

Abstract
Background:
Venous thrombosis (VT) and pulmonary embolism (PE), collectively venous thromboembolism (VTE), cause high mortality and morbidity. Factor XIII (FXIII) crosslinks fibrin to enhance thrombus stability and consequently may influence PE risk. Elucidating mechanisms contributing to PE is limited by a lack of models that recapitulate human PE characteristics.
Objective:
Develop a mouse model that permits embolization of red blood cell (RBC)- and fibrin-rich VT and determine the contribution of FXIII to PE risk.
Methods and Results:
In a thrombin infusion PE model, F13a+/+, F13a+/−, and F13a−/− mice had similar incidence of lung thrombi; however, thrombi were small, with low RBC content (≤7%), unlike human PEs (~70%). To identify a model producing PE consistent with histological characteristics of human PE, we compared mouse femoral vein electrolytic injury, femoral vein FeCl3 injury, and infrarenal vena cava (IVC) stasis models of VT. Electrolytic and FeCl3 models produced small thrombi with few RBCs (5% and 4%, respectively), whereas IVC stasis produced large thrombi with higher RBC content (68%) that was similar to human PEs. After IVC stasis and ligature removal (de-ligation) to permit thrombus embolization, compared to F13a+/+ mice, F13a+/− and F13a−/− mice had similar and increased PE incidence, respectively.
Conclusions:
Compared to thrombin infusion-, electrolytic injury-, and FeCl3-based models, IVC stasis produces thrombi that are most histologically similar to human thrombi. IVC stasis followed by de-ligation permits embolization of existing RBC- and fibrin-rich thrombi. Complete FXIII deficiency increases PE incidence, but partial deficiency does not.
Keywords: Venous thrombosis, pulmonary embolism, mouse, factor XIII, red blood cell
INTRODUCTION
Venous thrombosis (VT) with or without pulmonary embolism (PE), collectively venous thromboembolism (VTE), affects 1–2/1000 people annually.1,2 VTE has ~30% mortality within the first 30 days of presentation, typically associated with PE.3 PE occurs when part or all of the thrombus detaches, travels through the vasculature, and obstructs the pulmonary arterial network. Anticoagulants inhibit thrombus propagation but do not dissolve existing thrombi and are associated with bleeding risk. Thrombolytics dissolve thrombi but are associated with bleeding risk. There is a need to better understand molecular mechanisms that promote PE and develop therapies that safely prevent VTE.
Factor XIII (FXIII) is the zymogen of the transglutaminase FXIIIa that crosslinks fibrin. Crosslinking protects clots from mechanical disruption and biochemical dissolution (fibrinolysis). Crosslinking also promotes fibrin-mediated RBC retention in contracted thrombi.7–9 Previous studies using FeCl3 to induce femoral vein thrombosis suggested FXIII-deficient mice on a mixed 129Ola/CBACa background10 have increased embolic events compared to wildtype C57Bl/6 mice.11,12 Furthermore, following FeCl3-induced thrombosis, mice expressing fibrinogen with mutated crosslinking sites (FGG3X) have increased PE, suggesting FXIII stabilizes thrombi via fibrin γ-chain crosslinking.13 Accordingly, compared to patients with suspected but excluded PE, patients with acute PE have lower circulating FXIII-A subunit.14 However, reduced FXIII in these patients is associated with decreased fibrinogen and increased D-dimer, suggesting this relationship is due to consumption of FXIII during VTE rather than low FXIII levels prior to the event.14 Notably, a meta-analysis examining the prevalence of PE in patients with congenital coagulation disorders associated PE with deficiency in factors VII, VIII, IX, or XI, but not FXIII, raising questions about the role of FXIII in PE.15 Although congenital FXIII deficiency is rare, partial FXIII deficiency secondary to ongoing disease has been reported in patients with inflammatory bowel disease, sepsis, major surgery, trauma, and disseminated intravascular coagulation.16 One study found 21% of hospitalized adults and 52% of hospitalized children have a plasma FXIII concentration <70 U dL−1 (normal range ~70–140 U dL−1).17,18 Notably, hospitalized patients also have increased VTE risk.2,3 Thus, defining the effects of complete and partial FXIII deficiency on PE risk is needed.
A major characteristic of VT in humans is their high red blood cell (RBC) content (so-called “red clots”), which differentiates these thrombi from platelet-rich thrombi (so-called “white clots”) that form in high shear.4–6 Elucidating PE pathophysiology has been limited by a lack of small animal models that recapitulate the initial slow formation of these RBC-rich thrombi in situ and their subsequent embolization.2 In addition to the FeCl3 model which rapidly generates platelet-rich thrombi via free radical formation19–21, several other models have been used to study PE. Deployment of clots generated ex vivo22–27 allows control over thrombus composition and timing of embolization but does not recapitulate in situ formation of the initial thrombus. Intravenous infusion of platelet agonists (e.g., adenosine diphosphate28, collagen plus epinephrine28–30) or coagulation activators (e.g., thrombin31–34, tissue factor/thromboplastin28,35–37) produces thrombi that form in flowing blood and subsequently lodge in the lung microvasculature. Okano et al used femoral vein ligation plus fluorescent light to induce RBC- and fibrin-rich VT38; however, this model induced rapid thrombus formation with embolization ~30 minutes after de-ligation, which may not recapitulate the slow formation of human VT. Development of mouse models that better approximate VTE in humans is necessary to identify molecular mechanisms that contribute to PE pathophysiology.
Herein, we developed a novel VTE model in mice that produces RBC- and fibrin-rich thrombi that are histologically similar to human PE and permits subsequent embolization. Using this model, we evaluated the contribution of complete and partial FXIII deficiency to PE risk.
METHODS
Materials.
Alexa Fluor® 647 protein labeling kits were from Thermofisher (Waltham, MA) and IRDye 800CW was from LICOR (Lincoln, NE). Bio-Spin 30 chromatography columns were from Bio-Rad (Hercules, CA). Fibrinogen (FXIII-free peak 1) was from Enzyme Research Laboratories (South Bend, IN). Anti-GPIX antibody (Clone Xia.B4) was from Emfret analytics (Eibelstadt, Germany). Hematoxylin and eosin (H&E, Cat: 26043-05) was from Electron Microscopy Services (Hatfield, PA). Target retrieval solution (Cat: 00-4955-58) was from Invitrogen (Carlsbad, CA) and mouse-on-mouse blocking reagent, protein concentrate, and biotinylated anti-mouse IgG kit reagents (Cat: BMK-2202) were from Vector Laboratories (Burlingame, CA). Monoclonal anti-fibrin antibody (59D8) was a gift from Dr. Charles T. Esmon, Oklahoma Medical Research Foundation (Oklahoma City, OK). DAB (3,3′-Diaminobenzidine) chromogenic substrate and buffer (Cat: K3467) was from Dako (Glostrup, Denmark). Lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 mg/mL leupeptin) was from Cell Signaling (Danvers, MA).
Human PE.
PEs were obtained from the University of North Carolina (UNC) autopsy service with approval from the UNC Institutional Review Board. PE 1 was from a 50-year-old male with a history of coronary artery disease, hypertension, obesity, and tobacco use who presented on autopsy with saddle PE, multiple peripheral PEs, and pulmonary infarct. PE 2 was from a 17-year-old female with a history of pelvic germ cell tumor with metastasis to the lung, liver, and bone, and stage 2 kidney disease; femoral VT was detected by Doppler and D-dimer, and PE was found at autopsy. PE 3 was from a 63-year-old male with a history of coronary artery disease, myocardial infarction, hypertension, and Crohn’s disease; PE was found at autopsy. PE 4 was from a 46-year-old female with a history of lymphedema, hypertension, renal failure, obesity, and undifferentiated metastatic carcinoma likely of gynecologic origin. The patient presented with shortness of breath; autopsy revealed a large thrombus occluding the left pulmonary artery and smaller thrombi occluding the initial branches of the right pulmonary artery. Clots were fixed with 10% buffered formalin for 24–72 hours and stored in 70% ethanol at 4°C.
Mice.
Procedures were approved by the UNC Institutional Animal Care and Use Committee. F13a+/+, F13a+/−, and F13a−/− mice were backcrossed 6 generations on a C57BL/6J background.39 Additional C57BL/6N mice were obtained from Charles River Laboratories and maintained by homozygous breeding. Mice were maintained on a 12-hour light cycle and fed 2920X irradiated regular feed or 2919X irradiated breeder feed (Envigo, Huntingdon, UK). Male mice (8–12 weeks old) were used for all experiments and were anesthetized with 1.5% isoflurane in oxygen (2 L/min) or ketamine/xylazine (100/15 mg/kg).
Thrombin infusion PE model.
Human plasma-derived fibrinogen (FXIII-free peak 1) was labeled with an Alexa Fluor® 647 protein labeling kit, per manufacturer instructions. Anti-GPIX antibodies were incubated with IRDye 800CW for 1 hour at room temperature and eluted on a Bio-Spin 30 chromatography column. F13a+/+, F13a+/−, and F13a−/− mice were anesthetized with isoflurane and then injected retro-orbitally with Alexa Fluor® 647-labeled fibrinogen (0.15 mg/mL, final plasma concentration) and 800-labeled anti-GPIX antibodies (1 μg/g body weight), followed by thrombin injection (1250 U/kg) via tail vein. Mice were then removed from anesthesia and monitored for 5 minutes. Symptoms of thrombotic shock were recorded as severe (impaired mobility, irregular respiration, and/or failure to regain consciousness), moderate (lethargy and/or shallow respiration), or none. Lungs were perfused with 10 mL of 1X phosphate buffered saline (3 mL/min) and harvested for imaging and histology.
VT models.
VT models were compared using wild-type C57Bl/6N mice. Electrolytic injury was induced via 3-minute application of 3-V direct current by touching the femoral vein surface with the blunt end of a 25-gauge needle connected to the anode and completing the circuit by contacting local subdermal tissue with the cathode, as described.40 FeCl3 injury was induced by exposing the dried femoral vein to 4% FeCl3 (1.0 × 2.0-mm filter paper) for 5 minutes. For the infrarenal vena cava (IVC) stasis model, the IVC was separated from the aorta just distal to the left renal vein by blunt dissection. Lumbar branches were cauterized and the IVC and side branches were ligated, as described.41 Thrombi from all 3 models were harvested at 24 hours. For electrolytic and FeCl3 models, thrombi were collected with the vein wall and weighed together. For the IVC stasis model, the thrombi were separated from the vein wall and weighed separately. Not all mice were analyzed by all methods for each model.
Thrombus analysis by western blot.
Thrombi from F13a+/+, F13a+/−, and F13a−/− mice were retrieved 24 hours after IVC stasis, and thrombus lysates were analyzed by immunoblotting, as described.8 Briefly, thrombi were homogenized in lysis buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (MP Biomedicals, Solon, OH). Fibrin was pelleted and dissolved in 50 mM dithiothreitol, 12.5 mM EDTA, and 8 M urea at 60°C overnight, diluted in 6X reducing SDS sample buffer (Boston BioProducts, Ashland, MA), boiled for 5 minutes, separated on 10% tris-glycine polyacrylamide gels (Bio-Rad, Hercules, CA), and transferred to polyvinylidene difluoride membranes (Invitrogen, Carlsbad, CA). Membranes were blocked (Odyssey Blocking Buffer, LICOR Biosciences, Lincoln, NE) and then incubated with rabbit anti-human fibrinogen polyclonal antibody (Dako A008002-2, Dako/Agilent, Santa Clara, CA) and IRDye 800CW-labeled goat anti-rabbit antibody (LICOR Biosciences, Lincoln, NE), each for 1 hour at room temperature. Bands were quantified by densitometry using ImageJ (v1.48), and band intensity was reported as total arbitrary fluorescence units (AFU).
Ligature removal (de-ligation) PE model.
Anesthetized mice were injected retro-orbitally with Alexa Fluor® 647-labeled fibrinogen and 800-labeled anti-GPIX antibodies and then subjected to IVC stasis. After 24 hours, a second surgery was performed to remove the ligature. Mice were allowed to recover and observed for symptoms of thrombotic shock. After an additional 24 hours (48 hours after IVC stasis was induced), lungs were perfused via the left ventricle and harvested. Thrombi were separated from the vein wall, weighed, fixed in 10% buffered formalin for 24 hours, and stored in 70% ethanol.
Visualization of PE.
Lungs were imaged on a GE Typhoon FLA-9000 Imager (GE Healthcare, Pittsburgh, PA) or LI-COR Odyssey FC Scanner (LICOR Biosciences). The lungs were then fixed in 10% buffered formalin for 24 hours and stored in 70% ethanol. Formalin-fixed lungs were dehydrated and paraffin-embedded. Consecutive, 5-μm sections were cut and mounted. Every 4th slide was stained with H&E at the UNC Animal Histopathology and Lab Medicine Core. For immunohistochemistry, antigen retrieval was performed in a 95°C Target Retrieval Solution water bath. Slides were blocked with mouse IgG blocking reagent and incubated with primary antibody (59D8, 1:1000, 1 hour, room temperature). Negative controls were stained in the absence of primary antibody. Slides were incubated with a biotinylated anti-mouse IgG secondary antibody (10 minutes) and horseradish peroxidase-conjugated streptavidin (10 minutes) before development with DAB chromogenic substrate.
Thrombus and PE analysis.
PE burden was characterized by incidence, size, and composition. Whole lung fluorescent area was quantified using Image J; total pixels of fluorescent areas corresponding to platelets and fibrin or fibrinogen (collectively fibrin[ogen]) were reported as percentage of total lung pixel area. PEs were also identified by histology and differentiated from clots formed post-mortem by defined criteria: 1) presence in pulmonary arteries and 2) layers of platelets, fibrin, and RBCs. PE incidence was confirmed by a blinded pathologist. RBC composition was quantified using Image J; pixels of demarcated RBC area(s) were reported as percentage of total thrombus/PE pixel area. Fibrin composition was quantified by immunohistochemistry staining of thrombi/PEs that were scored by 5 independent, blinded observers on a scale of 0 to 3. Scores of 1, 2, and 3 were considered weak, moderate, or strong, respectively.
Statistical methods.
Descriptive statistics (mean, standard deviation, median, and interquartile range) were calculated for each experiment. Normality of data was assessed by Shapiro-Wilk tests. Experiments with normally-distributed data were analyzed by ordinary 1-way analysis of variance with Dunnett’s post-hoc tests for between-group comparisons. Experiments with non-normally-distributed data were analyzed by Kruskal-Wallis testing with Dunn’s post-hoc test for between-group comparisons. Fibrin high molecular weight band intensity was analyzed by analysis of variance testing with Brown-Forsythe and Welch post-hoc testing to accommodate different standard deviations. P<0.05 was considered significant.
RESULTS
FXIII deficiency does not alter PE incidence in the thrombin infusion model.
We first characterized PE incidence, size, and composition in a classical model triggered by intravenous thrombin infusion.31–34 We infused mice with fluorescently labeled FXIII-free fibrinogen and anti-GPIX antibodies, and then infused the mice with thrombin (Figure 1A). In the 5-minute observation period post-injection, F13a+/+, F13a+/−, and F13a−/− mice showed similar survival and signs of thrombotic shock (Figure 1B, C); the mouse that exhibited severe shock did not survive. Fluorescence imaging indicated similar platelet and fibrin(ogen) content in perfused lungs from thrombin-infused F13a+/+, F13a+/−, and F13a−/− mice (Figure 1D, E). These data from the thrombin-infusion model of PE suggested FXIII does not alter PE risk.
Figure 1. In the thrombin infusion model, F13a+/+, F13a+/−, and F13a−/− mice have similar PE incidence.
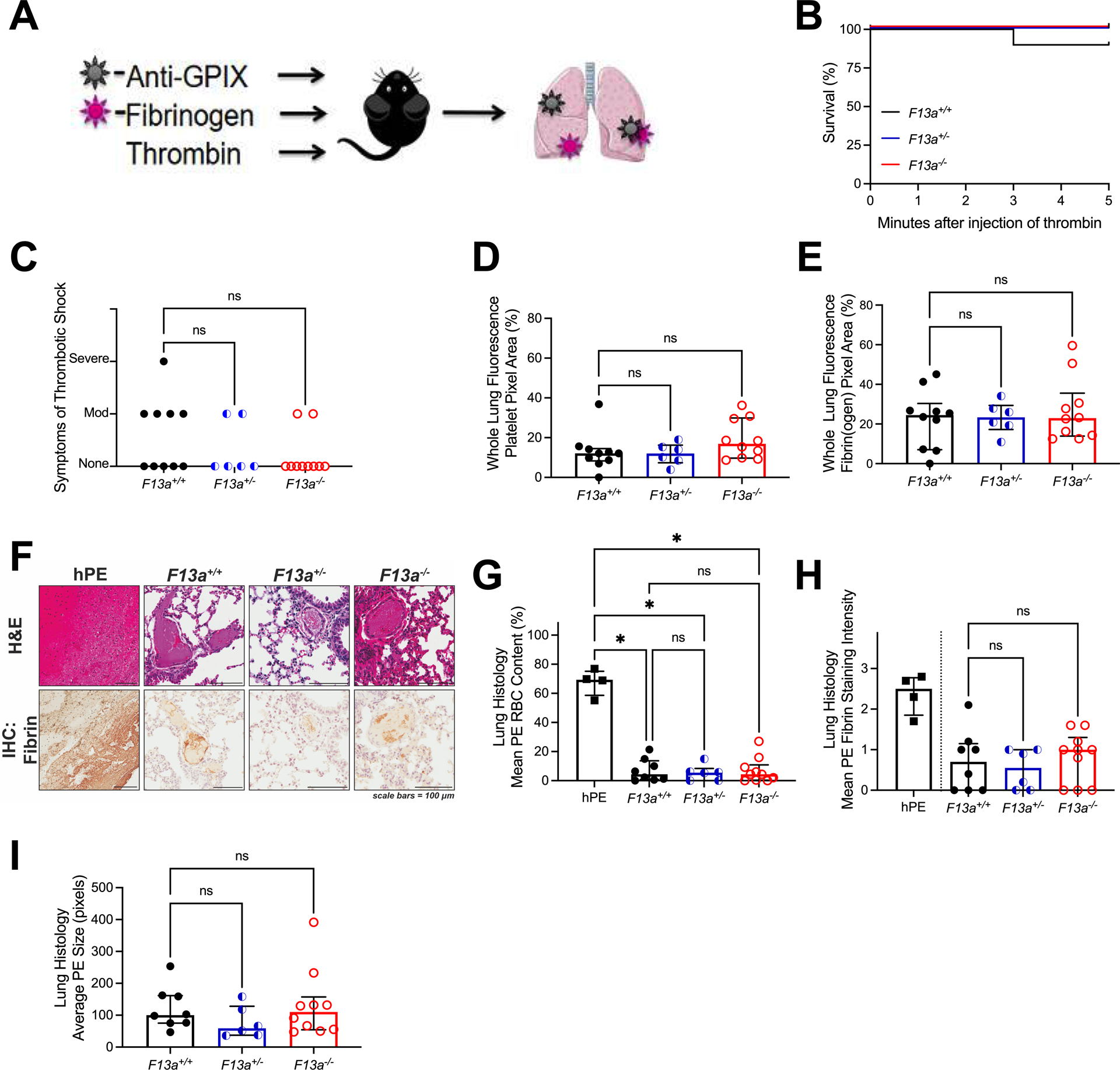
(A) F13a+/+ (N=10), F13a+/− (N=6), and F13a−/− (N=10) were injected retro-orbitally with Alexa Fluor-647 labeled fibrinogen and Alexa Fluor-800 labeled anti-GPIX antibodies and then injected intravenously with thrombin and observed. (B) Survival and (C) symptoms of shock. Mice were perfused after the observation period and the lungs were harvested. Fluorescent signal from (D) Alexa Fluor-800-anti-GPIX antibody and (E) Alexa Fluor-647-fibrin(ogen) in perfused lungs. Sections of human PE (hPE) and lung from thrombin-infused mice were analyzed by (F) H&E staining and immunohistochemistry for fibrin (brown staining); scale bars indicate 100 μm. (G) RBCs and (H) fibrin quantified from histology; fibrin staining in hPE is presented for illustrative purposes but not directly compared to mouse PE. (I) Average size of PE in thrombin-infused mice. Statistical comparisons were made by Kruskal-Wallis testing with Dunn’s post-hoc test. Dots represent individual mice; columns and error bars indicate median and interquartile range. *P<0.05; ns, non-significant
Thrombin infusion produces PEs histologically dissimilar to human PEs.
Previous studies have shown that VT and PE isolated from humans are characteristically rich in RBCs and fibrin.4–6 Accordingly, analysis of PE retrieved from human subjects demonstrated substantial RBC content (Figure 1F, G) and fibrin staining (Figure 1F, H). H&E staining of lungs from thrombin-infused F13a+/+, F13a+/−, and F13a−/− mice revealed small thrombi scattered throughout the lung vasculature that did not differ between genotypes in RBC content (Figure 1G), fibrin staining (Figure 1H), or size (Figure 1I). Given slight differences in antibody recognition of human and mouse proteins, fibrin staining of human and mouse thrombi could not be directly compared. However, for all genotypes, RBC content was significantly lower than that seen in human PE (Figure 1G). These data suggest PEs generated by thrombin infusion poorly recapitulate the cellular composition of human PEs.
IVC stasis produces thrombi that have similar RBC content as human PEs.
A major limitation of the thrombin infusion model is that the PEs do not originate from existing VT, but instead derive from microthrombi that form in circulation and become lodged in the lung microvasculature. To develop a model in which emboli arise from existing VT, we first generated thrombi in wild-type mice using 3 commonly-used mouse VT models: electrolytic/femoral vein, FeCl3/femoral vein, and IVC stasis.21 Both electrolytic and FeCl3 injury trigger thrombus formation within minutes, whereas thrombus formation initiated by IVC stasis takes place over several hours.20,21 We harvested thrombi from all 3 models at 24 hours for comparison to human PE. As anticipated20,21, thrombi produced by the femoral vein electrolytic and FeCl3 models arose quickly and were relatively small (3.3±2.2 and 2.5±1.7 mg, respectively), whereas thrombi produced by IVC stasis grew more slowly but were larger (12.0±7.9 mg) at harvest. Although thrombi produced by electrolytic and FeCl3 injury had significantly lower RBC content than human PE, RBC content of thrombi produced by IVC stasis was similar to that seen in human PE (Figure 2A, B). Analysis by immunohistochemistry showed similar fibrin staining in thrombi produced by all three mouse models (Figure 2A, C). Differences in the RBC content of thrombi generated by wild-type mice subjected to these experimental models of VT suggest IVC stasis better reproduces the RBC-rich composition of human VT.
Figure 2. Femoral vein electrolytic and FeCl3 models produce small thrombi with low RBC- and fibrin-content, whereas IVC stasis produces large, RBC-rich thrombi.
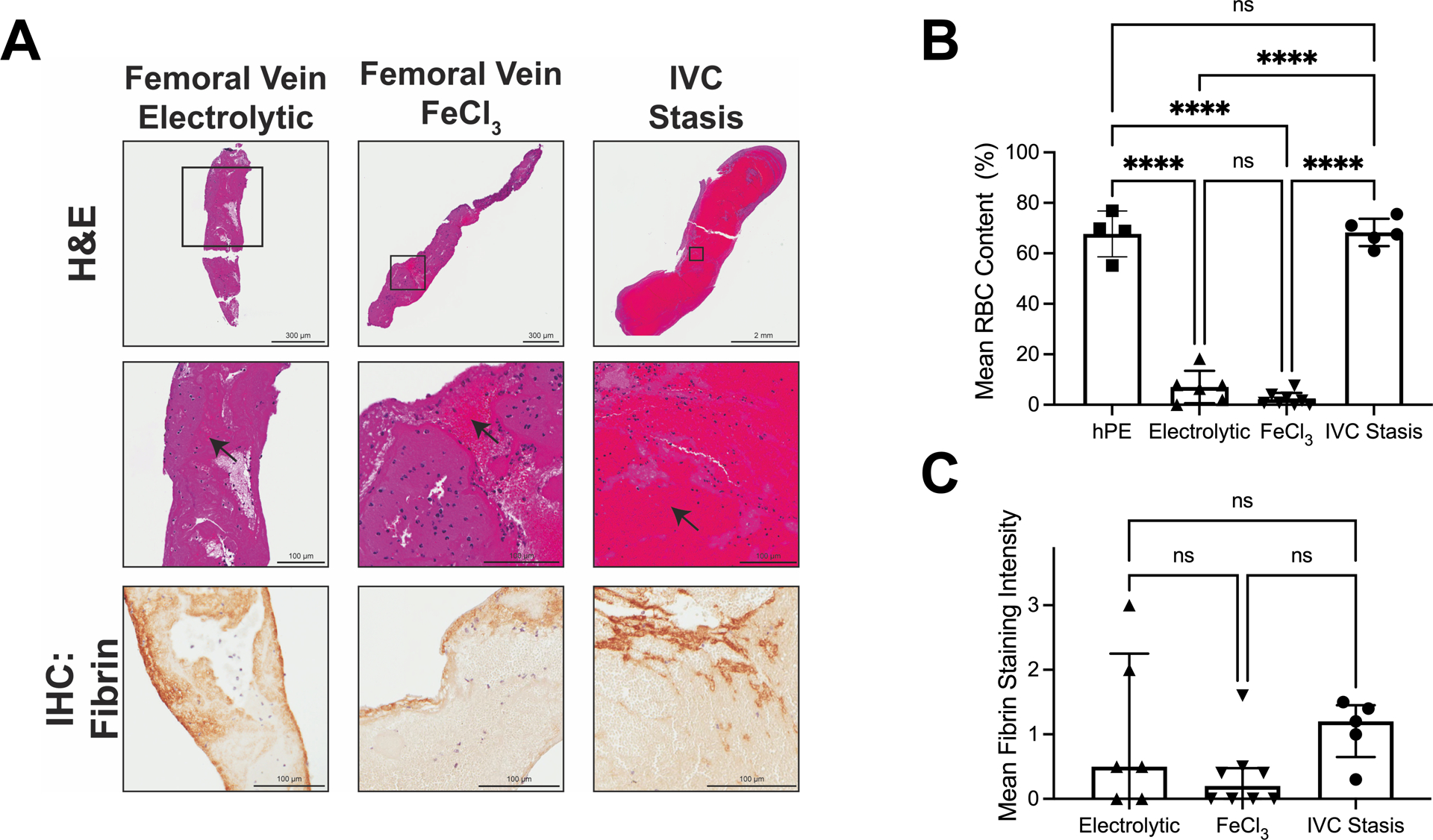
Thrombosis was induced using femoral electrolytic injury (N=6), femoral FeCl3 injury (N=8), and IVC stasis (N=5). (A) Representative whole thrombi (top row) and enlarged images (middle row) stained by H&E, and fibrin staining (brown, bottom row). Black arrows indicate areas rich in RBCs. (B) RBC content of thrombi from each model; data on human PE (hPE) are reproduced from Figure 1G. (C) Fibrin staining intensity of thrombi from each model. RBC data were normally distributed and compared using one-way ANOVA. The fibrin intensity data were not normally distributed and were compared using Kruskal-Wallis testing. Dots represent individual mice; columns and error bars indicate mean and standard deviation for RBCs, and median and interquartile range for fibrin. ****P<0.0001; ns, non-significant
De-ligation enables thrombus embolization, and complete FXIII deficiency increases incidence of PEs but partial FXIII deficiency does not.
We next developed the IVC stasis thrombosis model into a model of VTE. We first subjected mice to IVC stasis for 24 hours. As anticipated8,42, visual inspection of the IVC at 24 hours suggested thrombi were present in all F13a genotypes. We then removed the ligature and allowed mice to recover. After an additional 24 hours (48 hours after IVC ligation), mice were sacrificed, residual thrombus in the IVC was isolated and weighed, and lungs were perfused, harvested, and processed for fluorescence and histological analysis (Figure 3A). Compared to F13a+/+ and F13a+/− mice, F13a−/− mice had significantly decreased overall survival (Figure 3B), but no obvious signs of wound hemorrhage or bleeding around the IVC. In the 60-minute observation period after de-ligation, 4/23 (17%) F13a+/+, 5/17 (29%) F13a+/−, and 8/16 (50%) F13a−/− mice exhibited lethargy and irregular respiration consistent with clinical signs of thromboembolic events (Figure 3C) and all mice exhibiting severe shock died. Residual thrombus was apparent in the IVC of F13a+/+ and F13a+/− mice but not F13a−/− mice (Figure 3D), and some thrombi appeared attached to the vessel wall. Fluorescence imaging of whole perfused lungs indicated that compared to F13a+/+ and F13a+/− mice, F13a−/− mice had significantly increased fluorescence associated with platelets (Figure 3E) and a trend towards increased fluorescence associated with fibrin(ogen) (P=0.086, Figure 3F). Histological examination of perfused lungs confirmed this finding and showed that compared to F13a+/+ and F13a+/− mice, F13a−/− mice had increased PE incidence (25, 12, and 64%, respectively, Figure 3G). Thrombi were found in the peripheral and central lung regions (data not shown). The low incidence of PE in F13a+/− mice limited our ability to statistically compare these data to those from F13a+/+ and F13a−/− mice. However, PEs in F13a−/− mice were larger than PEs found in F13a+/+ mice (Figure 3H), suggesting most or all thrombus present in the IVC of F13a−/− mice at 24 hours embolized to the lungs upon de-ligation. H&E staining indicated that PEs in F13a+/+ mice had substantial RBC content (38±16%), whereas PEs in F13a+/− and F13a−/− mice showed proteinaceous material but few RBCs present (Figure 3I, J), consistent with previous findings that FXIII deficiency decreases RBC retention in VT.8,9,42 Immunohistochemical analysis suggested fibrin content of PEs did not differ significantly between genotypes (Figure 3I, K).
Figure 3. Following IVC stasis and de-ligation, complete FXIII deficiency increases embolization of RBC-rich PE, but partial FXIII deficiency does not.
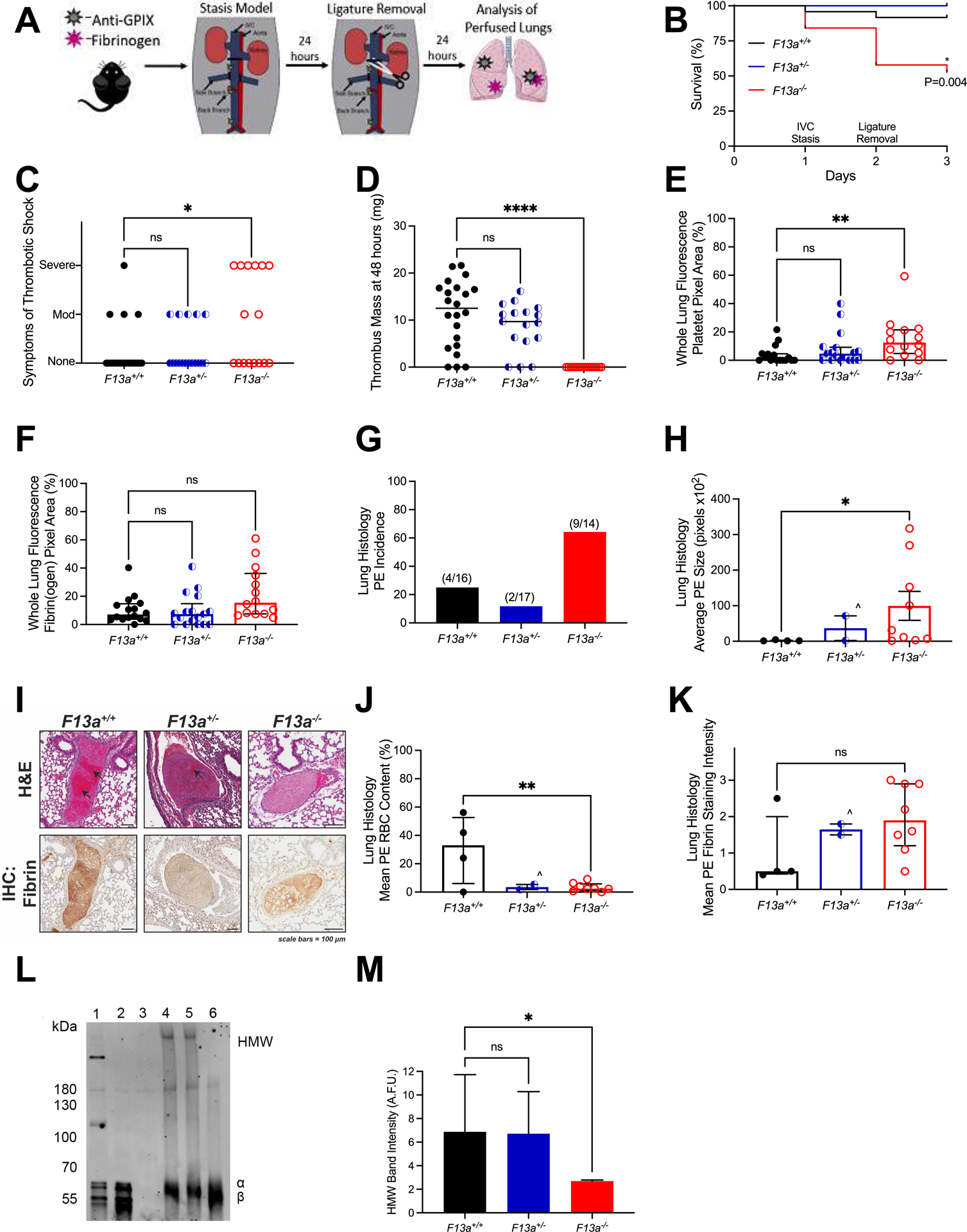
(A) F13a+/+ (N=24), F13a+/− (N=17), and F13a−/− (N=19) mice were injected retro-orbitally with Alexa Fluor-647 labeled fibrinogen and Alexa Fluor-800 labeled anti-GPIX antibodies and subjected to IVC stasis (day 1) followed by de-ligation (day 2). Mice were observed during recovery from anesthesia. (B) Survival and (C) symptoms of shock (days 2–3). Mice were perfused after the observation period and lungs were harvested. Fluorescent signal from (D) Residual IVC thrombus mass 24 hours after de-ligation. (E) Alexa Fluor-800-anti-GPIX antibody and (F) Alexa Fluor-647-fibrin(ogen) was quantified in perfused lungs. (G) PE incidence and (H) size from histology. (I) H&E staining and immunohistochemistry for fibrin (brown staining); black arrows indicate areas rich in RBCs. (J) Mean RBC content and (K) fibrin staining intensity. Statistical comparisons were made by one-way ANOVA or Kruskal-Wallis testing with Dunn’s post-hoc test, as appropriate. Dots represent individual mice; columns and error bars indicate median and interquartile range. For H, J, and K, ^ indicates that given N=2 PE for F13+/− mice, statistical comparison to this genotype was not performed. (L) Thrombi in a separate cohort of mice were harvested 24 hours after stasis induction, dissolved, and analyzed for fibrin crosslinking by SDS-PAGE and immunoblotting. Representative blot is shown. Lanes show platelet-poor plasma clot lysates from 1) F13a+/+ (positive fibrin crosslinking control), 2) F13a−/− (negative fibrin crosslinking control), and 3) Fgn−/− (negative control) mice, and thrombus lysates from 4) F13a+/+, 5) F13a+/−, and 6) F13a−/− mice. HMW, high molecular weight crosslinked fibrin species. (M) Densitometry of HMW band intensity. Bars show median and interquartile range for N=8, 5, and 3 for F13a+/+, F13a+/−, and F13a−/− mice, respectively. *P<0.05; ** P<0.01; ns, non-significant
Compared to F13a+/+ clots, plasma clots formed from F13a+/− mice have delayed crosslinking in vitro; however, F13a+/− clots become completely crosslinked over time.42 To measure thrombus crosslinking in F13a+/− mice in vivo, we harvested thrombi from a separate cohort of mice 24 hours after ligation and measured crosslinking by fibrin immunoblot. Whereas thrombi from F13a−/− mice showed no evidence of crosslinking, thrombi from both F13+/+ and F13+/− mice showed similar fibrin crosslinking 24 hours after ligation (Figure 3L, M), indicating partial (50%42) FXIII is sufficient to fulfill crosslinking function. Collectively, these data suggest IVC stasis followed by de-ligation permits embolization of pre-existing VT. Moreover, although complete FXIII deficiency (F13a−/−) increased PE incidence, partial deficiency (F13a+/−) did not.
DISCUSSION
Elucidating PE pathophysiology and improving strategies to identify individuals with increased PE risk is paramount. Previous epidemiological and biochemical studies have associated abnormal fibrin(ogen)43–49 and/or FXIIIa activity11,12,14 with abnormal thrombus stability and PE incidence. However, extension of these observations to the in vivo setting has been limited by a relative lack of animal models that recapitulate key characteristics of human VTE: embolization of existing RBC-rich VT to the lungs. Herein, we developed a new mouse model of VTE in which thrombosis is initiated by venous blood stasis in situ, and subsequent embolization of these RBC- and fibrin-rich thrombi to the lungs is permitted by de-ligation. Our study advances studies of VTE in several ways. First, our analysis provides a head-to-head comparison of common mouse models of VT and reveals that thrombi generated by electrolytic or FeCl3 injury differ from the composition of human PEs, whereas thrombi generated by IVC stasis are histologically more similar to human PEs. Second, our data show that de-ligation after IVC thrombus formation permits thrombus embolization and illustrate the feasibility of this approach as a mouse model of PE. Third, using this model, we show that whereas complete FXIII deficiency increases PE incidence, partial deficiency does not. These findings confirm earlier findings that complete FXIII deficiency increases embolization from FeCl3-induced thrombi in mice11,12 and extend knowledge to the broader and more common setting of partial FXIII deficiency.
A growing body of evidence shows a critical role for the type of experimental model in defining molecular mediators of hemostasis and thrombosis. For example, compared to FXIII-sufficient mice (F13+/+), FXIII-deficient mice (F13a−/−) show increased bleeding following tail transection, but not saphenous vein puncture.42 Moreover, mice with reduced FXIII antigen (F13a+/− or F13a−/−) or delayed FXIII activation (Fibγ390−396A) have normal arterial thrombus formation and no measurable reflow events following FeCl3 injury50,51, but reduced stasis- or stenosis-induced VT8,42. Collectively, these and other studies of FXIII function (8,11–13,42,50,52–54 and present work) suggest FXIII has non-overlapping roles in venous and arterial thrombosis. Thus, a major aspect of the current work investigating VTE is the use of a mouse model that recapitulates key aspects of VTE in humans. This model has several features. First, the de-ligation model is based on RBC- and fibrin-rich venous thrombi formed in situ via blood stasis. This setting better reflects cellular interactions that occur during the accrual of platelets, plasma proteins, and RBCs during VT than models in which normal flow is present. Although IVC stenosis also produces RBC- and fibrin-rich thrombi8, the higher reproducibility offered by IVC stasis compared to stenosis provides a more robust method for standardizing thrombus formation.21 Second, unlike PE models that involve intravenous infusion of platelet or coagulation activators that promote formation of microthrombi in circulation, PE in this de-ligation model originates from pre-existing thrombus. This scenario is consistent with the majority of patients with PE; although a subset of patients with PE do not demonstrate residual thrombus in a distal vein55–57, most PE present with evidence of DVT.58 A third advantage of the de-ligation model is control over the relative timing of thrombus formation and the start of embolization; the ability to synchronize these events facilitates comparisons between mice.
The finding that PE is increased in mice with complete FXIII-deficiency (11,12 and present study) but not in humans with congenital FXIII deficiency15 is unexpected and raises questions about the occurrence and outcome of VT in FXIII-deficient individuals. Since congenital FXIII deficiency is rare, affecting only 1 in ~3 million people worldwide16, the lack of reported PE in FXIII-deficient patients may reflect the small population of affected individuals. Alternatively, the lack of fibrin crosslinking in FXIII deficiency may enable rapid dissolution via endogenous fibrinolytic mechanisms (urokinase and/or tissue plasminogen activator22) in the lung, wherein any VT (with or without PE) that arise are transient and asymptomatic. Indeed, inhibition of crosslinking facilitates lysis of ex vivo-formed plasma clots deployed in mice and ferrets26,59, and a case report describing VT in a FXIII-deficient patient (3% FXIII) detected rapid thrombus resolution after initiation of treatment with low molecular weight heparin60. This effect may be due to decreased fibrin crosslinking and/or decreased crosslinking of antifibrinolytic molecules (e.g., α2-antiplasmin) to fibrin61. Given differences in thrombus composition, enhanced PE in F13a−/− mice subjected to FeCl3-mediated thrombosis11–13 or stasis-induced VT (present study) strengthens evidence supporting a specific role of FXIII in PE. Longer-term studies of thrombus resolution in FXIII-deficient mice are needed to characterize the ultimate fate of VT and PE and reconcile apparent discrepancies in PE incidence between FXIII-deficient humans and mice.
In both humans and mice, FXIII is present in plasma and cells.62 Although FXIII(a) appears to protect against PE, the relative contributions of plasma and platelet FXIII to clot stabilization have not yet been defined. Mitchell et al63 characterized the exposure of functional FXIII(a) on the surface of activated platelets, demonstrating its ability to crosslink fibrin. Interestingly, Gosk-Bierska et al64 detected reduced platelet FXIII mRNA in embolized thrombi compared to non-embolized thrombi in patients with atrial fibrillation, suggesting platelet FXIII mediates thromboembolic propensity. In this regard, it is interesting to note the increased fluorescence signal associated with platelets in the lungs of F13a−/− mice (Figure 3E), suggesting these RBC-depleted PE are composed primarily of platelets. Future application of this de-ligation model may help define the FXIII compartment that mediates PE risk, as well as other PE risk factors, including abnormal fibrin structure. Although the de-ligation model is sensitive to variables that increase PE risk, evaluation of VT associated with paradoxically decreased PE risk (e.g., factor V Leiden65) may be more challenging given the relatively low incidence of PE in wild-type mice.
This study has potential limitations. First, although IVC ligation produces an occlusive thrombus with histological characteristics of human VT and PE, it does not recapitulate all aspects of VT pathogenesis in humans. Notably, whereas venous thrombi in humans are thought to arise in valve pockets and extend in the direction of blood flow5, thrombi produced by IVC ligation initiate at the ligature site and extend distally, against blood flow. However, comparable to the situation in valve pockets, the IVC ligation creates an area of vortical flow that promotes hypoxia at the anatomical origin of the thrombus. IVC ligation followed by de-ligation was recently used by Li and colleagues to investigate time-restricted effects of blood flow restoration on VT and vein wall injury.66 Although their study did not investigate implications for PE, the observation that early, but not late, restoration of blood flow can improve experimental VT resolution was consistent with findings in human patients enrolled in the Acute Venous Thrombosis: Thrombus Removal with Adjunctive Catheter-Directed Thrombosis (ATTRACT) trial, suggesting this mouse model is clinically relevant. Second, although anti-fibrin antibody 59D8 was raised against human fibrin, we and others have shown it can also detect mouse fibrin.67–69 However, we have observed slightly higher staining intensity for mouse fibrin than human fibrin by immunoblot.69 Thus, we are cautious when comparing staining intensity in human and mouse thrombi. In addition, we have detected slight cross-reactivity against fibrinogen by immunoblot (unpublished observation) that may reflect conformational flexibility at the fibrinopeptide cleavage site, so some staining may indicate fibrinogen. However, fibrin(ogen) did not differ significantly between mouse genotypes. Third, embolic events in humans are spontaneous, whereas the de-ligation model requires a second surgery to permit embolism. Fourth, in humans, clinical data suggest some PEs are associated with residual thrombus in the veins (consistent with structural instability), whereas others present without detectable residual thrombus (suggesting complete thrombus detachment from the vessel wall or perhaps thrombosis in situ).3,55,57 It is unclear whether this model reflects structural instability and/or a failure of vessel wall adherence. The observation that some thrombi appeared attached to the vessel wall following de-ligation in F13a+/+ and F13a+/− mice but not in F13a−/− mice suggests embolization in F13a−/− mice may reflect abnormal adherence of the thrombus to the vessel wall. It has been proposed that transglutaminase-mediated crosslinking and oligomerization of fibrinogen αC domains contribute to adhesion of clots to endothelial cell integrins.70,71 It is also possible that residual thrombus in F13a−/− mice lysed prior to the harvest. Future studies using the de-ligation model may test the role of these events and differentiate between structural instability versus poor endothelial adherence in PE risk.
In summary, we have developed a VTE model that is based on the in situ formation of thrombi with histological characteristics of human VT, and that permits subsequent embolization of thrombi to the lungs. Using this model, we show that complete FXIII deficiency increases PE incidence, but partial deficiency does not. Further, like VT from F13a−/− mice, PE from these mice also demonstrate reduced RBC content. Our data advance understanding of FXIII in VTE. We anticipate this model is an adaptable tool that can be used to evaluate the contribution of other factors to PE risk, as well as long-term PE progression and treatment.
ESSENTIALS.
Thrombin infusion and vein electrolytic and FeCl3 injury produce thrombi that are unlike human PE
IVC stasis followed by de-ligation permits embolization of pre-existing, RBC-rich thrombi
Complete FXIII deficiency increases PE incidence, but partial FXIII deficiency does not
The de-ligation model is an adaptable tool to evaluate PE risk factors in mice
Acknowledgements:
We thank the UNC Animal Surgery Core Laboratory, UNC Animal Histopathology and Lab Medicine Core, and UNC Translational Pathology Laboratory. We also thank Dr. Lori A. Holle for technical assistance and Dr. Matthew J. Flick for thoughtful suggestions.
Funding:
This study was supported by funding from the National Institutes of Health (Integrative Biology Training Program [T32HL69768 to UNC/SK], F31HL139100 to SK, and R01HL126974 and R01HL147894 to ASW).
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
REFERENCES
- 1.Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J. Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost. 2007;5(4):692–699. [DOI] [PubMed] [Google Scholar]
- 2.Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, Mackman N. Venous thrombosis. Nat Rev Dis Pri. 2015;1(1):15006. [DOI] [PubMed] [Google Scholar]
- 3.Huisman MV, Barco S, Cannegieter SC, Le Gal G, Konstantinides SV, Reitsma PH, Rodger M, Noordegraaf AV, Klok FA. Pulmonary embolism. Nat Rev Dis Pri. 2018;4:18028. [DOI] [PubMed] [Google Scholar]
- 4.Undas A, Stepien E, Rudzinski P, Sadowski J. Architecture of a pulmonary thrombus removed during embolectomy in a patient with acute pulmonary embolism. J Thorac Cardiovasc Surg. 2010;140(3):e40–41. [DOI] [PubMed] [Google Scholar]
- 5.Sevitt S The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27(7):517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chernysh IN, Nagaswami C, Kosolapova S, Peshkova AD, Cuker A, Cines DB, Cambor CL, Litvinov RI, Weisel JW. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep. 2020;10(1):5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muszbek L, Yee VC, Hevessy Z. Blood coagulation factor XIII: structure and function. Thromb Res. 1999;94(5):271–305. [DOI] [PubMed] [Google Scholar]
- 8.Aleman MM, Byrnes JR, Wang J-G, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124(8):3590–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam WA, Meijers JC, Ni H, Ariens RA, Wolberg AS. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015;126(16):1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauer P, Metzner HJ, Zettlmeissl G, Li M, Smith AG, Lathe R, Dickneite G. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88(6):967–974. [PubMed] [Google Scholar]
- 11.Shaya SA, Gani DM, Weitz JI, Kim PY, Gross PL. Factor XIII prevents pulmonary emboli in mice by stabilizing deep vein thrombi. Thromb Haemost. 2019;119(6):992–999. [DOI] [PubMed] [Google Scholar]
- 12.Shaya SA, Saldanha LJ, Vaezzadeh N, Zhou J, Ni R, Gross PL. Comparison of the effect of dabigatran and dalteparin on thrombus stability in a murine model of venous thromboembolism. J Thromb Haemost. 2016;14(1):143–152. [DOI] [PubMed] [Google Scholar]
- 13.Duval C, Baranauskas A, Feller T, Ali M, Cheah LT, Yuldasheva NY, Baker SR, McPherson HR, Raslan A, Bailey MA, Cubbon RM, Connell SD, Ajjan RA, Philippou H, Naseem KM, Ridger VC, Ariens RAS. Elimination of fibrin g-chain cross-linking by FXIIIa increases pulmonary emboism arising from murine inferior vena cava thrombi. Proc Natl Acad Sci USA. 2021;118(27):e2103226118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kucher N, Schroeder V, Kohler HP. Role of blood coagulation factor XIII in patients with acute pulmonary embolism. Correlation of factor XIII antigen levels with pulmonary occlusion rate, fibrinogen, D-dimer, and clot firmness. Thromb Haemost. 2003;90(3):434–438. [DOI] [PubMed] [Google Scholar]
- 15.Girolami A, Cosi E, Tasinato V, Peroni E, Girolami B, Lombardi AM. Pulmonary embolism in congenital bleeding disorders: intriguing discrepancies among different clotting factors deficiencies. Blood Coagul Fibrinolysis. 2016;27(5):517–525. [DOI] [PubMed] [Google Scholar]
- 16.Karimi M, Bereczky Z, Cohan N, Muszbek L. Factor XIII deficiency. Semin Thromb Hemost. 2009;35(4):426–438. [DOI] [PubMed] [Google Scholar]
- 17.Lawrie AS, Green L, Mackie IJ, Liesner R, Machin SJ, Peyvandi F. Factor XIII--an under diagnosed deficiency--are we using the right assays? J Thromb Haemost. 2010;8(11):2478–2482. [DOI] [PubMed] [Google Scholar]
- 18.Appel IM, Grimminck B, Geerts J, Stigter R, Cnossen MH, Beishuizen A. Age dependency of coagulation parameters during childhood and puberty. J Thromb Haemost. 2012;10(11):2254–2263. [DOI] [PubMed] [Google Scholar]
- 19.Walton BL, Lehmann M, Skorczewski T, Holle LA, Beckman JD, Cribb JA, Mooberry MJ, Wufsus AR, Cooley BC, Homeister JW, Pawlinski R, Falvo MR, Key NS, Fogelson AL, Neeves KB, Wolberg AS. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129(18):2537–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz JA, Obi AT, Myers DD Jr., Wrobleski SK, Henke PK, Mackman N, Wakefield TW. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(3):556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz JA, Saha P, Cooley B, Palmer OR, Grover SP, Mackman N, Wakefield TW, Henke PK, Smith A, Lal BK. Choosing a mouse model of venous thrombosis: a consensus assessment of utility and application. J Thromb Haemost. 2019;17(4):699–707. [DOI] [PubMed] [Google Scholar]
- 22.Bdeir K, Murciano JC, Tomaszewski J, Koniaris L, Martinez J, Cines DB, Muzykantov VR, Higazi AA. Urokinase mediates fibrinolysis in the pulmonary microvasculature. Blood. 2000;96(5):1820–1826. [PubMed] [Google Scholar]
- 23.Murciano JC, Harshaw D, Neschis DG, Koniaris L, Bdeir K, Medinilla S, Fisher AB, Golden MA, Cines DB, Nakada MT, Muzykantov VR. Platelets inhibit the lysis of pulmonary microemboli. Am J Physiol Lung Cell Mol Physiol. 2002;282(3):L529–539. [DOI] [PubMed] [Google Scholar]
- 24.Butte AN, Houng AK, Jang IK, Reed GL. Alpha 2-antiplasmin causes thrombi to resist fibrinolysis induced by tissue plasminogen activator in experimental pulmonary embolism. Circulation. 1997;95(7):1886–1891. [DOI] [PubMed] [Google Scholar]
- 25.Kirchhof K, Welzel T, Zoubaa S, Lichy C, Sikinger M, de Ruiz HL, Sartor K. New method of embolus preparation for standardized embolic stroke in rabbits. Stroke. 2002;33(9):2329–2333. [DOI] [PubMed] [Google Scholar]
- 26.Singh S, Houng A, Reed GL. Releasing the brakes on the fibrinolytic system in pulmonary emboli: unique effects of plasminogen activation and alpha2-antiplasmin inactivation. Circulation. 2017;135(11):1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schultz J, Andersen A, Gade IL, Ringgaard S, Kjaergaard B, Nielsen-Kudsk JE. A porcine in-vivo model of acute pulmonary embolism. Pulm Circ. 2018;8(1):2045893217738217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth SS, Reis ED, Vaananen H, Zhang W, Coller BS. Variable protection of beta 3-integrin--deficient mice from thrombosis initiated by different mechanisms. Blood. 2001;98(4):1055–1062. [DOI] [PubMed] [Google Scholar]
- 29.Konstantinides S, Schafer K, Neels JG, Dellas C, Loskutoff DJ. Inhibition of endogenous leptin protects mice from arterial and venous thrombosis. Arterioscler Thromb Vasc Biol. 2004;24(11):2196–2201. [DOI] [PubMed] [Google Scholar]
- 30.Beviglia L, Poggi A, Rossi C, McLane MA, Calabrese R, Scanziani E, Cook JJ, Niewiarowski S. Mouse antithrombotic assay. Inhibition of platelet thromboembolism by disintegrins. Thromb Res. 1993;71(4):301–315. [DOI] [PubMed] [Google Scholar]
- 31.Gresele P, Momi S, Berrettini M, Nenci GG, Schwarz HP, Semeraro N, Colucci M. Activated human protein C prevents thrombin-induced thromboembolism in mice. Evidence that activated protein c reduces intravascular fibrin accumulation through the inhibition of additional thrombin generation. J Clin Invest. 1998;101(3):667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumada T, Dittman WA, Majerus PW. A role for thrombomodulin in the pathogenesis of thrombin-induced thromboembolism in mice. Blood. 1988;71(3):728–733. [PubMed] [Google Scholar]
- 33.Paul W, Gresele P, Momi S, Bianchi G, Page CP. The effect of defibrotide on thromboembolism in the pulmonary vasculature of mice and rabbits and in the cerebral vasculature of rabbits. Br J Pharmacol. 1993;110(4):1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbash IM, Schenke WH, Halabi M, Ratnayaka K, Faranesh AZ, Kocaturk O, Lederman RJ. Experimental model of large pulmonary embolism employing controlled release of subacute caval thrombus in swine. J Vasc Interv Radiol. 2011;22(10):1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellery PE, Maroney SA, Cooley BC, Luyendyk JP, Zogg M, Weiler H, Mast AE. A balance between TFPI and thrombin-mediated platelet activation is required for murine embryonic development. Blood. 2015;125(26):4078–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leon C, Freund M, Ravanat C, Baurand A, Cazenave JP, Gachet C. Key role of the P2Y(1) receptor in tissue factor-induced thrombin-dependent acute thromboembolism: studies in P2Y(1)-knockout mice and mice treated with a P2Y(1) antagonist. Circulation. 2001;103(5):718–723. [DOI] [PubMed] [Google Scholar]
- 37.Weiss EJ, Hamilton JR, Lease KE, Coughlin SR. Protection against thrombosis in mice lacking PAR3. Blood. 2002;100(9):3240–3244. [DOI] [PubMed] [Google Scholar]
- 38.Okano M, Hara T, Nishimori M, Irino Y, Satomi-Kobayashi S, Shinohara M, Toh R, Jaffer FA, Ishida T, Hirata KI. In Vivo imaging of venous thrombus and pulmonary embolism using novel murine venous thromboembolism model. JACC Basic Transl Sci. 2020;5(4):344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Souri M, Koseki-Kuno S, Takeda N, Degen JL, Ichinose A. Administration of factor XIII B subunit increased plasma factor XIII A subunit levels in factor XIII B subunit knock-out mice. Int J Hematol. 2008;87(1):60–68. [DOI] [PubMed] [Google Scholar]
- 40.Cooley BC. In vivo fluorescence imaging of large-vessel thrombosis in mice. Arterioscler Thromb Vasc Biol. 2011;31(6):1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aleman MM, Walton BL, Byrnes JR, Wang JG, Heisler MJ, Machlus KR, Cooley BC, Wolberg AS. Elevated prothrombin promotes venous, but not arterial, thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013;33(8):1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kattula S, Byrnes JR, Martin SM, Cooley BC, Flick MJ, Wolberg AS. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood Advances. 2018;2(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Undas A, Zawilska K, Ciesla-Dul M, Lehmann-Kopydlowska A, Skubiszak A, Ciepluch K, Tracz W. Altered fibrin clot structure/function in patients with idiopathic venous thromboembolism and in their relatives. Blood. 2009;114(19):4272–4278. [DOI] [PubMed] [Google Scholar]
- 44.Morris TA, Marsh JJ, Chiles PG, Auger WR, Fedullo PF, Woods VL Jr. Fibrin derived from patients with chronic thromboembolic pulmonary hypertension is resistant to lysis. Am J Respir Crit Care Med. 2006;173(11):1270–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris TA, Marsh JJ, Chiles PG, Magana MM, Liang NC, Soler X, Desantis DJ, Ngo D, Woods VL Jr. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood. 2009;114(9):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zabczyk M, Plens K, Wojtowicz W, Undas A. Prothrombotic fibrin clot phenotype is associated with recurrent pulmonary embolism after discontinuation of anticoagulant therapy. Arterioscler Thromb Vasc Biol. 2017;37(2):365–373. [DOI] [PubMed] [Google Scholar]
- 47.Marsh JJ, Chiles PG, Liang NC, Morris TA. Chronic thromboembolic pulmonary hypertension-associated dysfibrinogenemias exhibit disorganized fibrin structure. Thromb Res. 2013;132(6):729–734. [DOI] [PubMed] [Google Scholar]
- 48.Martinez MR, Cuker A, Mills AM, Crichlow A, Lightfoot RT, Chernysh IN, Nagaswami C, Weisel JW, Ischiropoulos H. Enhanced lysis and accelerated establishment of viscoelastic properties of fibrin clots are associated with pulmonary embolism. Am J Physiol Lung Cell Mol Physiol. 2014;306(5):L397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Undas A Prothrombotic fibrin clot phenotype in patients with deep vein thrombosis and pulmonary embolism: a new risk factor for recurrence. Biomed Res Int. 2017;2017:8196256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang Z, Kattula S, Holle LA, Cooley BC, Lin FC, Wolberg AS. Factor XIII deficiency does not prevent FeCl3-induced carotid artery thrombus formation in mice. Res Pract Thromb Haemost. 2020;4(1):111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113(11):1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shebuski RJ, Sitko GR, Claremon DA, Baldwin JJ, Remy DC, Stern AM. Inhibition of factor XIIIa in a canine model of coronary thrombosis: effect on reperfusion and acute reocclusion after recombinant tissue-type plasminogen activator. Blood. 1990;75(7):1455–1459. [PubMed] [Google Scholar]
- 53.Kattula S, Bagoly Z, Toth NK, Muszbek L, Wolberg AS. The factor XIII-A Val34Leu polymorphism decreases whole blood clot mass at high fibrinogen concentrations. J Thromb Haemost. 2020;18(4):885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duval C, Ali M, Chaudhry WW, Ridger VC, Ariens RA, Philippou H. Factor XIII A-Subunit V34L Variant Affects Thrombus Cross-Linking in a Murine Model of Thrombosis. Arterioscler Thromb Vasc Biol. 2016;36(2):308–316. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz T, Hingorani A, Ascher E, Marks N, Shiferson A, Jung D, Jimenez R, Jacob T. Pulmonary embolism without deep venous thrombosis. Ann Vasc Surg. 2012;26(7):973–976. [DOI] [PubMed] [Google Scholar]
- 56.Lindblad B, Eriksson A, Bergqvist D. Autopsy-verified pulmonary embolism in a surgical department: analysis of the period from 1951 to 1988. Br J Surg. 1991;78(7):849–852. [DOI] [PubMed] [Google Scholar]
- 57.Diebold J, Lohrs U. Venous thrombosis and pulmonary embolism. A study of 5039 autopsies. Pathol Res Pract. 1991;187(2–3):260–266. [DOI] [PubMed] [Google Scholar]
- 58.Girard P, Musset D, Parent F, Maitre S, Phlippoteau C, Simonneau G. High prevalence of detectable deep venous thrombosis in patients with acute pulmonary embolism. Chest. 1999;116(4):903–908. [DOI] [PubMed] [Google Scholar]
- 59.Reed GL, Houng AK. The contribution of activated factor XIII to fibrinolytic resistance in experimental pulmonary embolism. Circulation. 1999;99(2):299–304. [DOI] [PubMed] [Google Scholar]
- 60.Almeida A, Khair K, Hann I, Liesner R. Unusual presentation of factor XIII deficiency. Haemophilia. 2002;8(5):703–705. [DOI] [PubMed] [Google Scholar]
- 61.Singh S, Houng AK, Reed GL. Venous stasis-induced fibrinolysis prevents thrombosis in mice: role of alpha2-antiplasmin. Blood. 2019;134(12):970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91(3):931–972. [DOI] [PubMed] [Google Scholar]
- 63.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124(26):3982–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gosk-Bierska I, McBane RD, Wu Y, Mruk J, Tafur A, McLeod T, Wysokinski WE. Platelet factor XIII gene expression and embolic propensity in atrial fibrillation. Thromb Haemost. 2011;106(1):75–82. [DOI] [PubMed] [Google Scholar]
- 65.van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol. 2008;28(10):1872–1877. [DOI] [PubMed] [Google Scholar]
- 66.Li W, Kessinger CW, Orii M, Lee H, Wang L, Weinberg I, Jaff MR, Reed GL, Libby P, Tawakol A, Henke PK, Jaffer FA. Time-restricted salutary effects of blood flow restoration on venous thrombosis and vein wall injury in mouse and human subjects. Circulation. 2021;143(12):1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hui KY, Haber E, Matsueda GR. Monoclonal antibodies to a synthetic fibrin-like peptide bind to human fibrin but not fibrinogen. Science. 1983;222(4628):1129–1132. [DOI] [PubMed] [Google Scholar]
- 68.Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S, Weiler H. Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood. 2004;103(5):1728–1734. [DOI] [PubMed] [Google Scholar]
- 69.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117(18):4953–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dallabrida SM, Falls LA, Farrell DH. Factor XIIIa supports microvascular endothelial cell adhesion and inhibits capillary tube formation in fibrin. Blood. 2000;95(8):2586–2592. [PubMed] [Google Scholar]
- 71.Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Transglutaminase-mediated oligomerization of the fibrin(ogen) alphaC domains promotes integrin-dependent cell adhesion and signaling. Blood. 2005;105(9):3561–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]