

Abbreviations
- AD
autosomal dominant
- AR
autosomal recessive
- CoA
coenzyme A
- CSF
cerebrospinal fluid
- DLD
dihydrolipoamide dehydrogenase
- FFT
failure to thrive
- GRACILE
growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death
- MDS
mitochondrial depletion syndrome
- MMA
methylmalonic acid
- MNGIE
mitochondrial neurogastrointestinal encephalomyopathy
- MRI
magnetic resonance imaging
- mtDNA
mitochondrial DNA
- PMIM
Phenotype Mendelian Inheritance in Man
- tRNA
transfer RNA
Pretest
What clinical features should increase suspicion for underlying mitochondrial hepatopathy?
How are mitochondrial hepatopathies diagnosed?
How are mitochondrial hepatopathies treated?
Mitochondria play critical roles in energy, calcium, iron, and reduction/oxidation homeostasis, as well as regulation of apoptosis. They are the only organelle that contains its own circular genomes (mitochondrial DNA [mtDNA]). Maternally inherited mtDNA houses 37 genes encoding mitochondrial transfer RNAs (tRNAs), ribosomal RNA, and 13 proteins that exclusively function as subunits of the oxidative‐phosphorylation machinery. Additional proteins critical to mitochondrial structure and function are encoded by the nuclear genome.
Mitochondrial disorders include defects in oxidative‐phosphorylation complexes, mtDNA maintenance, and mtDNA transcription and translation and can result from mitochondrial or nuclear mutations and yield disease involving virtually every organ system, including the liver. Mitochondrial hepatopathies are heterogenous and individually rare, but collectively they comprise an important cause of early liver failure. In two studies of infants with acute liver failure, about 20% of cases were attributable to mitochondrial pathology. 1 , 2
Clinical Features
Mitochondrial liver disease may manifest as liver failure (acute, chronic, or recurrent), cholestasis, liver fibrosis, or elevated transaminases (Table 1). Presentation is typically pediatric. A mitochondrial etiology is especially likely in cases with multisystem involvement but is also associated with isolated hepatopathy. Low birth weight and intrauterine growth restriction have been associated. 3 , 4 Lactic acidosis and hypoglycemia are common biochemical features 5 , 6 and could lead to the misdiagnosis of other conditions, such as glycogen storage disease type I. 6
TABLE 1.
Mitochondrial Disorders Associated With Hepatopathy, Including Their Molecular Basis and Clinical, Pathological, and Biochemical Features
| Disease | PMIM # | Gene Associated | Cytogenetic Location | Gene Product | Function | Inheritance | Liver Phenotype | Pathological Features | Mitochondrial Features | Biochemical Features | Additional Features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Transient infantile liver failure | 613070 | TRMU | 22q13.31 | tRNA 5‐methylaminomethyl‐2‐thiouridylate methyltransferase | Mitochondrial translation | AR | Liver failure: acute, transient | Liver: fibrosis, macrovesicular steatosis | Deficiency of complex IV | Lactic acidosis | Disease resolution in patients surviving infantile liver failure event |
| Muscle: normal | Cardiomyopathy | ||||||||||
| mtDNA depletion syndrome 3: hepatocerebral type | 251880 | DGUOK | 2p13.1 | Deoxyguanosine kinase | mtDNA maintenance | AR | Liver failure: progressive, hepatocellular carcinoma | Liver: variable | mtDNA depletion, variable deficiency of complexes I, III, and IV | Lactic acidosis, elevated transferrin saturation and ferritin, elevated alpha‐fetoprotein | Hypotonia, ophthalmoplegia, nystagmus |
| May also present as noncirrhotic portal hypertension (PMIM # 617068) | |||||||||||
| mtDNA depletion syndrome 4A: Alpers type | 203700 | POLG | 15q26.1 | DNA polymerase gamma | mtDNA maintenance | AR | Liver failure: early, progressive | Brain: gray matter neuronal loss | mtDNA depletion, variable deficiency of complexes I, III, and IV | 3‐Methylglutaconic aciduria, elevated CSF protein | Epilepsy, psychomotor impairment, migraine headaches, cortical blindness |
| Liver: reactive astrocytosis, cirrhosis | |||||||||||
| mtDNA depletion syndrome 4B: MNGIE type | 174763 | POLG | 15q26.1 | DNA polymerase gamma | mtDNA maintenance | AR | Liver failure: early, progressive | Muscle: ragged red fibers, cytochrome c oxidase negative fibers | mtDNA depletion, variable deficiency of complexes I, III, and IV | Gastrointestinal manifestations: dysmotility, pseudoobstruction, malabsorption; peripheral neuropathy, ophthalmoplegia, weakness | |
| Liver: fibrosis | |||||||||||
| mtDNA depletion syndrome 6: hepatocerebral type (also: Navajo neurohepatopathy) | 256810 | MPV17 | 2p23.3 | Mitochondrial inner membrane protein MPV17 | mtDNA maintenance | AR | Liver failure: progressive; cholestatic liver disease | Liver: steatosis, cirrhosis | mtDNA depletion, variable deficiency of complexes I, III, and IV | Lactic acidosis, hypoglycemia | Neuropathy, psychomotor impairment or regression, growth failure |
| Combined oxidative phosphorylation deficiency 1 | 609060 | GFM1 | 3q25.32 | Mitochondrial elongation factor G1 | Mitochondrial translation | AR | Liver failure | Liver: necrosis, severe mitochondrial deficiency | Variable deficiency of complexes I, III, and IV | Growth failure, microcephaly, structural brain anomalies, encephalopathy, cardiomyopathy, early death | |
| Combined oxidative phosphorylation deficiency 2 | 610498 | MRPS16 | 10q22.2 | Mitochondrial ribosomal protein S16 | Mitochondrial transcription | AR | Liver failure | Variable deficiency of complexes I, III, and IV | Lactic acidosis | Congenital brain anomalies, hypotonia, early death | |
| Combined oxidative phosphorylation deficiency 3 | 610505 | TSFM | 12q14.1 | Mitochondrial translation elongation factor Ts | Mitochondrial translation | AR | Liver failure | Muscle: ragged red fibers, cytochrome c oxidase negative fibers | Variable deficiency of complexes I, III, and IV | Hypotonia, rhabdomyolysis, cardiomyopathy, Leigh syndrome, optic neuropathy | |
| Heart: lipid deposition, fibrosis | |||||||||||
| Combined oxidative phosphorylation deficiency 4 | 610678 | TUFM | 16p11.2 | Mitochondrial translation elongation factor Tu | Mitochondrial translation | AR | Liver failure | Variable deficiency of complexes I, III, and IV | Lactic acidosis, hyperammonemia | Brain imaging abnormalities (leukoencephalopathy), hypotonia | |
| mtDNA depletion syndrome 8B: MNGIE type | 612075 | RRM2B | 8q22.3 | Ribonucleotide reductase subunit M2B | Mitochondrial transcription | AR or AD | Liver failure: rapidly progressive | Muscle: cytochrome c oxidase negative fibers, lipid deposition | mtDNA depletion, variable deficiency of complexes I, III, and IV | Lactic acidosis, amino aciduria, elevated creatine kinase | Growth failure, weakness, hypotonia, epilepsy |
| mtDNA depletion syndrome 7: hepatocerebral type | 271245 | TWNK | 10q24.13 | Twinkle mtDNA helicase | mtDNA maintenance | AR | Liver failure: progressive | Liver: variable, including cirrhosis, steatosis, and hemosiderosis | mtDNA depletion, variable deficiency of complexes I, III, and IV | Anemia, electrolyte abnormalities | Encephalopathy, movement disorders, hypotonia, renal impairment/tubulopathy |
| mtDNA depletion syndrome 9: encephalomyopathic type with MMA | 245400 | SUCLG1 | 2p11.2 | Succinate‐CoA ligase, alpha subunit | mtDNA maintenance | AR | Liver failure | Muscle: variable fiber size with atrophy, lipid accumulation, ragged red fibers, cytochrome c oxidase negative fibers | Abnormal cristae on electron microscopy, variable deficiency of complexes I, III, and IV | Lactic acidosis, methylmalonic aciduria, elevated C3 on newborn screening | Hypertrophic cardiomyopathy, cognitive impairment, dystonia, sensorineural hearing loss, epilepsy, early death |
| GRACILE syndrome or mitochondrial complex III deficiency: nuclear type 1 | 603358 or 12400 | BCS1L | 2q35 | BCS1 homolog, ubiquinol‐cytochrome c reductase complex chaperone | Oxidative phosphorylation | AR | Cholestatic liver disease, hepatitis | Hemosiderosis | Deficiency of complex III | Lactic acidosis | Growth failure, amino aciduria, iron overload, early death |
| Pearson marrow‐pancreas syndrome | 557000 | Multiple | Contiguous mtDNA deletion syndrome | Spontaneous | Cholestatic liver disease, liver failure | Bone marrow: vacuolization | mtDNA depletion, variable deficiency of complexes I, III, and IV | 3‐Methylglutaconic aciduria | Sideroblastic anemia, pancytopenia, exocrine pancreatic dysfunction, endocrinopathies; can progress to Kearns‐Sayre syndrome | ||
| Mitochondrial complex IV deficiency: nuclear type 4 | 619048 | SCO1 | 17p13.1 | Synthesis of cytochrome c oxidase 1 | Oxidative phosphorylation; copper homeostasis | AR | Cholestatic liver disease | Liver: lipid vacuoles, steatosis | Deficiency of complex IV | Ketoacidosis, lactic acidosis | Encephalopathy, cardiomyopathy |
| Muscle: lipid accumulation | |||||||||||
| Combined oxidative phosphorylation deficiency 19 | 615595 | LYRM4 | 6p25.1 | LYR motif‐containing protein 4 | Iron homeostasis | AR | Cholestatic liver disease | Deficiency of complexes I, II, and III | Lactic acidosis | Severe in the neonatal period with potential for full recovery | |
| 3‐Methylglutaconic aciduria with deafness, encephalopathy, and Leigh‐like syndrome | 614739 | SERAC1 | 6q25.3 | Serine active site‐containing protein 1 | Phosphatidylglycerol remodeling | AR | Liver failure, cholestatic liver disease: infantile onset | Liver: steatosis, cirrhosis, fibrosis | Mild mtDNA depletion, variable deficiency of complexes I, III, and IV | 3‐Methylglutaconic aciduria, hyperammonemia | Dystonia, hypoacusis, hypotonia, poor feeding, short stature, myopathy |
| DLD deficiency | 246900 | DLD | 7q31.1 | DLD | Subunit for dehydrogenase enzymes | AR | Recurrent fulminant liver failure | Liver: Reye‐like appearance, mild fibrosis and/or increased glycogen deposition | Variable deficiency of complexes I, III, and IV | Metabolic decompensation with urinary alpha‐ketoacids, lactic acidosis, presence of alloisoleucine, elevated citrulline on newborn screen (variable) | Early‐onset neurological involvement, hepatomegaly, myopathy |
Mitochondrial depletion syndromes (MDSs) feature decreased mtDNA copy number secondary to defects in mtDNA replication. MDSs may present with infantile hepatocerebral syndrome with acute or chronic liver failure. Mortality is high. Hepatocellular carcinoma is a complication in survivors (DGUOK and MPV17). 4 , 5 Affected infants typically present with growth failure, feeding difficulty, developmental delay, and hypotonia. Although brain involvement is usually prominent, in rare cases DGUOK may cause isolated liver failure. In POLG disease, brain involvement may not be immediately apparent, but developmental regression and epilepsia partialis continua are inevitable. Valproate triggers fulminant liver failure in MDS 7 and is contraindicated. Pediatric valproate‐induced liver failure should be assumed to be MDS. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) secondary to thymidine phosphorylase deficiency is an adolescent‐onset MDS that may have cirrhosis, as well as gastrointestinal dysmotility, peripheral neuropathy, and asymptomatic leukoencephalopathy. 8
Single large‐scale mtDNA deletions cause a spectrum of phenotypes, including Pearson syndrome, a severe disease characterized by sideroblastic anemia and exocrine pancreatic insufficiency. More than 33% of patients have liver involvement (hepatomegaly, cholestasis, and/or progressive liver failure). Kearns‐Sayre syndrome develops in childhood in survivors. Point mutations in mtDNA almost never cause liver disease. 9
Mitochondrial translation disorders are also characterized by early‐onset liver failure with frequent neurological manifestations. Early diagnosis and management are critical because recovery is possible with appropriate medical support, for example, acute infantile liver failure caused by mutations in TRMU. 6 Mitochondrial hepatopathy is also associated with deficiency of individual oxidative‐phosphorylation complexes, for example, BCS1L mutations cause infantile cholestasis with liver iron overload and multisystemic features. 4
Work‐up/Diagnosis
Diagnostic work‐up for suspected mitochondrial hepatopathy includes full phenotyping, biochemical studies, functional studies, and genetic confirmation. 10 Cardiac, neuromuscular, retinal, or auditory involvement may raise suspicion for mitochondrial disease generally, but specific features may be diagnostic. Peripheral neuropathy (MPV17), pili torti (BCS1L), rotary nystagmus (DGUOK), and sideroblastic anemia (mtDNA deletion) can be observed (Fig. 1). Biochemical laboratory testing, including amino acids, organic acids, and lactate/pyruvate and acylcarnitine profiles, are not sensitive but can strongly increase suspicion for mitochondrial disease.
FIG 1.
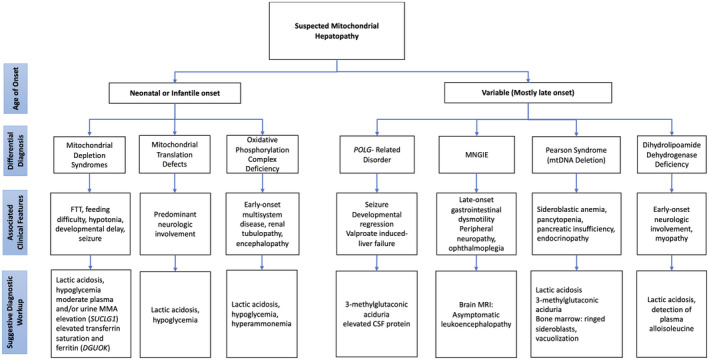
Diagnostic algorithm for mitochondrial hepatopathies.
Pathological examination of liver tissue plays a key role in confirming a clinical suspicion of mitochondrial hepatopathy and directing future testing (Fig. 2). Light microscopic findings are variable and depend on genetic lesion, patient age, and disease severity at time of biopsy. Classical findings beyond early infancy include nonzonal mixed (macrovesicular and microvesicular) steatosis, variable fibrosis, and presence of oncocytic hepatocytes with granular, hypereosinophilic cytoplasm. Electron microscopy shows this oncocytic change reflects proliferation of structurally abnormal mitochondria, characterized by lack of cristae, tubular cristae, and/or electron‐dense matrix granules. The light microscopic differential diagnosis includes other metabolic disorders, Wilson disease, and secondary mitochondrial dysfunction as a result of drug toxicity; ultrastructural changes, including mitochondrial proliferation, are more specific for primary mitochondrial disorders.
FIG 2.
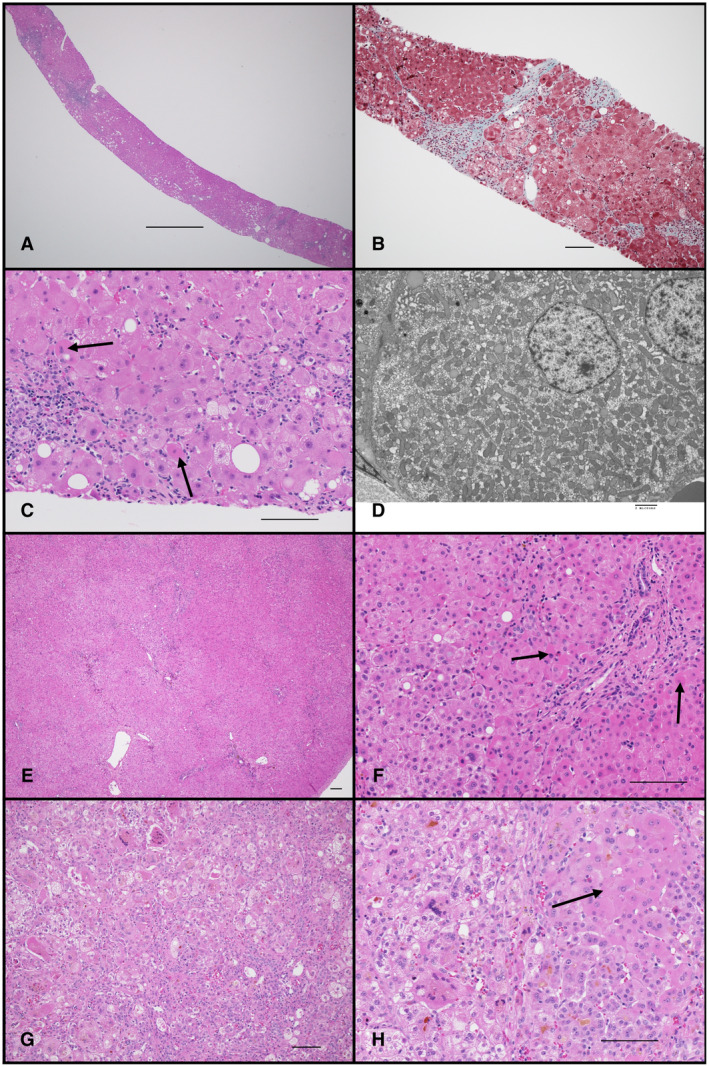
Pathological features of mitochondrial hepatopathy. (A‐D) MPV17 mutation in 18‐year‐old. (A) Liver needle biopsy showing patchy nonzonal steatosis and inflammation. (B) Gomori trichrome stain highlights patchy portal and sinusoidal fibrosis. (C) Higher‐magnification image illustrating macrovesicular and microvesicular steatosis and oncocytic hepatocytes (arrows). (D) Electron micrograph of hepatocyte with proliferation of mitochondria lacking normal cristae. (E, F) POLG mutation in 9‐month‐old. Liver wedge biopsy with intact architecture, minimal macrovesicular steatosis, and numerous oncocytic hepatocytes (two highlighted by arrows). (G, H) DGUOK mutation, 7‐week‐old with acute liver failure. Liver explant with severe neonatal hepatitis pattern, characterized by giant cell transformation, hepatocellular and canalicular cholestasis, hepatocellular necrosis, and rare foci of oncocytic hepatocytes (arrow). Scale bars: 1000 μm (A); 100 μm (B, C, E‐H); 2 μm (D).
Mitochondrial hepatopathy is also a significant causative factor for congenital liver failure, where the histological picture may be of neonatal (giant cell) hepatitis, or even so‐called neonatal hemochromatosis: hepatic parenchymal collapse with massive ductular proliferation, cholestasis, and evidence of siderosis both in liver and extrahepatic tissues. In such cases, classic histological and ultrastructural features may be absent; ancillary testing of rapidly procured postmortem tissue is required to establish the diagnosis. The differential diagnosis in these cases includes gestational alloimmune liver disease, congenital infection, or other metabolic disorders.
Acquiring tissue samples enables functional mitochondrial testing. Electron transport enzymology is sensitive for BCS1L (complex III) and SCO1 (complex IV) deficiency. BN‐PAGE can suggest mitochondrial translation defects; mtDNA quantification is the diagnostic gold standard for MDS diagnosis. Functional testing is most sensitively performed on liver tissue. Because there is a high degree of phenotype overlap, whole‐exome sequencing is often the most effective way to reach a definitive diagnosis. Analysis of mtDNA to rule out deletion is also warranted.
Management
Evidence‐based management guidelines for mitochondrial hepatopathies are lacking, and curative treatments are unavailable. Supportive measures led by a multidisciplinary team are the mainstay of therapy. Early introduction of enteral feeding optimizes mitochondrial function and minimizes lactic acidosis. Feeding tube placement may be required to ensure adequate nutritional support for patients. Avoidance of fasting and/or frequent feedings can prevent hypoglycemia. Specific therapies are indicated for some diagnoses: branched‐chain amino acid restriction and riboflavin supplementation for dihydrolipoamide dehydrogenase (DLD) deficiency, and cysteine supplementation for TRMU deficiency. 6
Liver transplantation for mitochondrial hepatopathy is controversial. Good outcomes have been reported in patients with mild MPV17 and DGUOK without neurological involvement. 3 , 5 However, posttransplant disease worsening has been observed in patients with MDS with any neurological involvement, especially in POLG. Generally, liver transplant is not contraindicated, but risks and benefits should be carefully evaluated. Posttransplant management is challenging because some immunomodulators are mitotoxic. Hematopoietic stem cell transplant has resulted in clinical improvement in some patients with MNGIE. 8
Mitochondrial dysfunction is associated with impaired redox homeostasis and glutathione depletion. N‐acetylcysteine may ameliorate oxidative stress and is a potential therapy for mitochondrial hepatopathies. 6 Gene therapy has shown experimental success for several monogenic liver disorders. The liver is amenable to gene therapy: it is easily targeted by adeno‐associated virus vectors and is a lifelong replicating tissue. 11 Therefore, it holds future promise as a therapeutic option for mitochondrial hepatopathies.
Conclusions
Mitochondrial hepatopathies are an important cause of infantile/pediatric liver failure. They should be suspected in patients with neonatal‐onset liver dysfunction, steatosis, fulminant or acute disease, or in individuals with neuromuscular or multiorgan involvement.
Posttest
1. What are clinical features that should increase suspicion for underlying mitochondrial hepatopathy?
Mitochondrial hepatopathy should be suspected in cases of neonatal‐onset liver dysfunction, steatosis, fulminant, or acute disease, or in individuals with neuromuscular or multiorgan involvement.
2. How are mitochondrial hepatopathies diagnosed?
Diagnosis of mitochondrial hepatopathies is multifaceted and includes biochemical and genetic testing, pathological examination of liver tissue, and functional mitochondrial enzymology.
3. How are mitochondrial hepatopathies treated?
Treatment for mitochondrial hepatopathy is largely supportive. N‐acetylcysteine may be helpful. Mitochondrial hepatopathy is not an absolute contraindication for consideration of transplantation.
This work was supported by R.D.G. consults for Minovia Therapeutics and received grants from National Institute of Diabetes and Digestive and Kidney Diseases (DK113250). Potential conflict of interest: Nothing to report.
REFERENCES
- 1. McKiernan P, Ball S, Santra S, et al. Incidence of primary mitochondrial disease in children younger than 2 years presenting with acute liver failure. J Pediatr Gastroenterol Nutr 2016;63:592‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Helbling D, Buchaklian A, Wang J, et al. Reduced mitochondrial DNA content and heterozygous nuclear gene mutations in patients with acute liver failure. J Pediatr Gastroenterol Nutr 2013;57:438‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jankowska I, Czubkowski P, Rokicki D, et al. Acute liver failure due to DGUOK deficiency—is liver transplantation justified? Clin Res Hepatol Gastroenterol 2021;45:101408. [DOI] [PubMed] [Google Scholar]
- 4. Visapää I, Fellman V, Vesa J, et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet 2002;71:863‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. El‐Hattab AW, Li F, Schmitt E, et al. MPV17‐associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations. Mol Genet Metab 2010;99:300‐308. [DOI] [PubMed] [Google Scholar]
- 6. Murali CN, Soler‐Alfonso C, Loomes KM, et al. TRMU deficiency: a broad clinical spectrum responsive to cysteine supplementation. Mol Genet Metab 2021;132:146‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stumpf JD, Saneto RP, Copeland WC. Clinical and molecular features of POLG‐related mitochondrial disease. Cold Spring Harb Perspect Biol 2013;5:a011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Filosto M, Cotti Piccinelli S, Caria F, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE‐MTDPS1). J Clin Med 2018;7:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wild KT, Goldstein AC, Muraresku C, et al. Broadening the phenotypic spectrum of Pearson syndrome: five new cases and a review of the literature. Am J Med Genet A 2020;182:365‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molleston JP, Sokol RJ, Karnsakul W, et al. Evaluation of the child with suspected mitochondrial liver disease. J Pediatr Gastroenterol Nutr 2013;57:269‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moscoso CG, Steer CJ. The evolution of gene therapy in the treatment of metabolic liver diseases. Genes (Basel) 2020;11:915. [DOI] [PMC free article] [PubMed] [Google Scholar]