

Abstract
Measurement of the location of molecules in tissues is essential for understanding tissue formation and function. Previously, we developed Slide-seq, a technology that enables transcriptome-wide detection of RNAs with a spatial resolution of 10 μm. Here, we report Slide-seqV2, which combines improvements in library generation, bead synthesis, and array indexing to reach an RNA capture efficiency of ~50% of single cell RNA sequencing data (~10x greater than Slide-seq) approaching the detection efficiency of droplet-based single-cell RNA-seq techniques. First, we leverage the detection efficiency of Slide-seqV2 to identify dendritically localized mRNAs in neurons of the mouse hippocampus. Second, we integrate the spatial information of Slide-seqV2 data with single-cell trajectory analysis tools to characterize the spatiotemporal development of the mouse neocortex, identifying underlying genetic programs that were poorly sampled with the Slide-seq. The combination of near-cellular resolution and high transcript detection efficiency makes Slide-seqV2 useful across many experimental contexts.
The ab initio identification of spatially defined gene expression patterns can provide insights into the development and maintenance of complex tissue architectures, and the molecular characterization of pathological states. We recently developed Slide-seq1, a spatial genomics technology that quantifies expression genome-wide with 10-micron spatial resolution. While recent developments in imaging-based transcriptomics have enabled the identification of tens to hundreds of pre-selected genes in fixed specimens2,3,4,5, array-based approaches6,7,1 such as Slide-seq critically decouple the imaging from molecular sampling, while simultaneously allowing for transcriptome-wide identification of molecular patterns in diverse tissue sections6,7,1,. In Slide-seq, densely barcoded bead arrays, termed “pucks,” are fabricated by split-pool phosphoramidite synthesis, and indexed up front using a sequencing by ligation strategy. Once the arrays are indexed, Slide-seq assays are performed with equipment found in a standard molecular biology laboratory, enabling the facile reconstruction of 3D tissue volumes that are tens or even hundreds of cubic millimeters in size.
However, Slide-seq’s low transcript detection sensitivity limited the range of biological problems to which the technology could be applied. Through improvements to the barcoded bead synthesis, the array sequencing pipeline, and the enzymatic processing of cDNA, we report here a version of Slide-seq with an order of magnitude higher sensitivity. With our new protocol, termed Slide-seqV2, we demonstrate a range of new analytical possibilities by leveraging its improved capture efficiency, including the identification of process-localized genes in neurons, and the analysis of developmental trajectories in situ.
We increased the yield of Slide-seq capture by improving the array generation pipeline as well as the library preparation strategy (Figure 1a). First, we developed a novel strategy to spatially index barcoded bead arrays using a monobase encoding scheme with sequencing by ligation using sequential interrogation by offset primers8,9 (Supplementary Figure 1a–c, Methods). We were motivated to develop the monobase encoding scheme for two reasons: first, SOLiD di-base encoding utilizes proprietary cleavage chemistry that is not commercially available; and second, computational matching between dibase sequenced barcodes and Illumina sequencing requires conversion between color and base space and is not error robust. Our open-source monobase sequencing strategy, which uses only readily available reagents, performed equivalently to SOLiD in array indexing (Supplementary Figure 1d,e). In addition, we optimized the conditions for split-pool synthesis of the 10 μm polystyrene barcoded beads (Methods), which improved the clonality of our barcodes (Supplementary Figure 2). Together, these strategies enabled more efficient recovery of gene expression on Slide-seqV2 arrays per Illumina read.
Figure 1: Highly improved mRNA detection sensitivity in Slide-seqV2.
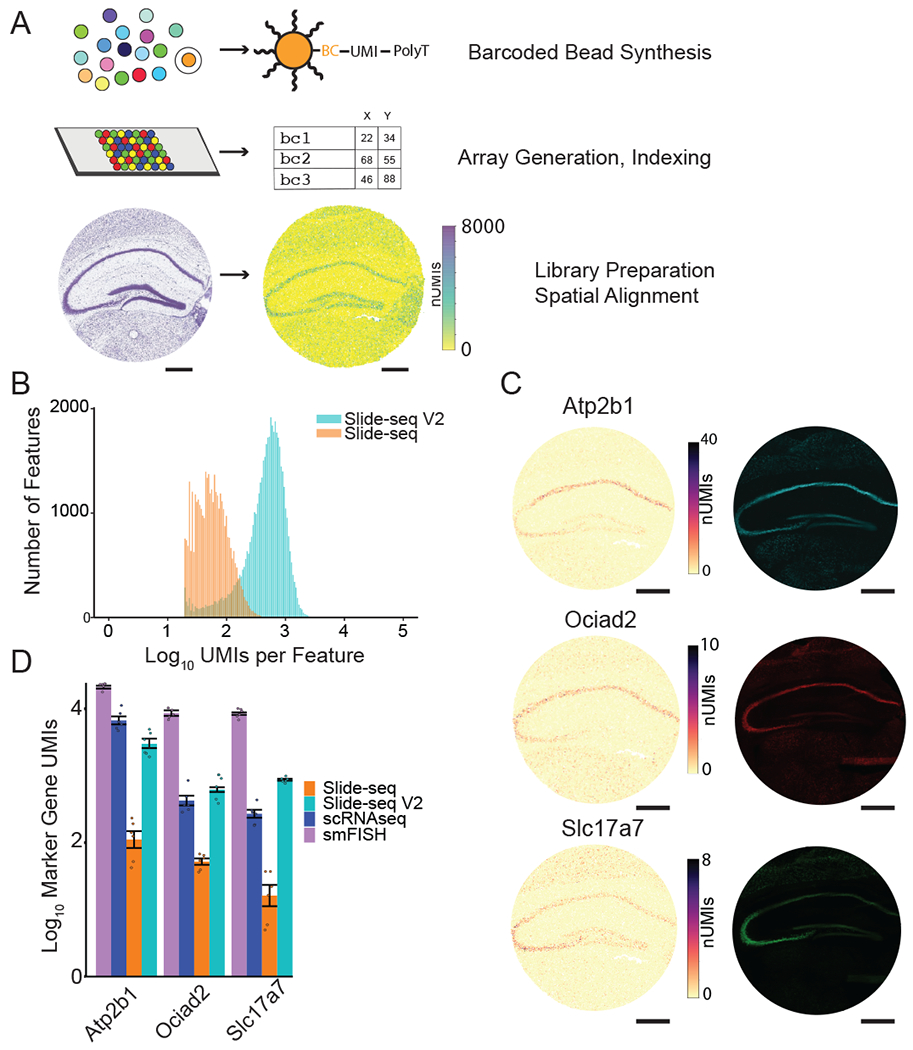
A) Overview of the Slide-seq method. An example array of mouse hippocampus generated with Slide-seqV2, with each bead colored by the number of UMIs.
B) Histogram of number of UMIs per bead for Slide-seq (red) versus Slide-seqV2 (blue) on serial mouse embryo sections.
C) Images of marker genes of hippocampus in Slide-seqV2 (left column) versus HCR FISH images (right column, N=1 HCR experiment on serial section of Slide-seq data shown).
D) Comparison of marker gene counts in mouse hippocampus CA1 across four modalities (N = 6 measurements per modality, mean ∓ sd reported in Supplementary Table 2). For smFISH, Slide-seqV2 and Slide-seq data, all transcript counts within a fixed area of CA1 were summed together; for scRNA-seq, we summed the counts for the number of CA1 pyramidal cells counted within this area.
(All scale bars 500 μm)
Next, we optimized the enzymatic library preparation steps of Slide-seqV2. We hypothesized that, due to the tissue’s inhibitory presence during reverse transcription, the template-switching reaction that adds a 3’ priming site for whole-transcriptome amplification was inefficient. We therefore added another second strand synthesis step10 after reverse transcription to increase the number of cDNAs that can be amplified by PCR. We performed Slide-seqV2 on E12.5 mouse embryos, obtaining ~9.3x more transcripts (UMIs) per bead, compared to the original Slide-seq protocol (Figure 1b, median 550 UMIs Slide-seqV2, 59 UMIs Slide-seq). Similarly, in the adult mouse hippocampus, we observed an 8.9x increase in the number of UMIs per bead, with the majority of the improvement (4.6x, Supplementary Table 1), attributable to the additional second strand synthesis step, and the remaining improvement largely due to improvements in bead barcode synthesis. In the mouse hippocampus, the capture efficiency of Slide-seqV2 was higher than that of a recently released commercial spatial transcriptomics (ST) technology (mean UMIs: Slide-seqV2 = 45,772, Visium = 27,952, for equal feature size, Supplementary Figure 3a–d) while maintaining 30x improved spatial resolution (30.25x by area per feature, 10 μm feature size for Slide-seqV2 compared to 55 μm feature size for Visium data). We also compared the sensitivity of Slide-seqV2 to HDST, another ST technology with high spatial resolution7. We found that in the mouse olfactory bulb, Slide-seqV2 recovered significantly more transcripts per 10 μm feature than HDST (44.9-fold mean difference; 494 UMIs Slide-seqV2 versus 11.5 UMIs for HDST; Supplementary Figure 3e,f).
Next, we sought to quantify the absolute sensitivity of Slide-seqV2 relative to other molecular technologies that measure RNA content in cells and tissues. We compared counts of CA1 marker genes (Atp2b1, Ocaid2, Slc17a7) in an equal number of cells, measured by: (1) Slide-seqV2; (2) Drop-seq, a high-throughput scRNA-seq method11,12; and (3) smFISH5,13,14 (Methods). We found that Slide-seqV2 detected similar patterns to smFISH (Figure 1C, Supplementary Figure 4, Supplementary Figure 5a) as well similar numbers of UMIs when compared to Drop-seq for the three genes measured (equivalent area in Slide-seqV2 to cells taken from Drop-seq) (mean +/− std. scRNAseq = 33.5±1.4, 2.1±1.5,1.2±1.5, Slide-seqV2 = 15.7±1.5, 2.3±2.4, 1.9±2.6, Figure 1D, N = 6, Supplementary Table 2). To more thoroughly characterize the sensitivity of Slide-seqV2, we compared the total UMI counts per gene for all genes detected in CA1 excitatory neurons in Drop-seq11 to an equivalent number of CA1 cells by area in Slide-seqV2. We found that, genome-wide, Slide-seqV2 detects approximately 44% ± 26% of the counts of Drop-seq (median +/− MAD, Supplementary Figure 5b) demonstrating that Slide-seqV2 capture efficiency approaches that of modern single cell technologies. Lastly, we found Slide-seqV2 to be highly reproducible between replicates (⍴=.98, Supplementary Figure 5c).
We next applied Slide-seqV2 to gain insight into biological problems where higher capture sensitivity is important. Neurons actively transport specific mRNAs to dendrites and postsynaptic densities, where they play critical roles in synaptic development and plasticity15–17. Previous studies have explored dendritic enrichment through physical microdissection or cell culture, but none has systematically identified the distribution of dendritically localized transcripts in situ. Dendritic mRNAs constitute only a tiny fraction of neuronal transcripts18, necessitating higher sensitivity methods for their detection. To identify dendritically localized mRNAs from our mouse hippocampal Slide-seqV2 dataset, we took advantage of the stereotyped architecture of the CA1 neuropil to reduce the spatial localization of transcripts to a 1D profile perpendicular to the CA1 soma layer (from stratum oriens (s.o.) to stratum pyramidale (s.p.) across stratum radiatum (s.r.), Figure 2a,b). For each gene detected in Slide-seqV2 (N=4 sections), we calculated the spatial expression as a function of distance from the soma (representative spatial expression profiles shown in Figure 2b, bottom).
Figure 2: Slide-seqV2 reveals spatial patterning of dendritically enriched mRNAs.
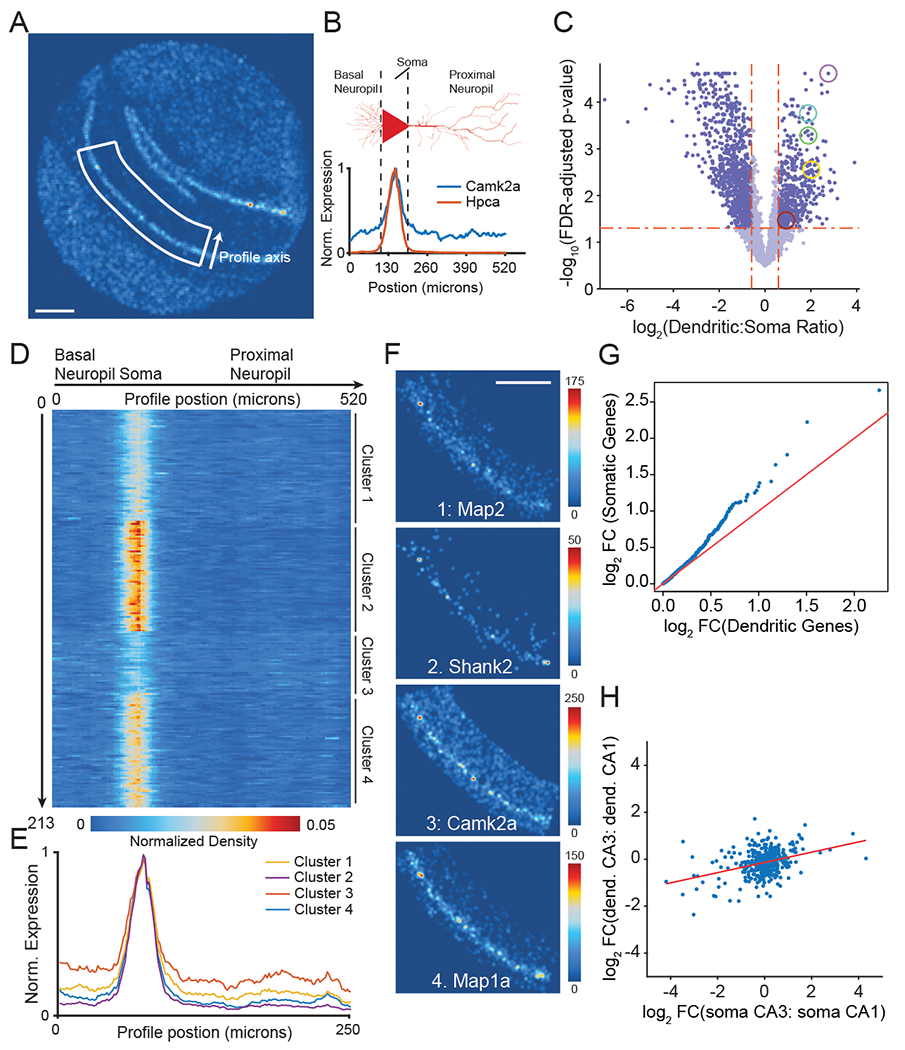
A) Spatial heatmap of number of UMIs for a hippocampal Slide-seqV2 dataset.
B) (top) Schematic of linear spatial profiling across CA1 soma and dendrites. (bottom) Spatial profiles of a CA1 marker (Hpca, red), and a classically dendritically localized gene (Camk2a, blue) are shown.
C) Differentially expressed genes in soma versus proximal dendrites. Highlighted are genes with FDR-corrected p-value <0.05 and fold change >2. Several classically known dendritically expressed genes are circled: (Camk2a, (FDR adjusted p = 2.8×10−3, Yellow), Ef1a (FDR adjusted p = 5×10−4, Green), Prkcz (FDR adjusted p = .034, Red), Map2 (FDR adjusted p = 1.7×10−4, Teal) and Ddn (FDR adjusted p = 2.×10−5, Purple). Two tailed, two sample t-test (N=5 tissue sections).
D) Expression heatmap of 237 dendritically enriched RNAs across the neuronal profile axis. Genes are shown clustered by their spatial profile (k-means clustering, 4 clusters). Rows are normalized and sum to 1.
E) Average spatial expression profile of each of the four gene clusters identified in D across CA1.
F) Slide-seqV2 reconstruction images of one synaptic protein-encoding gene from each of the four clusters in D. Scale bars are 500 μm for all Slide-seqV2 reconstructions. Color bar represents the total number of UMIs detected for gene.
G) Quantile-quantile plot of the log2 fold-change (log2 FC) between CA1 and CA3/dentate pyramidal cell types (defined by scRNA-seq12) of dendritic (x-axis), compared with somatic (y-axis) gene sets defined by the analysis in C.
H) Ratio of expression between CA3 and CA1 regions in soma and dendrites for Slide-seqV2 data. Linear fit shown in red (slope = 0.22, R2 = 0.13).
To select for dendritically localized mRNA, we performed differential expression analysis, comparing the proximal neuropil (s.r.) to the soma (s.p.). The CA1 neuropil contains glial cell types (i.e. microglia and astrocytes) that also contribute RNA and interfere with analysis; we therefore included only genes expressed in CA1 pyramidal cells (>0.5 TPM in CA1 pyramidal neurons) and excluded those that are markers of non-neuronal cell types (Online Methods, Supplementary Table 3), based on existing scRNA-seq data of the hippocampus11. After filtering, differential expression between the proximal neuropil and the soma revealed 213 significant genes with greater than 2-fold dendritic enrichment (Figure 2c, unpaired t-test, N =4 sections, Supplementary Table 3). These genes overlapped significantly (p<10−16, hypergeometric test, Supplementary Figure 6a) with lists of dendritically enriched RNAs from two previous studies19,20, suggesting Slide-seqV2 can discover dendritically enriched genes.
Next, we asked whether functionally related genes showed similarities in their dendritic enrichment. First, we grouped dendritically enriched genes according to their 1D spatial expression profile (Figure 2d). Using unsupervised clustering, we identified 4 clusters of spatial expression of dendritically localized genes in CA1 neuropil, with clusters having different degrees of dendritic enrichment (Figure 2e, Supplementary Table 3). To identify whether this observed spatial diversity in localization was related to protein function, we used gene ontology (GO) to determine the cellular components of each spatial cluster (Supplementary Figure 6b, Methods). We found that each cluster was enriched for ontologically distinct groups of genes. Specifically, the first 2 clusters were enriched for components of the cellular respiration machinery, as well as ubiquitin ligases, while clusters 3 and 4 were enriched for ribosomal subunits. Slide-seqV2’s genome-wide capture allowed us to visualize the heterogeneity in dendritic trafficking across two synaptic and two cytoskeletal genes chosen from each cluster (Figure 2f, spatial reconstructions of all 213 genes are shown in Supplementary Dataset 1). Taken together, these data demonstrate Slide-seqV2’s ability to characterize process-localized mRNAs, which appear to display significant heterogeneity amongst the various trafficked synaptic mRNA components.
The specificity of dendritically enriched genes for specific cell types (e.g. CA1 versus other pyramidal cells) has not been widely examined, in part because traditional approaches have measured dendritic trafficking in vitro or only from a single in vivo cell type. To explore this question, we integrated Slide-seqV2 data, which spans multiple hippocampal fields, with single-cell RNA-seq data from the same tissue. From an existing hippocampal single cell dataset11, we computed differential expression between CA1 and other hippocampal pyramidal cells for all genes (Methods). The dendritically localized set identified by Slide-seqV2 showed a significant depletion of differentially expressed genes relative to somatically enriched genes (p < 0.05, Wilcoxon rank sum test; Figure 2g). These results suggest that in the hippocampus, dendritically localized transcripts are more likely to be broadly expressed genes, rather than markers of specialized neuronal cell types.
Dendritically enriched genes are expressed and trafficked from the soma compartment; we therefore additionally asked whether cell-type specific expression changes are reflected in the dendritic compartment. When dendritically enriched genes are examined in Slide-seqV2 in both CA3 and CA1 neurons, the variance in fold change in the soma for dendritic genes is much larger than the variance in fold change observed for dendrites (Figure 2h, Two sample F-test, p=3 × 10−9). Furthermore, soma fold change only explains 13% of the variance in dendritic expression between CA3 and CA1 dendrites. These results indicate expression in dendrites is relatively buffered from the soma, suggesting the existence of distinct regulatory mechanisms in these two neuronal compartments.
During development, dynamic changes in gene expression across time and space help give rise to complex tissue architectures and terminally differentiated cell types. An array of computational strategies have been developed to identify and explore developmental trajectories from scRNA-seq data21–24, based upon similarities in gene expression between individual profiles. More recently, an additional approach called RNA velocity was developed that dynamically models expression trajectories by the relative quantities of spliced and unspliced transcripts for each gene25. Inspired by this work, we reasoned that the combination of Slide-seqV2’s enhanced capture efficiency--which approaches that of scRNA-seq technologies--and its near-single-cell resolution--may allow us to exploit these powerful algorithms directly on our spatial data to learn how developmental processes are proceeding across a tissue section.
In the embryonic mouse neocortex, neuronal development progresses along a radial axis that begins in the Ventricular Zone (VZ) and moves through the Subventricular Zone (SVZ), Intermediate Zone (IZ), and finally the Cortical Plate (CP), where neurons integrate into cortical layers in a birthdate-dependent manner26. We wondered whether Slide-seqV2 data could be used to successfully recover this highly spatially organized developmental trajectory27. We first applied unsupervised clustering28 to Slide-seqV2 data from an embryonic day 15 (E15.5) developing mouse brain to characterize gene expression gradients in the neocortex. We annotated clusters corresponding to cell types in different developing brain regions, including cortex and striatum (Figure 3a, Supplementary Figure 7a–b). Segregating just the radially developing cortex (Figure 3a, black box), we reclustered the beads to reveal populations representing the VZ, SVZ, IZ, CP, early cortical layers (L5/6) and Cajal Retzius cells (CR) (Figure 3a).
Figure 3: Slide-seqV2 of developing mouse cortex reconstructs spatial developmental trajectories.
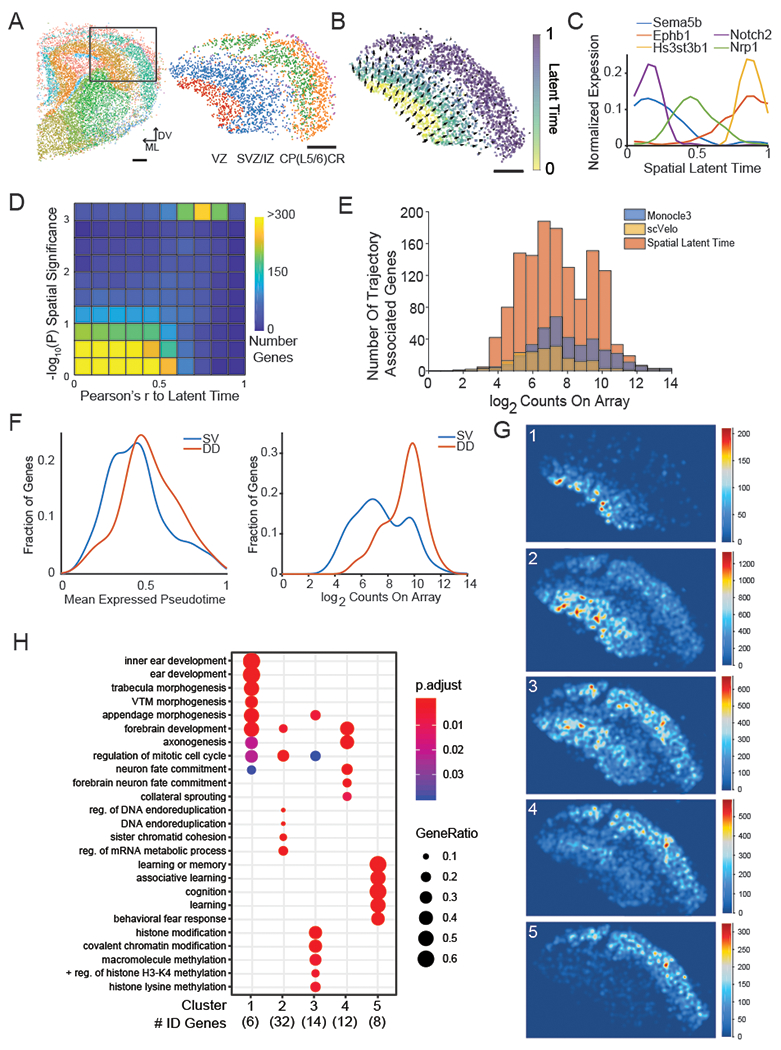
A) Left: Unsupervised cluster analysis of Slide-seqV2 data obtained from a section of E15 mouse brain. Black box delineated the region used in the analysis. (Scale bar, 200 μm, ML: medial/lateral axis, DV: dorsal ventral axis). Right: Beads present within black-box inset from top, colored by their annotated cluster identities, subsetted by clusters of cortical identity. Red = Ventricular Zone (VZ), Blue/Purple = Subventricular Zone/ Intermediate Zone, Green/ Orange = Cortical Plate/ Layer 5 / 6, Pink = Cajal Retzius Cells (CR cells). These reflect the layers present in the mouse cortex at this time point.
B) Beads within the anatomical region of developing cortex, colored by their assigned latent time metric from scVelo. Arrow size and direction correspond to the direction and magnitude of the spatial derivative of the latent time in physical space.
C) Expression profiles of sample genes jointly identified by Slide-seqV2, scVelo and Monocle3, across the Slide-seqV2-generated spatial latent time axis.
D) Two-dimensional density plot quantifying the relationship between a gene’s correlation with scVelo latent time (x-axis)and spatial significance (Permutation test, one-sided, y-axis), see Methods). Each square is colored by the number of genes found in that bin.
E) Stacked histogram of the number of genes associated to the developmental trajectory by Monocle3 (blue), scVelo (yellow), and spatial latent time (red), binned by expression level .
F) left: density plot of all spatial latent time genes (SV) compared to DD latent time genes (DD) across mean expressed latent time value; right: density plot of all spatial latent time genes (SV) compared to DD latent time genes (DD) for summed gene expression across array.
G) Slide-seqV2 reconstruction images of metagenes associated with each spatial cluster of DD genes (Methods).
H) Gene-ontology classifications using over-representation analysis (Methods) for biological process terms for each spatial cluster in G (Hypergeometric test, FDR-corrected p-value).
To determine whether Slide-seqV2 data can identify developmental trajectories, we first applied scVelo29, a recently developed trajectory inference method that leverages splicing information25, to order our beads along a predicted latent time (LT). Projection of each bead’s LT value onto spatial coordinates successfully recapitulated the established radial developmental axis of the neocortex (Figure 3b). A very similar trajectory was recovered using the pseudotime ordering generated by Monocle322,30(Supplementary Figure 7c).
During the course of a developmental process, each stage of maturation can proceed at a different rate. We wondered whether Slide-seqV2’s spatial information could be exploited to identify the relative rates of differentiation across the radial axis of neocortical development. To accomplish this, we took the spatial derivative of the scVelo-generated LT (Methods), recovering regions where LT changes most dramatically (Figure 3b, magnitude of arrows representing magnitude of the derivative). We found that the spatial rate of change was most pronounced at the earlier stages of the trajectory, decreasing as cells progress from VZ to SVZ/IZ, and largely terminating in the cortical plate.
Since each bead’s physical position is strongly predictive of its LT value, we reasoned that combining spatial and LT information could give us considerably greater statistical power to identify gene expression changes across this developmental process. The scVelo method was able to identify 179 genes with significant loading on LT, while the Monocle3 approach identified 377 genes. We previously demonstrated that we could leverage the spatial dimension of Slide-seq to systematically discover non-random spatial gene expression patterns1. Leveraging this, we identified 1349 spatially varying genes in the developing neocortex (P<0.005, Methods, Supplementary Table 4), spatial expression plots of all genes are in Supplementary Dataset 2). Among these were genes that are known to be involved in cortical development and are shared among Slide-seq and the trajectory inference methods including Sema5b and Nrp1, both of which encode proteins involved in axonal guidance31,32 (Figure 3c). We noted that these genes correlated strongly with the spatial LT axis. Thus, to systematically find genes that varied along this axis, we correlated the expression of these 1349 nonrandom genes with a spatial LT axis that was created by fitting a surface to the LT values in physical space (Methods). Of the 1349 spatially variable genes, 1043 correlated significantly with LT (pFdr <0.005), while very few of the non-spatially variable genes showed significant LT relationship (Figure 3d). In addition, the 1043 genes were highly overlapping with the trajectory inference methods across the range of expression levels (Figure 3e): amongst these were 76.5% of the scVelo-identified genes (137/179, Supplementary Figure 8, Supplementary Table 5), and 75.6% of the Monocle3-defined genes (285/377, Supplementary Figure 8, Supplementary Table 5). These results gave us confidence that the 1043 genes found using our spatial LT approach were truly associated with neocortical development.
We applied this spatial latent time approach to the embryonic eye at E12.5, a critical period of cell differentiation and migration33. We recovered the radial axis of ocular lens development, with strong spatial trajectories from the lens placode inward to the developing lens (Supplementary Figure 7d–e). Similarly to the cortex, we found spatial latent time provides a highly sensitive method to detect developmental genes along this trajectory. With this approach we identified >1000 genes as uniquely spatially variable (Supplementary Figure 7f;S8). Many genes recovered are important in ocular development, including genes essential to the development of the lens, and associated structures such as the lens placode (Pax6)34 and the primordial optic cup (Vax2)35. This list also includes many from the crystallin (Crybb3)36 and aldehyde dehydrogenases (Aldh1a1)37,38 gene families whose products form the fiber network of the lens36 and pattern signalling networks across the optic area37,38. Additionally, we found genes (Aldh1a3, Col9a1) identified as genetic drivers of disorders of ocular development39,40 that are spatially enriched, and whose protein products differentially pattern the lens placode, giving rise to distinct structures within the eye 33 (Supplementary Figure 7g). These results suggest spatial latent time, in combination with Slide-seqV2, can add spatial context to understanding the molecular signatures of genetic drivers of disease.
Inspired by the spatial enrichment of molecular signatures of genetic diseases in the developing eye, we next focused on neurodevelopmental disorders (DD), a class of diseases frequently caused by pathogenic mutations in protein coding genes41 that often disrupt the normal process of neocortical development. We asked how a set of 299 DD-associated genes, recently discovered by exome sequencing of DD parent-offspring trios42, distributed on our spatial LT trajectory. A total of 74 of the 299 DD-associated genes were found in the spatial LT gene set (1.87-fold enrichment, p = 3.2×10−8). These genes were expressed later in average LT compared with all spatial LT genes (Figure 3f, left). Interestingly, the average expression of the 74 DD-associated spatial LT genes was much higher than all spatial LT genes (Figure 3f, right). These 74 genes could be clustered into five groups based on their spatial expression patterns (Figure 3g, Methods). The individual clusters were enriched for distinct GO functional terms, suggesting that these genes participate in distinct developmental processes and pathways (Figure 3h), ranging from chromatin modification to establishment of neuronal states. Once additional phenotypic data becomes available about the relative clinical differences amongst these DD-associated genetic disorders, it will be revealing to understand how such phenotypes differentially load onto the spatial LT axis.
Here, we describe Slide-seqV2, a highresolution spatial genomics technology with nearly an order of magnitude higher sensitivity compared to the original Slide-seq protocol. In particular, we demonstrated how the higher capture efficiency of Slide-seqV2 significantly expands the scope of possible analyses, including the discovery of genes with distinct patterns of subcellular localization, and the tracing of developmental programs involved in fate specification through space.
To facilitate adoption of the technology, we have generated a streamlined pipeline for image processing and merging of short read sequencing and imaging data which is available on our Github repository (https://github.com/MacoskoLab/slideseq-tools)(Supplementary Figure 9, Methods). This pipeline provides statistics on the alignment of imaging and short read data, in addition to the gene expression matrix and spatial locations of each barcode, with minimal user intervention. The combination of efficient molecular biology workflows, open-source sequencing chemistry for array indexing, and easy-to-use software for merging imaging and sequencing data should support wide application of Slide-seq. We anticipate that the technical and computational improvements here will significantly accelerate the adoption of Slide-seqV2 across the academic community.
Online Methods
Barcoded beads
Bead barcodes were either synthesized by the Chemgenes Corporation or in-house on an Akta Oligopilot 10 on one of two polystyrene supports (Agilent PLRP-S-1000A 10 μm particles or 10 μm custom polystyrene from AMBiotech). Oligonucleotide synthesis was performed as described below. Beads were used with one of the two following sequences:
Chemgenes corporation beads:
5’-TTTTTTTTCTACACGACGCTCTTCCGATCTJJJJJJJJTCTTCAGCGTTCCCGAGAJJJJJJJNNNNNNNNT30
Custom synthesis beads:
5′-TTT_PC_GCCGGTAATACGACTCACTATAGGGCTACACGACGCTCTTCCGATCTJJJJJJJJTCTTCAGCGTTCCCGAGAJJJJJJJTCNNNNNNNNT25 (vs1)
5′-TTT_PC_GCCGGTAATACGACTCACTATAGGGCTACACGACGCTCTTCCGATCTJJJJJJJJTCTTCAGCGTTCCCGAGAJJJJJJNNNNNNNVVT30 (vs2)
“PC” designates a photocleavable linker; “J” represents bases generated by split-pool barcoding, such that every oligo on a given bead has the same J bases; “N” represents bases generated by mixing, so every oligo on a given bead has different N bases; and “TX” represents a sequence of X thymidines. “V” represents bases which contain A,C,G and no T.
Bead synthesis
PLRP-S resin (~10 μm mean particle diameter) from Agilent Technologies were functionalized with a non-cleavable linker by Chemgenes Corp. The functionalized beads were then used as a solid support for reverse-direction phosphoramidite synthesis (5’ to 3’) on an Akta OligoPilot 10 using standard solid-phase DNA synthesis protocol. 5’-CE (b-cyanoethyl) phosphoramidites were purchased from Glen Research and were dissolved in anhydrous acetonitrile to obtain a concentration of 0.1M. Successive phosphoramidites were coupled for 5 minutes using 5-Benzylmercaptotetrazole (0.30 M in acetonitrile) as an activator. Oxidation of phosphite backbone to phosphate backbone was achieved using iodine. Failure sequences were capped using acetic anhydride. Dichloroacetic acid was used as detritylation reagent. For split-pool synthesis cycles, beads were suspended in acetonitrile and were divided into 4-equal portions. These bead aliquots were then placed in 4 separate synthesis columns and were reacted with either dG, dC, dT, or dA phosphoramidites. After each cycle, beads were pooled, suspended in acetonitrile and aliquoted into 4 equal portions. The split pool procedure was repeated 15 times in total (two blocks of 8 and 7 cycles) to obtain 415 (~109) unique barcode sequences. After completion of the synthesis, the protecting groups from the nucleobases and phosphate backbone were removed by treating beads with 30% Ammonium Hydroxide containing 10% diethylamine for 40 h at room temperature. The beads were centrifuged and supernatant was discarded. Following this, beads were washed with 1% acetone in acetonitrile (3 times), water (3 times) and a buffer consisting of 10 mM tris,1mM EDTA (pH = 8, 3 times).
Puck Preparation
Puck preparation was performed as described previously1, with the following modification:
Beads were pelleted and resuspended water+ 10% DMSO at a concentration between 20,000 and 50,000 beads/μL , and 10μL of the resulting solution was pipetted into each position on the gasket. The coverslip-gasket filled with beads centrifuged at 40C, 850g for at least 30 minutes until the surface was dry.
Puck Sequencing
Puck sequencing was performed in a Bioptechs FCS2 flow cell using a RP-1 peristaltic pump (Rainin), and a modular valve positioner (Hamilton MVP). Flow rates between 1 mL/min and 3 mL/min were used during sequencing. Imaging was performed using a Nikon Eclipse Ti microscope with a Yokogawa CSU-W1 confocal scanner unit and an Andor Zyla 4.2 Plus camera. Images were acquired using a Nikon Plan Apo 10x/0.45 objective. After each ligation, images were acquired in the following channels: 488nm excitation with a 525/36 emission filter (MVI, 77074803); 561 nm excitation with a 582/15 emission filter (MVI, FF01-582/15-25); 561 nm excitation with a 624/40 emission filter (MVI, FF01-624/40-25); and 647 nm excitation with a 705/720 emission filter (MVI, 77074329). The final stitched images varied in size depending on the size of the Slide-seq array.
Pucks were sequenced using a sequencing-by-ligation approach, both with the SOLiD dibase-encoding strategy previously described1,45 and with a monobase-encoding strategy developed for this work. Fluorescent oligonucleotides were synthesized on an Akta OligoPilot 10 or obtained from IDT (Supplementary Table 6). A total of 8 fluorescent oligonucleotides were used and are referred to as 5(base) or 3(base) to indicate the corresponding mode of ligation and identity of the interrogated base. Each sequencing oligonucleotide interrogates the +2 base from the ligation junction, with each base identity corresponding to a fluorescent channel: A: FAM, C: Cy3, T: Cy5, G: Texas Red or AqP593.
The monobase sequencing strategy interrogates 14 split-pool bases using 3 modes of sequencing by ligation. This strategy is motivated by the need to eliminate the use of proprietary cleavage reagents from SOLID and allow for sequencing using commercially available oligonucleotides. The overall sequencing strategy (Supplementary Figure 1A) consists of a ligation to interrogate a split-pool base, followed by dehybridization using formamide before moving onto the next ligation. The 3 ligation modes are: 5’ ligation (ligation at the 5’ end of a hybridized sequencing primer), 3’ Ligation (ligation at the 3’ end of a hybridized sequencing primer), and SEDAL (ligation with a degenerate primer in solution).
On each bead sequence, there are two primer binding sites: Truseq primer site (T), and Universal Primer (UP) (Supplementary Figure 1B). All primer sequences are listed in Supplementary Table 6. Sequencing starts with 5’ ligation on the Truseq primer. First, the T-1 primer (Primer T shortened by 1 base on the 5’ end) is hybridized, and a ligation is performed to interrogate the first J. After stripping with formamide, the T primer is hybridized (Primer T), and the second J is interrogated. After stripping with formamide, the T+1 primer is hybridized (the + primers represent the T primer with an added N (representing all 4 bases) on the 5’ end). For the rest of the 5’ ligations, the same steps are repeated with T+2, UP-1, UP, UP+1 and UP+2, where UP primers follow the same conventions as Truseq.
Next, sequencing proceeds with 3’ ligations using the 3UP primer series and 3(base) sequencing oligonucleotides. These ligations are performed for 3UP+1, 3UP, and 3UP-1 (3UP+1 is UP plus an N base on the 3’ end, 3UP is the UP primer, and 3UP-1 is primer UP shorter by 1 base on the 3’ end.
Lastly, sequencing is performed with SEDAL, which utilizes degenerate primers in solution with the 5’ sequencing oligonucleotides. As the number of N bases added to the end of the hybridized primer increases, the sequencing efficiency decreases. Empirically, we find that we cannot use pre-hybridized + primers beyond 2 bases (T+2 and UP+2). To overcome this, we include shortened primers with additional N bases (+3 and +4) in solution with the fluorescent sequencing oligonucleotides. We perform 3 bases of SEDAL with 3 seperate primers (T+3, UP+3, and UP+4).
This sequencing approach is outlined schematically in Supplementary Figure 1B. For both 5’ and 3’ ligation modes, a primer was injected into the flow cell at 5 μM concentration in 4x SASC for 40 minutes. Subsequently, the flow cell was washed in 5 mL of wash buffer (50 mM Tris-Acetate + 0.05% Triton-X 100). Ligation mix (recipes below) was then flowed into the chamber and allowed to sit for 40 minutes, at which point flow was reversed to return the ligation mix to its original reservoir. Ligation mix was reused for a complete sequencing run before being replenished. After a subsequent wash, pucks were imaged as described above and then stripped using 10 mL of 80% formamide for 20 minutes. For SEDAL ligations, a primer was added at 5 μM concentration to the ligation mix and this mixture was flowed into the chamber and allowed to sit for 2 hours.
Bead barcodes consisted of 15 “J” bases, of which 14 were used. In order to sequence these barcodes, we performed 3 rounds of SEDAL, 8 rounds of 5’ ligation, and 3 rounds of 3’ ligation (Supplementary Figure 1A,B). The 14 primers necessary for this process were obtained from IDT (Supplementary Table 6). The ligation mix recipes are given below:
5’ Ligation mix:
1x T4 DNA Ligase Buffer (NEB)
6 U/μL T4 DNA Ligase (NEB)
20 μM each of 5T, 5A, 5G, and 5C oligonucleotides
3’ Ligation mix:
1x T4 DNA Ligase Buffer (NEB)
6 U/μL T4 DNA Ligase (NEB)
20 μM each of 3T, 3A, 3G, and 3C oligonucleotides
SEDAL Ligation mix:
1x T4 DNA Ligase Buffer (NEB)
6 U/μL T4 DNA Ligase (NEB)
5 μM primer
5 μM each of 5T, 5A, 5G, and 5C oligonucleotides
Microscopy
Imaging was performed using a Nikon Eclipse Ti microscope with a Yokogawa CSU-W1 confocal scanner unit and an Andor Zyla 4.2 Plus camera. Images were acquired using a Nikon Plan Apo 10x/0.45 objective. After each ligation, we acquired four images: one using a 488 nm laser and a 525/36 emission filter (MVI, 77074803); one using a 561 nm laser and a 582/15 emission filter (MVI, FF01-582/15-25); one using a 561nm laser and a 624/40 emission filter (MVI, FF01-624/40-25); and one using a 647nm laser and a 705/72 emission filter (MVI, 77074329). The final stitched images were 6030 pixels by 6030 pixels.
Image Processing and Basecalling
Image processing was performed as previously described, and we have made an easy to use image processing and base calling Matlab package that has been deposited into https://github.com/MacoskoLab/PuckCaller/. Input images are 4 channel sequencing images for each puck for each timepoint of sequencing. The outputs are, for each bead, a sequence string for the bead barcode. For monobase imaging the images are directly convertible to basespace rather than colorspace thus we omit the step of conversion of illumina reads to colorspace prior to comparison to the in situ indexing data as previously described. Metadata on all pucks used are shown in Supplementary Table 7.
Slide-seq tools
We developed the Slide-seq tools pipeline for processing Slide-seq data. The scripts, documentations and example data are available at https://github.com/MacoskoLab/slideseq-tools. The Slide-seq tools include several analysis steps and the workflow is illustrated in Supplementary Figure 9:
1) Extract Illumina barcodes: this step runs run_barcodes2sam.py and calls the ExtractIlluminaBarcodes function in Picard tools (https://github.com/broadinstitute/picard) to extract the barcode for each read in an Illumina lane from Illumina BCL files.
2) Convert Illumina basecalls to bam: this step runs run_processbarcodes.py and calls the IlluminaBasecallsToSam function in Picard tools to collect, demultiplex and sort reads across all of the tiles of a lane by barcode to produce an unmapped BAM file.
3) Pre-Alignment: this step runs run_alignment.py, calls the functions of TagBamWithReadSequenceExtended, FilterBam, TrimStartingSequence and PolyATrimmer in the Drop-seq tools (https://github.com/broadinstitute/Drop-seq) to tag unmapped bam files with a bead barcode (XC) and a molecular barcode (a.k.a. UMI, marked by tag XM), filter low-quality reads, trim reads with starting sequence and polyA tail, and calls the SamToFastq function in Picard tools to convert bam files to fastq files.
4) Align reads to genome reference: this step runs run_alignment.py and calls the STAR aligner44 to align reads in the fastq to a reference genome.
5) Post-Alignment: this step runs run_alignment.py to call SortSam and MergeBamAlignment in Picard tools to sort aligned bam files and to merge unmapped bam and aligned bam files, and calls the functions of TagReadWithInterval and TagReadWithGeneFunction in Drop-seq tools to tag reads with interval and gene identity.
6) Generate alignment reports and plots: this step runs generate_plots.py and calls the CollectRnaSeqMetrics function in Picard tools, and the functions of BamTagHistogram, BaseDistributionAtReadPosition and GatherReadQualityMetrics in Drop-seq tools, to generate a few reports based on the aligned bam file, such as read quality and mapping rate, base distribution across the reads, and data on the composition and quality of the bead nad molecular barcodes.
7) Select top cells by the number of transcripts: this step runs run_analysis_spec.py and calls the SelectCellsByNumTranscripts function in the Drop-seq tools to select top cells by the number of transcripts that the user specifies when submitting a request.
8) Match Illumina barcodes to bead barcodes: this step runs cmatcher.cpp to calculate hamming distances between each Illumina barcode and all of the bead barcodes from in situ sequencing. The list of uniquely matched Illumina barcodes with hamming distance <= 1, along with the matched bead barcodes, are outputted.
9) Generate reports and plots on matched barcodes: this step runs generate_plots_cmatcher.py and calls the CollectRnaSeqMetrics function in Picard tools and the functions DigitalExpression, BamTagHistogram, BaseDistributionAtReadPosition, GatherReadQualityMetrics and SingleCellRnaSeqMetricsCollector in Drop-seq tools to generate the digital gene expression matrix, along with quality metrics that include a histogram of hamming distance between Illumina and bead barcode matches, a color-scaled number of UMIs per bead, and other reports.
Supplementary Table 8 shows running time of the Slide-seq tools on four libraries: 190926_01, 190926_02, 190926_03 and 190926_06. The Illumina platform is NovaSeq, and there are two lanes in the experiment. Reads were aligned to GRCm38.81 genome sequence. The read base quality for alignment was set to 10. The minimum number of transcripts per cell for selecting top cells was set to 10 as well. Reads aligned to both exons and introns were involved in the gene expression analysis. In order to speed up the process, the Slide-seq tools split each lane of NovaSeq data into 10 slices, parallel ran the alignment steps on the slices and combined the alignment outputs together.
Slide-seqV2 library preparation
RNA Hybridization:
Pucks in 1.5 mL tubes were immersed in 200 μL of hybridization buffer (6x SSC with 2 U/μL Lucigen NxGen RNAse inhibitor) for 30 minutes at room temperature to allow for binding of the RNA to the oligos on the beads.
First Strand Synthesis
Subsequently, first strand synthesis was performed by incubating the pucks in RT solution for 1.5 hours at 52 C.
RT solution:
115 μL H2O
40 μL Maxima 5x RT Buffer (Thermofisher, EP0751)
20 μL 10 mM dNTPs (NEB N0477L)
5 μL RNase Inhibitor (Lucigen 30281)
10 μL 50 μM Template Switch Oligo (Qiagen #339414YCO0076714)
10 μL Maxima H-RTase (Thermofisher, EP0751)
Tissue Digestion:
200 μL of 2x tissue digestion buffer was then added directly to the RT solution and the mixture was incubated at 37C for 30 minutes.
2x tissue digestion buffer:
200 mM Tris-Cl pH 8
400 mM NaCl
4% SDS
10 mM EDTA
32 U/mL Proteinase K (NEB P8107S)
Second Strand Synthesis:
The solution was then pipetted up and down vigorously to remove beads from the surface, and the glass substrate was removed from the tube using forceps and discarded. 200 μL of Wash Buffer was then added to the 400 μL of tissue clearing and RT solution mix and the tube was then centrifuged for 3 minutes at 3000 RCF. The supernatant was then removed from the bead pellet, the beads were resuspended in 200 μL of Wash Buffer, and were centrifuged again. This was repeated a total of three times. The supernatant was then removed from the pellet. The beads were then resuspended in 200μl of ExoI mix and incubated at 37 °C for 50mins.
Wash Buffer:
10 mM Tris pH 8.0
1 mM EDTA
0.01% Tween-20
ExoI mix:
170μl H20
20μl ExoI buffer
10μl ExoI (NEB M0568)
After ExoI treatment the beads were centrifuged for 3 minutes at 3000 RCF. The supernatant was then removed from the bead pellet, the beads were resuspended in 200 μL of Wash Buffer, and were centrifuged again. This was repeated a total of three times. The supernatant was then removed from the pellet. The pellet was then resuspended in 200μl of 0.1 N NaOH and incubated for 5 minutes at room temp. To quench the reaction 200μl of Wash Buffer was added and beads were centrifuged for 3 minutes at 3000 RCF. The supernatant was then removed from the bead pellet, the beads were resuspended in 200 μL of Wash Buffer, and were centrifuged again. This was repeated a total of three times. Second Strand Synthesis was then performed on the beads by incubating the pellet in 200μ of Second Strand Mix at 37 °C for 1 hour.
Second Strand Synthesis mix:
133 μl H2O
40 μl Maxima 5x RT Buffer
20 μl 10 mM dNTPs
2μl 1mM dN-SMRT oligo
5μl Klenow Enzyme (NEB M0210)
After Second Strand Synthesis 200μl of Wash Buffer was added and the beads were centrifuged for 3 minutes at 3000 RCF. The supernatant was then removed from the bead pellet, the beads were resuspended in 200 μL of Wash Buffer, and were centrifuged again. This was repeated a total of three times.
Library Amplification:
200μl of water was then added to the bead pellet and the beads were moved into a 200 μL PCR strip tube, pelleted in a minifuge, and resuspended in 200 μL of water. The beads were then pelleted and resuspended in library PCR mix and PCR was performed as outlined below:
Library PCR mix:
22 μL H2O
25 μL of Terra Direct PCR mix Buffer (Takara Biosciences 639270)
1μl of Terra Polymerase (Takara Biosciences 639270)
1 μL of 100 μM Truseq PCR handle primer (IDT)
1 μL of 100 μM SMART PCR primer (IDT)
PCR program:
95 °C 3 minutes
- 4 cycles of:
- 98 °C 20 s
- 65 °C 45 s
- 72 °C 3 min
- 9 cycles of:
- 98 °C 20 s
- 67 °C 20
- 72 °C 3 min
- Then:
- 72 °C 5 min
- 4 °C forever
PCR cleanup and Nextera Tagmentation:
The PCR product was then purified by adding 30 μL of Ampure XP (Beckman Coulter A63880) beads to 50 μL of PCR product. The samples were cleaned according to manufacturer’s instructions and resuspended into 50 μL of water and the cleanup was repeated resuspending in a final concentration of 10μl. 1 μL of the library was quantified on an Agilent Bioanalyzer High sensitivity DNA chip (Agilent 5067-4626). Then, 600 pg of PCR product was taken from the PCR product and prepared into Illumina sequencing libraries through tagmentation with Nextera XT kit (Illumina FC-131-1096). Tagmentation was performed according to manufacturer’s instructions and the library was amplified with primers Truseq5 and N700 series barcoded index primers. The PCR program was as follows:
72°C for 3 minutes
95°C for 30 seconds
12 cycles of:
95°C for 10 seconds
55°C for 30 seconds
72°C for 30 seconds
72°C for 5 minutes
Hold at 10°C
Samples were cleaned with AMPURE XP (Beckman Coulter A63880) beads in accordance with manufacturer’s instructions at a 0.6x bead/sample ratio (30 μL of beads to 50 μL of sample) and resuspended in 10μL of water. Library quantification was performed using the Bioanalyzer. Finally, the library concentration was normalized to 4nM for sequencing. Samples were sequenced on the Illumina NovaSeq S2 flowcell 100 cycle kit with 12 samples per run (6 samples per lane) with the read structure 42 bases Read 1, 8 bases i7 index read, 50 bases Read 2. Each puck received approximately 200-400 million reads, corresponding to 3,000-5,000 reads per bead.
Animal Handling
All procedures involving animals at the Broad Institute were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals under protocol number 0120-09-16. All procedures involving animals at Harvard University were handled according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Harvard University (protocol number 11-03) and followed the guidelines set forth in the National Institute of Health Guide for the Care and Use of Laboratory Animals. Wild-type C57Bl/6 mice (from Charles River Laboratories) were housed in a 12:12 light-dark cycle with ad libitum access to food and water. We set harem breeding cages and defined morning of plug detection as E0.5.
Transcardial Perfusion
C57Bl/6 mice were anesthetized by administration of isoflurane in a gas chamber flowing 3% isoflurane for 1 minute. Anesthesia was confirmed by checking for a negative tail pinch response. Animals were moved to a dissection tray and anesthesia was prolonged via a nose cone flowing 3% isoflurane for the duration of the procedure. Transcardial perfusions were performed with ice cold pH 7.4 HEPES buffer containing 110 mM NaCl, 10 mM HEPES, 25 mM glucose, 75 mM sucrose, 7.5 mM MgCl2, and 2.5 mM KCl to remove blood from brain and other organs sampled. The appropriate organs were removed and frozen for 3 minutes in liquid nitrogen vapor and moved to −80C for long term storage.
Tissue Handling
Fresh frozen tissue was warmed to −20 °C in a cryostat (Leica CM3050S) for 20 minutes prior to handling. Tissue was then mounted onto a cutting block with OCT and sliced at a 5° cutting angle at 10 μm thickness. Pucks were then placed on the cutting stage and tissue was maneuvered onto the pucks. The tissue was then melted onto the puck by moving the puck off the stage and placing a finger on the bottom side of the glass. The puck was then removed from the cryostat and placed into a 1.5 mL eppendorf tube. The sample library was then prepared as below. The remaining tissue was re-deposited at −80 °C and stored for processing at a later date.
Diffusion Analysis
Determination of diffusion was determined as previously described by measuring features across CA1 mouse hippocampus1 for Slide-seqV2 data (Puck 200115_08).
Comparison of counts for Slide-seq, Slide-seqV2, FISH, and scRNAseq
For Slide-seq and Slide-seqV2 we subsetted a region of CA1 and took the total number of counts for each of the marker genes.
For the scRNAseq data, we used the mouse brain scRNAseq data from 12 and pulled an equal number of cells found from the Slide-seq data from the cluster representing the hippocampal CA1 neurons.
For the smFISH data we generated the data by using HCRV3.0 with probes sets against each of the genes chosen in 488nm, 594nm, and 647nm (Slc17a7, Ociad2, and Atp2b1 respectively). Following recommendations from the manufacturer we used the suggested number of probes per gene for HCRv3, namely 20 probe pairs per gene (40 probes total). Probe sequences were designed by Molecular Instruments. We stained the tissue with DAPI for segmentation purposes and for counting nuclei. We performed counting of smFISH data using a custom pipeline implemented using the Starfish package(https://github.com/spacetx/starfish) in python.
Comparison of Slide-seqV2 to 10x Visium technology and HDST
To compare Slide-seqV2 to 10x Visium data (https://www.10xgenomics.com/solutions/spatial-gene-expression/ ) we downloaded available coronal mouse hippocampus data and plotted the number of UMIs per spatial feature. We next binned Slide-seqV2 data from the same region to equivalent feature size (11 μm), merging the counts of Slide-seqV2 beads (10μm original) within each of the larger features generated (110μm).
To compare Slide-seqV2 to HDST we first obtained HDST data from supplementary data provided in the original HDST publication7. We used the data at 10 μm feature size (5x binned) and compared to Slide-seqV2 data collected from an equivalent region (mouse olfactory bulb). We then just plotted the number of counts per feature and took the mean to obtain the difference in average counts between Slide-seqV2 and HDST.
Spatial Comparison of Slide-seqV2 to OsmFISH
A spatial profile was taken along the length of the cortex for OsmFISH and Slide-seqV2 data perpendicular to the expression of the layer marker Lamp5, i.e. going from Layer 6 to Layer 1 of the cortex. Each gene for both datasets was normalized along this profile. Genes in OsmFISH >50% coefficient of variation along the profile were selected for analysis (this enriches for cortical layer markers, other genes do not have stereotyped patterns along this spatial dimension and cannot be compared). Both datasets were downsampled to 50 μm, and aligned spatially along the profile by aligning the positions of Layer 1 and Layer 4. To analyze spatial correlation, the Pearson correlation of each gene’s spatial profile against all genes was calculated between Slide-seqV2 and OsmFISH, as well as OsmFISH and OsmFISH.
Hippocampal Slide-seqV2
Slide-seqV2 was performed on the mouse hippocampus (N=4 sections, 2 mice). A spline was fit along the pyramidal cells layer of CA1. Beads were averaged to a profile perpendicular to this spline ~100 μm into the Basal neuropil, and ~400 μm to the proximal neuropil to form a spatial profile of gene expression along the CA-1 neuropil axis.
Dendritic enrichment analysis
To test for dendritic enrichment, for each gene, the gene expression in the soma layer (defined as +/− 32.5 micron from the peak of the profile counts for all genes) was compared against the gene expression in the proximal dendrites (greater than 32.5 μm away from the peak of the CA1 layer). We leveraged existing scRNA-seq data12 to exclude marker genes from cell types outside of CA1 using differential expression. Specifically, all genes with 2-fold higher expression in cell-type clusters other than CA1 were excluded from the analysis. For each gene, the gene expression in the Soma layer was normalized to the total number of UMI-counts in the Soma layer, and the gene expression in the proximal dendrite layer was normalized to the total number of UMI-counts in the proximal dendrite layer. A two sample t-test was performed to identify differentially expressed genes, and pFDR was calculated as described previously45.
Spatial clustering of dendritically enriched genes
For the 213 genes identified to be dendritically enriched, we clustered genes by their spatial profile along the CA1-neuropil axis via k-means clustering. The gap-statistic was used to determine the optimal number of clusters (K=4).
GO analysis
For each cluster identified by spatial profiling, GO analysis was performed using the clusterProfiler46 package in R. Cellular components from the org.Mm.eg.db (http://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html). Genome wide annotation for Mouse in Bioconductor was used for the ontology database. For Figure 3, GO analysis was performed using Biological processes from org.Mm.eg.db.
Cell type specificity of dendritic genes
To explore the relationship between cell type specificity of gene expression and dendritic localization (Figure 2g), we first computed differential expression between CA1 and other hippocampal principal cells from an existing single cell dataset12 using the FindMarkers() function in Seurat. Next, we compared the log2 fold-change of the genes in the dendritic and somatic gene sets (see Dendritic Enrichment Analysis) with a wilcoxon rank sum test, and visualized the comparison using a quantile-quantile plot. To calculate dendritic and soma expression in CA3, the same procedure as in CA1 was carried out (see Dendritic Enrichment Analysis). Soma fold change was calculated as the ratio of counts in CA3 soma to CA1 soma after normalizing by the total number of UMIs in each compartment. Dendrite fold change was calculated as the ratio of counts in CA3 soma to CA1 soma after normalizing by the total number of UMIs in each compartment.
Embryo samples
Whole mount frozen embryos were obtained from a commercial source Zyagen (San Diego, CA). The pregnant mice (C57BL/6NCrl) were bred and maintained by Charles River Laboratories. The time-pregnant mice (day 10) were shipped to Zyagen (San Diego, CA) the same day. The mice were sacrificed on the day of arrival for embryo collection.
Trajectory analysis
Trajectory analysis was performed using the recently released method scVelo30. We first loaded intronic and exonic gene expression matrices, UMAP coordinates created in Seurat from the original clustering of the Slide-seqV2 data, cluster IDs, and spatial coordinates of each bead from Slide-seqV2 into a scanpy object using a custom python environment. We next applied the latent time method developed in scVelo to our Slide-seqV2 expression data and plotted each bead using the Slide-seqV2 coordinates with the shading defined by the latent time ordering. Plots of individual expression of genes over latent time were generated using plotting functions in scVelo plotting the expression of each individual gene over the latent time axis with coloring of each bead by cluster identity to the original clustering of the data. Plots showing expression for each of the genes on the puck were performed using a custom python script. The gene lists for latent time were called as velocity loading genes from the scVelo pipeline using standard parameters and a likelihood cutoff of >0.1.
Monocle323,31 was run on the data by importing the UMAP and PCA coordinates from Seurat into a Single Cell Experiment object. The analysis was performed in accordance with the monocle tutorial found at. http://cole-trapnell-lab.github.io/monocle-release/monocle3/. The q-value cutoff for gene selection was q<0.005.
Fitting a spatial surface to latent time
Latent time data scores generated from scVelo and spatial coordinates were taken as a 3D set of points (x,y, latent time score) and a surface was fit over the set of points for a region of the cortex. A grid was created (80μm x 80μm for cortex) and the spatial derivative was taken over the grid using Matlab’s differentiate function. The fx, fy of the surface were extracted from Matlab and imported into python. The plot for Figure 3b was generated using a custom python script where the magnitude of the arrows represents the magnitude of the derivative at each of the points in the grid. The position of the underlying beads is from Slide-seqV2 and the color scale is from scVelo’s latent time output.
Spatially non-random gene analysis:
Test for spatial non-randomness was performed as previously described1 with the following modifications:
Genes were identified as spatially non-random using a custom Matlab application. The set of pairwise Euclidean distances between all beads was calculated. Candidate genes for the statistical significance analysis were required to have at least one transcript on at least 10 beads. To determine whether a transcript had a significantly non-random spatial distribution within a particular set of beads, we compared the distribution of pairwise distances between the beads expressing at least one count of that transcript to the distribution of pairwise distances between an identical number of beads, sampled randomly from all mapped beads on the puck with probability proportional to the total number of transcripts on the bead. Specifically, we generated 1000 such random samples, and for each sample calculated the distribution of pairwise distances. We then calculated the average distribution of pairwise distances, averages pairwise across all 1000 samples. Finally, we calculated the L1 norm between the distribution of pairwise distances for each of the 1000 random samples and the average distribution, and the L1 norm between the distribution of pairwise distances for the true sample of beads and the average distribution. We defined p to be the fraction of random samples having distributions closer to the average distribution (under the L1 norm) than the true sample, and considered any genes with values p<=0.005.
Spatial correlation to latent time:
Spatially identified genes were binned along 20 spatial contours of the same latent time as fitted by the surface described above. Expression was normalized for each bin by the total number of counts observed. For each gene, the Pearson’s correlation coefficient and the p-value of the correlation between the binned expression in the spatial latent time axis was correlated with a linear function of slope 1. pFDR was calculated as described previously50.
Spatial clustering of DD genes
For the 74 genes identified to be involved in developmental disorders that load onto pseudotime, the spatial correlation between each gene was determined by convolving the spatial expression of each gene with an integralBoxFilter of size 70 microns, and then the 2-D cross-correlation for each gene against each other gene was calculated with the Matlab function corr2. The spatial cross-correlation matrix was clustered using k-means, the gap-statistic was used to determine the optimal number of clusters (k=6).
Supplementary Material
Supplementary Data 1 Plots of all genes dendritically enriched in Slide-seqV2
Supplementary Data 2 Plots of all genes called as Spatially Significant in Slide-seqV2
Supplementary Table 3 Dendritically enriched gene-sets Sheet 1: Dendritically enriched RNAs identified by Slide-seqV2 along with Fold-Change enrichment, and FDR corrected q-value.
Sheet 2: Differentially expressed genes between CA1 and other scRNA-seq clusters which were removed from the dendritic analysis.
Sheet 3: Grouping of dendritically enriched genes by spatial clusters.
Sheet 4: List of genes which overlap with Tushev et al., 2018, and Ainsley et al., 2016 as well as genes uniquely identified by Slide-seqV2.
Supplementary Table 4 List of all genes called as Spatially Significant for Slide-seqV2 data in the developing cortex (Sheet 1) and eye (Sheet 2)
Supplementary Table 5 List of genes unique to each method regarding the trajectory inference:
Sheet 1: Genes unique to Slide-seqV2
Sheet 2: Genes unique to monocle3
Sheet 3: Genes unique to scVelo
Acknowledgments
We thank Jordane Dimidschstein, and Gord Fishell for their helpful advice on the developmental trajectory analysis. This work was supported by an NIH New Innovator Award (DP2 AG058488-01 to E.Z.M.), an NIH Early Independence Award (DP5, 1DP5OD024583 to F.C.), the NHGRI (R01, R01HG010647 to E.Z.M. and F.C.), and the Burroughs Wellcome Fund CASI award (to F.C.) as well as the Schmidt Fellows Program at the Broad Institute and the Stanley Center for Psychiatric Research.
Footnotes
Competing Interests:
R.R.S., F.C., and E.Z.M. are listed as inventors on a pending patent application related to the development of Slide-seq.
Code Availability Statement:
Code related to this manuscript can be found at: https://github.com/MacoskoLab/slideseq-tools, https://github.com/rstickels/Slide_seqv2.
The following packages version numbers were used for data processing and associated analyses:
https://github.com/broadinstitute/Drop-seq (Drop-seq-tools-2.3.0)
https://broadinstitute.github.io/picard/ (picard-2.18.14),
https://github.com/alexdobin/STAR (STAR-2.5.2a),
https://github.com/theislab/scvelo (0.1.25),
https://github.com/cole-trapnell-lab/monocle3 (beta),
https://github.com/satijalab/seurat (2.3.4),
Matlab 2017a, R3.5.3 and Python 3.7 were used for processing data.
Data Availability Statement:
All data is available at: https://singlecell.broadinstitute.org/single_cell/study/SCP815/sensitive-spatial-genome-wide-expression-profiling-at-cellular-resolution#study-summary.
Main Text References:
- 1.Rodriques SG et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah S, Lubeck E, Zhou W & Cai L In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Codeluppi S et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Ståhl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Vickovic S et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drmanac R et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Ke R et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Hughes TK, Wadsworth MH, Gierahn TM & Do T Highly efficient, massively-parallel single-cell RNA-seq reveals cellular states and molecular features of human skin pathology. bioRxiv (2019). [Google Scholar]
- 11.Saunders A et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi HMT et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Govindarajan A, Israely I, Huang S-Y & Tonegawa S The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69, 132–146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richter JD & Klann E Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 23, 1–11 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Huber KM, Kayser MS & Bear MF Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Kosik KS Life at Low Copy Number: How Dendrites Manage with So Few mRNAs. Neuron 92, 1168–1180 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Ainsley JA, Drane L, Jacobs J, Kittelberger KA & Reijmers LG Functionally diverse dendritic mRNAs rapidly associate with ribosomes following a novel experience. Nat. Commun 5, 4510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tushev G et al. Alternative 3’ UTRs Modify the Localization, Regulatory Potential, Stability, and Plasticity of mRNAs in Neuronal Compartments. Neuron 98, 495–511.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Saelens W, Cannoodt R, Todorov H & Saeys Y A comparison of single-cell trajectory inference methods. Nature Biotechnology vol. 37 547–554 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Setty M et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 34, 637–645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welch JD, Hartemink AJ & Prins JF SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 17, 106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lodato S & Arlotta P Generating neuronal diversity in the mammalian cerebral cortex. Annu. Rev. Cell Dev. Biol 31, 699–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Telley L et al. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, (2019). [DOI] [PubMed] [Google Scholar]
- 28.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell (2019) doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergen V, Lange M, Peidli S, Alexander Wolf F & Theis FJ Generalizing RNA velocity to transient cell states through dynamical modeling. doi: 10.1101/820936. [DOI] [PubMed] [Google Scholar]
- 30.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature vol. 566 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruediger T et al. Integration of opposing semaphorin guidance cues in cortical axons. Cereb. Cortex 23, 604–614 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Polleux F, Giger RJ, Ginty DD, Kolodkin AL & Ghosh A Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science 282, 1904–1906 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Heavner W & Pevny L Eye development and retinogenesis. Cold Spring Harb. Perspect. Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashery-Padan R, Marquardt T, Zhou X & Gruss P Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 14, 2701–2711 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbieri AM et al. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc. Natl. Acad. Sci. U. S. A 96, 10729–10734 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andley UP Crystallins in the eye: Function and pathology. Prog. Retin. Eye Res 26, 78–98 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Niederreither K, Subbarayan V, Dollé P & Chambon P Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet 21, 444–448 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Fan X et al. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol 23, 4637–4648 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snead MP et al. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye 25, 1389–1400 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fares-Taie L et al. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am. J. Hum. Genet 92, 265–270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin HC et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science 362, 1161–1164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaplanis J et al. Integrating healthcare and research genetic data empowers the discovery of 49 novel developmental disorders. bioRxiv 797787 (2019) doi: 10.1101/797787. [DOI] [Google Scholar]
Online Methods References:
- 43.McKernan KJ et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Research vol. 19 1527–1541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storey JD A Direct Approach to False Discovery Rates. J. R. Stat. Soc. Series B Stat. Methodol 64, 479–498 (2002). [Google Scholar]
- 46.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.org.Mm.eg.db. Bioconductor http://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data 1 Plots of all genes dendritically enriched in Slide-seqV2
Supplementary Data 2 Plots of all genes called as Spatially Significant in Slide-seqV2
Supplementary Table 3 Dendritically enriched gene-sets Sheet 1: Dendritically enriched RNAs identified by Slide-seqV2 along with Fold-Change enrichment, and FDR corrected q-value.
Sheet 2: Differentially expressed genes between CA1 and other scRNA-seq clusters which were removed from the dendritic analysis.
Sheet 3: Grouping of dendritically enriched genes by spatial clusters.
Sheet 4: List of genes which overlap with Tushev et al., 2018, and Ainsley et al., 2016 as well as genes uniquely identified by Slide-seqV2.
Supplementary Table 4 List of all genes called as Spatially Significant for Slide-seqV2 data in the developing cortex (Sheet 1) and eye (Sheet 2)
Supplementary Table 5 List of genes unique to each method regarding the trajectory inference:
Sheet 1: Genes unique to Slide-seqV2
Sheet 2: Genes unique to monocle3
Sheet 3: Genes unique to scVelo
Data Availability Statement
All data is available at: https://singlecell.broadinstitute.org/single_cell/study/SCP815/sensitive-spatial-genome-wide-expression-profiling-at-cellular-resolution#study-summary.