

Abstract
Background
ACAN (OMIM 155760) is located on chromosome 15q26 and encodes the production of aggrecan. Aggrecan is a large chondroitin sulfate proteoglycan with a molecular weight of 254 kDa and contains 2530 amino acids. It is a critical structural component of the extracellular matrix of cartilage, including growth plate, articular, and intervertebral disk cartilage. It plays a key role in bone development.
Methods
Here, we describe two pedigrees with loss‐of‐function variants in ACAN. Whole exome sequencing was performed for the probands from each family. We illustrate the clinical variability associated with ACAN variants.
Results
The proband of pedigree A manifested short stature, relative macrocephaly, mild flat nasal bridge, low‐set ears, short neck, and short thumbs. The proband of pedigree B had short height, abnormal vertebral development, and central precocious puberty. By trio‐based whole exome sequencing and in silico analyses, we identified two de novo heterozygous variants of ACAN: NM_013227.4: c.116dupT, p.Arg40Glufs*51 and NM_013227.4: c.2367delC, p.Ser790Glnfs*20 (accession number: AC103982.10).
Conclusion
The clinical manifestations of ACAN gene variants are diverse. ACAN gene variants are important genetic factors for short stature and should be considered as the differential diagnosis of children with idiopathic short stature (ISS).
In this manuscript, we describe two pedigrees with novel ACAN gene variants, which were both discovered for the first time. We illustrate the clinical variability associated with ACAN variants.

1. INTRODUCTION
Short stature refers to people whose height is less than two standard deviations or less than the third percentile of people of the corresponding age, sex, and race. It represents one of the most frequent referrals to pediatric endocrinologists, with an incidence of 3%–5% (Stavber et al., 2020; Yang et al., 2018). Genetics and environment can both affect a person’s final height. Some short children can find clear causes, such as insufficient growth hormone (GH) secretion, hypothyroidism. Most children are diagnosed as ISS with unknown reason. Pathogenic SHOX gene variants are considered to be the most common monogenic cause accounting for 2%–15% of ISS cases (Plachy et al., 2019). Recent studies have shown that the prevalence of ACAN gene variants in children with ISS is 1.4%–6% (Hu et al., 2017; Hauer et al., 2017). Heterozygous ACAN gene variants may be the second leading cause of ISS.
Clinical manifestations of ACAN variants can be classified into three categories: spondyloepiphyseal dysplasia, Kimberley type (SEDK, OMIM 608361), short stature and advanced bone age, with or without early onset osteoarthritis and/or osteochondritis dissecans (OMIM 165800), and spondyloepimetaphyseal dysplasia, aggrecan type (SEMD, OMIM 612813). Gleghorn and colleagues first reported human SEDK and severe osteoarthritis associated with ACAN autosomal dominant inheritance mutation in 2005 (Gleghorn et al., 2005). Tompson and colleagues first reported SEMD, clinically manifested severe short stature and craniofacial deformities owing to ACAN autosomal recessive inheritance mutation in 2009 (Tompson et al., 2009). Most ACAN variants are autosomal dominant inheritance, while SEMD is autosomal recessive inheritance, including homozygous variants and compound heterozygous variants (Fukuhara et al., 2019; Tompson et al., 2009). Gender difference is not obvious in the incidence of ACAN gene variants.
Five Chinese pedigrees with ACAN gene variants have been reported (Hu et al., 2017; Zeng et al., 2018; Xu et al., 2018). This article reports another two pedigrees with short stature caused by novel ACAN gene mutations. The proband of pedigree A manifested short stature, relative macrocephaly, mild flat nasal bridge, low‐set ears, short neck, and short thumbs. The proband of pedigree B had short height, abnormal vertebral development, and central precocious puberty. The mutation sites of the two pedigrees were both discovered for the first time.
2. MATERIALS AND METHODS
2.1. Patients
We enrolled five members of two pedigrees from China in our study. Medical records including physical examination and biochemical tests were acquired.
2.2. Whole exome sequencing
Peripheral blood was collected from the patients and their family members and genomic DNAs were isolated using QIAamp DNA Blood Mini Kit (Qiagen). DNAs concentration was measured by Thermo Scientific NanoDrop 2000 (Thermo Fisher Scientific). Genomic DNAs were fragmented to 100–700 bp with a Covaris S220 ultrasonicator (Cole‐Parmer), and then fragments measuring of 150–200 bp were selected with magnetic beads. Then sequencing libraries were prepared; library preparation included end repair, adapter ligation and PCR enrichment was carried out as recommended by Illumina protocols (Illumina). The amplified DNA was captured using GenCap WES capture kit (MyGenostics). The enrichment libraries were sequenced on Illumina HiSeq X Ten sequencer. After sequencing, the raw sequencing data were filtered to remove low‐quality reads or probable artifacts. Then sequencing reads were aligned to a reference human genome (hg19) using BWA (http://bio‐bwa.sourceforge.net). Duplicated reads were removed using Picard tools (http://broadinstitute.github.io/picard) and mapping reads were used for variation detection. The variants of single nucleotide variation (SNP) and inserts and deletions (InDel) were detected by GATK Haplotype Caller (https://software.broadinstitute.org/gatk), and then GATK Variant Filtration was used to filter variants. Variants were further annotated by ANNOVAR (http://annovar.openbioinformatics.org/en/latest) and associated with multiple databases, such as 1000 Genomes Project (http://www.1000genomes.org), dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), HGMD (http://www.hgmd.cf.ac.uk), and predicted by SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster (http://www.mutationtaster.org), GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html).
2.3. Sanger sequencing
Filtered candidate variants were confirmed by PCR and Sanger sequencing. PCR primers were designed by Primer 3.0 online software (http://primer3.ut.ee). Amplified PCR products were analyzed on ABI 3730 Genetic Analyzer (Thermo Fisher Scientific). The final determined variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov).
3. RESULTS
3.1. Pedigree A
The proband is a Chinese boy of non‐consanguineous parents. He was referred to us because of growth retardation for more than 4 years. He was born prematurely at 35 weeks of gestation with birth weight of 2.2 kg, birth length of 45 cm (−3.07 SDS), and unknown head circumference. He went to pediatric endocrine clinic due to short stature at 4 years of age. The peak level of GH was 5.47 µg/L, indicating partial GH deficiency. After 1 year of GH treatment, his height increased by 9–10 cm. His father was 178.7 cm (+1.00 SDS), his mother was 149 cm (−2.16 SDS), and his maternal grandfather was 150 cm (−3.76 SDS). His mother’s sister also showed short stature. The proband’s mother and grandfather both have macrocephaly and short neck.
His height was 109.3 cm (−4.14 SDS), his weight was 21.8 kg, and his head circumference was 54 cm at 8 years 8 months. Other observed anomalies included flat nasal bridge, low‐set ears, short neck, short thumbs, and pectus excavatum (Figure 1a). Bone age (BA) was 5 years 6 months by x‐ray imaging of the left hand (Figure 1b).
FIGURE 1.
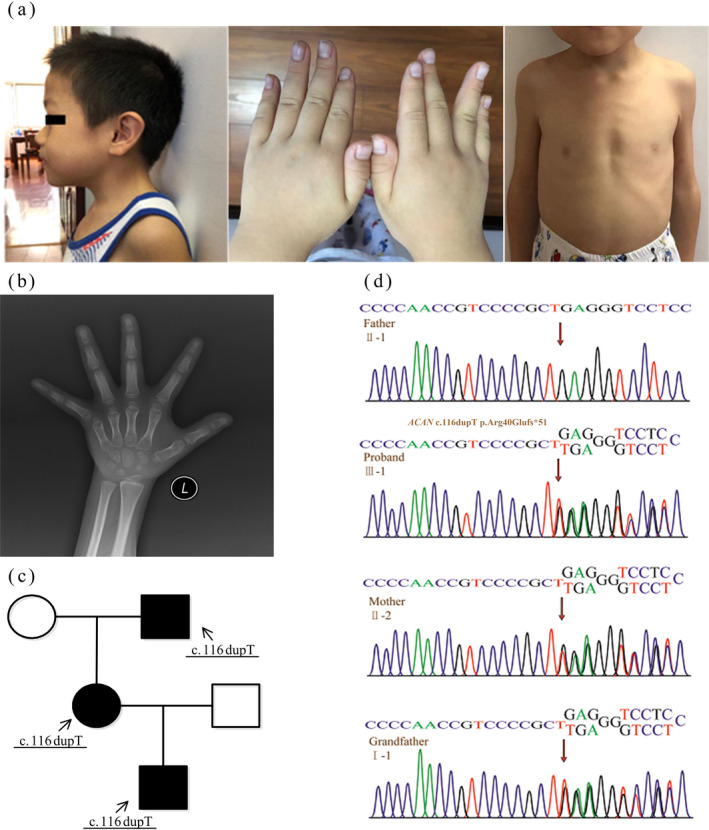
Clinical presentation and genetic analysis of pedigree A. (a) The proband of pedigree A: the proband manifested relative macrocephaly, mild flat nasal bridge, low‐set ears, short neck, short thumbs, and pectus excavatum. (b) BA was 5 years and 6 months by x‐ray imaging of the left hand (Chronological age: 8 years and 8 months). (c) Pedigrees of the family: the proband and his mother, maternal grandfather had the same gene mutation. (d) The gene map of pedigree A: the proband (Ⅲ‐1) and his mother (Ⅱ‐2), maternal grandfather (Ⅰ‐1) had the same heterozygous frameshift mutation c.116dupT (p.Arg40Glufs*51) in exon 3 of the ACAN gene, while the father (Ⅱ‐1) was normal
Next‐generation sequencing (NGS) was performed in the proband and identified a novel heterozygous frameshift mutation c.116dupT (p.Arg40Glufs*51) in exon 3 of the ACAN gene (Figures 1d and 3a), which was inherited from his mother (Figure 1c), while his father was normal (Figure 1d). Variants were subsequently validated in the same site of his maternal grandfather by Sanger sequencing (Figure 1d). According to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines, the variant was determined as a suspected pathogenic variant (PVS+PM2).
FIGURE 3.
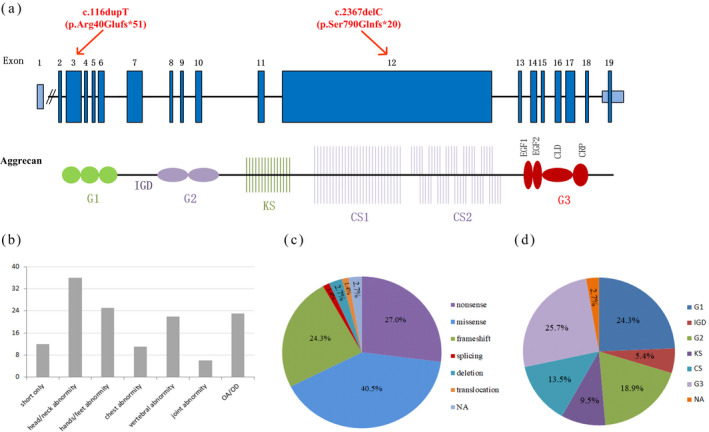
The structure of ACAN, different phenotypes, and the proportion of mutation types and domains. (a) The structure of ACAN and the locations of pathogenic sequence variants. Structure of ACAN is shown in the upper row with exon numbers making in the blue blocks. Structure of the aggrecan protein is shown in the lower row with crucial domains drawing approximately to scale. (G, globular domain; IGD, interglobular domain; KS, keratan sulfate; CS, chondroitin sulfate; EGF, epidermal growth factor‐like domain; CLD, C‐type lectin domain; CRP, complement regulatory‐like domain). (b) Number of ACAN mutations with different phenotypes. (c) The proportion of ACAN mutation types. (d) The proportion of ACAN mutation domains
3.2. Pedigree B
The proband is a 7‐year and 9‐month‐old girl and she is the first child of unrelated Chinese couple. Her bilateral breasts had been developing for half a year when we encountered. She was born at 41 weeks of gestation via cesarean section delivery. Her birth weight was 2.65 kg. Her father was 146.3 cm (−4.38 SDS), her mother was 140 cm (−3.84 SDS), her maternal grandfather was 150 cm (−3.76 SDS), and her maternal grandmother was 159 cm (−0.30 SDS). Her mother had menarche around 12 years old.
In the last evaluation, her height was 120.1 cm (+1.06 SDS) and her weight was 30 kg. She was in Tanner stage B4PH2.
Laboratory investigations revealed no abnormality in the complete blood count, liver function, renal function, serum electrolytes, blood gas analysis, and thyroid function. GH peak value was normal (13.1 ng/ml) at 90 minutes, luteinizing hormone (LH) basal value was 3.45 mU/ml, LH peak value was 50.7 mU/ml at 15 minutes, follicle‐stimulating hormone (FSH) basal value was 8.23 mU/ml, FSH peak value was 21.95 mU/ml at 30 minutes, the ratio of LH/FSH peak was 2.31, indicating precocious puberty. BA was 10 years old on the basis of x‐ray imaging. As shown in Figure 2a, lateral radiograph: the lumbar spine was slightly lateralized. The physiological curvature of the cervical spine became straight, cervical 4 and 5 were partially fused, and the intervertebral space between them was slightly narrow. Abdominal ultrasound revealed an intrahepatic bile duct stones (0.3 × 0.3 cm). Cardiac ultrasound and double kidney ultrasound were normal. Ultrasound of uterine and ovary showed that the inner diameter of the uterine section was 3.8 × 2.4 × 2.9 cm; the left ovary was 3.0 × 1.6 cm, the right ovary was 2.6 × 1.3 cm; and the left follicle was 0.5 × 0.5 cm, the right follicle was 0.6 × 0.6 cm, indicating pubertal development. Pituitary MRI showed that the height of the adenohypophysis was 7.3 mm and the upper edge of the pituitary gland was slightly raised as the physiological environment changed. The patient’s karyotype was 46, XX, 9qh+.
FIGURE 2.
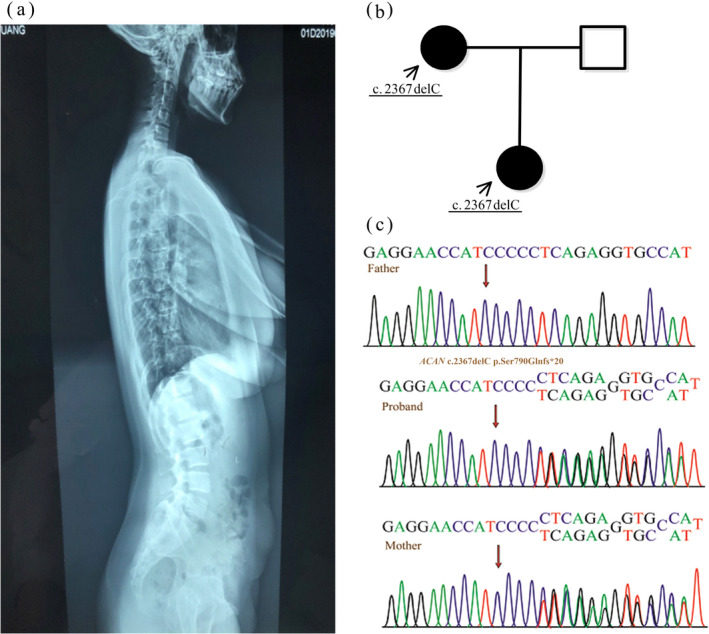
Lateral radiograph of the proband and genetic analysis of pedigree B. (a) Lateral radiograph: the lumbar spine was slightly lateralized. The physiological curvature of the cervical spine became straight, cervical 4 and 5 were partially fused, and the intervertebral space between them was slightly narrow. (b) Pedigrees of the family: the proband and his mother had the same gene mutation. (c) The gene map of pedigree B: the proband and her mother had the same heterozygous frameshift mutation c.2367delC (p.Ser790Glnfs*20) in exon 12 of the ACAN gene, while the father was normal
We identified a novel heterozygous frameshift variant c.2367delC (p.Ser790Glnfs*20) in exon 12 of ACAN gene of the proband (Figures 2c and 3a), which was from her mother (Figure 2b). No variant was found in her father (Figure 2c). The variant c.2367delC was determined as a suspected pathogenic variant (PVS+PM2).
The patient had been treated with gonadotropin‐releasing hormone analog (GnRHa) for 1 year owing to precocious puberty. Her height increased by 3 cm in this year. The latest examination showed LH was 0.42 mU/ml, FSH was 1.55 mU/ml, and bone age was 11 years (chronological age 8‐year and 9‐month old), and she started to get GH treatment recently achieving a growth rate of 3.7 cm for 5 months.
4. DISCUSSION
Aggrecan is a large chondroitin sulfate proteoglycan. The core protein of aggrecan contains three globular domains (N‐terminal G1 and G2 domains, C‐terminal G3 domain), interglobular domain (IGD), keratan sulfate domain (KS), and chondroitin sulfate domain (CS; Figure 3a). G1 domain interacts with hyaluronan. G3 domain binds to tenascins and fibulins through its C‐type lectin domain (CLD). G2 domain is highly conserved during evolution, while its biology function is still unclear (Gkourogianni et al., 2017).
Patients with ACAN variants have a wide spectrum of clinical phenotypes. Relevant studies were searched from PubMed, Web of Science, Human Gene Mutation Database (HGMD), and Online Mendelian Inheritance in Man (OMIM). Seventy‐four ACAN variants including ours are summarized in Table 1. As Table 1 shows, all patients with ACAN variants showed short stature, 16% (12/74) ACAN variants only manifested short stature without other symptoms. Head and/or neck deformities are the second common manifestations (Figure 3b), and manifested in 70% (7/10) of children born small for gestational age (SGA). About 40.5% (30/74) of ACAN variants were missense variants, 27.0% (20/74) were nonsense variants, and 24.3% (18/74) were frameshift variants (Figure 3c). Most variants occurred in the G1 and G3 domains (Figure 3d). About 56.5% (13/23) of the variants that occurred in IGD and G3 domain exhibited osteoarthritis/osteochondritis (OA/OD), while no OA/OD was observed in KS domain. Variants in CLD can affect articular cartilage and cause early onset arthritis (Nilsson et al., 2014), which is consistent with the highest number of OA/OD in G3 mutation in our summary. Most children with ACAN gene variants had advanced BA, some children had normal BA or delayed BA. As our Table 1 indicates, all frameshift variants in KS, CS, and G3 domain presented advanced BA, while variants in G2 mainly presented normal BA. In this study, the ACAN gene mutations were frameshift mutations in the G1 domain and CS, respectively (Figure 3a), and obviously delayed BA was observed in the male proband of pedigree A, while advanced BA was observed in the female proband of pedigree B.
TABLE 1.
Summary of ACAN genetic results including nucleotide change, mutation site, mutation type, and clinical phenotype
| Nucleotide change | Exon/intron | Domain | Clinical phenotype | Reference | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | Body parts involved | OA/OD | Others | BA | GH deficient | SGA | |||||||||
| He/N | Ha/F | C | V | J | |||||||||||
| Nonsense variants (n = 20, 27%) | |||||||||||||||
| 1 | c.61G>T | E2 | G1 | Y | Advanced | Gkourogianni et al. (2017) | |||||||||
| 2 | c.301C>T | E3 | G1 | NA | Stavber et al. (2020) | ||||||||||
| 3 | c.492C>G | E4 | G1 | Y | Y | Advanced | Gkourogianni et al. (2017) | ||||||||
| 4 | c.515del | E4 | G1 | Y | Y | Y | Delay | Hauer et al. (2017) | |||||||
| 5 | c.532A>T | E4 | G1 | Y | Advanced | Y | Gkourogianni et al. (2017) | ||||||||
| 6 | c.1047_1048dellinsAC | E6 | G1 | Y | Y | NA | Gkourogianni et al. (2017) | ||||||||
| 7 | c.1180C>T | E7 | IGD | Y | Y | Y | Y | Delay | Hauer et al. (2017) | ||||||
| 8 | c.1411C>T | E7 | IGD | Y | Y | Y | Advanced | Liang et al. (2020) | |||||||
| 9 | c.1443G>T | E7 | IGD | Y | Y | Y | NA | Gkourogianni et al. (2017) | |||||||
| 10 | c.1526C>A | E8 | G2 | Y | NA | Gkourogianni et al. (2017) | |||||||||
| 11 | c.1608C>A | E9 | G2 | Y | Y | Y | Absent left kidney | Normal/advanced | Y | van der Steen et al. (2017) | |||||
| 12 | c.1762C>T | E10 | G2 | Y | Y | Delay | Liang et al. (2020) | ||||||||
| 13 | c.1774C>T | E10 | G2 | Y | Y | Normal | Hauer et al. (2017) | ||||||||
| 14 | c.2099G>A | E11 | KS | Y | NA | Stavber et al. (2020) | |||||||||
| 15 | c.2369C>G | E12 | KS | Y | Advanced | Y | Sentchordi‐Montané et al. (2018) | ||||||||
| 16 | c.4657G>T | E12 | CS | Y | Y | Y | NA | Gkourogianni et al. (2017) | |||||||
| 17 | c.4852C>T | E12 | CS | Y | Y | Delay | Y | Tatsi et al. (2017) | |||||||
| 18 | c.5597C>A | E12 | CS | Y | Y | Y | Advanced | Hauer et al. (2017) | |||||||
| 19 | c.7090C>T | E12 | G3 | Y | Y | Y | Y | Advanced | Y | van der Steen et al. (2017) | |||||
| 20 | c.7203G>A | E16 | G3 | Y | Y | NA | Gkourogianni et al. (2017) | ||||||||
| Missense variants (n = 30, 40.5%) | |||||||||||||||
| 1 | c.151T>G | E3 | G1 | Y | Y | Advanced | Y | Hauer et al. (2017) | |||||||
| 2 | c.223T>C | E3 | G1 | Y | Y | Advanced | Gkourogianni et al. (2017) | ||||||||
| 3 | c.371G>A | E3 | G1 | Delay | Sentchordi‐Montané et al. (2018) | ||||||||||
| 4 | c.742G>A | E5 | G1 | Y | Y | Normal | Gkourogianni et al. (2017) | ||||||||
| 5 | c.903G>C | E6 | G1 | Y | Y | Y | Y | Normal | Gkourogianni et al. (2017) | ||||||
| 6 | c.916A>T | E6 | G1 | Y | Advanced | Gkourogianni et al. (2017) | |||||||||
| 7 | c.1046A>G | E6 | G1 | Y | NA | Hattori et al. (2017) | |||||||||
| 8 | c.1598C>T | E9 | G2 | Y | Normal | Sentchordi‐Montané et al. (2018) | |||||||||
| 9 | c.1702G>A | E9 | G2 | Y | Y | Y | Delay | Hauer et al. (2017) | |||||||
| 10 | c.1817C>A | E10 | G2 | Y | Y | Y | Café‐au‐lait Spots | Delay | Liang et al. (2020) | ||||||
| 11 | c.1930G>A | E10 | G2 | Y | Normal | Sentchordi‐Montané et al. (2018) | |||||||||
| 12 | c.1948G>A | E10 | G2 | Y | Delay | Sentchordi‐Montané et al. (2018) | |||||||||
| 13 | c.1979C>T | E10 | G2 | Y | NA | Hattori et al. (2017) | |||||||||
| 14 | C.2164C>G | E11 | KS | Precocious puberty | Advanced | Wang et al. (2020) | |||||||||
| 15 | c.2218A>T | E11 | KS | Y | Normal | Sentchordi‐Montané et al. (2018) | |||||||||
| 16 | c.2266G>C | E11 | KS | Y | Y | Y | Advanced | Liang et al. (2020) | |||||||
| 17 | c.4138G>T | E12 | CS | Y | Y | Y | Hypertension | NA | Fukuhara et al. (2019) | ||||||
| 18 | c.5061T>A | E12 | CS | Y | Y | Y | Hypertension | NA | Fukuhara et al. (2019) | ||||||
| 19 | c.6142C>G | E12 | CS | Y | Normal | Y | Sentchordi‐Montané et al. (2018) | ||||||||
| 20 | c.6799G>A | NA | G3 | Y | Y | Y | Y | Y | NA | Tompson et al. (2009) | |||||
| 21 | c.6907G>A | NA | G3 | Y | NA | Stattin et al. (2010) | |||||||||
| 22 | c.6970T>C | NA | G3 | Y | Y | Y | Delay | Florio et al. (2019) | |||||||
| 23 | c.7064T>C | E12 | G3 | Y | Y | Y | Y | Normal | Nilsson et al. (2014) | ||||||
| 24 | c.7069A>T, | E12 | G3 | Y | NA | Stavber et al. (2020) | |||||||||
| 25 | c.7153G>A | E15 | G3 | Y | Y | NA | Gkourogianni et al. (2017) | ||||||||
| 26 | c.7276G>T | E16 | G3 | Y | Advanced | Y | Gkourogianni et al. (2017) | ||||||||
| 27 | c.7276G>A | E16 | G3 | Y | Normal | Y | Sentchordi‐Montané et al. (2018) | ||||||||
| 28 | c.7429G>A | E16 | G3 | Y | NA | Gkourogianni et al. (2017) | |||||||||
| 29 | c.7465T>C | NA | G3 | Y | Delay | Xu et al. (2018) | |||||||||
| 30 | c.7469G>A | E18 | G3 | Y | Y | Y | Delay | Liang et al. (2020) | |||||||
| Frameshift variants (n = 18, 24.3%) | |||||||||||||||
| 1 | c.6_13delCACTTTAC | E2 | G1 | Y | Delay | Y | Hu et al. (2017) | ||||||||
| 2 | c.116dupT | E3 | G1 | Y | Y | Y | Delay | This article | |||||||
| 3 | c.272delA | E3 | G1 | Diabetes | Advanced | Y | Nilsson et al. (2014) | ||||||||
| 4 | c.410_418delinsTGGA | E3 | G1 | NA | Stavber et al. (2020) | ||||||||||
| 5 | c.661delT | E5 | G1 | Acanthosis nigricans | Normal | Y | Hu et al. (2017) | ||||||||
| 6 | c.1117_1120delCAGA | E7 | IGD | Y | Normal | Y | Hu et al. (2017) | ||||||||
| 7 | c.1425delA | E8 | G2 | Y | NA | Gkourogianni et al. (2017) | |||||||||
| 8 | c.1733‐1G>A | I9 | G2 | Y | Normal | Liang et al. (2020) | |||||||||
| 9 | c.1744delT | E10 | G2 | Y | Y | Y | Advanced | Y | Dateki et al. (2017) | ||||||
| 10 | c.2367delC | E12 | KS | Y | Pre cocious puberty | Advanced | This article | ||||||||
| 11 | c.2535_2536 insTTCA | E12 | KS | Y | NA | Hattori et al. (2017) | |||||||||
| 12 | c.4762_4765del | E12 | CS | Y | Advanced | Y | van der Steen et al. (2017) | ||||||||
| 13 | c.5391delG | E12 | CS | Y | Advanced | Quintos et al. (2015) | |||||||||
| 14 | c.6193delC | E12 | CS | Y | Advanced | ZENG et al. (2018) | |||||||||
| 15 | c.6404delC | E12 | CS | Y | Advanced | Y | Tatsi et al. (2017) | ||||||||
| 16 | c.7041delG | E12 | G3 | Y | NA | Stavber et al. (2020) | |||||||||
| 17 | c.7222dupA | E16 | G3 | Y | Advanced | Yang et al. (2018) | |||||||||
| 18 | c.7269delG | E15 | G3 | Y | Advanced | Sentchordi‐Montané et al. (2018) | |||||||||
| Splicing variants (n = 1, 1.4%) | |||||||||||||||
| 1 | c.2026+1G>A | I10 | G2 | Y | Y | Y | Normal | Nilsson et al. (2014) | |||||||
| Deletions (n = 2, 2.7%) | |||||||||||||||
| 1 | c.71_1051del | E12 | G3 | Y | Y | Y | Y | NA | Stavber et al. (2020) | ||||||
| 2 | c.7093_7095 delGAG | E15 | G3 | Y | NA | Hattori et al. (2017) | |||||||||
| Translocation (n = 1, 1.4%) | |||||||||||||||
| 1 | t (10; 15) (q22.3; q26.1) | I1 | NA | Y | Normal | Crippa et al. (2018) | |||||||||
| Unknown (n = 2, 2.7%) | |||||||||||||||
| 1 | c‐7‐2A>C* | I1 | NA | Y | Delay | Y | Sentchordi‐Montané et al. (2018) | ||||||||
| 2 | c.7342G>A | E17 | G3 | Y | Normal | Sentchordi‐Montané et al. (2018) | |||||||||
Abbreviations: BA, bone age; C, chest abnormity, including pectus excavatum/barrel chests/rib valgus et al; Ha/F, hands/feet abnormity, including short thumbs/short metacarpal bones/broad great toes/flat feet et al; He/N, head/neck abnormity, including macrocephaly/midface hypoplasia/prominent forehead/broad forehead/flat nasal bridge/low‐set ears/posteriorly rotated ears/short neck et al; J, joint abnormity, including cubitus valgus/internal rotation of elbow/patellar (sub) luxation et al; NA, not available; OA/OD, osteoarthritis/osteochondritis; S, short only; SGA, small for gestational age; V, vertebral abnormity, including lumbar lordosis/scoliosis/spine deformity/lumbar disk herniation et al.
Short stature is the most common phenotype, mostly are family inheritance, some are sporadic. In a genetic testing study of 428 idiopathic short European children, 6 children had ACAN gene variants (Hauer et al., 2017). A study of 218 Chinese children with familial short stature revealed that 3 children had ACAN gene variants (Hu et al., 2017). These studies suggest that ACAN pathogenic variants are common causes of ISS. Some children also showed OA/OD and protrusion of intervertebral disk. Florio et al. reported a boy with ACAN gene variant, presented septic arthritis, which further suggested that ACAN variants are associated with the joint involvement (Florio et al., 2019). Genetic testing is recommended to perform for patients whose height below −3.00 or −2.50 SDS, with congenital abnormalities, facial abnormalities, and abnormal bone development (Quintos et al., 2015).
Wang et al. reported the first patient of ACAN variant with precocious puberty (Wang et al., 2020). Aggrecan is a critical structural component of growth plate encoded by ACAN, the rate of growth plate chondrogenesis is regulated by multiple hormones such as GH, thyroid hormone, and sex hormones. Abnormally elevated sex hormones may occur in children with precocious puberty. In our study, the female proband of pedigree B showed breast development at around 7 years old, and bilateral breast was at stage B4 when she was 7 years 9 months. Both GnRH stimulation test and uterine ovarian ultrasound supported central precocious puberty. After 1 year of GnRHa treatment, her sex hormones decreased to normal levels. But her height increased slowly, and her BA matured by 1 year. She started to get GH treatment recently. This is the second patient of ACAN variant with precocious puberty, which enriched the possible clinical phenotypes of ACAN gene variants though the potential pathogenesis was not clear.
GH treatment or combined with GnRHa treatment is used to improve the final height of patients at present. Most children have benefited from GH treatment, but some patients showed poor GH treatment effect (Xu et al., 2018). In our study, the male child of pedigree A was treated with GH for 1 year from 4 years old, and his height increased by 9–10 cm, which is satisfactory. The female child of pedigree B was treated with GH combined with GnRHa, for 5 months and her height increased by 3.7 cm.
In conclusion, our study revealed two novel variants of ACAN in children with short stature, and enriched the genotype and possible clinical phenotypes of ACAN variants. Future research would clarify the potential role of GH therapy or combination with GnRHa in a large sample of people.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
M.W. designed the study and contributed to the data analysis and interpretation, the drafting of the article. Z.L. and Y.Y. were the study coordinators, responsible for the collection of clinical data. Y.W. contributed to the data analysis and interpretation, the drafting of the article. X.L. was the team leader, secured funding for this project, and approved the submitted paper. All authors read and approved the final manuscript.
ETHICAL COMPLIANCE
This study was approved by the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Approval number: TJ‐IRB20201016) and adhered to the tenets of the Declaration of Helsinki. Informed written consent was obtained from the patients and their parents. Participants were also informed that all derived data would be used only for scientific and noncommercial purposes. All clinical information and the medical histories were collected at Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Hubei Wuhan.
ACKNOWLEDGMENTS
The authors acknowledge all the patients and their family members for their participation.
Wei, M. , Ying, Y. , Li, Z. , Weng, Y. , & Luo, X. (2021). Identification of novel ACAN mutations in two Chinese families and genotype–phenotype correlation in patients with 74 pathogenic ACAN variations. Molecular Genetics & Genomic Medicine, 9, e1823. 10.1002/mgg3.1823
Funding information
This research was supported by the National Key R&D Program of China (No. 2018YFC1002400) and Special Science and Technology Major Project of Hubei Province (ZDZX2020000020).
REFERENCES
- Crippa, M. , Giangiobbe, S. , Villa, R. , Bestetti, I. , De Filippis, T. , Fatti, L. , Taurino, J. , Larizza, L. , Persani, L. , Bellini, F. , Finelli, P. , & Bonati, M. T. (2018). A balanced reciprocal translocation t(10;15)(q22.3;q26.1) interrupting ACAN gene in a family with proportionate short stature. Journal of Endocrinological Investigation, 41(8), 929–936. 10.1007/s40618-017-0819-3 [DOI] [PubMed] [Google Scholar]
- Dateki, S. , Nakatomi, A. , Watanabe, S. , Shimizu, H. , Inoue, Y. , Baba, H. , Yoshiura, K. I. , & Moriuchi, H. (2017). Identification of a novel heterozygous mutation of the Aggrecan gene in a family with idiopathic short stature and multiple intervertebral disc herniation. Journal of Human Genetics, 62(7), 717–721. 10.1038/jhg.2017.33 [DOI] [PubMed] [Google Scholar]
- Florio, A. , Papa, R. , Caorsi, R. , Consolaro, A. , Gastaldi, R. , Gattorno, M. , & Picco, P. (2019). A child with a novel ACAN missense variant mimicking a septic arthritis. Italian Journal of Pediatrics, 45(1), 148. 10.1186/s13052-019-0719-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara, Y. , Cho, S. Y. , Miyazaki, O. , Hattori, A. , Seo, J. H. , Mashima, R. , Kosuga, M. , Fukami, M. , Jin, D. K. , Okuyama, T. , & Nishimura, G. (2019). The second report on spondyloepimetaphyseal dysplasia, aggrecan type: A milder phenotype than originally reported. Clinical Dysmorphology, 28(1), 26–29. 10.1097/MCD.0000000000000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkourogianni, A. , Andrew, M. , Tyzinski, L. , Crocker, M. , Douglas, J. , Dunbar, N. , Fairchild, J. , Funari, M. F. A. , Heath, K. E. , Jorge, A. A. L. , Kurtzman, T. , LaFranchi, S. , Lalani, S. , Lebl, J. , Lin, Y. , Los, E. , Newbern, D. , Nowak, C. , Olson, M. , … Dauber, A. (2017). Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. The Journal of Clinical Endocrinology and Metabolism, 102(2), 460–469. 10.1210/jc.2016-3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleghorn, L. , Ramesar, R. , Beighton, P. , & Wallis, G. (2005). A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. American Journal of Human Genetics, 77(3), 484–490. 10.1086/444401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori, A. , Katoh‐Fukui, Y. , Nakamura, A. , Matsubara, K. , Kamimaki, T. , Tanaka, H. , Dateki, S. , Adachi, M. , Muroya, K. , Yoshida, S. , Ida, S. , Mitani, M. , Nagasaki, K. , Ogata, T. , Suzuki, E. , Hata, K. , Nakabayashi, K. , Matsubara, Y. , Narumi, S. , Tanaka, T. , … Fukami, M. (2017). Next generation sequencing‐based mutation screening of 86 patients with idiopathic short stature. Endocrine Journal, 64(10), 947–954. 10.1507/endocrj.EJ17-0150. [DOI] [PubMed] [Google Scholar]
- Hauer, N. N. , Sticht, H. , Boppudi, S. , Büttner, C. , Kraus, C. , Trautmann, U. , Zenker, M. , Zweier, C. , Wiesener, A. , Jamra, R. A. , Wieczorek, D. , Kelkel, J. , Jung, A. M. , Uebe, S. , Ekici, A. B. , Rohrer, T. , Reis, A. , Dörr, H. G. , & Thiel, C. T. (2017). Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Scientific Reports, 7(1), 12225. 10.1038/s41598-017-12465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Gui, B. , Su, J. , Li, H. , Li, N. , Yu, T. , Zhang, Q. , Xu, Y. , Li, G. , Chen, Y. , Qing, Y. , Li, C. , Luo, J. , Fan, X. , Ding, Y. , Li, J. , Wang, J. , Wang, X. , Chen, S. , & Shen, Y. (2017). Novel pathogenic ACAN variants in non‐syndromic short stature patients. Clinica Chimica Acta, 469, 126–129. 10.1016/j.cca.2017.04.004 [DOI] [PubMed] [Google Scholar]
- Liang, H. , Miao, H. , Pan, H. , Yang, H. , Gong, F. , Duan, L. , Chen, S. , Wang, L. , & Zhu, H. (2020). Growth promoting therapies maybe useful in short stature patients with non‐specific skeletal abnormalities caused by Acan heterozygous mutations: six Chinese cases and literature review. Endocrine Practice, 26(11), 1255–1268. 10.4158/EP-2019-0518 [DOI] [PubMed] [Google Scholar]
- Nilsson, O. , Guo, M. H. , Dunbar, N. , Popovic, J. , Flynn, D. , Jacobsen, C. , Lui, J. C. , Hirschhorn, J. N. , Baron, J. , & Dauber, A. (2014). Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. The Journal of Clinical Endocrinology and Metabolism, 99(8), E1510–E1518. 10.1210/jc.2014-1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachy, L. , Strakova, V. , Elblova, L. , Obermannova, B. , Kolouskova, S. , Snajderova, M. , Zemkova, D. , Dusatkova, P. , Sumnik, Z. , Lebl, J. , & Pruhova, S. (2019). High prevalence of growth plate gene variants in children with familial short stature treated with GH. The Journal of Clinical Endocrinology and Metabolism, 104(10), 4273–4281. 10.1210/jc.2018-02288 [DOI] [PubMed] [Google Scholar]
- Quintos, J. B. , Guo, M. H. , & Dauber, A. (2015). Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. Journal of Pediatric Endocrinology & Metabolism, 28(7–8), 927–932. 10.1515/jpem-2014-0450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentchordi‐Montané, L. , Aza‐Carmona, M. , Benito‐Sanz, S. , Barreda‐Bonis, A. C. , Sánchez‐Garre, C. , Prieto‐Matos, P. , Ruiz‐Ocaña, P. , Lechuga‐Sancho, A. , Carcavilla‐Urquí, A. , Mulero‐Collantes, I. , Martos‐Moreno, G. A. , Del Pozo, A. , Vallespín, E. , Offiah, A. , Parrón‐Pajares, M. , Dinis, I. , Sousa, S. B. , Ros‐Pérez, P. , González‐Casado, I. , & Heath, K. E. (2018). Heterozygous aggrecan variants are associated with short stature and brachydactyly: Description of 16 probands and a review of the literature. Clinical Endocrinology, 88(6), 820–829. 10.1111/cen.13581 [DOI] [PubMed] [Google Scholar]
- Stattin, E. L. , Wiklund, F. , Lindblom, K. , Onnerfjord, P. , Jonsson, B. A. , Tegner, Y. , Sasaki, T. , Struglics, A. , Lohmander, S. , Dahl, N. , Heinegård, D. , & Aspberg, A. (2010). A missense mutation in the aggrecan C‐type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. American Journal of Human Genetics, 86(2), 126–137. 10.1016/j.ajhg.2009.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavber, L. , Hovnik, T. , Kotnik, P. , Lovrečić, L. , Kovač, J. , Tesovnik, T. , Bertok, S. , Dovč, K. , Debeljak, M. , Battelino, T. , & Avbelj Stefanija, M. (2020). High frequency of pathogenic ACAN variants including an intragenic deletion in selected individuals with short stature. European Journal of Endocrinology, 182(3), 243–253. 10.1530/EJE-19-0771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsi, C. , Gkourogianni, A. , Mohnike, K. , DeArment, D. , Witchel, S. , Andrade, A. C. , Markello, T. C. , Baron, J. , Nilsson, O. , & Jee, Y. H. (2017). Aggrecan mutations in nonfamilial short stature and short stature without accelerated skeletal maturation. Journal of the Endocrine Society, 1(8), 1006–1011. 10.1210/js.2017-00229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompson, S. W. , Merriman, B. , Funari, V. A. , Fresquet, M. , Lachman, R. S. , Rimoin, D. L. , Nelson, S. F. , Briggs, M. D. , Cohn, D. H. , & Krakow, D. (2009). A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C‐type lectin domain of aggrecan. American Journal of Human Genetics, 84(1), 72–79. 10.1016/j.ajhg.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Steen, M. , Pfundt, R. , Maas, S. , Bakker‐van Waarde, W. M. , Odink, R. J. , & Hokken‐Koelega, A. (2017). ACAN gene mutations in short children born SGA and response to growth hormone treatment. The Journal of Clinical Endocrinology and Metabolism, 102(5), 1458–1467. 10.1210/jc.2016-2941 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Ge, J. , Ma, J. , Qiao, L. , & Li, T. (2020). Short stature with precocious puberty caused by aggrecan gene mutation: A case report. Medicine, 99(34), e21635. 10.1097/MD.0000000000021635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, D. , Sun, C. , Zhou, Z. , Wu, B. , Yang, L. , Chang, Z. , Zhang, M. , Xi, L. , Cheng, R. , Ni, J. , & Luo, F. (2018). Novel aggrecan variant, p. Gln2364Pro, causes severe familial nonsyndromic adult short stature and poor growth hormone response in Chinese children. BMC Medical Genetics, 19(1), 79. 10.1186/s12881-018-0591-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Zhang, C. , Wang, W. , Wang, J. , Xiao, Y. , Lu, W. , Ma, X. , Chen, L. , Ni, J. , Wang, D. , Shi, J. , & Dong, Z. (2018). Pathogenic gene screening in 91 Chinese patients with short stature of unknown etiology with a targeted next‐generation sequencing panel. BMC Medical Genetics, 19(1), 212. 10.1186/s12881-018-0730-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, T. , Liao, L. , Li, N. , Wang, J. , Peng, J. , Guo, Y. , & Li, H. (2018). Familial short stature caused by ACAN gene mutation: a familial case report. Journal of Clinical Pediatrics, 36(6), 463–466. (in Chinese). [Google Scholar]