

Abstract
Obesity is a chronic condition associated with dyslipidemia and insulin resistance. Here, we show that the offspring of obese mothers are dyslipidemic and insulin resistant from the outset. Maternal and cord blood and placental tissues were collected following C-section at term. Patients were grouped as being normal weight (NW, BMI = 18–24.9) or obese (OB, BMI ≥ 30), and separated by fetal sex. We measured plasma lipids, insulin, and glucose in maternal and cord blood. Insulin resistance was quantified using the HOMA-IR. Placental markers of lipid and energy metabolism and relevant metabolites were measured by western blot and metabolomics, respectively.
For OB women, total cholesterol was decreased in both maternal and cord blood, while HDL was decreased only in cord blood, independent of sex. In babies born to OB women, cord blood insulin and insulin resistance were increased. Placental protein expression of the energy and lipid metabolism regulators PGC1α, and SIRT3, ERRα, CPT1α, and CPT2 decreased with maternal obesity in a sex-dependent manner (P < 0.05). Metabolomics showed lower levels of acylcarnitines C16:0, C18:2, and C20:4 in OB women’s placentas, suggesting a decrease in β-oxidation. Glutamine, glutamate, alpha-ketoglutarate (αKG), and 2-hydroxyglutarate (2-HG) were increased, and the glutamine-to-glutamate ratio decreased (P < 0.05), in OB placentas, suggesting induction of glutamate into αKG conversion to maintain a normal metabolic flux.
Newly-born offspring of obese mothers begin their lives dyslipidemic and insulin resistant. If not inherited genetically, such major metabolic perturbations might be explained by abnormal placental metabolism with potential long-term adverse consequences for the offspring’s health and wellbeing.
Keywords: Maternal obesity, lipid profile, placental function, metabolomics, insulin resistance
Introduction
In the adult US population, cardiac and metabolic diseases have a respective prevalence of 35% and 48% (21). Traditionally, the risk of metabolic syndrome was believed to be determined by interactions among genetic and lifestyle factors. However, it has become well established that exposure to an adverse in utero environment also increases the risk of metabolic disease, a phenomenon called ‘developmental programming’. Accordingly, the intrauterine environment is now considered one of the key determinants of cardiac and metabolic health in offspring.
Pregnancy per se is a distinct metabolic milieu characterized by careful optimization of substrate availability to meet the fetus’s growing demands. As pregnancy progresses, embryonic and placental requirements for energy substrates increase to maintain growing metabolic activity; these substrates include glucose, fatty acids, and amino acids.1 In addition, the maternal insulin resistance that develops during later stages of pregnancy2 ensures an optimal supply of carbohydrates when fetal growth is highest.3 However, the natural progression of this metabolic regime is disrupted by maternal obesity, leading to adverse outcomes for both mother and fetus.4 Specifically, maternal obesity programs the offspring to experience major metabolic challenges in adulthood – including obesity and cardiovascular and metabolic diseases (1–5). More than 65% of women who become pregnant in the USA are overweight or obese, making this a critical concern.
Maternal–fetal metabolism is regulated by the placenta, which controls crosstalk between mother and offspring. The placenta produces an array of signaling molecules (hormones, cytokines, for example) broader than that of any other organ except the brain.5 Maternal obesity has been reported to dysregulate placental metabolism, leading to excessive production of reactive oxygen species,6 accumulation of inflammation,7 epigenetic modifications,8 and defective autophagy.9
This study aimed to understand the effect of maternal obesity on fetal metabolism. We found that babies born to obese mothers are insulin resistant, exhibit abnormal plasma lipid profiles, and dysregulation of placental energy metabolism. Thus, in the offspring of obese mothers, dyslipidemia and dysregulated lipid metabolism appear at birth and, if left untreated, put the offspring at high risk of obesity and cardiac and metabolic diseases.
Materials and methods
Ethical approval of study participants
Maternal blood, cord blood, and placentas were collected from labor and delivery units at the University of Texas Health San Antonio and Oregon Health & Science University under protocols approved by the respective Institutional Review Boards and with informed consent from the patients.
Collection and processing of placental tissue
Placentas were collected immediately following cesarean section from term pregnancies with the absence of labor. Placental tissues for this study were collected from subjects who: (1) Had either normal or high prepregnancy body mass index (BMI), respectively, grouped as normal weight (NW; BMI = 18.5–24.9) or obese (OB; BMI = 30–45); (2) Had an uncomplicated singleton pregnancy, and (3) Delivered by C-section. Exclusion criteria included multifetal gestation, gestational diabetes, preeclampsia, chronic inflammatory diseases, tobacco use, illicit drugs, or both; and recent bariatric surgery. The placentas were randomly sampled as described previously.10
Plasma collection
Maternal blood was collected from fasting patients before C-section. Cord blood was collected and placed in EDTA-containing collection tubes. Plasma was immediately separated from whole blood by centrifugation at 2000 g for 10 min at 4°C, then flash-frozen in liquid nitrogen and stored at −80°C for further analyses.
Materials
The following primary antibodies were used: Sirt3 (Cell Signaling Technology Cat# 2627, RRID:AB_2188622), PGC1α (Abcam Cat# ab54481, RRID:AB_881987), phospho (S571)-PGC1α (R and D Systems Cat# AF6650, RRID:AB_10890391), NRF1, NRF2, and TFAM (Cell Signaling Technology, Cat # 46743, 12721, and 8076, respectively); ERRα (NOVUS Cat# NBP2–16380), CPT1α (Cell Signaling Technology Cat# 12252, RRID:AB_2797857), CPT2 (Thermo Fisher Scientific Cat# PA5–12217, RRID:AB_2292215), and β-actin (Sigma-Aldrich Cat# A2228, RRID:AB_476697). The following secondary antibodies were used: anti-rabbit IgG, HRP-linked (Cell Signaling Technology Cat# 7074, RRID:AB_2099233) and anti-mouse IgG, HRP-linked (Cell Signaling Technology Cat# 7076, RRID:AB_330924).
Homeostasis Model Assessment of Insulin Resistance (HOMA-IR)
Fasting maternal and cord plasma glucose concentrations were determined using the Bayer Contour Next One glucometer following manufacturer instructions. Corresponding insulin concentrations were determined using human insulin ELISA kits (ThermoFisher Scientific Cat# KAQ1251) according to manufacturer instructions. Insulin resistance was then calculated using the following formula: fasting insulin (micro U/l) × fasting glucose (mg/dl)/405 as described before.11
Lipid measurements
Plasma lipids were measured using standard techniques.12 Low-density lipoprotein cholesterol (LDL-c) was calculated using the Friedewald equation: total cholesterol – (HDL-c VLDL-cholesterol/5). Lipid levels are reported in mg/dl.
Metabolite quantification
Metabolites were measured from placental villous tissue as described previously.9 Briefly, lipids were extracted from tissue (~100 mg) using 200 μl of water and 800 μl of ice-cold chloroform: methanol (2:1). Samples were homogenized with a lyser from BioSpec (Bartlesville, OK, USA) for 2 min, with a 1 min break after the first minute, and then maintained on ice for 30 min. After centrifugation at 13,800 g for 10 min, the chloroform layer was removed, dried in vacuo, and reconstituted in 50% isopropanol. HPLC-tandem-MS analysis was conducted on a Thermo Fisher Q Exactive fitted with a PicoChip nanospray source (New Objective, Woburn, MA, USA). HPLC conditions were: column, PicoChip (Waters Atlantis dC18; 150 μm × 105 mm; 3-μm particle); mobile phase A, acetonitrile/water (40:60) containing 10 mM ammonium acetate; mobile phase B, acetonitrile:isopropanol (10:90) containing 10 mM ammonium acetate; gradient (55 min total), 10% B for 7 min (sample loading), 10–99% B over 33 min, and 99% B for 15 min; flow rate, 1 μl/min. Data-dependent analyses were conducted using one full MS scan (70,000 resolution, m/z 200) followed by six tandem-MS scans using the following parameters: detection, positive ion, and dynamic exclusion, 10 s. The raw data files were processed with SIEVE (Thermo Fisher, Waltham, MA USA). Peak alignment and integration were performed, and the relative abundance of each lipid was determined between the two experimental groups. Metabolite concentrations were normalized to 4-C-13-malate.
Protein isolation
Approximately 100 mg of placental tissue was homogenized in 300 μl of Radioimmunoprecipitation assay (150 mM NaCl, 5 mM EDTA pH 8.0, 50 mM Tris pH 8.0, 1.0% NP-40 (IGEPAL CA-630), 0.5% Sodium Deoxycholate, 0.1% SDS) buffer containing protease/phosphatase inhibitors (Thermo Scientific Cat# A32959). Total protein concentrations were determined using the Pierce BCA Protein assay (Thermo Scientific Cat# 23223) according to manufacturer protocol. Samples were aliquoted and stored at −80°C to avoid freeze–thaw cycles.
Western blots
Approximately 30 μg of protein was separated on hand-cast 12% SDS PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes using a Mini-PROTEAN tetra cell electrophoresis chamber (BioRad Cat# 1658004). Membranes were blocked with 5% (w/v) nonfat milk in TBS 0.1% Tween 20 (TBST) and then simultaneously incubated with primary antibodies to protein of interest and β-actin (ACTB). Afterward, the membranes were washed with TBST and then incubated with HRP-conjugated secondary antibodies. Finally, the membranes were incubated with Supersignal West Pico Plus ECL Substrate (Thermo Scientific Cat# 34578) for 5 min and imaged using the GBOX system (Syngene). Western blots were then washed with TBST, probed with a secondary antibody to ACTB, and analyzed using Genetools software (Syngene).
Statistical analysis
The Shapiro–Wilk test was used to test for normal distribution in all data sets. Significant differences between the groups in normally distributed data were tested as an interaction between fetal gender (M, F) and pregnancy complications such as maternal obesity within a two-way ANOVA followed by Student’s t-test. For non-parametric data, the Kruskal–Wallis test was applied for the same factors (fetal sex and maternal adiposity), followed by the Mann–Whitney U post hoc test. Resulting P-values < 0.1 are reported as statistically significant. Data are presented as box-and-whisker plots: the bottom and top of the boxes indicate the 25th and 75th percentiles, respectively, while the whiskers indicate the 5th and 95th percentiles.
Results
Study participants
The clinical characteristics of the study participants are presented in Table 1. Indications for cesarean section included only elective repeat or breech presentation. No differences in indications were observed between normal weight and obese groups. By experimental design, the NW and OB groups differed significantly in terms of BMI. No such differences were observed in terms of maternal and gestational age, birth weight, or placental weight. Maternal weight gain was significantly lower in the OB group for both male and female offspring.
Table 1.
Clinical characteristics of study participants. Samples were separated by fetal sex and grouped as NW (BMI 18–25) or OB (BMI ≥ 30) and data are expressed as mean (range)
| NW |
OB |
|||
|---|---|---|---|---|
| Fetal sex | Male (n = 22) | Female (n = 25) | Male (n = 27) | Female (n = 24) |
| Prepregnancy BMI (kg/m2) | 22.3 (18.2–24.7) |
22.6 (18.3–25.0) |
37.7* (29.2–60.0) |
36.3* (29.9–44.1 |
|
| ||||
| Maternal age (years) | 30 (20–40) |
29.8 (21–41) |
30.0 (20–38) |
29.2 (22–39) |
|
| ||||
| Gestational age (weeks) | 39.0 (36.0–40.0) |
39.2 (37.4–41.0) |
38.9 (36.3–40.3) |
39.1 (37.1–39.6) |
|
| ||||
| Fetal weight (grams) | 3508 (2845–4325) |
3450 (2975–4975) |
3387 (2220–4402) |
3369 (2920–3980) |
|
| ||||
| Placental weight (grams) | 546.9 (424.5–649.9) |
558.0 (359.4–758.6) |
544.4 (367.9–637.4) |
484.3 (389.0–595.2) |
|
| ||||
| Weight gain (kilograms) | 19.3 (−3 to 41.0) |
19.4 (6.8–38.0) |
12.4* (−5.5 to 39.3) |
14.1* (−3.0 to 44.9) |
|
| ||||
| Ethnicity (Hispanic/non-Hispanic) | 10/12 | 12/13 | 12/14 | 17/7 |
P < 0.05, NW versus OB.
Insulin resistance in neonates born to obese mothers
We found that insulin and glucose in fasting maternal and cord plasma collected at term did not differ with fetal sex. Therefore, we pooled data from males and females (Table 2, data separated by fetal sex are available in Supplemental Table S1). In contrast, OB neonates exhibited higher insulin and HOMA-IR cord blood values, increased by 1.6- (P = 0.006) and 1.7-fold (P = 0.01), respectively. Cord blood insulin also showed a strong positive correlation with maternal BMI (r = 0.5, P = 0.01). No between-group differences were detected in maternal or cord plasma glucose levels.
Table 2.
Fasting plasma insulin, glucose, and HOMA-IR. Data were pooled for males and females. Values are presented as median (range)
| Maternal plasma (n = 25) |
Cord plasma (n = 25) |
|||
|---|---|---|---|---|
| NW (n = 12) | OB (n = 13) | NW (n = 12) | OB (n = 13) | |
| Plasma insulin (μIU/ml) | 12.96 (1.94–43.46) |
16.33 (1.8–38.17) |
6.38 (2.45–10.45) |
10.51* (3.03–21.56) |
|
| ||||
| Plasma glucose (mg/dl) | 78.00 (44–94) |
77.5 (45–99) |
92.5 (69–106) |
82.00 (38–115) |
|
| ||||
| HOMA-IR | 2.16 (0.21–8.37) |
3.21 (0.44–7.26) |
1.32 (0.75–2.54) |
2.21* (1.08–5.32) |
P < 0.05, NW versus OB.
Changes in maternal and fetal lipid metabolism
Previous studies have documented a robust linear relationship between dyslipidemia and circulating insulin independent of obesity.13 We, therefore, measured total and high-density lipoprotein cholesterol (HDL-c) and triglycerides in fasting maternal and cord blood plasma. We calculated LDL-c levels using the Friedewald equation. We found no sex-dependent differences in lipid profiles and accordingly pooled data from male and female offspring (Table 3, data separated by fetal sex are available in Supplemental Table S2). Surprisingly, OB women showed decreased total cholesterol levels in both maternal (P = 0.03) and cord (P = 0.06) plasma (Fig. 1a, d). In addition, cord blood HDL content was decreased by 30% in OB women (P = 0.01), and overall inversely correlated with maternal BMI (r = −0.50, P = 0.01, Fig. 1c, f). No changes were observed in maternal levels of HDL-c (Fig. 1b, e), LDL-c, or triglycerides (Table 3).
Table 3.
Lipid profile of maternal and cord blood plasma
| Maternal plasma (n = 32) |
Cord plasma (n = 25) |
|||
|---|---|---|---|---|
| Lipids mg/dl | NW (n = 17) |
OB (n = 15) |
NW (n = 13) |
OB (n = 12) |
| TC | 252 (197–329) |
230* (149–282) |
58 (48–70) |
50† (33–68) |
|
| ||||
| HDL-c | 70 (28–101) |
59 (29–92) |
27 (19–34) |
19* (13–31) |
|
| ||||
| LDL-c | 144 (75–210) |
118† (75–184) |
28 (22–432) |
25 (17–34) |
|
| ||||
| TG | 211 (120–538) |
194 (108–382) |
21 (16–37) |
27 (16–56) |
TC, total cholesterol; HDL-c, high-density lipoprotein cholesterol; LDL-c, low-density lipoprotein cholesterol; TG, triglycerides.
Data are presented as median (range).
P < 0.05, NW versus OB.
P < 0.1 NW versus OB.
Fig. 1.
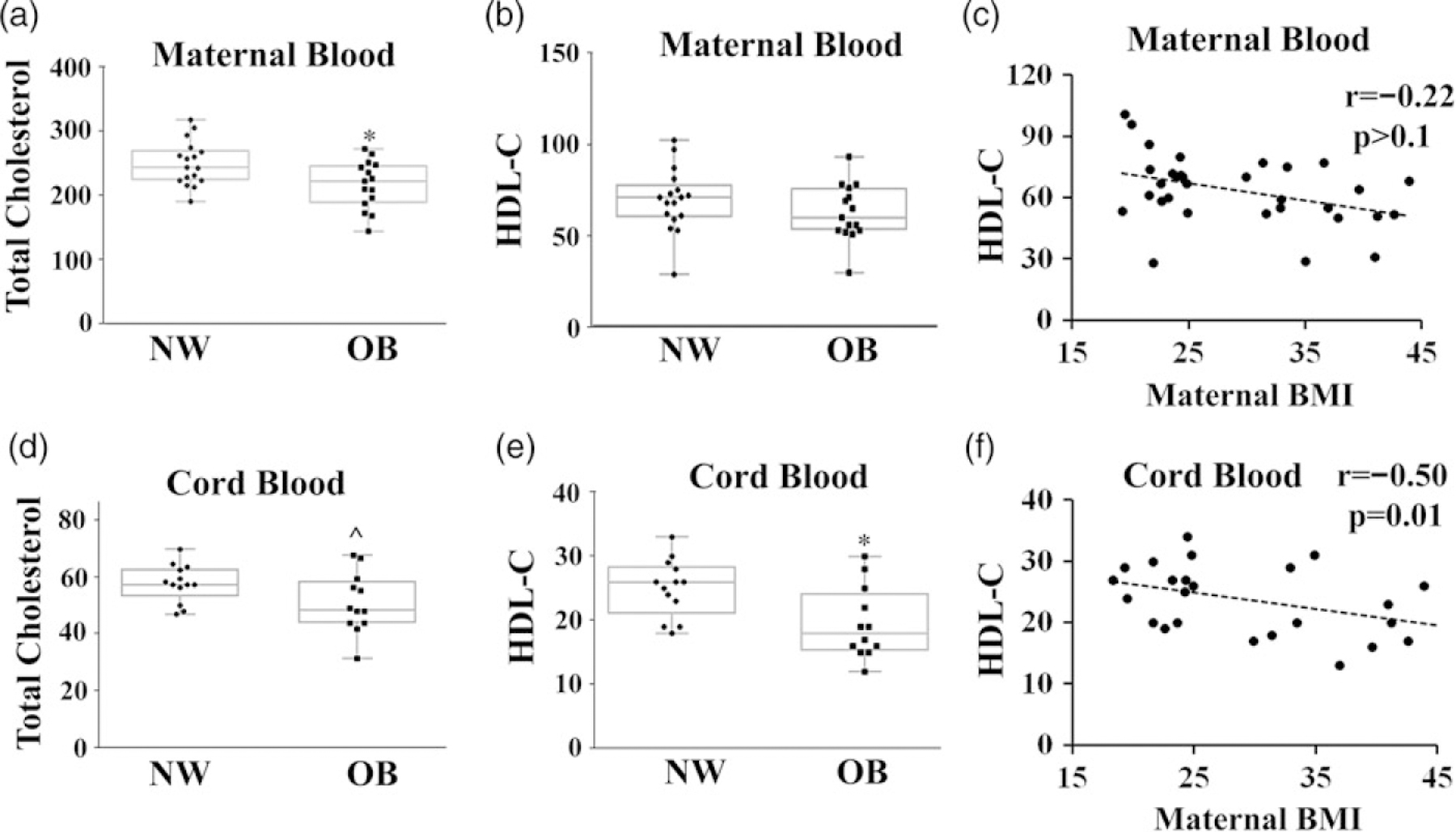
Lipid profiles in maternal and cord blood. Quantification of total and HDL cholesterol concentrations (mg/dl) in maternal (a, b) and cord blood (d, e). Correlation between maternal (c) and cord (f) plasma HDL cholesterol and maternal BMI. Sample size is shown in Table 3.
*P < 0.05, NW versus OB; ^P < 0.1, NW versus OB.
Placental lipid uptake
We next hypothesized that reductions in cord blood total cholesterol and HDL are associated with decreased placental lipid uptake. We measured placental protein expression of the two receptors involved in lipid uptake, LDL receptor (LDLR), and Scavenger receptor B1 (SR-B1) (Fig. 2). While we found no changes in SR-B1 expression (Fig. 2a, c), LDLR expression decreased by 40% in the placentas of male but not female offspring of obese women (P < 0.05, Fig. 2a, b).
Fig. 2.
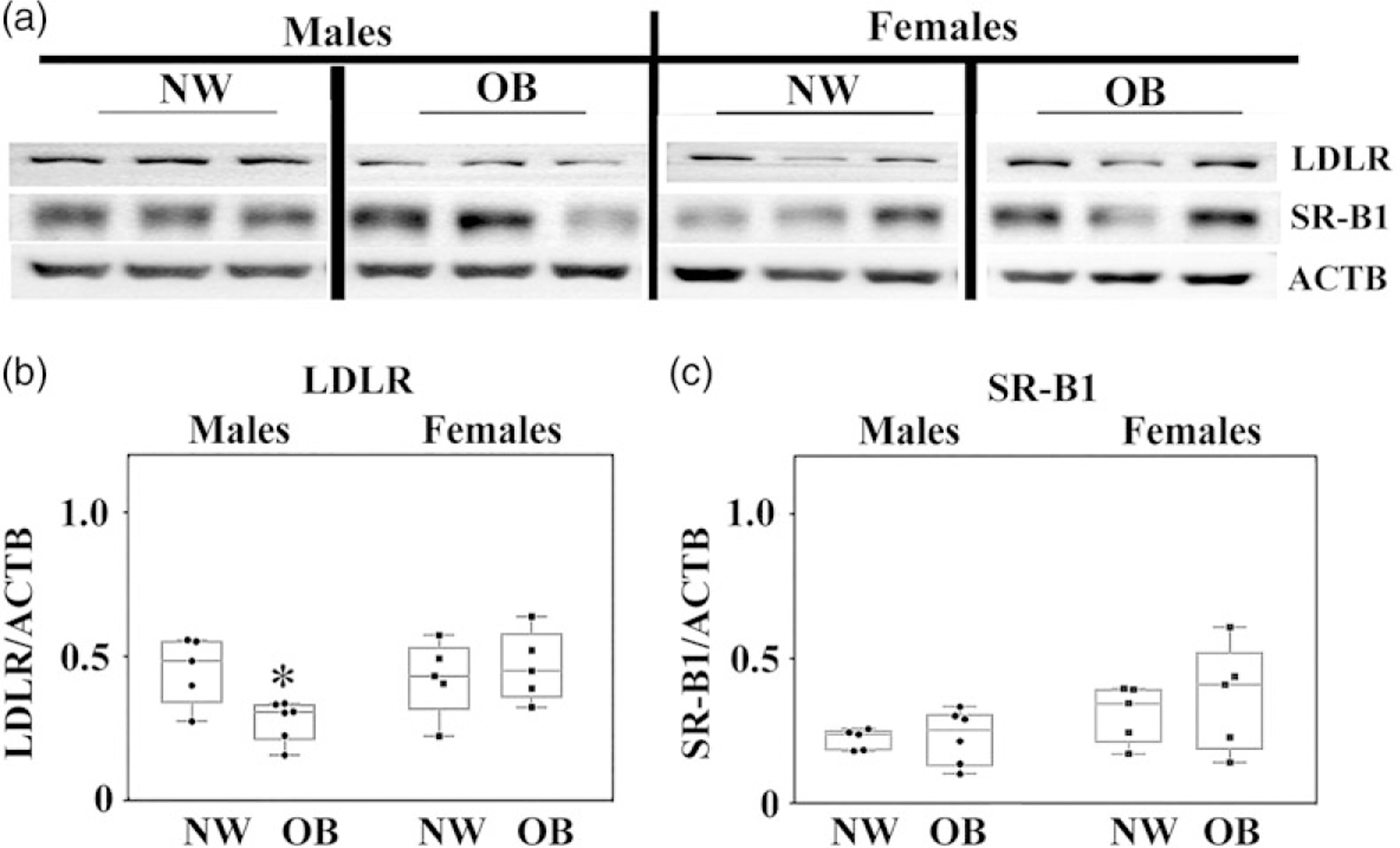
Expression of LDL and HDL (SR-B1) receptors in the placentas of NW and OB women. (a) Representative western blots; (b–c) quantification data for (b) LDL and (c) SR-B1. N = 6/group by BMI/sex.
*P < 0.05, NW versus OB.
Placental lipid and energy metabolism is affected by maternal obesity
Prolonged exposure to insulin has been previously shown to suppress mitochondrial biogenesis14 and cause mitochondrial dysfunction.15 Next, we examined the changes in regulators of placental lipid metabolism and mitochondrial biogenesis using western blots. A key regulator of mitochondrial metabolism is peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α),16 the expression of which dropped significantly in the placentas of male but not female offspring from OB women (P = 0.02, Fig. 3a, b). Prior work has shown that insulin inhibits PGC1α via protein kinase B (AKT)-mediated phosphorylation of Ser 570.17 Notably, the ratio of phospho (p)-PGC1α to total (t-)PGC1α (P = 0.02, Fig. 3a, c) was increased in the placentas of males born to OB mothers indicating inactivation of PGC1α.
Fig. 3.
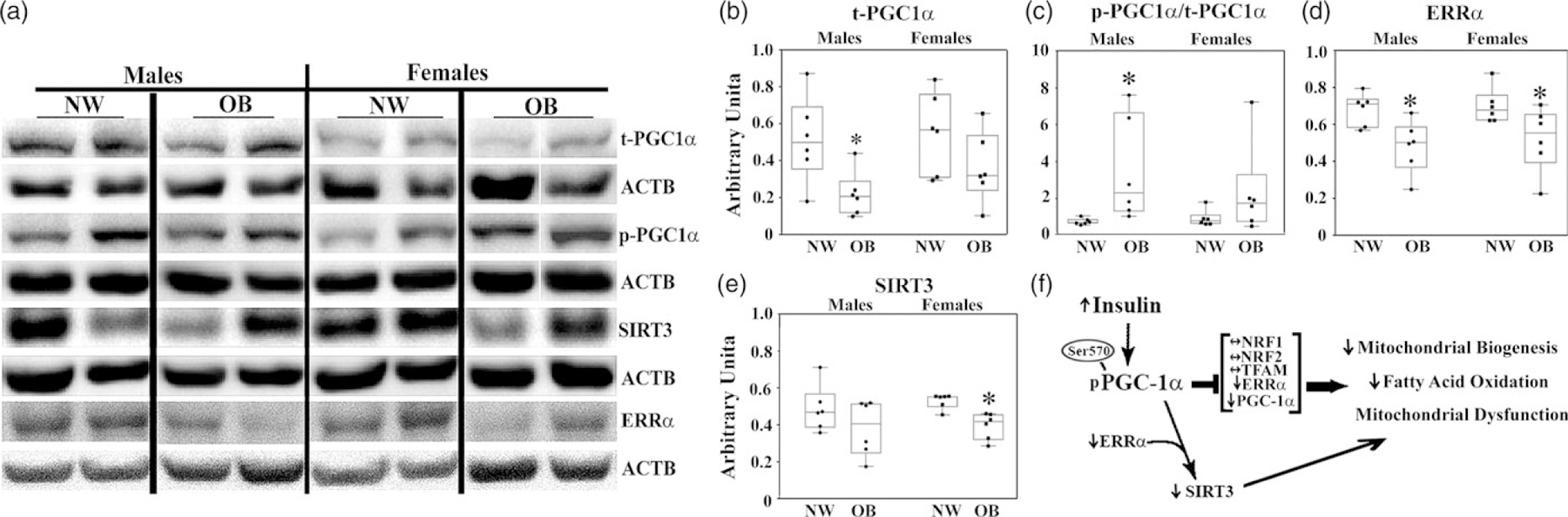
Effect of maternal obesity on the expression of key placental mitochondrial proteins. Representative images (a); and quantification data for t-PGC1α (b); p-PGC1α/t-PGC1α (c); ERRα (d); SIRT3 (e). f. Simplified diagram showing the effect of maternal obesity on the action of PGC1α and its target genes. N = 6/sex/group by BMI.
*P < 0.1, in OB group versus NW.
PGC-1α activates the expression of numerous target genes by recruitment to promoter regions and interaction with transcription factors. We next analyzed the expression of PGC-1a target genes involved in mitochondrial biogenesis and mitochondrial function, such as nuclear respiratory factors 1 and 2 (NRF1, NRF2), a mitochondrial transcription factor A (TFAM), estrogen-related receptor alpha (ERRα), and its downstream target sirtuin 3 (SIRT3) by western blot. Importantly, ERRα is activated by cholesterol18 and plays a major role in fatty acid oxidation.19 While the placental expressions of NRF1, NRF2, and TFAM were not significantly affected by maternal obesity (Supplemental Fig 1), there was sexual dimorphism in expression of NRF1 in NW group and NRF2 in both NW and OB groups with females showing significantly higher levels of NRF1 and NRF2 than males. In contrast, the expression of ERRα was affected by maternal obesity. It showed a significant reduction in expression in the placentas of OB compared with NW women, regardless of fetal sex (Fig. 3a, d). SIRT3 that activates proteins crucial for oxidative phosphorylation, the tricarboxylic acid (TCA) cycle, and fatty acid oxidation,20 was significantly decreased in OB women with female offspring (P = 0.003, Fig. 3a, e), though with no significant change in males.
The carnitine system is a key regulator of lipid metabolism and metabolic flexibility, intertwining different pathways, factors, and metabolites that supply an energetic and biosynthetic demand of cells.21,22 It consists of enzymes and transport proteins, which carry out fatty acids, imports them into mitochondria, and facilitate β-oxidation (Fig. 4a). Using metabolomics analysis, we found that placentas from obese women with male fetuses have significantly lower levels of acylcarnitines: C16:0, C18:2, and C20:4 (Fig. 4b–d) compared with placentas of NW women. Sex-specific effects were also observed in protein expression of Carnitine palmitoyl transferases 1α and 2 (CPT1α and CPT2), that facilitate the oxidation and transport of fatty acids from the plasma membranes to the mitochondria.23 OB women with female but not male babies expressed lower placental levels of these proteins (Fig. 4a, e–g). Remarkably, female offspring of NW women had a significantly higher placental expression of CPT1α and CPT2 than their male counterparts P < 0.05).
Fig. 4.
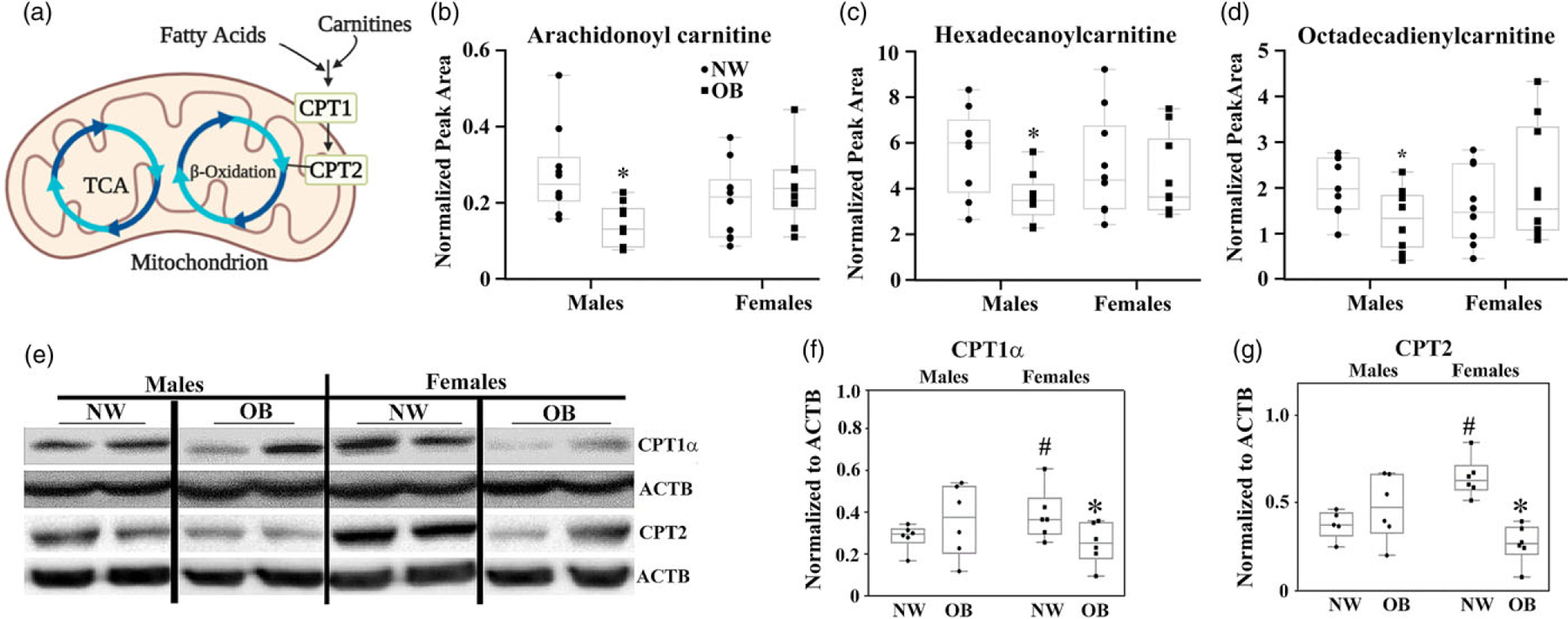
Maternal obesity decreases placental fatty acid metabolism. (a) Schematic of carnitine system. (b–d) Targeted metabolomics in the placentas of NW and OB women with male and female fetuses. Levels of acylcarnitines, arachidonoyl carnitine (C20:4, b), hexadecanoylcarnitine (C16:0, c), and octadecadienylcarnitine (C18:2, d), measured by mass spectrometry. N = 10 placentas/per group of adiposity/fetal sex, P < 0.05 versus NW group. (e–g) Western blot for CPT1α and CPT2. Representative images (e), and quantification graphs for CPT1α (f), and CPT2 (g). N = 6/group/sex.
*, P < 0.1 OB versus NW. #P < 0.1 females versus males within the same maternal BMI group.
Next, we studied the effect of maternal obesity on placental energy metabolism using a metabolomics approach. We found the concentrations of glutamine, glutamate, alpha-ketoglutarate (α-KG), and 2-hydroxyglutarate (2-HG) to be significantly increased in the placentas of both males and females from OB mothers (Fig. 5a, b, d, e, g). These changes suggest activation of glutamine oxidation, a metabolic shunt that converts glutamine into α-KG and 2-HG for subsequent introduction into the Krebs cycle.24 We also examined the ratio of glutamine to glutamate, an indicator of glutamine synthase activity. This ratio was reduced significantly (P = 0.0002) only in the placentas of males born to OB women (Fig. 5c). Prior research showed that α-KG could undergo a reverse reductive carboxylation with a subsequent synthesis of citrate,24 a process that serves as a precursor for fatty acid biosynthesis and has been reported to occur in low-oxygen environments such as inflamed tissue and tumors.25 Consistent with this, we found increased citrate levels in the placentas of males and female fetuses born to OB women (1.6-fold and twofold, respectively, P < 0.1) (Fig. 5f). We also observed a baseline sexual dimorphism in placental levels of glutamate, α-KG, and 2-HG, with males born to NW women showing significantly higher values than females (Fig. 5). No sex differences, however, were observed in the levels of these metabolites in the OB group.
Fig. 5.
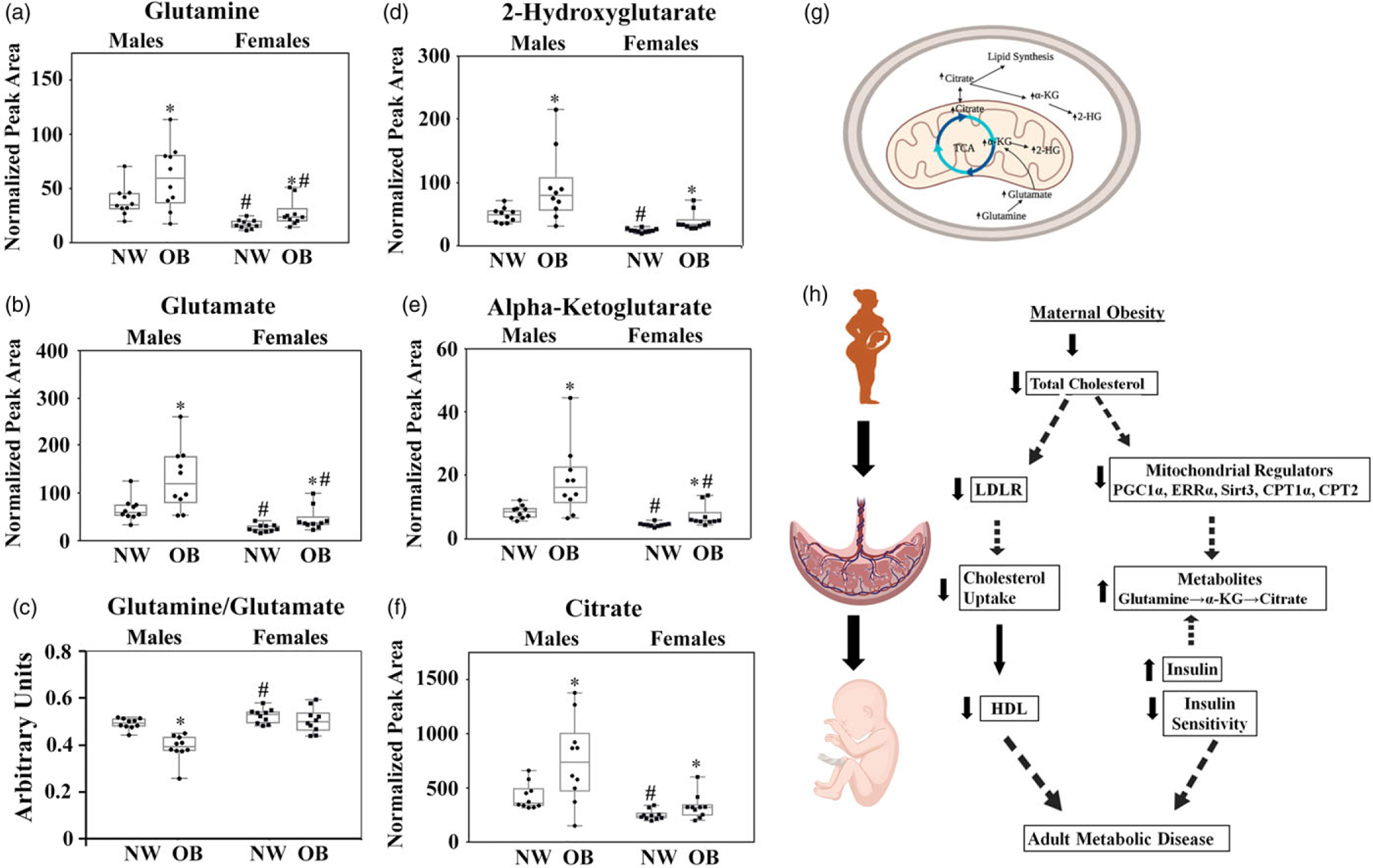
a–f, Metabolic perturbations in the placentas of obese women. Placental levels of glutamine (a), glutamate (b), glutamine-to-glutamate ratio (c), 2-HG (d), α-KG (e), and citrate (f) in NW and OB women with male and female fetuses. Metabolite concentrations were normalized to 4-C-13-malate. N = 40 (10/group/sex).
*P < 0.1, NW versus OB; #P < 0.1, females versus males within the same maternal BMI group. g, TCA cycle scheme summarizing findings from the metabolomics analysis. h, graphic summary of the findings.
Discussion
Our data suggest an association between exposure to obesogenic maternal environment and fetal metabolic dysregulations (Fig. 5h). Here, we reveal perturbations in fetal insulin sensitivity, lipid metabolism, and placental energy metabolism. These early abnormalities put children of obese mothers at high risk of becoming obese and developing metabolic diseases later in life. This could potentially create a trajectory of obesity, and metabolic dysfunction established in utero and perpetuated across generations.
Several factors have been previously linked to insulin resistance, including genetic predisposition, inflammation, and impaired mitochondrial function.26–29 Maternal insulin level is considered as the strongest predictor of offspring insulin resistance,30 however, maternal insulin sensitivity was not affected by obesity in our data. One explanation could be that our study was carried out at term, and thus the changes in fetal insulin sensitivity could represent a result of processes occurring earlier on. Two previous studies31,32 found no changes in cord blood glucose and insulin levels between neonates born to lean and obese women,32 or obese women before and after sustainable weight loss by bariatric surgery.31 In contrast, our data show that fetuses born to obese mothers have significantly higher levels of insulin and HOMA-IR. Different findings might be explained either by mixed mode of delivery in both studies, vaginal, emergency C-section, and elective C-section, versus only elective C-section in our study suggesting that delivery itself might affect the cord blood levels of insulin and glucose independent of maternal BMI.
The exact mechanisms driving hyperinsulinemia and insulin resistance in the offspring of obese mothers are not known. Insulin does not cross the placenta, however, the placenta expresses insulin receptors, and responds to high levels of insulin by a decrease in genes implicated in cholesterol utilization and mitochondrial metabolism.33 Our data show a sex-dependent reduction in the expression of genes responsible for mitochondrial biogenesis and function, and lipid uptake, and utilization. These results are also in line with evidence by our group and others suggesting that obesity induces a state of mitochondrial dysfunction in the term human placenta.6,34 Accumulation of lipid metabolites, and mitochondrial dysfunction, in turn, contribute to insulin resistance35 suggesting activation of adverse feed-forward mechanisms. Increase in offspring insulin resistance could be also explained by the rise in placental amino acid transport seen in placentas from obese women,36 which has been shown to stimulate insulin production.37
The theory of developmental programming establishes a link between maternal obesity and the risk of metabolic diseases in offspring later in life.38 Our data conclude that adverse metabolic perturbations in the offspring of obese mothers occur even before birth indicating that fetuses of obese mothers already represent a group at high risk for adult metabolic diseases.
Obesity in pregnancy is associated with dysregulated lipid profiles in both maternal and fetal circulation
In the nonpregnant state, obesity is associated with elevated levels of circulating lipids.39 However, as lipids are not routinely measured in pregnant women, little is known about how pregnancy affects the lipid profile.40 Cholesterol is essential for embryonic and fetal development,41,42 and while a fetus can synthesize its cholesterol, it nonetheless relies on the mother’s supply.42 We found total cholesterol to be decreased in both maternal and cord blood plasma. Our data are in agreement with Vahratian et al.43 and Meyer et al.,44 which reported high levels of cholesterol during the first trimester of pregnancy and their gradual decrease by the third trimester, with obese women having lower cholesterol levels than lean women.
To supply the fetus, maternal LDL-c and HDL-c are taken up by the placenta and transported across the trophoblasts and endothelial cells into the fetal circulation.41 The placenta expresses receptors for both HDL and LDL, which are SR-B1 and LDLR, respectively; however, there is evidence that fetal HDL is excessively bound to apolipoprotein E (apoE), a ligand for LDLR, thus allowing for HDL uptake by LDL receptors as well.45 While we found no change in the expression of placental SR-B1 with maternal obesity, LDLR levels were significantly decreased in the placentas of obese women with male fetuses. Our data are in agreement with previous reports showing a reduction in placental LDLR but not SR-B1 expression in overweight and obese women, independent of the level of maternal cholesterol.46 Notably, dysregulation of the LDLR pathway has been linked to activation of the mTOR pathway, hyperglycemia, and chronic inflammation.47 The long-term effects of low fetal HDL has not been studied in the settings of maternal obesity. However, it has been shown that patients with Tangier disease and Familial HDL deficiency, disorders associated with low levels of HDL cholesterol, have approximately four- to sixfold increased risk of atherosclerotic cardiovascular disease, compared with age-matched controls.48,49 If abnormal lipoprotein profile persists through childhood, it will lead to the progression of cardiovascular diseases in children born to obese mothers.50
Maternal obesity and placental mitochondrial dysfunction
Insulin resistance and obesity have been previously linked to impaired mitochondrial function.27,29 Petersen et al. have shown that insulin resistance in the skeletal muscle of healthy, young, lean but insulin-resistant offspring of patients with type 2 diabetes is associated with dysregulation of intramyocellular fatty acid metabolism.51 Accumulated evidence indicates a suppression of β-oxidation,52 carnitine deficiency,53,54 and decreased expression of CPT1α and CPT2 transporters55–57 in the settings of obesity and type 2 diabetes. Interestingly, we found that placental β-oxidation responds to maternal obesity in a sex-dependent manner. Male offspring present carnitine deficiency, a common feature of insulin-resistant states such as advanced age, genetic diabetes, and diet-induced obesity,54 whereas females show a reduction in CPT1 and CPT2 transporters. Decreased lipid oxidation in obese women’s placentas eventually leads to metabolic imbalance, causing excessive production of reactive oxygen species and suppression of oxidative phosphorylation, both events being previously reported by our team6 and others.58 In turn, oxidative stress could contribute to the development of insulin resistance by impairing mitochondrial function,27 thereby creating a feedforward progression of mitochondrial dysfunction and insulin resistance. The expression of PGC-1α, a transcription factor that plays a key role in mitochondrial biogenesis, supposed to be stimulated by oxidative stress;59,60 however, data from this study and reports from other groups61 suggest a decrease in PGC-1α content in the settings of obesity and diabetes, and data from this study suggest a decrease in PGC-1α content. It has been previously reported that insulin phosphorylates and inhibits PGC-1α via the intermediary protein kinase Akt2/protein kinase B (PKB)-b mechanism,17 and our data supports this finding. Phosphorylation prevents the recruitment of PGC-1a to the cognate promoters, impairing its ability to induce expression of target genes and promote gluconeogenesis and fatty acid oxidation.17 PGC1α induces expression of numerous transcription factors involved in the regulation of metabolic activity: Nuclear respiratory factors NRF1 and 2, sirtuins (SIRTs), estrogen-related receptors (ERRs), and carnitine palmitoyltransferases (CPTs).62 Recently, SIRT3 has gained attention for its roles in mitochondrial function63 and the inhibition of ROS production.64 Meanwhile, ERRα plays a pivotal role in energy metabolism due to controlling many nuclear genes associated with mitochondrial function, and nearly half of all mitochondrial DNA genes.65 Interestingly, when co-activated by PGC1α, ERRα can bind to ERRα-binding motifs in its own promoter and regulate its own transcription.66 Our data indicate that maternal obesity significantly reduces placental levels of both SIRT3 and ERRα.
Carnitine palmitoyltransferases 1α and 2 (CPT1α and CPT2) are responsible for shuttling long-chain fatty acids from the cytosol into the mitochondrial matrix for subsequent β-oxidation.67 We found expression of CPT1α and CPT2 to be reduced, but only in female-carrying placentas. Kim et al. reported that obese individuals have reduced activity of CPT1α, which directly correlates with a reduced ability of their mitochondria to oxidize long-chain fatty acids.68
Maintaining adequate metabolic flux
Metabolomics analysis of placental tissue from NW and OB women showed an accumulation of glutamine, glutamate, α-KG, 2-HG, and citrate. Glutamine plays a central role in fetal carbon and nitrogen metabolism, and in humans exhibits one of the highest fetal/maternal plasma ratios of all amino acids. Glutamine is taken up from the fetal circulation by the fetal liver, converted into glutamate via glutaminase, and then released back into the fetal circulation, where it is then taken up by the placenta and converted back into glutamine via glutamine synthetase.69 Glutamate is also converted into α-KG by glutamate dehydrogenase, located in the mitochondria; this α-KG feeds directly into the Krebs cycle, where it undergoes a series of oxidation-reduction reactions to produce the high-energy intermediates NADH and FADH2. A decrease in the glutamine-to-glutamate ratio was previously linked to childhood obesity,70 and high risks of insulin resistance and diabetes in adults;71 and our study found this ratio to be significantly reduced in obese women’s placentas.
Thus, the placentas from obese women recapitulate metabolic events typical for obese individuals: mitochondrial dysfunction and changes in lipid metabolism. As a multifunctional organ, the placenta represents a unique information source that sheds light on factors related to pregnancy and the intrauterine environment. Furthermore, it provides insights into future clinical issues (obesity, diabetes, metabolic syndrome) that can become evident years after delivery. Our study’s design does not allow us to distinguish between the programming effect of the maternal environment and genetic effects. Thus, since many of the changes we observe in obese mothers’ fetuses will be present in obese adults, some of these effects may be also inherited genetically.
Our data show that maternal weight gain in the obese group was lower than in the group of normal-weight women, and was consistent with current recommendations of the Institute of Medicine.72 Also, we found no differences in birth weight. We can speculate that gaining the recommended amount of weight in the obese group may have prevented excessive fetal growth. Our data are consistent with a previously published review from the Healthy Start Study, showing that maternal prepregnancy BMI, and gestational weight gain were positively and independently associated with neonatal adiposity but not birth weight.73 This review suggests that higher maternal weight gain during pregnancy, regardless of prepregnancy BMI, is directly related to offspring adiposity at birth. Another systematic review that includes 35 studies shows evidence of associations between excessive gestational weight gain and increased birth weight and fetal growth, again, independently of prepregnancy BMI.74 Another review that focused primarily on maternal obesity and perinatal outcomes found no increase in the occurrence of macrosomic babies in offspring of either obese (BMI = 30–39.9), morbidly obese (BMI = 40–49.9), or superobese (BMI > 50) women.75 Interestingly, in overweight and obese women weight loss or gain ≤5 kg was associated with increased risk of small for gestational age babies, decreased lean body mass, and head circumference.76 However, low maternal weight gain did not associate with better placental function and fetal metabolic outcomes such as fetal lipid profile or insulin sensitivity, suggesting that prepregnancy obesity is sufficient to affect the placenta and the fetus. Our cohort found no correlation between gestational weight gain and fetal lipid profile, fetal insulin sensitivity, and placental function (data not shown).
Sexual dimorphism in placental response to maternal obesity
While no sex differences were observed in lipid profiles or insulin sensitivity of babies born to obese mothers, the placental adaptation to maternal obesity was fetal sex-dependent. We found fetal sex-dependent changes in proteins responsible for placental lipid uptake (LDLR, Fig. 2), mitochondrial function and biogenesis (PGC-1a, and SIRT3, Fig. 3), and fatty acid oxidation (acylcarnitines, CPT1α, and CPT2, Fig. 4). Sex-dependent differences in placental response to maternal obesity have been shown before.6,9,77–79 Studies from our group and others demonstrated sexual dimorphism in placental thickness,80 mitochondrial functions,6,78 autophagy,9 expression of inflammatory markers,78,81 and epigenetic modifications.82–84 The mechanisms underlying the sexually dimorphic effect of maternal obesity remain poorly understood. One potential explanation can be the fetal sex-dependent differences in the levels of hormonal or metabolic milieu. For example, estrogen-dependent decrease in SIRT3 expression has been previously reported in female aging hearts,85 and in the brain.64,86 Male-specific reduction in the brain expression of PGC-1α was reported in the mouse model of chronic high-fat diet consumption, and has been linked to the accumulation of sphingolipids and saturated fatty acids in the brain of male mice.87 Consistent with these data, we found a decrease in expression of PGC-1α in male placentas from OB women, which could be at least partially explained by the placental accumulation of ceramides and dihydroceramides that we have reported before.9 Thus, whether it is the presence of a Y chromosome, an extra X chromosome, metabolic or epigenetic changes, or a difference in the levels of hormones, the sex differences in the placenta are remarkably reflected in the placental adaptation to adverse intrauterine environment.
In summary, we show that both male and female offspring of obese mothers display altered plasma lipid profiles and early signs of insulin resistance. Children gestated in an adverse intrauterine environment are predisposed to become obese later and are at high risk of developing metabolic diseases. This creates a trajectory of obesity that begins in utero and is perpetuated across generations.
Supplementary Material
Highlights.
Maternal obesity reduces the transfer of cholesterol from mother to fetus.
Neonates born to obese mothers are dyslipidemic and insulin resistant.
Placental lipid metabolism is affected by maternal obesity.
Glutamine oxidation is activated in the placentas of obese women as a compensatory mechanism.
Acknowledgments.
The authors thank the women who donated their placentas for this study. We also thank the Labor and Delivery Department at OHSU and the Maternal and Fetal Research Team for coordinating the collection of placentas.
Financial support.
This work was funded by NIH HD097398 (AM), HD076259 (AM, LM), American Heart Association GRNT29960007 (AM), and CTSA grant (UL1TR001120) (AM).
Footnotes
Conflicts of interest. The authors declare no conflict of interest.
Supplementary material. To view supplementary material for this article, please visit https://doi.org/10.1017/S2040174420001026.
References
- 1.Hay WW Jr. Placental-fetal glucose exchange and fetal glucose metabolism. Trans Am Clin Climatol Assoc. 2006; 117, 321–340. [PMC free article] [PubMed] [Google Scholar]
- 2.Sonagra AD, Biradar SM, Dattatreya K, Murthy DSJ. Normal pregnancy – a state of insulin resistance. J Clin Diagn Res. 2014; 8, CC01–CC3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Homko CJ, Sivan E, Reece EA, Boden G. Fuel metabolism during pregnancy. Semin Reprod Endocrinol. 1999; 17, 119–125. [DOI] [PubMed] [Google Scholar]
- 4.Grieger JA, Bianco-Miotto T, Grzeskowiak LE, et al. Metabolic syndrome in pregnancy and risk for adverse pregnancy outcomes: a prospective cohort of nulliparous women. PLoS Med. 2018; 15, e1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter AM. Evolution of placental function in mammals: the molecular basis of gas and nutrient transfer, hormone secretion, and immune responses. Physiol Rev. 2012; 92, 1543–1576. [DOI] [PubMed] [Google Scholar]
- 6.Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab. 2014; 307, E419–E425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008; 29, 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal P, Morriseau TS, Kereliuk SM, Doucette CA, Wicklow BA, Dolinsky VW. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit Rev Clin Lab Sci. 2018; 55, 71–101. [DOI] [PubMed] [Google Scholar]
- 9.Muralimanoharan S, Gao X, Weintraub S, Myatt L, Maloyan A. Sexual dimorphism in activation of placental autophagy in obese women with evidence for fetal programming from a placenta-specific mouse model. Autophagy. 2016; 12, 752–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon B, Bucher M, Maloyan A. A primary human trophoblast model to study the effect of inflammation associated with maternal obesity on regulation of autophagy in the placenta. J vis Exp. 2017; 127, 56484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song DK, Hong YS, Sung YA, Lee H. Insulin resistance according to beta-cell function in women with polycystic ovary syndrome and normal glucose tolerance. PloS One. 2017; 12, e0178120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minicocci I, Santini S, Cantisani V, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013; 54, 3481–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reaven GM, Lerner RL, Stern MP, Farquhar JW. Role of insulin in endogenous hypertriglyceridemia. J Clin Invest. 1967; 46, 1756–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu HY, Yehuda-Shnaidman E, Hong T, et al. Prolonged exposure to insulin suppresses mitochondrial production in primary hepatocytes. J Biol Chem. 2009; 284, 14087–14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei S, Gu H, Yang X, Guo H, Liu Z, Cao W. Prolonged exposure to insulin induces mitochondrion-derived oxidative stress through increasing mitochondrial cholesterol content in hepatocytes. Endocrinology. 2012; 153, 2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metabol. 2005; 1, 361–370. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007; 447, 1012–1016. [DOI] [PubMed] [Google Scholar]
- 18.Casaburi I, Chimento A, De Luca A, et al. Cholesterol as an endogenous ERRalpha agonist: a new perspective to cancer treatment. Front Endocrinol. 2018; 9, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirebeau-Prunier D, Le Pennec S, Jacques C, et al. Estrogen-related receptor alpha and PGC-1-related coactivator constitute a novel complex mediating the biogenesis of functional mitochondria. FEBS J. 2010; 277, 713–725. [DOI] [PubMed] [Google Scholar]
- 20.Brenmoehl J, Hoeflich A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion. 2013; 13, 755–761. [DOI] [PubMed] [Google Scholar]
- 21.Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013; 62, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metabol. 2017; 25, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: thematic review series: genetics of human lipid diseases. J Lipid Res. 2012; 53, 4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang CV. Links between metabolism and cancer. Genes Develop. 2012; 26, 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearce EJ, Pearce EL. Immunometabolism in 2017: driving immunity: all roads lead to metabolism. Nat Rev Immunol. 2018; 18, 81–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008; 582: 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martins AR, Nachbar RT, Gorjao R, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis. 2012; 11, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karaderi T, Drong AW, Lindgren CM. Insights into the genetic susceptibility to type 2 diabetes from genome-wide association studies of obesity-related traits. Curr Diab Rep. 2015; 15, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Meo S, Iossa S, Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol. 2017; 233, R15–R42. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Twinn DS, Gascoin G, Musial B, et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci Rep. 2017; 7, 44650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maric T, Kanu C, Johnson MR, Savvidou MD. Maternal, neonatal insulin resistance and neonatal anthropometrics in pregnancies following bariatric surgery. Metabolism. 2019; 97, 25–31. [DOI] [PubMed] [Google Scholar]
- 32.Tinius RA, Cahill AG, Strand EA, Cade WT. Altered maternal lipid metabolism is associated with higher inflammation in obese women during late pregnancy. Integr Obes Diabetes. 2015; 2, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lassance L, Haghiac M, Leahy P, et al. Identification of early transcriptome signatures in placenta exposed to insulin and obesity. Am J Obstetr Gynecol. 2015; 212, 647.e1–647.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassance L, Haghiac M, Minium J, Catalano P, Hauguel-de Mouzon S. Obesity-induced down-regulation of the mitochondrial translocator protein (TSPO) impairs placental steroid production. J Clin Endocrinol Metabol. 2015; 100, E11–E18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008; 102, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly AC, Powell TL, Jansson T. Placental function in maternal obesity. Clin Sci. 2020; 134, 961–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Javed K, Fairweather SJ. Amino acid transporters in the regulation of insulin secretion and signalling. Biochem Soc Trans. 2019; 47, 571–590. [DOI] [PubMed] [Google Scholar]
- 38.Segovia SA, Vickers MH, Gray C, Reynolds CM. Maternal obesity, inflammation, and developmental programming. BioMed Res Int. 2014; 2014, 418975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feingold KR. Obesity and dyslipidemia. In Endotext (eds. Feingold KR, Anawalt B, Boyce A, et al. ), 2000. MDText.com, Inc., South Dartmouth, MA. [Google Scholar]
- 40.Geraghty AA, Alberdi G, O’Sullivan EJ, et al. Maternal and fetal blood lipid concentrations during pregnancy differ by maternal body mass index: findings from the ROLO study. BMC Pregnancy Childbirth. 2017; 17, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woollett L, Heubi JE. Fetal and neonatal cholesterol metabolism. In Endotext (eds. Feingold KR, Anawalt B, Boyce A, et al. ), 2000. MDText.com, Inc., South Dartmouth, MA. [Google Scholar]
- 42.Woollett LA. Fetal lipid metabolism. Front Biosci. 2001; 6, D536–D545. [DOI] [PubMed] [Google Scholar]
- 43.Vahratian A, Misra VK, Trudeau S, Misra DP. Prepregnancy body mass index and gestational age-dependent changes in lipid levels during pregnancy. Obstetr Gynecol. 2010; 116, 107–113. [DOI] [PubMed] [Google Scholar]
- 44.Meyer BJ, Stewart FM, Brown EA, et al. Maternal obesity is associated with the formation of small dense LDL and hypoadiponectinemia in the third trimester. J Clin Endocrinol Metab. 2013; 98, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Augsten M, Hackl H, Ebner B, et al. Fetal HDL/apoE: a novel regulator of gene expression in human placental endothelial cells. Physiol Genomics. 2011; 43, 1255–1262. [DOI] [PubMed] [Google Scholar]
- 46.Ethier-Chiasson M, Duchesne A, Forest JC, et al. Influence of maternal lipid profile on placental protein expression of LDLr and SR-BI. Biochem Biophys Res Commun. 2007; 359, 8–14. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Ma KL, Ruan XZ, Liu BC. Dysregulation of the low-density lipoprotein receptor pathway is involved in lipid disorder-mediated organ injury. Int J Biol Sci. 2016; 12, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rust S, Rosier M, Funke H, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999; 22, 352–355. [DOI] [PubMed] [Google Scholar]
- 49.Brooks-Wilson A, Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999; 22, 336–345. [DOI] [PubMed] [Google Scholar]
- 50.Blackmore HL, Ozanne SE. Maternal diet-induced obesity and offspring cardiovascular health. J Dev Orig Health Dis. 2013; 4, 338–347. [DOI] [PubMed] [Google Scholar]
- 51.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004; 350, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serra D, Mera P, Malandrino MI, Mir JF, Herrero L. Mitochondrial fatty acid oxidation in obesity. Antioxid Redox Signal. 2013; 19, 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seiler SE, Martin OJ, Noland RC, et al. Obesity and lipid stress inhibit carnitine acetyltransferase activity. J Lipid Res. 2014; 55, 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noland RC, Koves TR, Seiler SE, et al. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem. 2009; 284, 22840–22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Becker C, Kukat A, Szczepanowska K, et al. CLPP deficiency protects against metabolic syndrome but hinders adaptive thermogenesis. EMBO Rep. 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hulver MW, Berggren JR, Cortright RN, et al. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003; 284, E741–E747. [DOI] [PubMed] [Google Scholar]
- 57.Fujiwara N, Nakagawa H, Enooku K, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018; 67, 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hastie R, Lappas M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta. 2014; 35, 673–683. [DOI] [PubMed] [Google Scholar]
- 59.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006; 127, 397–408. [DOI] [PubMed] [Google Scholar]
- 60.Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008; 451, 1008–1012. [DOI] [PubMed] [Google Scholar]
- 61.Crunkhorn S, Dearie F, Mantzoros C, et al. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007; 282, 15439–15450. [DOI] [PubMed] [Google Scholar]
- 62.Supruniuk E, Mikłosz A, Chabowski A. The implication of PGC-1α on fatty acid transport across plasma and mitochondrial membranes in the insulin sensitive tissues. Front Physiol. 2017; 8, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013; 48, 634–639. [DOI] [PubMed] [Google Scholar]
- 64.Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. 2017; 16, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ranhotra HS. Estrogen-related receptor alpha and mitochondria: tale of the titans. J Recept Signal Transduct Res. 2015; 35, 386–390. [DOI] [PubMed] [Google Scholar]
- 66.Mootha VK, Handschin C, Arlow D, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004; 101, 6570–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016; 7, e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000; 279, E1039–E1044. [DOI] [PubMed] [Google Scholar]
- 69.Watford M Glutamine and glutamate: nonessential or essential amino acids? Anim Nutr. 2015; 1, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Isganaitis E, Rifas-Shiman SL, Oken E, et al. Associations of cord blood metabolites with early childhood obesity risk. Int J Obes. 2015; 39, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng S, Rhee EP, Larson MG, et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation. 2012; 125, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rasmussen KM, Catalano PM, Yaktine AL. New guidelines for weight gain during pregnancy: what obstetrician/gynecologists should know. Curr Opin Obstet Gynecol. 2009; 21, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Starling AP, Brinton JT, Glueck DH, et al. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study. Am J Clin Nutr. 2015; 101, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siega-Riz AM, Viswanathan M, Moos MK, et al. A systematic review of outcomes of maternal weight gain according to the Institute of Medicine recommendations: birthweight, fetal growth, and postpartum weight retention. Am J Obstetr Gynecol. 2009; 201, 339.e1–339.e14. [DOI] [PubMed] [Google Scholar]
- 75.Marshall NE, Guild C, Cheng YW, Caughey AB, Halloran DR. Maternal superobesity and perinatal outcomes. Am J Obstetr Gynecol. 2012; 206, 417.e1–417.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Catalano PM, Mele L, Landon MB, et al. Inadequate weight gain in overweight and obese pregnant women: what is the effect on fetal growth? Am J Obstetr Gynecol. 2014; 211, 137.e1–137.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leon-Garcia SM, Roeder HA, Nelson KK, et al. Maternal obesity and sex-specific differences in placental pathology. Placenta. 2016; 38, 33–40. [DOI] [PubMed] [Google Scholar]
- 78.Muralimanoharan S, Guo C, Myatt L, Maloyan A. Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int J Obesity. 2015; 39, 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rosenfeld CS. Sex-specific placental responses in fetal development. Endocrinology. 2015; 156, 3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mando C, Calabrese S, Mazzocco MI, et al. Sex specific adaptations in placental biometry of overweight and obese women. Placenta. 2016; 38, 1–7. [DOI] [PubMed] [Google Scholar]
- 81.Aye IL, Lager S, Ramirez VI, et al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014; 90, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Houde AA, St-Pierre J, Hivert MF, et al. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J Dev Orig Health Dis. 2014; 5, 132–141. [DOI] [PubMed] [Google Scholar]
- 83.Houde AA, Guay SP, Desgagne V, et al. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics. 2013; 8, 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lesseur C, Armstrong DA, Paquette AG, Li Z, Padbury JF, Marsit CJ. Maternal obesity and gestational diabetes are associated with placental leptin DNA methylation. Am J Obstetr Gynecol. 2014; 211, 654.e1–654.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barcena de Arellano ML, Pozdniakova S, Kuhl AA, Baczko I, Ladilov Y, Regitz-Zagrosek V. Sex differences in the aging human heart: decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging. 2019; 11, 1918–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lejri I, Grimm A, Eckert A. Mitochondria, Estrogen and female brain aging. Front Aging Neurosci. 2018; 10, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morselli E, Criollo A, Rodriguez-Navas C, Clegg DJ. Chronic high fat diet consumption impairs metabolic health of male mice. Inflamm Cell Signal. 2014; 1, e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.