

Abstract
BACKGROUNDS AND AIMS:
Increased permeability is implicated in the pathogenesis of intestinal disease. In vitro and in vivo studies have linked downregulation of the scaffolding protein ZO-1, encoded by the TJP1 gene, to increased tight junction permeability. This has not, however, been tested in vivo. Here, we assessed the contributions of ZO-1 to epithelial barrier function and mucosal homeostasis.
METHODS:
Public GEO data sets and biopsies from inflammatory bowel disease (IBD) patients and healthy controls were analyzed. Tjp1f/f; vil-CreTg mice with intestinal epithelial-specific ZO-1 knockout (ZO-1KO.IEC) mice and Tjp1f/f mice littermates, without Cre expression, were studied using chemical and immune-mediated models of disease as well as colonic stem cell cultures.
RESULTS:
ZO-1 transcript and protein expression were reduced in IBD patient biopsies. Despite mildly increased intestinal permeability, ZO-1KO.IEC mice were healthy and did not develop spontaneous disease. ZO-1KO.IEC mice were hypersensitive to mucosal insults and displayed defective repair. Further, ZO-1-deficient colonic epithelia failed to upregulate proliferation in response to damage in vivo or Wnt signaling in vitro. ZO-1 associated with centrioles in interphase cells and mitotic spindle poles during division. In the absence of ZO-1, mitotic spindles failed to correctly orient, resulting in mitotic catastrophe and abortive proliferation. ZO-1 is, therefore, critical for upregulation of epithelial proliferation and successful completion of mitosis.
CONCLUSION:
ZO-1 makes critical, tight junction-independent contributions to Wnt signaling and mitotic spindle orientation. As a result, ZO-1 is essential for mucosal repair. We speculate that ZO-1 downregulation may be one cause of ineffective mucosal healing in IBD patients.
Keywords: inflammatory bowel disease, mitosis, mitotic spindle, Wnt, intestinal permeability
Lay summary:
Downregulation of the tight junction protein ZO-1 in disease causes only mild increases in tight junction permeability but profoundly impairs mucosal healing.
Graphical Abstract
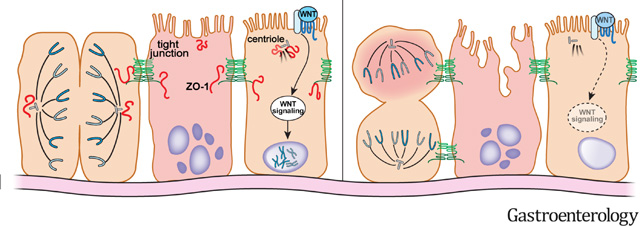
Introduction
Compromised intestinal barrier function, i.e., increased intestinal permeability, has been hypothesized to be a Crohn’s disease risk factor for over 40 years. This was based on the observation that, among Crohn’s disease patients in remission, increased permeability was associated with relapse1 as well as identification of a subset of healthy first-degree relatives of Crohn’s disease patients with increased intestinal permeability.2 A recent study assessed intestinal permeability in healthy first-degree relatives of Crohn’s disease patients and found that, of those who developed Crohn’s disease or ulcerative colitis, intestinal permeability was 50% greater than the relatives who remained disease-free.3 Thus, intestinal barrier dysfunction is a biomarker of Crohn’s disease risk. Despite the central role of the tight junction in barrier regulation, the potential contributions of specific tight junction proteins in disease development, progression, and remission have primarily been inferred by correlating expression changes in small groups of patients with in vitro studies of cultured monolayers.4–7
To screen tight junction protein expression in an unbiased manner, we interrogated public datasets from IBD patients and controls. We found consistent downregulation of the tight junction scaffolding protein zonula occludens-1 (ZO-1) and confirmed this immunohistochemically. Further study using genetically-modified mice identified previously unrecognized, yet critical, tight junction-independent ZO-1 contributions to Wnt-β-catenin signaling, mitotic spindle orientation, and effective epithelial repair.
Materials and Methods
Mouse studies followed protocols approved by Institutional Animal Care and Use Committees and used vil-mRFP1-ZO-1 (Tg(Vil1-mRFP1/TJP1)#Tjr, MGI:5584023), Tjp1f/f (Tjp1tm2c(KOMP)Wtsi, MGI:6272009), vil-Cre (B6.Cg-Tg(Vil1-cre)20Syr, MGI:3053819), vil-CreERT2 (B6.Cg-Tg(Vil1-cre/ERT2)23Syr/J, MGI:6278020), H2B-mCherry (R26-H2B-mCherry, CDB:CDB0239K), and GFP-β-actin (Pfn1tm2.1(GFP/ACTB)Wit, MGI:5568700) mice. Tjp1f/f littermates were used as WT controls for Tjp1f/f-vil-Cre (ZO-1KO.IEC) mice. All experiments using mice were performed at least 3 times with similar results. Further experimental details are available in the Supplemental Methods.
Results
ZO-1 expression is downregulated in inflammatory bowel disease
To comprehensively assess tight junction protein transcription in IBD, we screened the GDS3268 RNA microarray data set, which includes biopsies from 67 ulcerative colitis patients and 31 healthy control subjects.8 This confirmed published data showing increased CLDN1 and CLDN2 and reduced OCLN, CLDN4, and CLDN8 expression in ulcerative colitis (Fig 1A).6, 7 These changes were generally consistent across five additional ulcerative colitis and four Crohn’s disease data sets (Suppl. Table 1).
Figure 1. ZO-1 transcripts and protein are downregulated in ulcerative colitis, Crohn’s disease, and experimental immune-mediated IBD.

A. Relative transcript numbers for eleven tight junction proteins and E-cadherin are shown for 67 ulcerative colitis patients (orange triangles) and 31 healthy control subjects (purple triangles) from data set GDS3268. B, C. Quantitative immunofluorescence showing reduced ZO-1 (green) expression in colon biopsies from twenty ulcerative colitis patients (orange triangles) relative to thirteen healthy controls (purple triangles) and ileal biopsies from thirteen Crohn’s disease patients (orange triangles) relative to ten healthy controls (purple triangles). D. Quantitative immunofluorescence showing reduced ZO-1 expression (green) in colonic tissue from fifteen mice with adoptive transfer colitis relative to seventeen littermate controls that did not receive T cell transfer (blue circles). E-cadherin (white) and nuclei (blue) are shown for orientation in all micrographs. Bars = 20 μm and 10 μm (high magnification). Student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also found that TJP1 mRNA transcripts, which encode ZO-1, were markedly reduced in five of six of ulcerative colitis 8 and four of five Crohn’s disease data sets (Fig. 1, Suppl. Fig. 1). Among these, two ulcerative colitis data sets included biopsies from patients in remission, of which one (GSE59071) demonstrated that ZO-1 downregulation persisted while the other (GSE128682) showed restoration of ZO-1 expression during remission (Suppl. Fig. 1). ZO-1 downregulation may, therefore, be both a consequence and a cause of disease.
The mRNA analyses are inconsistent with previous studies reporting that ZO-1 downregulation in IBD is limited to sites of neutrophil transmigration.5, 7 We therefore assessed ZO-1 protein expression in IBD using a newly-developed antibody validated for quantitative morphometry.9 Colonic biopsies from 18 ulcerative colitis patients and 13 healthy controls as well as ileal biopsies from 13 Crohn’s disease patients and 10 healthy controls were evaluated. ZO-1 protein expression was reduced 3.9±0.03-fold in ulcerative colitis (Fig. 1B), and 5.3±0.03-fold in Crohn’s disease (Fig. 1C), and 8.7±0.06-fold in mice with immune-mediated, T cell transfer colitis (Fig. 1D). ZO-1 transcription and protein expression are, therefore, significantly downregulated in ulcerative colitis, Crohn’s disease, and immune-mediated, experimental IBD. The adoptive transfer colitis data further demonstrate that ZO-1 protein expression can be reduced in response to mucosal inflammation. The data do not, however, exclude the possibility of a vicious cycle where ZO-1 downregulation can also drive mucosal inflammation.
Intestinal epithelial-specific ZO-1 knockout does not cause spontaneous disease
Analysis of Tjp1 knockout mice is not possible, as these mice are not viable.10 We therefore generated intestinal epithelial-specific Tjp1 knockout mice (Fig. 2A).11 Expected numbers of pups expressing the villin-Cre transgene were born when Tjp1f/f; vil-CreTg (ZO-1KO.IEC) mice were crossed to Tjp1f/f (WT) mice. Moreover, ZO-1KO.IEC mice grew and gained weight identically to their WT littermates (Suppl. Fig. 2A). Despite the absence of Tjp1 transcripts and ZO-1 protein (Fig. 2A) within small intestinal and colonic epithelium, transcription of other tight junction proteins was largely unaffected (Fig. 2B). Moreover, no changes in tight junction protein transcription were detected in brain, lung, heart, liver, and kidney (Fig. 2B). Finally, transmission electron microscopy, which is unbiased with respect to specific proteins, did not detect ultrastructural tight junction abnormalities in ZO-1KO.IEC mice (Fig. 2C), although the thickened cortical actin and disordered microvilli reported previously were confirmed.11
Figure 2. ZO-1 deletion in intestinal epithelial cells results in only mild increases in paracellular leak pathway permeability.
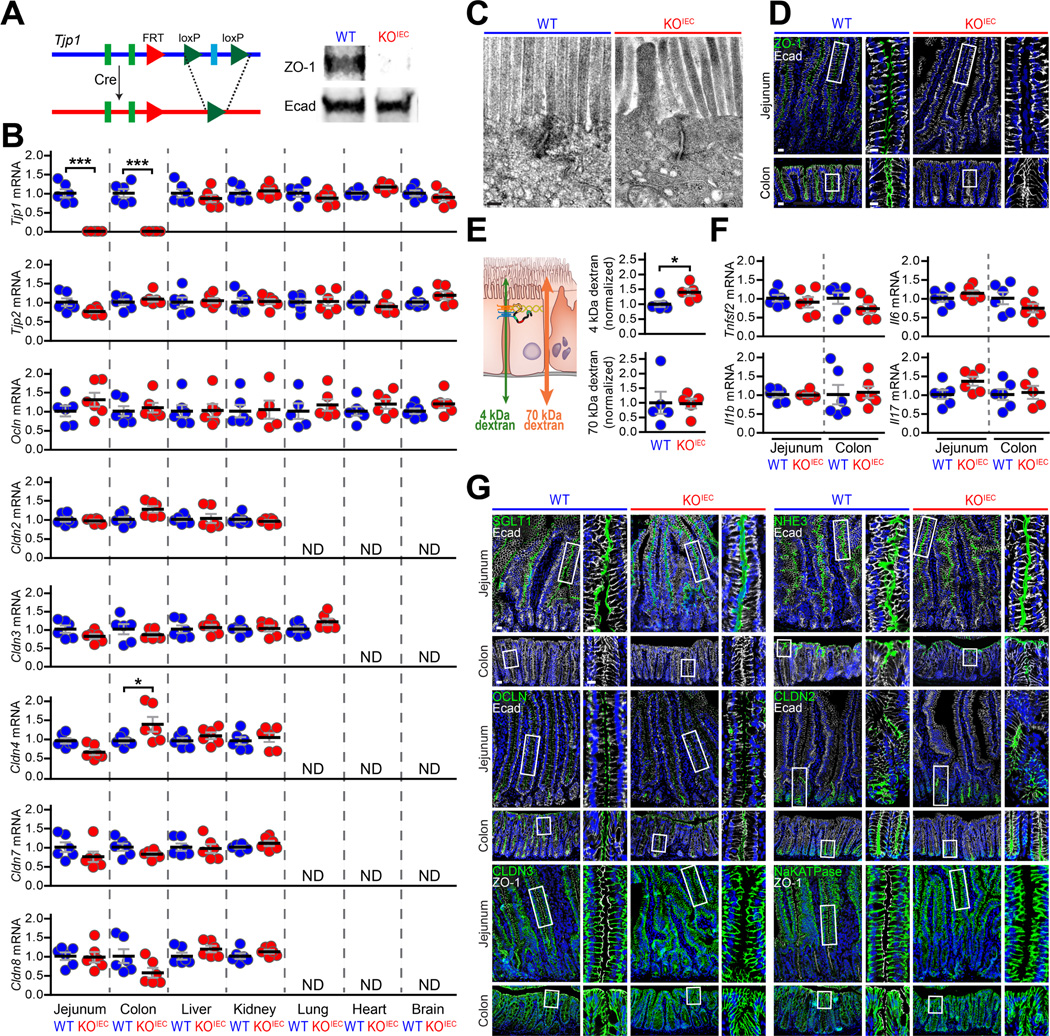
A. Targeting construct, deleted allele, and Western blots of isolated colonic epithelial cells from Tjp1f/f (WT) and Tjp1f/f; vil-CreTg (ZO-1KO.IEC) mice. B. qRT-PCR of transcripts from jejunal epithelial cells, colonic epithelial cells, kidney, lung, heart, and brain of ZO-1KO.IEC (red circles) and WT (blue circles) mice. Only Tjp1 (ZO-1) transcripts and, in the colon, Cldn4 (claudin-4) transcripts were affected. ND, not detected. n =6 per group. C. Representative transmission electron micrographs of jejunal epithelium from ZO-1KO.IEC and WT mice. There were no differences detected. D. Representative micrographs confirming loss of ZO-1 (green) expression in jejunum and colon of ZO-1KO.IEC mice. E. Permeability of the tight junction-dependent leak and tight junction-independent unrestricted pathway probes 4 kDa and 70 kDa dextran, respectively, demonstrating that ZO-1 deletion causes a small, but significant, increase in leak pathway permeability without affecting the unrestricted pathway. n =5 per group. F. qRT-PCR of mucosal transcripts show that ZO-1 deletion does not alter cytokine expression in jejunum or colon. n =6 per group. G. Representative immunofluorescence of indicated transport and tight junction proteins (green) in jejunum and colon of ZO-1KO.IEC and WT mice. E-cadherin (white) and nuclei (blue) are shown for orientation in all micrographs. Bars = 20 μm, 10 μm (high magnification), and 200 nm (electron micrographs). Student’s t test. *; P < 0.05; ***, P < 0.001.
Intestinal epithelial ZO-1 is not required for mucosal organization and barrier function
Conventional wisdom states that ZO-1 is essential for epithelial barrier function. We were therefore initially surprised at the absence of spontaneous disease in ZO-1KO.IEC mice. We first considered the possibility that some intestinal epithelial clones had escaped Tjp1 deletion, as has been reported previously in some Cre-mediated knockouts,12 but residual ZO-1 expression was not detected in small intestinal or colonic epithelium (Fig. 2D).
We next assessed in vivo intestinal barrier function of ZO-1KO.IEC mice. We focused on leak and unrestricted pathway permeabilities using 4 kDa dextran (28 Å diameter) and 70 kDa dextran (120 Å diameter), respectively,13, 14 because ZO-1 knockdown increases leak pathway permeability in vitro.15 Flux of 4 kDa dextran, but not 70 kDa dextran, was mildly increased in ZO-1KO.IEC mice (Fig. 2E), consistent with increased leak pathway, i.e., macromolecular, permeability in vivo. Notably, the absence of increased 70 kDa dextran flux indicates the absence of mucosal damage, i.e., unrestricted pathway upregulation.
Consistent with the absence of mucosal damage, transcripts for Tnfsf2, IL1β, Il6, and Il17 were unaffected by intestinal epithelial ZO-1 knockout (Fig. 2F). Further, expression and distribution of other epithelial transport and tight junction proteins were intact in ZO-1KO.IEC mice (Fig. 2G). ZO-1 deletion did, however, lead to reduced abundance of stem cells, enteroendocrine, Paneth, and tuft cells (Suppl. Fig. 2B, C) and mRNA transcripts marking these cell types (Suppl. Fig. 2D); Muc2 transcripts and goblet cells were similar in WT and ZO-1KO.IEC. Thus, despite mildly increased leak pathway permeability, intestinal epithelial ZO-1 knockout has only minor effects on mucosal function and, most importantly, is insufficient to cause disease.
Intestinal epithelial ZO-1 is critical for efficient mucosal repair following chemical injury
It is common for phenotypes of mice lacking critical genes, e.g., Rag1, Cldn2, and Ocln,4, 13, 16 to be revealed only after stress. ZO-1KO.IEC and WT littermates were therefore treated with 1% DSS for 6 days. This did not affect weight of WT mice but did induce a robust proliferative response within the crypts and transit-amplifying zone (Fig. 3A, B). In contrast, ZO-1KO.IEC mice displayed substantial weight loss and failed to significantly increase epithelial proliferation (Fig. 3A, B).
Figure 3. Mucosal repair is defective in ZO-1KO.IEC mice.
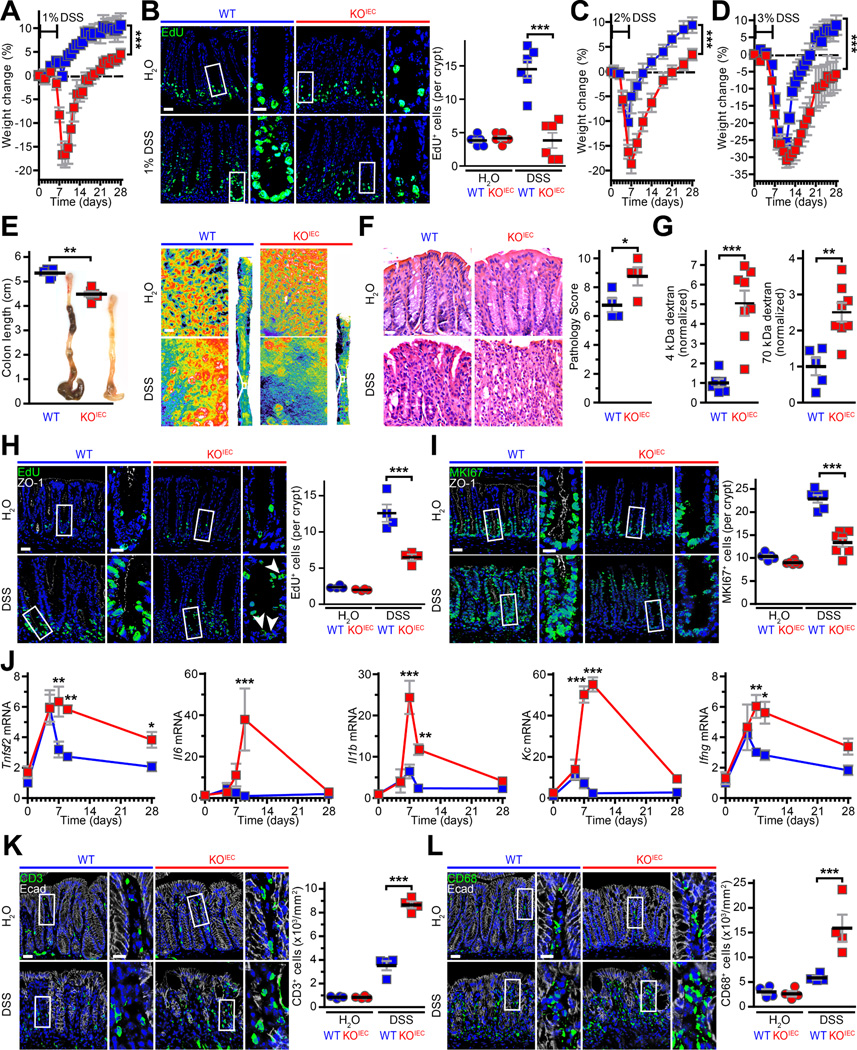
A. ZO-1KO.IEC (red squares) and WT (blue squares) mice were treated with 1% DSS for six days and then returned to normal water. Weight curves are shown. n = 4–5 per group. B. EdU (green) was administered 2hr before sacrifice. Treatment with 1% DSS markedly increased numbers of proliferating cells in WT, but not ZO-1KO.IEC, mice. n =4–5 per group. C, D. Weight curves of ZO-1KO.IEC and WT mice treated with 2% or 3% DSS for six days and allowed to recover. n = 6–7 per group. E. Colonic shortening after 2% DSS treatment is significantly greater in ZO-1KO.IEC relative to WT mice. Representative pseudocolor images of methylene blue-stained colons at low and high magnification show that crypts are completely lost in ZO-1KO.IEC but partially preserved in WT. n = 5–6 per group. F. Representative images and histopathology scores of ZO-1KO.IEC and WT mice without DSS exposure after (H2O) or after 6 days of 2% DSS treatment and one day of recovery (DSS). n = 4 per group. G. Permeability of both 4 kDa dextran and 70 kDa dextran is dramatically increased in 2% DSS-treated ZO-1KO.IEC relative to WT mice. This indicates greater mucosal damage, i.e., unrestricted pathway permeability, rather than tight junction leak pathway permeability increases. n =6–8 per group. H, I. EdU and MKI67 (green) stains shows similar numbers of proliferating cells in ZO-1KO.IEC and WT mice at baseline. However, only WT mice are able to significantly increase proliferation in response to 2% DSS. ZO-1 (white) and nuclei (blue) are shown for orientation. n =4–6 per group. J. qRT-PCR of mucosal transcripts before, after 6 days of 2% DSS treatment, wand during recovery shows both increased and prolonged cytokine elevation in ZO-1KO.IEC, relative to WT, mice. n =5 per group. K, L. Infiltration of CD3+ T cells and CD68+ macrophages (green) is markedly greater in 2% DSS-treated ZO-1KO.IEC, relative to WT, mice. E-cadherin (Ecad, white) and nuclei (blue) are shown for orientation. n =4 per group. Bars = 20 μm, 10 μm (high magnification), and 50 μm (pseudocolor gross image). Student’s t test (A-G) and ANOVA with Bonferroni’s correction (H-L). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To better understand their increased susceptibility, ZO-1KO.IEC mice and WT littermates were treated with 2% or 3% DSS (Fig. 3C, D). Treatment with 2% DSS both induced greater weight loss and delayed recovery in ZO-1KO.IEC, relative to WT, mice (Fig. 3C). Although the more intense insult of 3% DSS resulted in similar degrees of initial injury and weight loss in ZO-1KO.IEC and WT mice, ZO-1KO.IEC mice continued to demonstrate impaired recovery (Fig. 3D). Moreover, body weights of ZO-1KO.IEC mice never achieved those of DSS-treated WT littermates, even after prolonged periods of recovery. Taken together, the effects of 1%, 2%, and 3% DSS on ZO-1KO.IEC and WT mice demonstrate that epithelial ZO-1 deletion compromises mucosal repair.
As it encompassed features of 1% and 3% DSS, 2% DSS was selected for further analysis. After 2% DSS treatment colons of ZO-1KO.IEC mice were shorter, thickened, and more erythematous with greater areas of complete crypt loss and elevated histopathology scores relative to WT mice (Fig. 3E, F). Although 4 kDa dextran permeability increases were markedly greater in DSS-treated ZO-1KO.IEC mice relative to WT mice, this was due to flux across the unrestricted, rather than leak, pathway, as serum recovery of 70 kDa dextran was also far greater in ZO-1KO.IEC, relative to WT, mice (Fig. 3G) and both dextrans are able to traverse the unrestricted pathway (Fig. 2E).14 This contrasts with the specific leak pathway upregulation, indicated by increased flux of 4 kDa, not 70 kDa, dextran in unstressed ZO-1KO.IEC mice (Fig. 2E). As with 1% DSS, DSS (2%) failed to induce increases in DNA synthesis (Fig. 3H) or the MKI67 (Ki-67) proliferative index (Fig. 3I) in ZO-1KO.IEC mice. Moreover, mucosal inflammatory responses, measured as cytokines (Fig. 3J) as well as infiltration of CD3+ T cells (Fig. 3K) and CD68+ mononuclear cells, e.g., macrophages (Fig. 3L), were far greater in ZO-1KO.IEC mice.
Although the data demonstrate that ZO-1 is critical to mucosal healing and that ZO-1 loss leads to defective activation of repair processes. They do not, however, fully exclude the possibility that ZO-1 also limits sensitivity to DSS-induced injury. To address this, we exposed mice to TNBS, which induces damage via a different process than DSS. Weight loss, clinical scores, unrestricted pathway permeability increases, pathology scores, and cytokine production were all greater in ZO-1KO.IEC, relative to WT, mice and epithelial proliferative responses were markedly diminished (Figure S3). These similar effects of ZO-1 deletion in two distinct forms of chemically induced colitis that cause damage by different processes but share mechanisms of repair indicate that disruption of repair is the primary means by which ZO-1 loss enhances colitis severity.
Intestinal epithelial ZO-1 promotes mucosal healing after immune-mediated injury
DSS and TNBS cause severe colitis but are not representative of immune-mediated human disease. As a simple model of immune-mediated mucosal damage, disseminated T cell activation and cytokine storm were induced by treating mice with anti-CD3. This results in acute cytokine-driven diarrhea, without epithelial damage, that resolves by five hours after anti-CD3 administration.17 After a 12 to 18 hour asymptomatic interval, mice develop severe small intestinal mucosal damage and inflammatory diarrhea that remits by day 4. Despite similar damage 2 days after anti-CD3 administration (Fig. 4A), activation of both epithelial proliferation and repair were defective (Fig. 4B, Suppl. Fig. 4A), and epithelial apoptosis was increased (Suppl. Fig. 4B), in ZO-1KO.IEC mice relative to WT mice. This supports the conclusion, based on impaired recovery after 3% DSS treatment, that repair is the primary deficiency in ZO-1-deficient epithelial cells. Consistent with this, histologic damage as well as Il6, Ifng, and Il1b transcripts were greater in ZO-1KO.IEC mice than WT mice (Suppl. Fig. 4C). ZO-1 is, therefore, required for effective epithelial repair following immune-mediated mucosal damage.
Figure 4. Epithelial ZO-1 deletion delays and attenuates transcriptional responses to T cell activation-induced mucosal injury.

A. Representative images and histopathology scores of jejunum from ZO-1KO.IEC (red) and WT mice (blue) before, two days after, and four days after anti-CD3 treatment. Despite similar injury scores at 2 days, subsequent repair (at 4 days) was relatively ineffective in ZO-1KO.IEC mice. n = 5–7 per group. B. MKI67 (green) immunostains show similar numbers of proliferating cells in jejunal mucosal of ZO-1KO.IEC and WT mice at baseline. However, only WT mice are able to significantly increase proliferation at 2 days after anti-CD3 treatment. n = 5 per group. C. GSEA of RNAseq data from jejunal epithelia. Comparisons of ZO-1KO.IEC and WT transcripts before and 2 and 4 days after anti-CD3 treatment are shown (only comparisons between the genotypes under the indicated condition are shown). Activation of several repair pathways is markedly deficient in ZO-1KO.IEC mice. The green line in each graph shows, by rank, transcripts from the indicated pathway with increased (positive values) or reduced (negative values) expression. Vertical black lines indicate the location of individual genes on the green curves. Normalized p values and false discovery rate (FDR) q values are shown on each graph. D. qRT-PCR of representative transcripts before, two days after, and four days after anti-CD3 treatment confirm defective transcriptional activation in ZO-1KO.IEC jejunal epithelia. n = 4–8 per group. Bars = 20 μm and 10 μm (high magnification). ANOVA with Bonferroni’s correction. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To better understand the mechanisms responsible for defective repair responses in the absence of epithelial ZO-1 expression, jejunal epithelial cells were harvested from ZO-1KO.IEC and WT mice before as well as 2 and 4 days after anti-CD3 treatment. Gene set enrichment analysis (GSEA) of RNAseq data from isolated intestinal epithelium showed that, in the absence of exogenous stress, Wnt-β-catenin signaling was reduced in ZO-1KO.IEC relative to WT (Fig. 4C). These differences were confirmed by qRT-PCR analysis showing reduced expression of Wnt3, Lgr5, Olfm4, Ascl2, Bmi1, Hopx, and Notch1 transcripts in jejunal epithelial from ZO-1KO.IEC, relative to WT, mice (Suppl. Fig. 2D).
Comparisons between ZO-1KO.IEC and WT GSEA before and 2 and 4 days after anti-CD3 administration showed that activation of Wnt-β-catenin signaling, as well as mitotic spindle, G2/M checkpoint, DNA repair, oxidative phosphorylation, and glycolysis pathways, were defective in ZO-1KO.IEC at 2 days after anti-CD3 treatment (Fig. 4C). These differences were no longer present at day 4, primarily because the pathways were downregulated in WT (Fig. 4D); only minor activation was detected in ZO-1KO.IEC jejunal epithelial cells, even 4 days after anti-CD3 treatment. These data indicate that ZO-1 loss both delays and attenuates activation of repair responses.
ZO-1 knockout causes a cell-intrinsic proliferative defect
We turned to in vitro colonoid culture and 4-hydroxytamoxifen (4-OHT) inducible Cre to better characterize the proliferative defect in ZO-1-deficient epithelia. Colonic crypt cells from Tjp1f/f and Tjp1f/f; vil-Cre-ERT2Tg mice grew and proliferated indistinguishably in the absence of 4-OHT (Fig. 5A). 4-OHT extinguished ZO-1 expression in Tjp1f/f; vil-Cre-ERT2Tg colonoids (Fig. 5B) and markedly reduced growth and proliferation as measured by cross-sectional area and bud, i.e., crypt-like domain, numbers, respectively (Fig. 5C, D). In contrast, 4-OHT did not affect Tjp1f/f colonoids. Similar experiments comparing Tjp1f/f and Tjp1f/f; vil-CreTg colonoids yielded comparable results (Suppl. Fig. 5A).
Figure 5. ZO-1 deletion compromises epithelial proliferation in colonoid cultures.

A, B, C. Colonic epithelial stem cells were harvested from Tjp1f/f; vil-CreERT2Tg and Tjp1f/f mice. Two days after passage, 1 μM 4-hydroxytamoxifen (4-OHT) was added to the media as indicated. 4-OHT had no effect on Tjp1f/f colonoids but markedly reduced growth of Tjp1f/f; vil-CreERT2Tg colonoids. Representative images are shown. n = 18–20 per group. D. 4-OHT treatment effectively eliminated detectable ZO-1 (white) expression. Nuclei (blue) are shown for reference. E, F, G. Stem cells harvested from Tjp1f/f; vil-CreERT2Tg and Tjp1f/f mice were treated with 4-OHT, as indicated, and the GSK3 inhibitor CHIR99021 (5 μM). CHIR99021 enhanced growth and budding within Tjp1f/f; vil-CreERT2Tg and 4-OHT-treated Tjp1f/f colonoids, but was ineffective in 4-OHT-treated Tjp1f/f; vil-CreERT2Tg 2 colonoids. Representative images are shown. n = 18–22 per group. H, I. CHIR99021 enhanced EdU incorporation within Tjp1f/f; vil-CreERT2Tg and 4-OHT-treated Tjp1f/f colonoids, but was ineffective in 4-OHT-treated Tjp1f/f; vil-CreERT2Tg colonoids. n = 18–20 per group. J, K. CHIR99021 increase the fraction of cells in S-G2-M phases of the cell cycle within Tjp1f/f; vil-CreERT2Tg and 4-OHT-treated Tjp1f/f colonoids, but was ineffective in 4-OHT-treated Tjp1f/f; vil-CreERT2Tg colonoids. Representative histograms of flow cytometric data from propidium iodide (PI) stained cells are shown. n = 4–6 per group. Bars = 20 μm. ANOVA with Bonferroni’s correction. **, P < 0.01; ***, P < 0.001.
We attempted to rescue Wnt signaling and growth in ZO-1KO.IEC colonoids using the GSK3 inhibitor CHIR99021. This significantly increased growth and proliferation of Tjp1f/f; vil-Cre-ERT2Tg colonoids (without 4-OHT) and 4-OHT-treated Tjp1f/f colonoids but failed to stimulate 4-OHT-treated Tjp1f/f; vil-Cre-ERT2Tg colonoids (Fig. 5E, F, G). DNA synthesis (Fig. 5H, I) and participation in the cell cycle (Fig. 5J, K) were also defective in 4-OHT-treated Tjp1f/f; vil-Cre-ERT2Tg colonoids. Results using Tjp1f/f and Tjp1f/f; vil-Cre colonoids (Suppl. Fig. 5A) also demonstrated reduced expression of the stem and transit-amplifying cell markers Lgr5, Lrig1, Cdk2, and Pcna in ZO-1KO.IEC, relative to WT, colonoids (Suppl. Fig. 5B). Finally, in vitro analyses confirmed that ZO-1 deletion was responsible for the observed defects, as both basal and CHIR99021-augmented proliferation were rescued by transgenic mRFP1-ZO-1 expression in colonoids isolated from Tjp1f/f; vil-CreTg; vil-Cre-mRFP1-ZO1 mice (Suppl. Fig. 5C). ZO-1-deficiency, therefore, causes an epithelial-intrinsic proliferative deficiency that is insensitive to GSK3 inhibition and results in reduced numbers of stem and transit-amplifying cells.
ZO-1 deletion leads failure of mitotic spindle orientation, abortive proliferation, and apoptosis
We hypothesized that the relative health of unstressed ZO-1KO.IEC mice might reflect the ability of ZO-1-deficient epithelial cells to proliferate at near-normal basal levels despite being incapable of upregulating proliferation in response to injury. To assess this, EdU incorporation was monitored after 2 and 24 hours of continuous labeling. As expected, the number of labeled WT cells nearly doubled over this interval (Fig. 6A). In contrast, there was no significant change in the number of EdU-labeled ZO-1KO.IEC cells (Fig. 6A). Although less than half as many ZO-1KO.IEC cells, relative to WT, were labeled at 2 hours, the absence of any increase suggests that, in addition to a reduced proliferation rate, ZO-1KO.IEC cells were unable to progress and successfully complete the process of cell division. This defect is referred to as failure of mitotic progression and typically results in mitotic catastrophe, a form of apoptosis.18 Consistent with this, numbers of PO-PRO1-positive, 7-amino-actinomycin D-negative cells, i.e., apoptotic cells, were nearly 4-fold greater in ZO-1KO.IEC, relative to WT, colonoids (Fig. 6B). Apoptosis induced by mitotic catastrophe often involves caspase-3 activation; the number of cleaved caspase-3+ cells was more than 4-fold greater in ZO-1KO.IEC, relative to WT, colonoids (Fig. 6C).
Figure 6. ZO-1 is required for mitotic spindle orientation and productive mitosis.
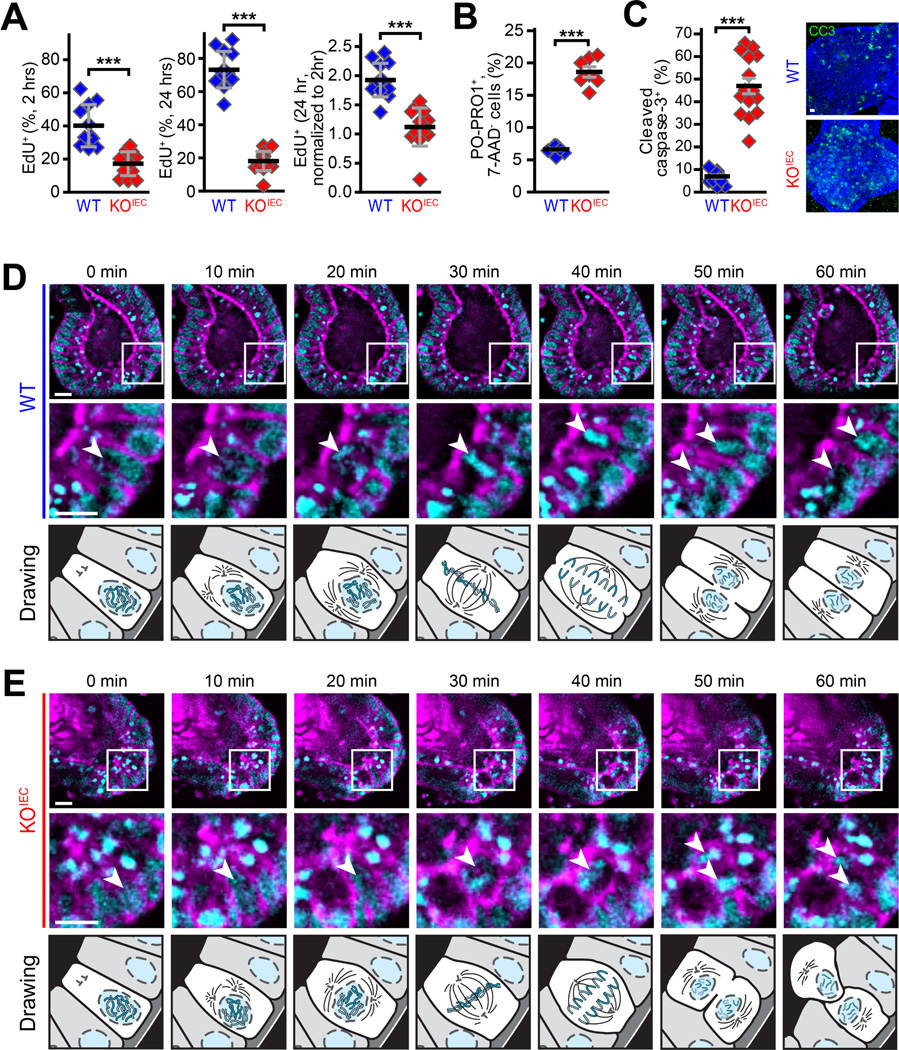
A. ZO-1KO.IEC and WT colonoids were treated with CHIR99021 and harvested two hours and twenty-four hours after EdU addition. Numbers of labeled cells increased markedly over this interval in WT but not ZO-1KO.IEC colonoids. n = 10–13 per group. B, C. Numbers of PO-PRO1+, 7-AAD− and cleaved caspase-3+ apoptotic cells are much greater in CHIR99021-treated ZO-1KO.IEC, relative to WT, colonoids n = 5–9 per group. D, E. Colonoids were grown from WT and ZO-1KO.IEC mice expressing EGFP-β-actin (magenta) and H2B-mCherry (cyan). Images were collected at ten minutes intervals. The mitotic spindle (arrows) is well oriented in WT (30 min) but not ZO-1KO.IEC (40 min) colonoids. The chromosomes separate according to the mitotic spindle planes 10 minutes later. This results in production of two daughter cells, both in contact with the basolateral matrix material, whose nuclei return to the monolayer in WT colonoids. In contrast, only one daughter cell maintains basolateral contact and returns to the monolayer in ZO-1KO.IEC; the second daughter cell is shed into the lumen. Mitotic events representative of the majority of cell divisions for each genotype are shown. The drawings below each set of images are presented to assist with simplify interpretation of the micrographs above. Bars = 10 μm. Student’s t test. *, P < 0.05; ***, P < 0.001.
The GSEA showing delayed and attenuated activation of mitotic spindle and G2/M checkpoint pathways in ZO-1KO.IEC suggested that ZO-1 might be required for mitotic spindle orientation. qRT-PCR of individual mRNA species confirmed delayed and attenuated transcription of centrosome and mitotic spindle components, including Tacc3, Aurka, Espl1, Nek2, and Plk1, and proteins involved in chromatid separation, identification of DNA damage sites, or mitotic progression, e.g., Top2a, Rad23b, Cdk1, Ube2c, and Cdc25b (Fig. 4D). We therefore considered the possibility that mitotic catastrophe could contribute to the failure of EdU+ ZO-1KO.IEC cells to accumulate over 24 hours of labeling.
To identify the stage at which cell division failed in ZO-1KO.IEC cells, colonoids expressing fluorescent-tagged β-actin and histone 2B were imaged during cell division. Mitotic events were easily recognized by apical nuclear displacement within the epithelial monolayer (Fig 6D). As the process matured, chromosomes segregated and WT cells oriented the spindle pole such that a line connecting the two centrioles was parallel to the basal surface of the monolayer. This was followed by formation of the metaphase plate and cell division in a plane perpendicular to the apical plasma membrane. The two daughter cells maintained contact with the basement membrane and their nuclei then returned to the monolayer.
Initial phases of mitosis in ZO-1KO.IEC colonoids were similar to WT. Mitotic spindle orientation was, however, nearly always defective in ZO-1KO.IEC colonoids (Fig. 6E). Mitotic spindles failed to orient correctly, with the metaphase plate was nearly perpendicular to the apical cell membrane, and, when cytokinesis completed, only one daughter nucleus returned to the monolayer (Fig. 6E). The second daughter cell lost contact with the basement membrane and the nucleus failed to return to the monolayer. This led to an accumulation of bright, compact nuclei consistent with the nuclear condensation that occurs during apoptosis (Fig. 6E). The data therefore indicate mitotic catastrophe and apoptosis occur in ZO-1KO.IEC cells due to defective spindle orientation. In sum, this abortive proliferative process fails to induce net cell number increases.
ZO-1 associates with the centriole and mitotic spindle and is required for mitotic spindle orientation in vivo
ZO-1 has been localized to the nucleus of actively-proliferating cells19 and is known to associate closely or bind directly to actomyosin, microtubules, and associated proteins,20 some of which are present at the mitotic spindle. To evaluate the potential association of ZO-1 with the mitotic apparatus during cell division, colonoids were prepared from transgenic mice that express mRFP-ZO-1 within the intestinal epithelium.21 Cytoplasmic foci of ZO-1 were frequently identified at the poles of dividing cells. Although these were difficult to recognize in individual z-sections because nuclei, spindles, and ZO-1 were typically in separate planes and colonoids actively move during culture in matrigel. ZO-1 spots at spindle poles processes were, however, easily detected using 3D reconstructions (Fig. 7A). These images revealed the appearance of 2 bright cytoplasmic ZO-1 foci as the nucleus of the dividing cell moved apically. One bright focus of ZO-1 remained associated with each newly-formed daughter nucleus as they moved back into the monolayer. ZO-1 therefore associates transiently with the mitotic apparatus during epithelial cell division.
Figure 7. ZO-1 associates with spindle poles in vitro and is required for effective mitosis in vivo.
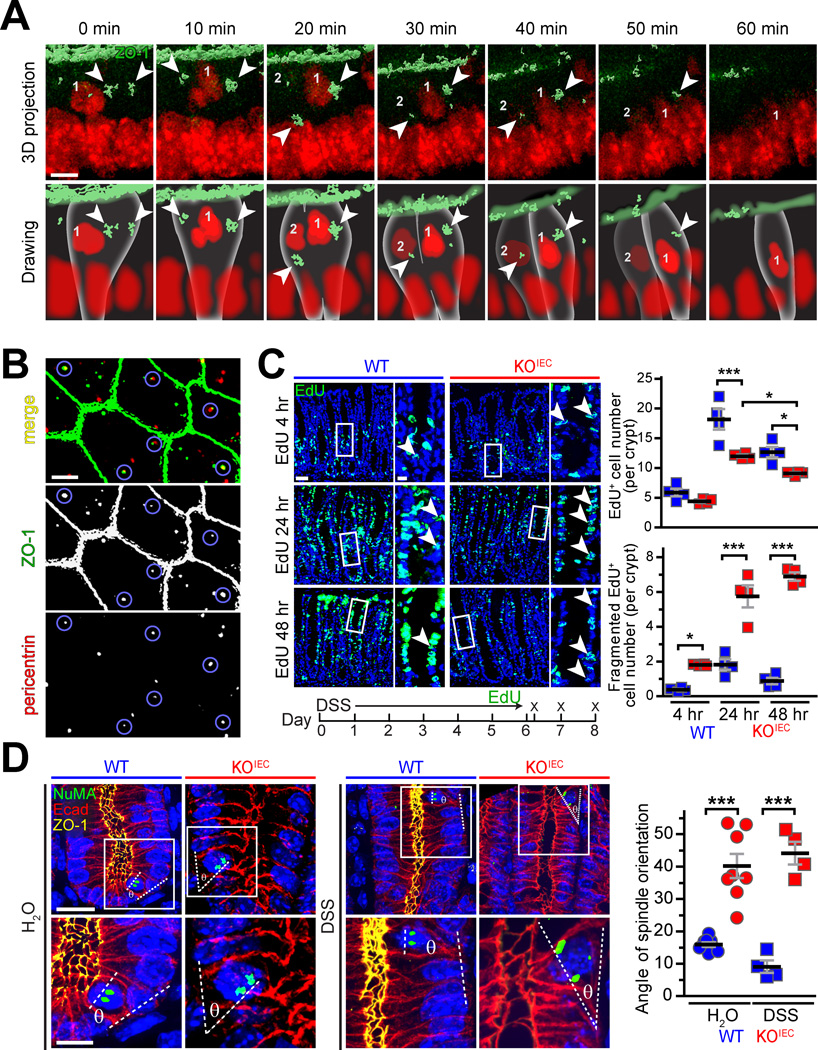
A. Colonoids from vil-mRFP1-ZO-1Tg mice were labeled with NucSpot (red). ZO-1 (green) surface contours were generated from 3-dimensional stacks. A representative mitotic event with ZO-1 localization at the spindle poles (arrows) is shown. The lower row of drawings is presented to assist with interpretation of the 3-dimensional image projections above. B. Immunostains of small intestinal epithelial cytopreps show ZO-1 (green) foci within the subapical cytoplasm. These overlap with pericentrin (red; sites of overlap are highlighted by blue circles), demonstrating that ZO-1 localizes to the centrole and pericentriolar apparatus in interphase intestinal epithelia. C. After six days of treatment with 2% DSS, mice were returned to water, injected with EdU (green), and sacrificed 4, 24, and 48 hours later. Increases in labeled cell numbers between 4 to 24 hours were far greater in WT, relative to ZO-1KO.IEC, colonic epithelium. Consistent with abortive proliferation, the number of apoptotic colonocytes with fragmented EdU staining increased markedly in ZO-1KO.IEC, but not WT, mice. n = 4 per group. D. Tissue sections from WT and ZO-1KO.IEC mice receiving water or 2% DSS were labeled for NuMa (green), E-cadherin (red), ZO-1 (yellow), and nuclei (blue). Mitotic events with two distinct NuMa foci showed apparent spindle misorientation in ZO-1KO.IEC (red) relative to WT (blue) mice. n = 4–8 per group. Bars = 5 μm, panel A; 5 μm, panel B; 40 μm and 10 μm (high magnification), panels C and D. ANOVA with Bonferroni’s correction. *, P < 0.05; ***, P < 0.001.
We have previously reported that, in transgenic mice, mRFP-ZO-1 is present both at tight junctions and in bright subapical foci.21 We considered the hypothesis that these might represent interphase centrioles, which are located within the subapical cytoplasm in the same z-plane as tight junction-associated ZO-1.22 To avoid potential artifacts due to fusion protein overexpression, intestinal epithelial cytopreps from wildtype (not Tjp1f/f) mice were fixed using −20°C methanol (to precipitate proteins in place); this allowed immunodetection of ZO-1 foci within the apical cytoplasm (Fig. 7B). These ZO-1 foci colocalized with pericentrin (Fig. 7B). Thus, in addition to associating with mitotic spindle poles during cell division, ZO-1 is present at interphase centrioles.
ZO-1 localization at mitotic spindles provides a potential explanation for failure of ZO-1KO.IEC mice to recover fully from mucosal injury. To determine whether abortive proliferation occurred in vivo, WT and ZO-1KO.IEC mice were injected with EdU immediately after ending 6 days of treatment with 2% DSS. Mice were sacrificed 4 hours, 24 hours, and 48 hours later. As expected, the numbers of EdU-positive colonocyte nuclei increased progressively in WT mice, with labeled nuclei throughout the expanded transit-amplifying zone at 24 hours (Fig. 7C). By 48 hours, numbers of labeled nuclei decreased and those that remained were concentrated within the upper one-third of the crypt, consistent with normal turnover (Fig. 7B). Numbers of EdU-positive nuclei also increased between 4 hours and 24 hours in ZO-1 KOIEC mice, albeit less robustly than in WT mice (Fig. 7C). However, labeled nuclei were scattered throughout the crypt and did not move to the surface as a wave in ZO-1 KOIEC mice (Fig. 7C). On closer examination, it was apparent that many of the labeled nuclei in ZO-1 KOIEC colons were fragmented nuclei, consistent apoptosis. The number of fragmented nuclei in ZO-1 KOIEC mice increased dramatically between 4 hours and 24 hours and remained elevated at 48 hours; this did not occur in WT mice (Fig. 7C). Thus, consistent with the in vitro observations, the in vivo data indicate that ZO-1 deletion results in abortive proliferation and apoptosis.
Finally, we asked if, similar to in vitro colonoid culture, spindle orientation was defective in ZO-1-deficient epithelia in vivo. To assess this, sections from control and DSS-treated WT and ZO-1KO.IEC mice were stained for ZO-1 and the nuclear mitotic apparatus protein (NuMA), an essential component of centrosomes.23 Spindle orientation was assessed by drawing a straight line connecting the NuMA-containing spindle poles and a second line along the basement membrane at that site. These lines should be nearly parallel, with an angle between them of less than ∼20°, if the spindle is correctly oriented.11 This was the case in WT epithelia, both without and after DSS treatment (Fig. 7D). In contrast, spindle orientation was frequently defective in ZO-1KO.IEC epithelia (Fig. 7D). Thus, as observed in vitro, ZO-1 deletion leads to defective orientation of the mitotic spindle in vivo. Taken as a whole, these data reveal a critical contribution of ZO-1 to mitotic spindle orientation in vivo and explain the means by which ZO-1 loss results in abortive proliferation and ineffective mucosal repair.
Discussion
ZO-1 was the first epithelial tight junction protein identified,24 but is also expressed in cells that do not form tight junctions, including blastomeres at the 8 cell stage of mammalian development.25 This contrasts with some other tight junction proteins whose expression is limited to epithelial and endothelial cells and whose knockout induces only subtle abnormalities.26–28 In contrast, knockout of Tjp1, which encodes ZO-1, leads to developmental defects as early as E8.5, extensive apoptosis of neuroectodermal tissues, failure of vascular development, and embryonic death.10 ZO-1 must, therefore, have critical functions separate from tight junctions. Consistent with this, ZO-1 knockdown or knockout in cell lines did not prevent tight junction assembly or development of barrier function. 15, 29, 30
ZO-1KO.IEC mice were born, grew, and gained weight normally, did not develop spontaneous disease, and displayed only a mild increase in leak pathway permeability. This suggests that mice are able to compensate for ZO-1 loss via mechanisms that do not depend on altered steady-state expression of other tight junction proteins. To unmask potentially critical ZO-1 functions in intestinal epithelia, ZO-1KO.IEC mice were challenged using 3 distinct approaches. The results, which were consistent across all three models, demonstrated enhanced mucosal damage, defective repair, and sustained inflammatory responses in ZO-1KO.IEC mice relative to WT littermates.
Our studies identified two separate, but synergistic, effects of ZO-1 loss. First, ZO-1-deficient epithelial cells were deficient in activation of proliferative, e.g., Wnt-β-catenin, and repair pathways following injury. This correlated with in vivo and in vitro proliferative defects in intestinal epithelial stem cells but contrasts sharply with previous in vitro studies showing accelerated growth of MDCK and embryonic stem cells after ZO-1 knockdown or knockout, respectively.29–31 Our studies suggest that these divergent results were not merely due to differences between in vivo and in vitro growth conditions. Moreover, we confirmed that the phenotypes observed are the result of ZO-1 loss, as transgenic mRFP1-ZO-1 expression rescued proliferation in ZO-1 knockout intestinal stem cells. ZO-1 loss therefore creates an epithelial-intrinsic proliferative deficit that is, at least in part, due to defective Wnt pathway activation. Although the molecular events responsible for this abnormality remain to be defined, the observations that ZO-1 can bind directly to α-catenin32 and that expression of a truncated ZO-1 protein lacking the α-catenin-binding GuK domain in MDCK cells, which causes epithelial-mesenchymal transition, activates β-catenin signaling33 provide some potential clues as to the underlying mechanism. In contrast, the in vivo defects observed here are distinct from the reported association of ZO-1 with MRCKβ at the leading edge of migrating non-polarized cells.34
At baseline, neither EdU incorporation nor MKI67 expression, both markers of dividing cells, were altered in ZO-1KO.IEC mice. This suggests that, in the absence of stress, intestinal epithelia lacking ZO-1 are able to proliferate at a level sufficient to maintain homeostasis despite reduced expression of Wnt pathway genes. However, ZO-1KO.IEC mice were unable to upregulate proliferation following injury. This also explains the in vitro growth defect observed in colonoids, as these cells are grown with media that contains growth factors including Wnt, Rspondin, EGF, and noggin.
Although profound, the proliferative failure could not explain increased apoptotic rates of ZO-1-deficient intestinal epithelia. Live imaging of colonoids demonstrated that ZO-1 deletion resulted in mitotic spindle misorientation. This caused one daughter cell to lose contact with the basement membrane and undergo anoikis or mitotic catastrophe-associated apoptosis. This resulted in an abortive proliferative process that failed to produce additional viable cells but did generate an excess of cleaved caspase-3-positive apoptotic cells.
Although the mechanisms by which DSS and TNBS induce colitis are incompletely defined, the protection afforded by reduced epithelial caspase-3 expression (in occludin knockout mice)4 indicates that caspase-3-dependent apoptosis is central to both. Moreover, the apoptosis of ZO-1-deficient intestinal epithelia induced by growth-promoting stimuli, both in vivo and in vitro, correlates with the massive apoptosis that occurs within rapidly-dividing neuroectodermal tissues of ZO-1 knockout embryos.10 Thus, although speculative, the data could support the idea that ZO-1 is primarily required under conditions where cell division must be accelerated. Delayed mitotic spindle orientation in ZO-1-deficient epithelia could therefore explain the consequences of ZO-1 loss in both unstressed and stressed mice.
The observed spindle misorientation is similar to our previous in vitro report of multi-lumen formation in ZO-1 knockdown MDCK cells grown in three-dimensional culture.30 We did not, however, detect multilumen enteroids or colonoids (not shown). One possible explanation could be that this reflects the difference between non-immortalized epithelial cells, such as those within colonoids, that undergo anoikis upon detachment from the basement membrane and immortalized cell lines are capable of anchorage-independent growth. In the latter, anchorage-independent outgrowths resulting from misoriented mitosis could be the origin of the septae that divide cysts into multiple lumens.
Our previous in vitro studies did not provide insight into the means by which ZO-1 directs mitotic spindle orientation. Here, we used live imaging to demonstrate a transient association between ZO-1 and spindle poles and immunostains to document the presence of ZO-1 within interphase centrioles. This was unexpected, as ZO-1 has not been previously reported to associate with the centrosome or bind directly to microtubules. Nevertheless, together with reports linking centrioles and primary cilia to Wnt, Hedgehog, and Notch signaling as well as microfilament organization (including microvilli),35–37 a potential regulatory function of ZO-1 at centrosomes could integrate the seemingly disparate phenotypes observed in ZO-1-deficient epithelia.
As a whole, these data should prompt reevaluation of studies in which a correlation between ZO-1 downregulation and increased intestinal permeability has led to the conclusion that there is a causal relationship. Our studies definitively demonstrate that ZO-1 is not required for barrier function and that, in the absence of stress, intestinal epithelia can compensate for ZO-1 loss. Nevertheless, ZO-1 is critical to epithelial repair and contributes to both Wnt-β-catenin signaling and mitotic spindle orientation. It is, therefore, likely that the ZO-1 downregulation in human and experimental inflammatory bowel disease compromises mucosal repair and, in doing so, promotes disease progression. Interventions that overcome these consequences of ZO-1 loss may, therefore, provide new means of enhancing mucosal healing and promoting resolution of active disease.
Supplementary Material
BACKGROUND AND CONTEXT
Intestinal barrier loss in patients with inflammatory bowel disease (IBD) is associated with reduced expression of the tight junction scaffold protein ZO-1, but the direct contributions of ZO-1 to barrier maintenance have not been studied in vivo.
NEW FINDINGS
ZO-1 expression was reduced in IBD patient biopsies. In mice, intestinal epithelial ZO-1 deletion modestly increases tight junction leak pathway permeability but does not cause profound barrier defects or spontaneous disease. Mucosal repair is, however, severely compromised in mice lacking intestinal epithelial ZO-1. This reflects defects in Wnt-β-catenin signaling and directs mitotic spindle orientation, both of which may be related to the new observation that ZO-1 accumulate around centrioles.
LIMITATIONS
These studies were performed using human tissue samples and genetically-modified mice. Further work is needed to define the mechanisms by which ZO-1 associates with centrioles, regulates mitotic spindle orientation, and promotes Wnt-β-catenin signaling. The impact of ZO-1 downregulation on mucosal healing in human disease also requires further study.
IMPACT
ZO-1 downregulation attenuates Wnt-β-catenin signaling and induces abortive epithelial proliferation to disrupt mucosal repair. Restoration of ZO-1 expression may promote healing.
Acknowledgments:
We thank Tiffany S. Davanzo (Slaybaugh Studios) for her beautiful illustrations and Heather Marlatt (Nationwide Histology) for her outstanding assistance with histologic staining and tissue microarray development.
Funding: This work was supported by National Institutes of Health grants R01DK61931 (JRT), R01DK68271 (JRT), R0DK099097 (CA), and P30DK034854 (the Harvard Digestive Disease Center) and by the National Natural Science Foundation of China grants 81800464 (LZ) and 82070548 (LZ).
Footnotes
Conflict of interest: GS is an employee of Abbvie, Inc. JRT is a founder and shareholder of Thelium Therapeutics and has served is a consultant for Entrinsic, Immunic, and Kallyope.
Resource sharing: Data, analytic methods, and study materials will be made available to academic investigators upon reasonable request.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993; 341:1437–9. [DOI] [PubMed] [Google Scholar]
- 2.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med 1986; 105:883–5. [DOI] [PubMed] [Google Scholar]
- 3.Turpin W, Lee SH, Raygoza Garay JA, et al. Increased Intestinal Permeability Is Associated With Later Development of Crohn’s Disease. Gastroenterology 2020; 159:2092–2100 e5. [DOI] [PubMed] [Google Scholar]
- 4.Kuo WT, Shen L, Zuo L, et al. Inflammation-induced Occludin Downregulation Limits Epithelial Apoptosis by Suppressing Caspase-3 Expression. Gastroenterology 2019; 157:1323–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kucharzik T, Walsh SV, Chen J, et al. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol 2001; 159:2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005; 129:550–64. [DOI] [PubMed] [Google Scholar]
- 7.Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007; 56:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noble CL, Abbas AR, Cornelius J, et al. Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut 2008; 57:1398–405. [DOI] [PubMed] [Google Scholar]
- 9.Nalle SC, Zuo L, Ong M, et al. Graft-versus-host disease propagation depends on increased intestinal epithelial tight junction permeability. J Clin Invest 2019; 129:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsuno T, Umeda K, Matsui T, et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell 2008; 19:2465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odenwald MA, Choi W, Kuo WT, et al. The scaffolding protein ZO-1 coordinates actomyosin and epithelial apical specializations in vitro and in vivo. J Biol Chem 2018; 293:17317–17335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kieckhaefer J, Lukovac S, Ye DZ, et al. The RNA polymerase III subunit Polr3b is required for the maintenance of small intestinal crypts in mice. Cell Mol Gastroenterol Hepatol 2016; 2:783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai PY, Zhang B, He WQ, et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017; 21:671–681 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanez-Paredes SD, Abtahi S, Kuo W-T, et al. Differentiating Between Tight Junction-Dependent and Tight Junction-Independent Intestinal Barrier Loss In Vivo. Methods in Molecular Biology: Springer US, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Itallie CM, Fanning AS, Bridges A, et al. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell 2009; 20:3930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raju P, Shashikanth N, Tsai PY, et al. Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice. J Clin Invest 2020; 130:5197–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clayburgh DR, Barrett TA, Tang Y, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest 2005; 115:2702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansilla S, Priebe W, Portugal J. Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle 2006; 5:53–60. [DOI] [PubMed] [Google Scholar]
- 19.Gottardi CJ, Arpin M, Fanning AS, et al. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc Natl Acad Sci U S A 1996; 93:10779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Itallie CM, Aponte A, Tietgens AJ, et al. The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks. J Biol Chem 2013; 288:13775–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchiando AM, Shen L, Graham WV, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol 2010; 189:111–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bacallao R, Antony C, Dotti C, et al. The subcellular organization of Madin-Darby canine kidney cells during the formation of a polarized epithelium. J Cell Biol 1989; 109:2817–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merdes A, Heald R, Samejima K, et al. Formation of spindle poles by dynein/dynactin-dependent transport of NuMA. J Cell Biol 2000; 149:851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevenson BR, Siliciano JD, Mooseker MS, et al. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol 1986; 103:755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming TP, McConnell J, Johnson MH, et al. Development of tight junctions de novo in the mouse early embryo: control of assembly of the tight junction-specific protein, ZO-1. J Cell Biol 1989; 108:1407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paschoud S, Bongiovanni M, Pache JC, et al. Claudin-1 and claudin-5 expression patterns differentiate lung squamous cell carcinomas from adenocarcinomas. Mod Pathol 2007; 20:947–54. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto K, Imasato M, Yamazaki Y, et al. Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology 2014; 147:1134–45 e10. [DOI] [PubMed] [Google Scholar]
- 28.Guillemot L, Schneider Y, Brun P, et al. Cingulin is dispensable for epithelial barrier function and tight junction structure, and plays a role in the control of claudin-2 expression and response to duodenal mucosa injury. J Cell Sci 2012; 125:5005–14. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Lim SB, Ng MY, et al. ZO-1 regulates Erk, Smad1/5/8, Smad2, and RhoA activities to modulate self-renewal and differentiation of mouse embryonic stem cells. Stem Cells 2012; 30:1885–900. [DOI] [PubMed] [Google Scholar]
- 30.Odenwald MA, Choi W, Buckley A, et al. ZO-1 interactions with F-actin and occludin direct epithelial polarization and single lumen specification in 3D culture. J Cell Sci 2017; 130:243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balda MS, Garrett MD, Matter K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol 2003; 160:423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh M, Nagafuchi A, Moroi S, et al. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J Cell Biol 1997; 138:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reichert M, Muller T, Hunziker W. The PDZ domains of zonula occludens-1 induce an epithelial to mesenchymal transition of Madin-Darby canine kidney I cells. Evidence for a role of beta-catenin/Tcf/Lef signaling. J Biol Chem 2000; 275:9492–500. [DOI] [PubMed] [Google Scholar]
- 34.Huo L, Wen W, Wang R, et al. Cdc42-dependent formation of the ZO-1/MRCKbeta complex at the leading edge controls cell migration. EMBO J 2011; 30:665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campos Y, Qiu X, Gomero E, et al. Alix-mediated assembly of the actomyosin–tight junction polarity complex preserves epithelial polarity and epithelial barrier. Nature Communications 2016; 7:11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyun M-L, Kim S-O, Lee HG, et al. Wnt3a Stimulation Promotes Primary Ciliogenesis through β-Catenin Phosphorylation-Induced Reorganization of Centriolar Satellites. Cell Reports 2020; 30:1447–1462.e5. [DOI] [PubMed] [Google Scholar]
- 37.Akhshi T, Trimble WS. A non-canonical Hedgehog pathway initiates ciliogenesis and autophagy. Journal of Cell Biology 2021; 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.