors resulted in a final diagnosis of sCJD.
Conclusion
Although extremely rare, sCJD can occur in young people and should be considered in the setting of rapidly progressive neuropsychiatric conditions. Postmortem examination is required to diagnose the type of prion disease and remains important to surveil for known and potentially novel acquired prion diseases.
The human prion disease, Creutzfeldt-Jakob disease (CJD), can occur in 3 forms: sporadic, genetic, and acquired. Sporadic CJD (sCJD), the most common form, represents approximately 85% of cases, with a median age at death of 67 years.1 CJD in persons <30 years of age is very rare and usually the result of an exogenous exposure or genetic mutation.1 We describe a person with sCJD who died at age 22 years.
The patient was a right-handed man who had symptom onset in July 2018 when he was 21 years and 4 months of age. He had no family history of neurodegenerative disease, no travel outside of the United States, and an unremarkable medical history that was absent of neuropsychiatric disorders and receipt of tissue transplants, blood transfusions, and cadaver-derived pituitary hormones. The only surgery he underwent was arthroscopic surgery of the knee approximately 7 years prior to symptom onset. The patient was not a hunter, and according to family members only consumed venison on 1 or 2 occasions, from deer harvested from an area where chronic wasting disease has not been found.
Initially, coworkers noticed that the patient's affect was uncharacteristically flat. Two months later, he started having memory problems and apraxia, and he became agitated and depressed. Shortly thereafter, he became paranoid that cars were following him, and 6 months following symptom onset, his gait became unsteady and bradykinetic. He developed significant insomnia, as well as parasomnias, new-onset hypertension, cold intolerance, and occasional myoclonus of the extremities. He became combative with caregiving tasks and would not allow physical examination. Limited examination 7 months after symptom onset demonstrated perseverative motor behaviors, ataxic and bradykinetic gait, spontaneous levitation of the left arm, mild bilateral upper extremity positional tremors, and a Medical Research Council Prion Rating Scale score of 7/20.2 The following month he developed double incontinence and dysphagia. Death occurred approximately 11 months after symptom onset.
A diagnostic workup for reversible etiologies was unrevealing. Brain MRI performed 3 months after onset demonstrated asymmetric hyperintensity in most cortical areas and bilateral caudate nuclei on diffusion-weighted imaging (Figure 1). An EEG completed 4 months after symptom onset demonstrated slowing and sharp wave forms. CSF analyses conducted at the National Prion Disease Pathology Surveillance Center (NPDPSC) 4 months following symptom onset revealed elevated 14-3-3 protein, total tau level of 7,817 pg/mL, and an indeterminate real-time quaking-induced conversion (RT-QuIC) result.3 RT-QuIC analyses did not meet the threshold for a positive result but consistently revealed abnormal prion protein seeding activity on 2 separate diagnostic runs.
Figure 1. Brain MRI 3 Months After Symptom Onset.
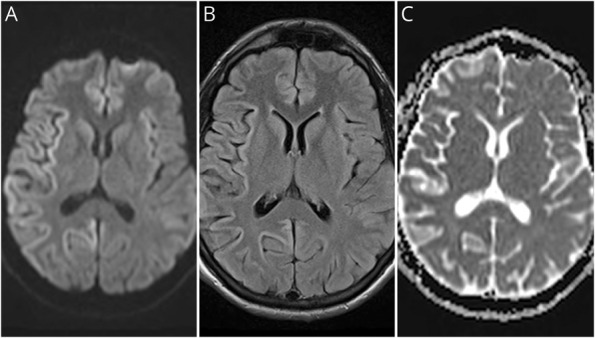
Hyperintensity is seen in most cortices and bilateral caudate heads on diffusion-weighted imaging (A) and fluid-attenuated inversion recovery (B) sequences. Attenuation of the signal is noted in some cortical areas on apparent diffusion coefficient (C).
Neuropathologic analyses were performed at the NPDPSC. Immunohistology and histologic analyses demonstrated features consistent with the VV1 subtype of sCJD (Figure 2).4 Prion protein gene (PRNP) analyses demonstrated no pathogenic mutations as well as valine homozygosity at codon 129. Western blot analyses confirmed the presence of type 1 prion protein.
Figure 2. Histopathology.
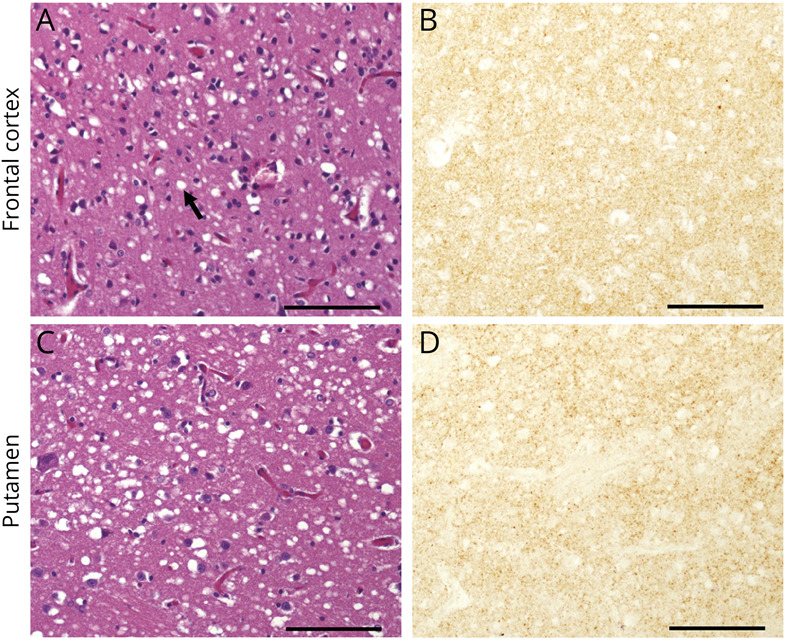
Hematoxylin & eosin (A and C) and immunohistochemistry (B and D) of prion protein. (A and C) Spongiform degeneration with medium size; vacuoles (arrow). (B and D) Faint PrP immunostaining with synaptic pattern. Antibody: 3F4; size bar: 100 µm.
The absence of known acquired prion disease risk factors in this very young patient is noteworthy, and the results of analyses performed at the NPDPSC offer confidence that this patient had a nongenetic, sporadic prion disease. Although the etiology of sporadic prion disease is unknown, possible explanations include posttranslational modification of the normal prion protein or somatic mutations. The indeterminate RT-QuIC result is consistent with a previous finding of lower diagnostic sensitivity of this assay for the VV1 sCJD subtype.3 Patients with prion disease of this subtype represent a small percentage of sporadic prion disease cases (∼2% of NPDPSC cases). They tend to be younger than patients with prion disease as a whole (Table),4 but the authors are aware of only 1 VV1 case reported worldwide in a patient who was younger (19 years at onset) than the patient in this report; an additional case reported, also in a patient 19 years of age at onset, is believed to be a duplicate.5-7
Table.

In recent years, concerns have been raised about the possibility of transmission of chronic wasting disease, a prion disease of cervids, to humans. Human patients with prion disease in the <30 years age group, while unusual, continue to be an important sentinel for acquired prion disease. Therefore, it is important for physicians to consider the possibility of prion disease in young patients and to take advantage of the diagnostic services provided by NPDPSC (cjdsurveillance.com) and other international prion disease surveillance centers, especially their autopsy coordination programs. It also remains crucial to determine the presence of known or possibly new risk factors for acquired disease, including possible exposure to chronic wasting disease.
Acknowledgment
The authors thank the patients, families, physicians, and National Prion Disease Pathology Surveillance Center staff.
Glossary
- CJD
Creutzfeldt-Jakob disease
- NPDPSC
National Prion Disease Pathology Surveillance Center
- RT-QuIC
real-time quaking-induced conversion
- sCJD
sporadic Creutzfeldt-Jakob disease
Appendix. Authors
Footnotes
Editorial, page 801
Study Funding
Centers for Disease Control and Prevention (1NU38CK000480) (B.S.A., M.C., I.C.) and NIH (K99AG068359) (I.C.).
Disclosure
Dr. Appleby reports research funding through CDC, NIH, and Ionis. Dr. Maddox, Dr. Schonberger, Dr. Cali, T. Hammett, Dr. Cohen, and Dr. Belay report no disclosures relative to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Maddox RA, Person MK, Blevins JE, et al. Prion disease incidence in the United States, 2003-2015. Neurology. 2020;94(2):e153-e157. [DOI] [PubMed] [Google Scholar]
- 2.Thompson AGB, Lowe J, Fox Z, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013;136(pt 4):1116-1127. [DOI] [PubMed] [Google Scholar]
- 3.Rhoads DD, Wrona A, Foutz A, et al. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology. 2020;95(8):e1017-e1026. [DOI] [PubMed] [Google Scholar]
- 4.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224-233. [PubMed] [Google Scholar]
- 5.Meissner B, Westner IM, Kallenberg K, et al. Sporadic Creutzfeldt-Jakob disease: clinical and diagnostic characteristics of the rare VV1 type. Neurology. 2005;65(10):1544-1550. [DOI] [PubMed] [Google Scholar]
- 6.Petzold GC, Westner I, Bohner D, Einhäupl KM, Kretzschmar HA, Valdueza JM. False-positive pulvinar sign on MRI in sporadic Creutzfeldt-Jakob disease. Neurology. 2004;62(7):1235-1236. [DOI] [PubMed] [Google Scholar]
- 7.Corato M, Cereda C, Cova E, Ferrarese C, Ceroni M. Young-onset CJD: age and disease phenotype in variant and sporadic forms. Funct Neurol. 2006;21(4):211-215. [PubMed] [Google Scholar]