

Abstract
BACKGROUND:
The authors previously reported the results of the nonpancreatic neuroendocrine neoplasm cohort of the SWOG S1609 DART (Dual Anti–CTLA-4 and Anti–PD-1 Blockade in Rare Tumors) trial, which permitted all histologic grades and had a 44% overall response rate (ORR) among patients with high-grade disease. Here they sought to validate their findings in a dedicated prospective cohort of high-grade neuroendocrine neoplasms within S1609.
METHODS:
A prospective, open-label, multicenter, phase 2 clinical trial of ipilimumab plus nivolumab was conducted across multiple rare tumor cohorts. The dedicated, high-grade neuroendocrine neoplasm cohort was examined here. The primary end point was the ORR according to version 1.1 of the Response Evaluation Criteria in Solid Tumors. Secondary end points included progression-free survival (PFS), overall survival (OS), and toxicity.
RESULTS:
Nineteen patients with high-grade neuroendocrine neoplasms (defined by local pathology review) were enrolled in this cohort of S1609. The most common primary sites were unknown primaries (21%), which were followed by the rectum, gastroesophageal junction, cervix, and pancreas (11%). The median number of lines of prior therapy was 1 (range, 0–3). All patients were microsatellite-stable. The median Ki-67 value was 80%. The ORR was 26% (95% confidence interval [CI], 11%–45%), and the clinical benefit rate (stable disease for ≥6 months plus partial responses plus complete responses) was 32% (95% CI, 13%–57%). The 6-month PFS rate was 32% (95% CI, 16%–61%) with a median PFS of 2.0 months (95% CI, 1.8 months to ∞) and a median OS of 8.7 months (95% CI, 6.1 months to ∞). The most common toxicities were fatigue (32%) and rash (26%), and the most common grade 3/4 immune-related adverse event was rash (15%); there were no events that required treatment discontinuation and no grade 5 events.
CONCLUSIONS:
Ipilimumab plus nivolumab demonstrated a 26% ORR in patients with high-grade neuroendocrine neoplasms, with durable responses seen in patients with refractory disease.
Keywords: Dual Anti–CTLA-4 and Anti–PD-1 Blockade in Rare Tumors (DART), high-grade neuroendocrine neoplasms, ipilimumab, S1609, nivolumab
INTRODUCTION
Cancer immunotherapy, particularly immune checkpoint blockade, has dramatically improved outcomes for patients with cancer, including rare tumors such as Merkel cell carcinoma and anal cancer.1,2 However, there is little information on the potential benefit from immune checkpoint blockade for the vast majority of rare tumors. To address this, we launched the SWOG S1609 DART (Dual Anti–CTLA-4 and Anti–PD-1 Blockade in Rare Tumors) trial, a federally funded basket immunotherapy study investigating multiple rare tumors and lower dose ipilimumab with nivolumab. The trial is investigating combinatorial immune checkpoint blockade across 52 rare tumor types and is open across the United States at 861 sites, with 734 accruals in the past 3 years; thus, it is dispelling the notion that rare tumor clinical trials are not feasible.
We have previously reported our results with ipilimumab and nivolumab across all grades of nonpancreatic neuroendocrine neoplasms with a 44% overall response rate (ORR) in patients with high-grade disease versus a 0% ORR in patients with low-or intermediate-grade tumors.3 The ORR by grade analysis was not prespecified; however, because of the magnitude of difference by grade as well as the durability of responses in heavily pretreated patients with metastatic disease, further study in a new, prospective cohort was pursued. We describe here the clinical activity of ipilimumab and nivolumab in a separate, dedicated cohort of high-grade neuroendocrine neoplasms within S1609.
MATERIALS AND METHODS
The trial was conducted by SWOG, and the investigational agents were provided by the Cancer Therapy Evaluation Program of the National Cancer Institute (NCI) under an NCI collaborative research and development agreement with Bristol-Myers Squibb. The protocol and all amendments were approved by SWOG, the NCI, the NCI central institutional review board, and the regulatory committees at the participating institutions. All study subjects provided their voluntary, written informed consent, and the study was conducted in accordance with the Declaration of Helsinki.
Rationale for Population
Rare cancers for the S1609 cohort design were identified on the basis of an incidence of fewer than 6 in 100,000 per year.4 The cohort of high-grade neuroendocrine neoplasms was opened on the basis of ipilimumab/nivolumab activity in the aforementioned subset analysis of a neuroendocrine neoplasm cohort within S1609. Notably, this high-grade neuroendocrine cohort was distinct from and was accrued independently of previously reported neuroendocrine cohorts in S1609.3 Local pathology review was used with pathology report review by the SWOG study team. Neuroendocrine neoplasm grading was based on 2010 World Health Organization criteria; however, no separate central pathology confirmatory review was performed. Lung primary neuroendocrine neoplasms were considered eligible if the mitotic rate or Ki-67 index was >20% with the exception of small cell lung cancers, which were excluded. The pancreas as the primary site was permitted for this cohort if the tumor was high-grade. Treatment-emergent small cell neuroendocrine prostate cancer was assigned to a separate cohort of S1609 because of its unique clinical and pathologic characteristics. Microsatellite instability status, Ki-67, and primary organ site information was collected as part of the study.
Patient Selection
Patients were required to be 18 years old or older and have an Eastern Cooperative Oncology Group performance status of 0 to 2 with an absolute neutrophil count ≥ 1000/μL, a platelet count ≥ 75,000/μL, a hemoglobin level ≥ 8 g/dL, a creatinine clearance ≥ 50 mL/min, a total bilirubin level ≤ 2.0 × the institutional upper limit of normal (IULN), aspartate aminotransferase and alanine aminotransferase levels ≤ 3.0 × IULN, a Thyroid Stimulating Hormone or free T4 serum level ≤ IULN, and an adrenocorticotropic hormone level ≤ IULN. Women of child-bearing potential were required to have a negative serum pregnancy test, and subjects were required to practice adequate birth control during protocol participation.
Treatment and Monitoring
Treatment consisted of intravenous ipilimumab (1 mg/kg) every 6 weeks with intravenous nivolumab (240 mg) every 2 weeks until disease progression, symptomatic deterioration, a treatment delay for any reason > 56 days, or unacceptable or immune-related toxicity with an inability to decrease prednisone to <10 mg daily or per patient request.
Patients were evaluated with a history and a physical and toxicity assessment at least every 6 weeks at the beginning of each cycle. Laboratory evaluations included a complete blood count, a comprehensive metabolic panel, thyroid-stimulating hormone, free thyroxine, adrenocorticotropic hormone, cortisol, and lipase. Imaging studies by computed tomography for disease assessment were performed before the study; at week 8, week 16, and week 24; and then every 12 weeks until progression.
Statistical Methods and Outcomes
The primary objective was to evaluate the ORR (confirmed complete responses and partial responses) according to version 1.1 of the Response Evaluation Criteria in Solid Tumors (RECIST) on the basis of local assessments. A two-stage design was used to distinguish between a true ORR ≤ 5% (null hypothesis: patients had failed all known active therapies) and a true ORR ≥ 30% (alternative hypothesis: a potentially clinically meaningful difference in tumor response in refractory solid tumors). The first stage sample size was 6 patients; if 1 or more had a response (a confirmed complete response or partial response), an additional 10 patients were to be accrued. The design specified that 2 or more responses out of 16 patients would reject the null hypothesis (1-sided α = 13%, power = 87%). SWOG trials typically enroll 5% to 10% more patients than the eligible sample size to account for ineligible patients. The accrual to this study was rapid, and only 1 patient was not eligible, so a total of 20 patients were accrued. There were 2 responses in the first 16 patients meeting the design criteria; we present P values and confidence intervals (CIs) with the full sample size.5 The secondary objectives were to estimate progression-free survival (PFS), overall survival (OS), ORRs by immune-related RECIST, and PFS by immune-related RECIST and to assess toxicity by the Common Terminology Criteria for Adverse Events (version 4.0). Data are as of July 27, 2020.
PFS was measured from the start of protocol therapy to the first date of progression by RECIST (version 1.1) or death by any cause, with patients last known to be alive without progression censored at the date of last contact. OS was measured from the date of study registration to the date of death by any cause, with patients last known to be alive censored at the date of last contact. PFS and OS estimates were calculated with the Kaplan-Meier method and compared with log-rank tests. CIs for medians were constructed with the method of Brookmeyer and Crowley,6 and CIs for point estimates (eg, 6-month PFS) were calculated with log-log transformation. CIs for the primary ORR analysis accounted for the 2-stage design and observed sample size5 with the R functions get_CI and get_p_KC from the OneArmPhaseTwoStudy package. Exact binomial CIs were calculated otherwise. A Fisher exact test was used to compare groups. All analyses were performed with R version 3.4.4.
RESULTS
Patient Characteristics
Twenty patients from 12 National Clinical Trial Network institutions were registered between October 2018 and August 2019, with 19 patients meeting the eligibility criteria and receiving protocol therapy (summarized in Table 1). The patients in this cohort were distinct from previously reported cohorts.3 The median age was 49 years (range, 30–80 years). Overall, 58% of the patients in this cohort were men. The performance status was 0 to 1 for 89% of the patients, and 11% of the patients had a Zubrod performance status of 2. The median number of prior lines of therapy was 1, and all patients had microsatellite-stable disease. The most common sites of primary tumors were unknown primaries (n = 4), which were followed by the cervix, gastroesophageal junction, pancreas (poorly differentiated), and rectum (all n = 2). There was 1 lung primary patient in this cohort without small cell features. The majority of tumors in this cohort were poorly differentiated (58%), with 32% having an unknown differentiation status. Well and moderately differentiated tumors each represented 5% of cases in this cohort according to local pathology review.
TABLE 1.
Patient Characteristics (n = 19)
| Characteristic | Summary |
|---|---|
| Age, median (range), y | 49 (30–80) |
| Sex, No. (%) | |
| Female | 8 (42) |
| Male | 11 (58) |
| Performance status, No. (%) | |
| 0 | 9 (47) |
| 1 | 8 (42) |
| 2 | 2 (11) |
| Ethnicity, No. (%) | |
| Hispanic | 0 (0) |
| Not Hispanic | 19 (100) |
| Race, No. (%) | |
| White | 18 (95) |
| Unknown race | 1 (5) |
| Primary site, No. (%) | |
| Cecum | 1 (5) |
| Cervix | 2 (11) |
| Colon | 1 (5) |
| Esophagus | 1 (5) |
| GE junction | 2 (11) |
| Larynx | 1 (5) |
| Lunga | 1 (5) |
| Nasal cavity | 1 (5) |
| Poorly differentiated pancreas | 2 (11) |
| Rectum | 2 (11) |
| Unknown primary | 4 (21) |
| Vulva | 1 (5) |
| Ki-67, median (range), % | 80 (20–99) |
| Microsatellite instability status, No. (%) | |
| MSS | 19 (100) |
| Prior lines of therapy, median (range) | 1 (0–3) |
| Differentiation, No. (%) | |
| Well differentiated | 1 (5) |
| Moderately differentiated | 1 (5) |
| Poorly differentiated | 11 (58) |
| Unknown | 6 (32) |
Abbreviations: GE, gastroesophageal; MSS, microsatellite stable.
Atypical lung neuroendocrine tumor (high-grade).
Toxicities
Adverse events are summarized in Table 2, with 84% of patients experiencing an adverse event during the study and 37% developing a grade 3/4 adverse event. The most common toxicities of any grade were fatigue (36%) and rash (26%), with the most common grade 3/4 toxicities being rash (15%) followed by nausea, lipase elevation, alkaline phosphatase elevation (all 5%). The most common immune-mediated toxicity was rash (26%), which was followed by aspartate aminotransferase elevation (21%) and alanine aminotransferase elevation (15%). The most common grade 3/4 immune-related adverse event was rash (15%), which was followed by lipase elevation (10%). No patients required permanent therapeutic discontinuation for management of their toxicity, and there were no treatment-related deaths.
TABLE 2.
Treatment-Related Adverse Events (n = 19)
| Any Grade, No. (%) | Grade 3–5, No. (%) | |
|---|---|---|
| Any | 16 (84.2) | 7 (36.8) |
| Serious | 6 (31.6) | 4 (21.1) |
| Led to discontinuation | 0 (0.0) | 0 (0.0) |
| Lead to death | 0 (0.0) | 0 (0.0) |
| >10% of patients | ||
| Fatigue | 7 (36.8) | 0 (0) |
| Rash, maculopapular | 5 (26.3) | 3 (15.8) |
| Nausea | 4 (21.1) | 1 (5.3) |
| Aspartate aminotransferase increased | 4 (21.1) | 0 (0) |
| Lipase increased | 3 (15.8) | 2 (10.5) |
| Alanine aminotransferase increased | 3 (15.8) | 0 (0) |
| Hypothyroidism | 3 (15.8) | 0 (0) |
| Pruritus | 3 (15.8) | 0 (0) |
| Vomiting | 3 (15.8) | 0 (0) |
| Anemia | 2 (10.5) | 1 (5.3) |
| Alkaline phosphatase increased | 2 (10.5) | 0 (0) |
| Chills | 2 (10.5) | 0 (0) |
| Pain | 2 (10.5) | 0 (0) |
| Immune-mediated | 13 (68.4) | 4 (21.1) |
| Rash, maculopapular | 5 (26.3) | 3 (15.8) |
| Aspartate aminotransferase increased | 4 (21.1) | 0 (0) |
| Lipase increased | 3 (15.8) | 2 (10.5) |
| Alanine aminotransferase increased | 3 (15.8) | 0 (0) |
| Hypothyroidism | 3 (15.8) | 0 (0) |
| Pruritus | 3 (15.8) | 0 (0) |
| Diarrhea | 1 (5.3) | 0 (0) |
| Hyperthyroidism | 1 (5.3) | 0 (0) |
| Infusion-related reaction | 1 (5.3) | 0 (0) |
| Serum amylase increased | 1 (5.3) | 0 (0) |
Outcomes
Among the 19 patients, 5 had confirmed partial responses (ORR, 26%; 95% CI, 11%–45%; P = .002 [the calculation accounted for the 2-stage design and the 19 patients analyzed]); with an additional patient (5%) with stable disease for >6 months, for a clinical benefit rate of 32% (Table 3). Response rates did not appear to vary by primary site of origin or number of prior lines of therapy, although the limited number of patients within each group restricted robust comparisons (Fig. 1). The primary sites for the 5 responders were poorly differentiated pancreatic, rectal, colon, esophageal, and lung. The overall 6-month PFS rate was 32% (16%–61%) with a median PFS of 2.0 months with ongoing responses and a median OS of 8.9 months (Fig. 2). The duration of response is shown in Figure 3, which demonstrates durable responses in some patients with metastatic disease. The durations of response for the 5 responders were 8, 8+, 11+, 12+, and 31 months (1 patient was incorrectly enrolled by the local site in a different cohort and was restratified to this appropriate cohort). We also assessed patients with immune-related RECIST, and there was no reclassification from their RECIST response (version 1.1).
TABLE 3.
Best Response Summary by Version 1.1 of RECIST (n = 19)
| Best RECIST Response | No. (%) |
|---|---|
| Confirmed partial response | 5 (26) |
| Clinical benefit (stable disease for ≥6 mo) | 1 (5) |
| Stable disease for <6 mo | 5 (26) |
| Progression | 7 (37) |
| Not assesseda | 1 (5) |
Abbreviation: RECIST, Response Evaluation Criteria in Solid Tumors.
The patient received palliative radiation therapy before the first scan and was not assessable for a RECIST response.
Figure 1.
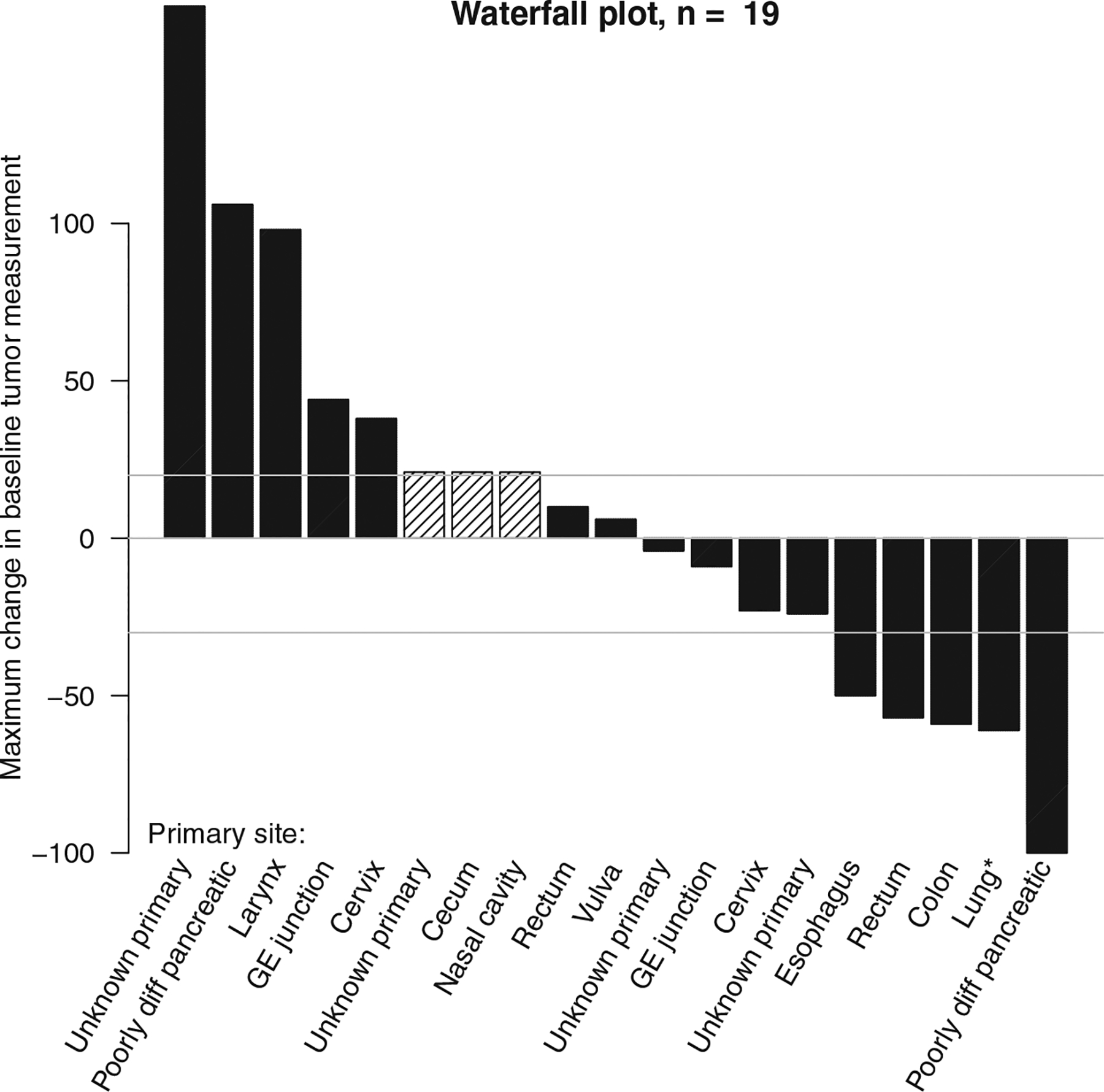
Waterfall plot. Crosshatching indicates that a patient did not have tumor measurements available because of progression due to new lesions at the first assessment (n = 2) or the receipt of radiation therapy before the first assessment (n = 1). *Atypical lung neuroendocrine tumor, high-grade. GE indicates gastroesophageal.
Figure 2.
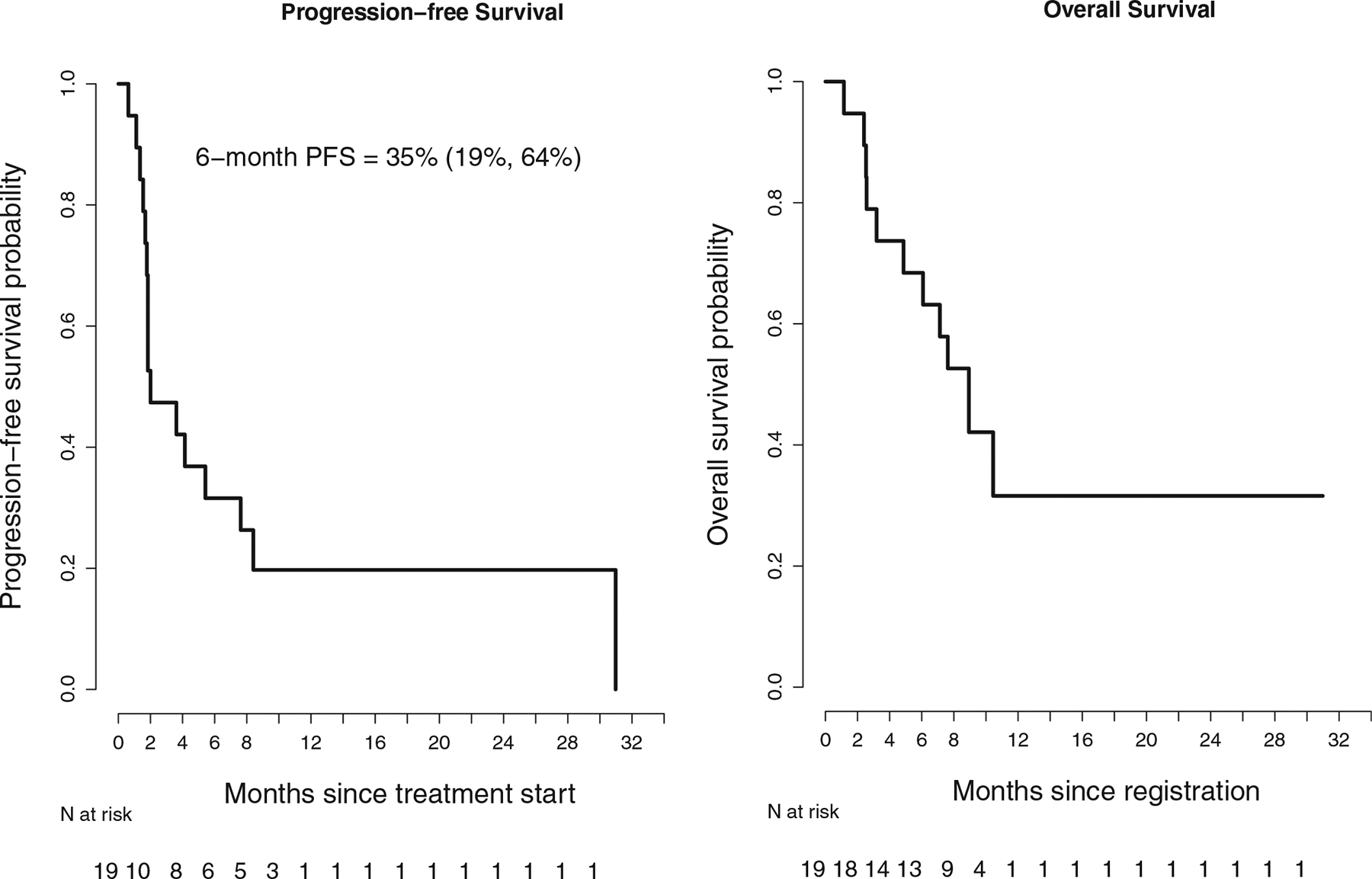
RECIST (version 1.1): PFS and overall survival. PFS indicates progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Figure 3.

RECIST (version 1.1): swimmer plot. Arrows indicate ongoing responses. *Atypical lung neuroendocrine tumor, high-grade. GE indicates gastroesophageal; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors.
DISCUSSION
Neuroendocrine neoplasms and, in particular, high-grade neuroendocrine neoplasms arise from multiple organ sites and are characterized by a poor prognosis.7 Historically, chemotherapy has remained the mainstay of treatment for high-grade neuroendocrine neoplasms, with transient responses for the majority of patients.8 These aggressive tumors are typically treated with platinum and etoposide combination chemotherapy; initial responses are seen in approximately two-thirds of patients, but the median survival is approximately 15 months, and longer term outcomes are poor.9,10
The advent of immune checkpoint blockade has resulted in long-term remissions in some patients with metastatic disease (eg, small cell lung cancer).11 However, the effect of immunotherapy in neuroendocrine cancers in particular is less clear.12,13 Prior studies of anti–PD-1 monotherapy have had limited activity across neuroendocrine neoplasms, a tumor type with a predilection for brain metastasis.14–18
Across the S1609 DART trial, the combination of low-dose ipilimumab and nivolumab was chosen to maximize the likelihood of an immunotherapeutic response with respect to monotherapy, and the dose of intravenous ipilimumab (1 mg/kg every 6 weeks) was chosen for a balance of toxicity and efficacy and on the basis of the results of CheckMate 227.19 This dosing combination of ipilimumab and nivolumab has also become one of the frontline standards of care for metastatic non–small cell lung cancer.
As previously reported, S1609 cohort 23 investigated ipilimumab and nivolumab across all grades of nonpancreatic neuroendocrine neoplasms and demonstrated a 44% ORR in patients with high-grade disease versus 0% in patients with low-to intermediate-grade disease.3 However, this was a post hoc analysis by grade, and the microsatellite instability status was not available for many patients. S1609 cohort 52, as reported here, prospectively studied low-dose ipilimumab plus nivolumab in a cohort of high-grade neuroendocrine neoplasms and demonstrated a 26% ORR, with an additional 5% of patients having stable disease for ≥6 months. This regimen was tolerable, with no toxicity-related treatment discontinuation required. The difference in the ORRs between the previously reported post hoc high-grade subgroup and this prospective cohort is likely related to statistical considerations related to small sample sizes and reaffirms the importance of prospective clinical evaluation of subgroup analyses, as performed here. Notably, from a safety stand-point, there were no treatment-related deaths in either cohort 23 (n = 32) or cohort 52 (n = 19).
Another recently published analysis of a basket trial with a plurality of lung neuroendocrine neoplasms (CA209–538) used ipilimumab with a different dosing schema (1 mg/kg every 3 weeks for 4 doses) in combination with nivolumab every 2 weeks.20 In that study, 4 of 13 patients with high-grade tumors, 3 of 13 patients with intermediate-grade tumors, and none of 3 patients with low-grade tumors had an objective radiographic response. The adverse event profile was broadly similar, though with more grade 3/4 immune hepatitis (14%) and colitis (7%) and with 1 case of myocarditis. The role of the dosing interval of ipilimumab (1 mg/kg)—every 3 weeks for 4 doses in CA209–538 versus every 6 weeks continuously in S1609 DART—in driving immune-related adverse event toxicity remains an ongoing question across studies featuring combinatorial immunotherapy, although there were no treatment-related deaths across CA209–538 or in any neuroendocrine cohort of S1609 reported to date. This study, similarly to the cohort reported here, permitted patients with pancreatic neuroendocrine tumors: 3 of 7 (43%), all with high-grade tumors (differentiation status unclear), responded to therapy, and they included a patient who had disease progression on prior anti–PD-1 treatment. Because of the historical response rates with anti–PD-1 monotherapy (on the order of 5%), the unique contribution of ipilimumab in high-grade disease is worthy of further investigation.
Weaknesses of this study include its lack of randomization, relatively small sample size, and lack of central pathology review. One of the patients in this cohort had an atypical lung carcinoid tumor; although ipilimumab and nivolumab are approved by the Food and Drug Administration for epithelial non–small cell lung cancer, outside small cell lung cancer, there are limited data concerning immunotherapeutic approaches for lung tumors with atypical neuroendocrine differentiation.11,19,21 Tumor grading was performed locally, most often with Ki-67 assessment. Local imaging response assessments were used for the primary end point of ORR with RECIST (version 1.1), and the lack of randomization limits interpretation of therapeutic benefit.
With a benefit in a subgroup of high-grade neuroendocrine neoplasms, the identification of predictive biomarkers to select patients who may preferentially respond to immune checkpoint blockade is crucial. Limited molecular profiling was previously performed on patients in this cohort. Among responders in the cohort, 1 had a POLE mutation with 93 mutations per megabase; however, the other patients in this cohort, all of whom were microsatellite-stable, did not have a recurrent molecular aberration or tumor mutation burden greater than 10 mutations per megabase. This reinforces the need for the translational biomarker analyses that are ongoing across S1609 with a focus on the tumor mutation burden as a key DNA biomarker, transcriptome to assess RNA, and PD-L1 protein expression by immunohistochemistry to assess the central dogma of cancer immunotherapeutic biomarkers across rare cancers.22,23 Clinically, a case of response to anti–PD-1 with radiation has been reported in high–mutational burden neuroendocrine neoplasms previously,24 and this suggests alternative potential synergies.
We have described here the dedicated high-grade neuroendocrine neoplasm cohort of SWOG S1609 assessing low-dose ipilimumab plus nivolumab, and our findings prospectively confirm benefit in patients with high-grade disease independently of the primary organ site. Future clinical studies should evaluate immunotherapy in combination with cytotoxic chemotherapy in the first-line setting. Additionally, predictive markers are needed to identify patients who may benefit from combinatorial immune checkpoint blockade.
FUNDING SUPPORT
This work was supported by the National Cancer Institute of the National Institutes of Health (grants CA180888, CA180819, CA180821, CA233331, CA233320, and CA180828) and in part by Bristol-Myers Squibb. The National Cancer Institute negotiated a collaborative research and development agreement with Bristol-Myers Squibb to provide nivolumab and ipilimumab for this clinical trial. Razelle Kurzrock reports funding from the Joan and Irwin Jacobs Fund and the National Cancer Institute (grant P30 CA023100). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST DISCLOSURES
Sandip Pravin Patel reports that his university receives research grants from Bristol-Myers Squibb, Eli Lilly, Incyte, AstraZeneca/MedImmune, Merck, Pfizer, Roche/Genentech, Xcovery, Fate Therapeutics, Genocea, and Iovance; he also reports personal fees from Amgen, AstraZeneca, BeiGene, Bristol-Myers Squibb, Certis, Eli Lilly, Genentech, Illumina, Merck, Novartis, Pfizer, Rakuten, and Tempus. Young Kwang Chae reports research grants from AbbVie, Bristol-Myers Squibb, Biodesix, Lexent Bio, and Freenome and consulting fees, payments, and/or honoraria from Roche/Genentech, AstraZeneca, Bristol-Myers Squibb, Foundation Medicine, Counsyl, Guardant Health, Boehringer Ingelheim, Biodesix, ImmuneOncia, Lilly Oncology, Merck, Pfizer, Tempus, Lunit, and Takeda. Bhavana Konda reports research funding to her institution from Bristol-Myers Squibb, Xencor, Merck, Eisai, and Eli Lilly & Co. Elad Sharon is a full-time employee of the National Cancer Institute. Christopher W. Ryan reports clinical trial support paid to his institution from Argos, Bristol-Myers Squibb, CytRx, Daiichi-Sankyo, Exelixis, Genentech, Novartis, Karyopharm, Merck, Nektar, Pfizer, TRACON, and Xynomic and consulting fees from AstraZeneca, Bristol-Meyer Squibb, Deciphera, Exelixis, Eisai, Janssen, and Daiichi-Sanyo. Razelle Kurzrock reports research funding from Boehringer Ingelheim, Debiopharm, Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Konica Minolta, Medimmune, Takeda, Top Alliance, and OmniSeq as well as consultant fees from LOXO, X-Biotech, Actuate Therapeutics, Roche, and NeoMed. She also serves as an advisor to Soluventis; receives speaker fees from Roche; has equity in IDbyDNA, CureMatch, and Soluventis; receives fees from Bicara Therapeutics, Biological Dynamics, Pfizer, TD2/Volastra, and Turning Point Therapeutics; serves on the boards of CureMatch and CureMetrix; and is a cofounder of CureMatch. The other authors made no disclosures.
We thank Ms. Marcia Horn, JD (SWOG patient advocate and president/chief executive officer, International Cancer Advocacy Network), Ms. Christy Klepetko (protocol coordinator, SWOG Operations Office), Heloisa P. Soares, MD, PhD (University of Utah), and Howard Streicher, MD (Investigational Drug Branch, Cancer Therapy Evaluation Program, National Cancer Institute).
The investigational new drug sponsor was the Division of Cancer Treatment and Diagnosis of the National Cancer Institute.
Footnotes
This trial was registered at ClinicalTrials.gov (NCT02834013) on July 15, 2016.
REFERENCES
- 1.Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med. 2016;374:2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris VK, Salem ME, Nimeiri H, et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel SP, Othus M, Chae YK, et al. A phase II basket trial of dual anti–CTLA-4 and anti–PD-1 blockade in rare tumors (DART SWOG 1609) in patients with non-pancreatic neuroendocrine tumors. Clin Cancer Res. 2020;26:2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeSantis CE, Kramer JL, Jemal A. The burden of rare cancers in the United States. CA Cancer J Clin. 2017;67:261–272. [DOI] [PubMed] [Google Scholar]
- 5.Koyama T, Chen H. Proper inference from Simon’s two-stage designs. Stat Med. 2008;27:3145–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookmeyer R, Crowley J. A k-sample median test for censored data. J Am Stat Assoc. 1982;77:433–440. [Google Scholar]
- 7.Heetfeld M, Chougnet CN, Olsen IH, et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2015;22:657–664. [DOI] [PubMed] [Google Scholar]
- 8.Kunz PL, Reidy-Lagunes D, Anthony LB, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42:557–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spigel DR, Hainsworth JD, Greco FA. Neuroendocrine carcinoma of unknown primary site. Semin Oncol. 2009;36:52–59. [DOI] [PubMed] [Google Scholar]
- 10.Hainsworth JD, Spigel DR, Litchy S, Greco FA. Phase II trial of paclitaxel, carboplatin, and etoposide in advanced poorly differentiated neuroendocrine carcinoma: a Minnie Pearl Cancer Research Network study. J Clin Oncol. 2006;24:3548–3554. [DOI] [PubMed] [Google Scholar]
- 11.Horn L, Mansfield AS, Szczęsna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–2229. [DOI] [PubMed] [Google Scholar]
- 12.Yao JC, Strosberg J, Fazio N, et al. 1308O: activity & safety of spartalizumab (PDR001) in patients (pts) with advanced neuroendocrine tumors (NET) of pancreatic (Pan), gastrointestinal (GI), or thoracic (T) origin, & gastroenteropancreatic neuroendocrine carcinoma (GEP NEC) who have progressed on prior treatment (Tx). Ann Oncol. 2018;29(suppl 8):viii467–viii468. [Google Scholar]
- 13.Fottner C, Apostolidis L, Ferrata M, et al. A phase II, open label, multicenter trial of avelumab in patients with advanced, metastatic high-grade neuroendocrine carcinomas NEC G3 (WHO 2010) progressive after first-line chemotherapy (AVENEC). J Clin Oncol. 2019;37:4103. [Google Scholar]
- 14.Mulvey C, Raj NP, Chan JA, et al. Phase II study of pembrolizumab-based therapy in previously treated extrapulmonary poorly differentiated neuroendocrine carcinomas: results of part A (pembrolizumab alone). J Clin Oncol. 2019;37:363.30576267 [Google Scholar]
- 15.Vijayvergia N, Dasari A, Deng M, et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: joint analysis of two prospective, non-randomised trials. Br J Cancer. 2020;122:1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strosberg J, Mizuno N, Doi T, et al. Efficacy and safety of pembrolizumab in previously treated advanced neuroendocrine tumors: results from the phase II KEYNOTE-158 study. Clin Cancer Res. 2020;26:2124–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krug S, Teupe F, Michl P, Gress TM, Rinke A. Brain metastases in patients with neuroendocrine neoplasms: risk factors and outcome. BMC Cancer. 2019;19:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tawbi HA, Forsyth PA, Algazi A, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non–small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein O, Kee D, Markman B, et al. Immunotherapy of ipilimumab and nivolumab in patients with advanced neuroendocrine tumours: a subgroup analysis of the CA209–538 clinical trial for rare cancers. Clin Cancer Res. 2020;26:4454–4459. [DOI] [PubMed] [Google Scholar]
- 21.Paz-Ares L, Dvorkin M, Chen Y, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394:1929–1939. [DOI] [PubMed] [Google Scholar]
- 22.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–856. [DOI] [PubMed] [Google Scholar]
- 23.Khagi Y, Kurzrock R, Patel SP. Next generation predictive biomarkers for immune checkpoint inhibition. Cancer Metastasis Rev. 2017;36:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharabi A, Kim SS, Kato S, et al. Exceptional response to nivolumab and stereotactic body radiation therapy (SBRT) in neuroendocrine cervical carcinoma with high tumor mutational burden: management considerations from the center for personalized cancer therapy at UC San Diego Moores Cancer Center. Oncologist. 2017;22: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]