

Abstract
Background
Hypohidrotic ectodermal dysplasia (HED) is mainly caused by ectodysplasin A (EDA) gene mutation. Fetus with genetic deficiency of EDA can be prenatally corrected. This study aimed at revealing the pathogenesis of two HED families and making a prenatal diagnosis for one pregnant female carrier.
Designs
Genomic DNA was extracted from two HED patients and sequenced using whole exome sequencing (WES). The detected mutations were confirmed in patients and family members using Sanger sequencing. The expression of soluble ectodysplasin A1 (EDA1) protein was studied by western blot. The transcriptional activity of NF‐κB pathway was tested by dual luciferase assay. The genomic DNA of fetus was extracted from shed chorion cells and EDA gene was screened through Sanger sequencing.
Results
We identified two novel EDA mutations: c.1136T>C (p.Phe379Ser) and c.[866G>C;868A>T] (p.[Arg289Pro;Ser290Cys]). Further examinations revealed that these two mutated EDA1 proteins showed completely impaired solubility, and the transcriptional NF‐κB activation induced by these missense mutant‐type EDA1 proteins was significantly reduced compared with wild‐type EDA1. Furthermore, the analysis of amniotic fluid samples from a pregnant heterozygote indicated that the fetus was a c.1136T>C mutation female carrier.
Conclusions
This study extended the mutation spectrum of X‐linked hypohidrotic ectodermal dysplasia (XLHED) and applied prenatal diagnosis for the pregnant carrier, which can be helpful in genetic counseling, prenatal diagnosis, and intervention for the XLHED family.
Keywords: ectodysplasin A, hypohidrotic ectodermal dysplasia, novel mutation, prenatal diagnosis, whole exome sequencing
The study identified two novel EDA gene mutations by whole exome sequencing and made a prenatal diagnosis for a pregnant female carrier.

1. INTRODUCTION
Ectodermal dysplasia (ED) is a genetic human disorder that affects the development of two or more ectodermal structures, including hair, teeth, nails, cornea, central nervous system, and certain glands (Wright et al., 2019). There are more than 200 different clinical types of ED, among which hypohidrotic ectodermal dysplasia (HED) is most common, with an incidence rate of ~1.6 in 100,000 (Nguyen‐Nielsen et al., 2013; Priolo & Laganà, 2001). The typical clinical manifestations of HED are sparse hair, tooth agenesis, and abnormal sweating function (Guazzarotti et al., 2015; Kere et al., 1996). The most severe complication is central hyperthermia, which is due to the absence of sweat glands and can result in the death of infants and children (Blüschke et al., 2010).
HED is a genetically heterogeneous group of diseases, which is caused by mutations in several genes that encode components of the EDA signaling pathway (Mikkola, 2009). The first genetic variation identified as causative for HED was EDA gene, other HED with similar phenotypes were found in defects of EDAR, EDARADD (Headon et al., 2001), and TRAF6 (Chassaing et al., 2006). Pathogenic variation in any of these genes can result in a similar clinical phenotype (J. Wright et al., 2019). More than 90% of HED carry pathogenic mutations in EDA (OMIM 300451), EDAR (OMIM 604095), EDARADD (OMIM 606603), or WNT10A (OMIM 606268), with X‐linked, autosomal dominant and autosomal recessive inheritance (Cluzeau et al., 2011; Guazzarotti et al., 2018). X‐linked hypohidrotic ectodermal dysplasia (XLHED, MIM 305100) is the most common form of HED and is caused by EDA gene mutation. The X‐chromosomal EDA contains eight exons with a variety of transcripts. The longest isoform is ectodysplasin A1 (EDA1), with 391 amino acid residues (Bayés et al., 1998). Ectodysplasin A1 is a trimeric type II membrane protein and is released from the cell surface by the cleavage of a furin‐like enzyme (Chen et al., 2001; Elomaa et al., 2001). Secreted ectodysplasin A1 consists of a short positively charged sequence that is required for interactions with heparan sulfate proteoglycans, a 19 Gly‐X‐Y repeat collagen domain that activates EDA1 by multimerization and a 150 amino acid residue long C‐terminal TNF homology domain (THD) which is responsible for interaction with receptor (Swee et al., 2009; Trzeciak & Koczorowski, 2016). The trimeric ligand EDA1 protein binds to the receptor EDAR and recruits the adaptor EDARADD to activate the downstream NF‐κB pathway (Clauss et al., 2008; Sadier et al., 2014). Mutations in these genes disturb the differentiation of ectodermal appendages.
HED can be identified by typical clinical characteristics: hypodontia, hypohidrosis, and hypotrichosis (Anbouba et al., 2020). The disease status of the fetus can be prenatal diagnosed through tooth germ sonography and DNA sequencing from chorion cells (Hammersen et al., 2019; Lin et al., 2017). In our study, we identified two novel EDA gene mutations and carried out a prenatal diagnosis for one pregnant heterozygous carrier.
2. MATERIALS AND METHODS
2.1. Subject
This study included two male HED patients in the Ninth People’s Hospital affiliated with Shanghai Jiao Tong University School of Medicine. The patients had the following clinical manifestations: congenital tooth agenesis, sparse hair, and sweat problems. All patients provided informed consent for this project. The study was conducted in accordance with the principles of the Declaration of Helsinki. This study was approved by the Ethics Committee of the Ninth People’s Hospital affiliated with Shanghai Jiao Tong University School of Medicine.
2.2. DNA sample collection
Blood samples were obtained from patients and family members, mixed with EDTA for anticoagulation, transferred into a labeled freezing tube, and placed at −80℃ for preservation.
2.3. Whole exome sequencing
Whole exome sequencing was performed by Novogene, Beijing, China. Total genomic DNA was extracted using the protocol recommended by the blood genomic extraction kit (Qiagen). The library was prepared using the Agilent SureSelect Human All Exon V6 Kit (Agilent Technologies). After quality control procedures, sequencing was performed using the Illumina platform with an average of 100X depth. After obtaining the sequence readings, SAMtools (GATK v3.7) was used to identify SNP and indel positions. To find the pathogenic mutations from a large number of variants, we needed to further analyze. The processes were as follows: (a) removed mutations with frequency >1% in more than one of four frequency databases: 1000 Genomes Project Database (www.1000genomes.org), dbSNP (http://www.bioinfo.org.cn), NHLBI Exome Variant Server (www.evs.gs.washington.edu), and Genome Aggregation Database (https://gnomad.broadinstitute.org); (b) reserved mutations in exons and splicing regions (splicing junction 10 bp); (c) removed synonymous mutations that were not predicted to alter amino acid by the software, and removed small (<10 bp) nonframeshift indel mutations in the repeat region; and (d) reserved mutations that were predicted as harmful according to the softwares of SIFT, PolyPhen, MutationTaster, and CADD (implemented in the dbNSFP database). The detected EDA (reference sequence NM_001399.5) mutations were confirmed in the patients and family members using Sanger sequencing as described previously (Liu et al., 2019).
2.4. Construction of EDA1 expression plasmids
Secreted EDA1 was cloned into the expression vector pcDNA3.1 (provided by Chen‐hui Huang), which contained an HA signal peptide, a Myc tag, a linker (GGSGGSGGSGGS), and amino acids 180–391 of EDA1. Mutations (c.1136T>C and c.[866G>C;868A>T]) were constructed by site‐directed mutagenesis as previously described (Wohlfart et al., 2016).
2.5. Generate stable HEK293T‐EDAR cells
Human embryonic kidney 293T (HEK293T) cells were seeded in 6‐wells plates before transfection. Then HEK293T cells were transfected with PCDH vector (provided by Chen‐hui Huang) encoding full‐length EDAR with a C‐terminal Flag tag. After 24 hr, we used 1 ug/ml of puromycin to select the cells that stably expressed EDAR protein. The expression of EDAR was confirmed by western blotting (Elomaa et al., 2001).
2.6. Expression of soluble EDA1
HEK293T cells were seeded in 12‐wells plates the day before transfection. One microgram of vectors (pcDNA3.1) encoding mutant soluble Myc‐tagged EDA1 protein was transfected into each well using Lipofectamine 2000 (Invitrogen), and vectors encoding wild‐type soluble Myc‐tagged EDA1 and vector pcDNA3.1 were used as positive and negative controls, respectively. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin, and 100 µg/ml streptomycin. Forty‐eight hours after the transfection, the cells and supernatants were harvested separately for western blot study, and the Myc‐tagged EDA1 proteins in cell lysates and supernatants were detected by western blots as described previously (Liu et al., 2019). For immunofluorescence studies, the cells were fixed with 4% paraformaldehyde for 20 min, then permeabilized before blocking in PBS with 5% horse serum for 1 hr and then stained overnight with mouse‐anti‐Myc (Thermo Fisher Scientific). Goat anti‐mouse 488 (Thermo Fisher Scientific) was used as a secondary antibody and DAPI (Thermo Fisher Scientific) was used for counterstaining. Images were captured using a microscope (Olympus IX83).
2.7. Luciferase assay
Stable HEK293T‐EDAR cells were seeded in 12‐wells plates dishes the day before transfection. Five hundred nanograms of pNF‐κB Luc plasmid (Promega), 10 ng of pRL‐TK Renilla reference plasmid (Promega), and the expression plasmid containing either wild‐type EDA1 or the mutated EDA1 were co‐transfected into each well using Lipofectamine 2000. Twenty‐four hours after the transfection, firefly luciferase activity in the cell lysates was measured and normalized with renilla luciferase activity using the dual luciferase assay system (Promega). Each experiment was performed in triplicate and repeated three times. Data were assessed by Student’s t test (p < .05).
2.8. Prenatal diagnosis
Patient A’s mother was a pregnant woman. After pathogenic gene was identified by whole exome sequencing and proved by functional studies, prenatal diagnosis was performed by KANGSO MEDICAL, Beijing, China. Amniotic fluid was obtained from the pregnancy carrier at a gestational age of 15 weeks with the help of ultrasound. Sanger sequencing was performed to diagnose fetal sex and analyze EDA gene mutations as described previously (Ma et al., 2018). The diagnosis of fetal sex was based on the amplification of the SRY gene. The primers for SRY genes were 5′‐GAATATTCCCGCTCTCCGGA‐3′ and 5′‐GCTGGTGCTCCATTCTTGAG‐3′, and short tandem repeat (STR) analysis was used to exclude maternal cell contamination.
3. RESULTS
3.1. Patient phenotypes
The two novel EDA mutation patients were males aged 20–30 years old, and all showed typical symptoms of HED: hypohidrosis, hypotrichosis, and oligodontia (Figure 1). All patients had severe tooth agenesis problems with more than 20 missing permanent teeth (excluding the third molars), and except the first molars, other teeth were missing. Patient A’s hair was slightly sparse, however his eyebrows were completely missing. The hair and eyebrows of patient B were completely missing. All patients reported that they had abnormal sweating function. Patients had no cleft lip or palate, intelligence, vision, or immune problems.
FIGURE 1.
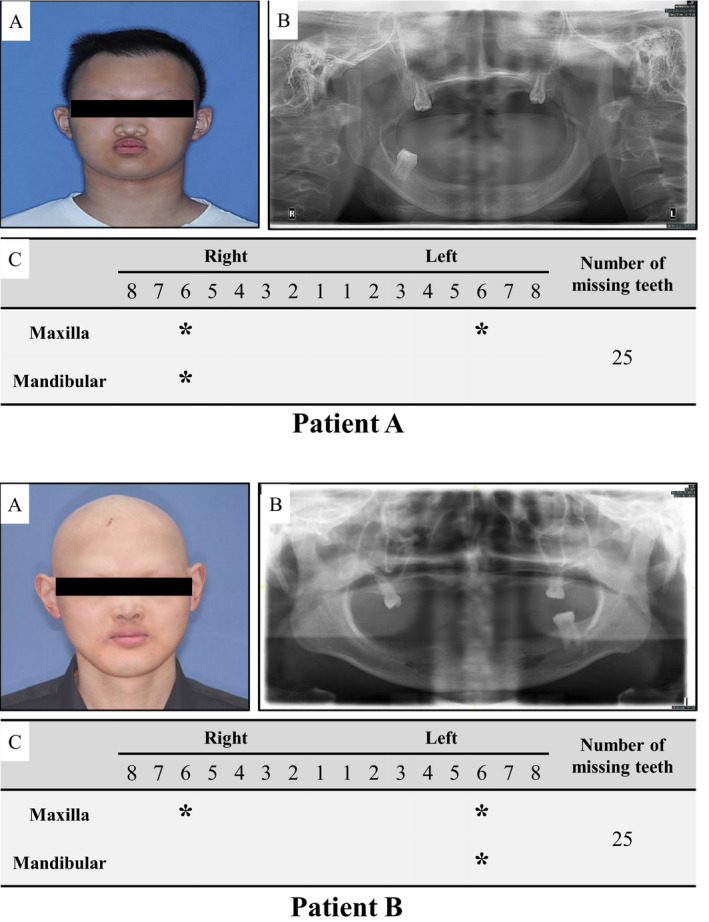
The clinical features of patients with EDA mutation. (a) Positive view of clinical photograph of patients; (b) Panoramic radiograph of patients; and (c) Summary of permanent teeth loss in patients. Patients all show sparse hair, absent eyebrows, and severe tooth agenesis
3.2. Mutation detection
We identified two novel EDA gene mutations: Patient A had a missense mutation (c.1136T>C, p.Phe379Ser) located in the TNF homology domain (Figure 2d). Sanger sequencing revealed that the mother of patient A was a pregnant heterozygous carrier with the same mutation (Figure 2c), while the father was unaffected (Figure 2b). Patient B had two missense mutation sites located in the THD region (c.[866G>C;868A>T]), which resulted in the change of amino acid 289 from Arg to Pro and amino acid 290 from Ser to Cys (Figure 2i). Sanger sequencing showed that the parents of patient B were normal (Figure 2g,h), indicating the mutation was de novo. All mutations were located in the TNF homology domain (Figure 3a) and were highly conserved during evolution (Figure 3b).
FIGURE 2.
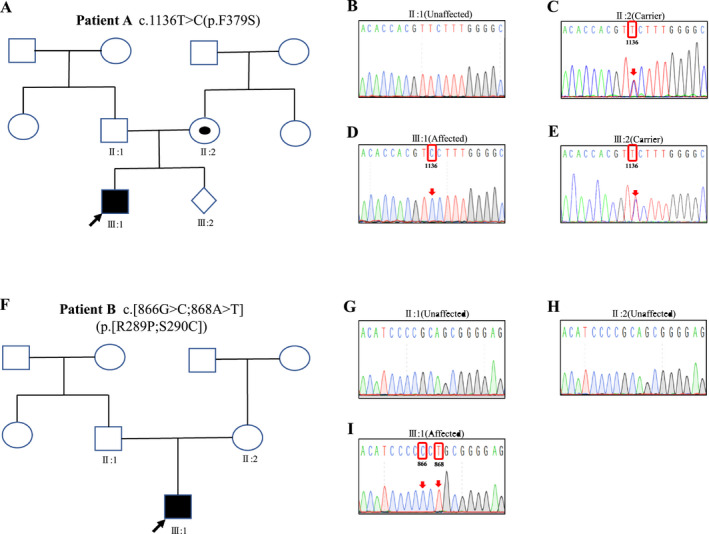
Pedigrees and Sanger sequencing of families with EDA mutation. (a) The pedigree of the patient A; (b) The EDA c.1136T>C mutation was not observed in healthy father of patient A; (c) The mother of the patient A was a pregnant heterozygous carrier with c.1136T>C missense mutation in EDA gene; (d) Mutation status of EDA c.1136T>C for patient A; (e) Prenatal diagnosis of a EDA c.1136T>C heterozygous missense mutation in the fetal EDA gene; (f) The pedigree of the patient B; (g,h) The parents of patient B were unaffected; and (i) Mutation status of EDA c.[866G>C;868A>T] for patient B. The mutant base is boxed. XLHED, X‐linked hypohidrotic ectodermal dysplasia; WT, wild type; Mut, mutation; □, normal male; ○, normal male; ◉, female XLHED carrier; ■, male XLHED patient and ⋄, fetus; the arrow shows the proband; pedigree with those affected shown in black filled symbols
FIGURE 3.
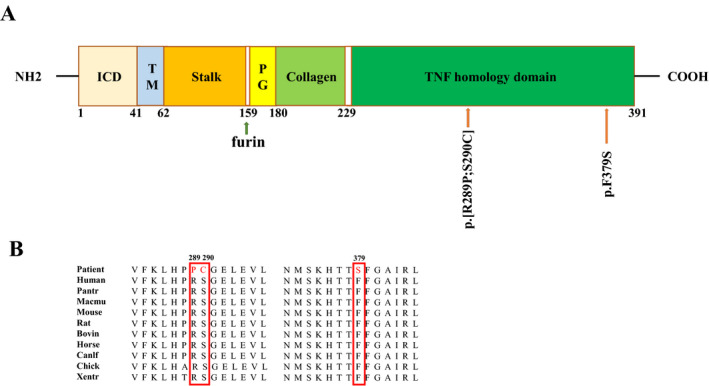
Location and conservation of EDA mutations. (a) Schematic diagram of EDA1 protein with the intracellular domain (ICD), transmembrane domain (TM), stalk region, furin site, proteoglycans‐binding domain (PG), collagen domain, TNF homology domain (THD); The amino acid sequence and location for affected individuals are shown with arrow. (b) Conservation analysis of affected amino acids in the EDA1 show that the affected residues are highly conserved among 10 different species, the affected amino acids are boxed
3.3. Mutant EDA1 protein expression
We detected the expression of mutant EDA1 protein. The results showed that p.Phe379Ser and p.[Arg289Pro; Ser290Cys] missense mutations in the THD region both significantly affected the expression of EDA1 soluble protein and were fully retained in the cytoplasm. The missense mutations also severely impaired the ability of producing intracellular EDA1 proteins (Figure 4).
FIGURE 4.
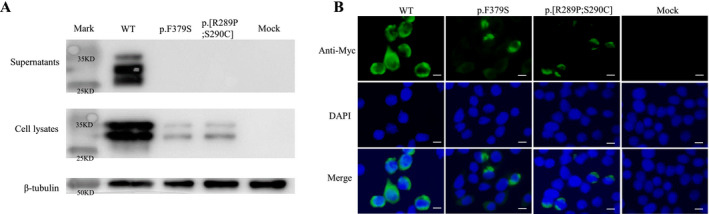
Protein expression of mutant EDA1 in transfected cell. (a) HEK293T cells were transfected with vectors encoding mutant or wild‐type soluble Myc‐tagged EDA1 protein, supernatants, and cell lysates were separately analyzed by western blotting, β‐tubulin was used as a loading control. The bands showed that wild‐type EDA1 can produce intracellular and extracellular proteins, the weaker bands of p.F379S and p.[R289P;S290C] mutations in the cell lysates showed decreased intracellular protein expression, and the lack of band of p.F379S and p.[R289P;S290C] mutations in the supernatant revealed these mutations fully impaired expression of soluble EDA1. (b) HEK293T cells were transfected with WT or mutant EDA1 protein vector contained an N‐terminal Myc tag. Nucleus was labeled with DAPI (blue), cells were stained with anti‐Myc antibody, followed by Alexa Fluor 488‐conjugated secondary antibody. The decreased fluorescence intensity of p.F379S and p.[R289P;S290C] mutations showed decreased intracellular EDA1 protein expression. Scale bar: 5 μm. WT, wild type. Data shown are representative of three experiments
3.4. Mutant EDA1 impairs the downstream NF‐κB activation
HEK293T‐EDAR cells were transfected with wild‐type EDA1 or mutant‐type EDA1. The result showed that the wild‐type EDA1 significantly upregulated the NF‐κB activity. While the mutant‐type EDA1 (c.1136T>C and c.[866G>C;868A>T]) severely impaired NF‐κB activation (Figure 5).
FIGURE 5.
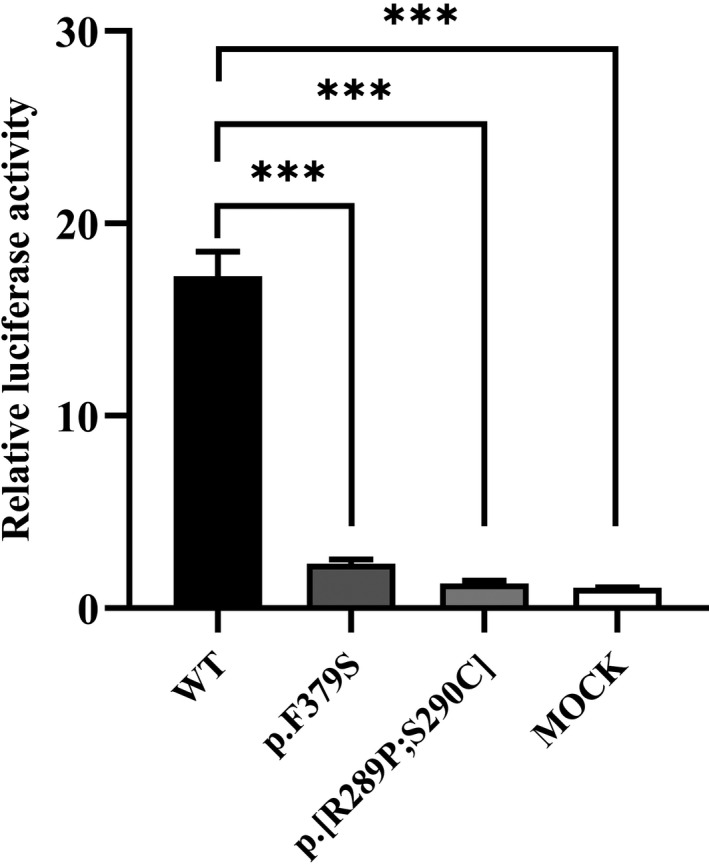
NF‐κB transcriptional activation of mutant EDA1. The mutant EDA1 protein completely abrogated transcriptional NF‐κB activation compared to wild‐type EDA1. ∗∗∗means p < .001 (Student t test)
3.5. Prenatal diagnosis of EDA gene mutation
We identified the pathogenic EDA gene mutation (c.1136T>C, p.Phe379Ser) in patient A. The mother of patient A was a pregnant heterozygous carrier. Sanger sequencing analysis of the amniotic fluid revealed that there was a c.1136T>C heterozygous missense mutation in the fetal EDA gene (Figure 2e), which indicated that the fetus was a female carrier of XLHED.
4. DISCUSSION
XLHED is the most common form of HED and is caused by EDA gene mutation. EDA1 is a TNF superfamily ligand consisting of a C‐terminal TNF homology domain, a domain that is responsible for binding with the receptor EDAR, activating the NF‐κB pathway, and regulating the development of ectodermal organs (Kowalczyk‐Quintas & Schneider, 2014). When the mutation occurs in this region, it may affect the secretion of EDA1, the formation of the trimer EDA1, or the receptor binding activity (Liu et al., 2019; Mues et al., 2010; Schneider et al., 2001). In our study, we identified two novel EDA gene mutations through clinical manifestation and gene sequencing. All mutations were located in the TNF homology domain. The pathogenesis of missense mutation was verified to affect the function of the EDA1 protein by molecular and cellular biology experiments. The missense mutations in patient A and patient B resulted in the secretory disturbance of mutant EDA1 protein outside the cell. The protein was fully retained in the cytoplasm, resulting in the complete absence of normal secreted EDA1 protein. The NF‐κB activation was severely impaired and the downstream pathway activation was totally abolished; therefore, all patients were presented with severe congenital tooth loss. Interestingly, the first molars were least affected, which was consistent with the previous finding that EDA mutation had minimal influence on first molars in XLHED (Zhang et al., 2011). However, in some XLHED patients with severe tooth agenesis, it is difficult to diagnose the agenesis of the first or second molars because of the abnormal position and morphology of teeth. Thus, further studies are needed to investigate the dental phenotype and elucidate the molecular mechanism.
HED seriously affects physical and mental health of patients and brings heavy economic burdens to family and society. In the past, the main treatments of HED were symptomatic approaches, including avoiding high temperature, prosthetic treatment, psychological treatment, and speech therapy to improve life quality of patients (Bildik et al., 2012; Ding et al., 2020; Schnabl et al., 2018). Fortunately, in 2018, Schneider treated three affected human fetuses through the intra‐amniotic delivery of a fusion protein that substitutes for the function of the affected EDA1 protein (Schneider et al., 2018). Although the sample size was small and the follow‐up of the live‐born infants was limited, the approach resulted in effective treatment of a genetic disorder and provided a new means of protein replacement therapy to correct XLHED. With the prenatal treatment of XLHED becoming a reality, the necessity to find and precisely diagnose XLHED has never been more important. In our study, Patient A’s mother was a pregnant woman, and she was identified as a carrier by genetic sequencing. After EDA gene mutation was identified by sequencing and proved by functional studies, the amniotic fluid samples were obtained from the pregnant mother of patient A and analyzed by Sanger sequencing according to the EDA gene mutation locus, and the fetus was predicted to be a heterozygous carrier. Our experience indicated that amniotic fluid is utilized for prenatal diagnosis in XLHED families, and we emphasize that it is critically important to find the real pathogenic gene of the patient.
5. CONCLUSIONS
Now, XLHED can be corrected by a new means of prenatal treatment with Fc‐EDA. Our study expands the known EDA mutation spectrum, and we applied prenatal diagnosis for a XLHED family, which could help in pregnancy counseling, fetal diagnosis, and prenatal correction in X‐linked hypohidrotic ectodermal dysplasia families.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest.
AUTHOR CONTRIBUTIONS
Kang Yu: the conception and design of the study, drafting the manuscript. Yihan Shen: acquisition of data, drafting the manuscript. Cailing Jiang: analysis and interpretation of data, drafting the manuscript. Wei Huang: the conception and design of the study, revising the manuscript critically for important intellectual content. Feng Wang: acquisition of data, drafting the manuscript. Yiqun Wu: the conception and design of the study, revising the manuscript critically for important intellectual content, funding acquisition. All authors gave final approval of the version to be submitted.
ETHICAL STATEMENT
The study protocol was reviewed and approved by the Ethics Committee of the Ninth People’s Hospital affiliated with Shanghai Jiao Tong University School of Medicine. All patients and family members provided written informed consent to participate in the study.
ACKNOWLEDGMENTS
The authors thank all hypohidrotic ectodermal dysplasia participants and their family members in this study. The authors also thank Professor. Huang (Shanghai Institute of Precision Medicine) for providing the equipment and technical support. We acknowledge the grants/awards of Clinical Research Plan of SHDC (SHDC2020CR3049B), CAMS Innovation Fund for Medical Sciences (CIFMS) (Project No. 2019‐I2M‐5‐037), “Multidisciplinary Team” Clinical Research Project of Ninth People’s Hospital affiliated to Shanghai Jiao Tong University, School of Medicine (2017‐1‐005), the Project of Biobank of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (YBKB202101), Natural Science Foundation of Shanghai (No. 21ZR1437700) and the Research Discipline fund (No. KQYJXK2020) from Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, and College of Stomatology, Shanghai Jiao Tong University.
Yu, K. , Shen, Y. , Jiang, C.‐L. , Huang, W. , Wang, F. , & Wu, Y.‐Q. (2021). Two novel ectodysplasin A gene mutations and prenatal diagnosis of X‐linked hypohidrotic ectodermal dysplasia. Molecular Genetics & Genomic Medicine, 9, e1824. 10.1002/mgg3.1824
Contributor Information
Feng Wang, Email: diana_wangfeng@aliyun.com.
Yi‐Qun Wu, Email: yiqunwu@hotmail.com.
REFERENCES
- Anbouba, G. M. , Carmany, E. P. , & Natoli, J. L. (2020). The characterization of hypodontia, hypohidrosis, and hypotrichosis associated with X‐linked hypohidrotic ectodermal dysplasia: A systematic review. American Journal of Medical Genetics. Part A, 182(4), 831–841. 10.1002/ajmg.a.61493 [DOI] [PubMed] [Google Scholar]
- Bayés, M. , Hartung, A. J. , Ezer, S. , Pispa, J. , Thesleff, I. , Srivastava, A. K. , & Kere, J. (1998). The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin‐A with deletion mutations in collagenous repeats. Human Molecular Genetics, 7(11), 1661–1669. 10.1093/hmg/7.11.1661 [DOI] [PubMed] [Google Scholar]
- Bildik, T. , Ozbaran, B. , Kose, S. , Koturoglu, G. , Gokce, B. , Gunaydin, A. , & Altintas, I. (2012). Hypohidrotic ectodermal dysplasia: A multidisciplinary approach. International Journal of Psychiatry in Medicine, 44(3), 225–240. 10.2190/PM.44.3.d [DOI] [PubMed] [Google Scholar]
- Blüschke, G. , Nüsken, K. D. , & Schneider, H. (2010). Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Human Development, 86(7), 397–399. 10.1016/j.earlhumdev.2010.04.008 [DOI] [PubMed] [Google Scholar]
- Chassaing, N. , Bourthoumieu, S. , Cossee, M. , Calvas, P. , & Vincent, M. (2006). Mutations in EDAR account for one‐quarter of non‐ED1‐related hypohidrotic ectodermal dysplasia. Human Mutation, 27(3), 255–259. 10.1002/humu.20295 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Molloy, S. S. , Thomas, L. , Gambee, J. , Bachinger, H. P. , Ferguson, B. , Zonana, J. , Thomas, G. , & Morris, N. P. (2001). Mutations within a furin consensus sequence block proteolytic release of ectodysplasin‐A and cause X‐linked hypohidrotic ectodermal dysplasia. Proceedings of the National Academy of Sciences of the United States of America, 98(13), 7218–7223. 10.1073/pnas.131076098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss, F. , Manière, M.‐C. , Obry, F. , Waltmann, E. , Hadj‐Rabia, S. , Bodemer, C. , Alembik, Y. , Lesot, H. , & Schmittbuhl, M. (2008). Dento‐craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): A review. Journal of Dental Research, 87(12), 1089–1099. 10.1177/154405910808701205 [DOI] [PubMed] [Google Scholar]
- Cluzeau, C. , Hadj‐Rabia, S. , Jambou, M. , Mansour, S. , Guigue, P. , Masmoudi, S. , Bal, E. , Chassaing, N. , Vincent, M.‐C. , Viot, G. , Clauss, F. , Manière, M.‐C. , Toupenay, S. , Le Merrer, M. , Lyonnet, S. , Cormier‐Daire, V. , Amiel, J. , Faivre, L. , de Prost, Y. , … Smahi, A. (2011). Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Human Mutation, 32(1), 70–72. 10.1002/humu.21384 [DOI] [PubMed] [Google Scholar]
- Ding, M. , Fan, Y. , Qin, M. , Claes, P. , Matthews, H. , Peng, H. , & Zhu, J. (2020). Facial morphological changes following denture treatment in children with hypohidrotic ectodermal dysplasia. Pediatric Dentistry, 42(4), 315–320. [PubMed] [Google Scholar]
- Elomaa, O. , Pulkkinen, K. , Hannelius, U. , Mikkola, M. , Saarialho‐Kere, U. , & Kere, J. (2001). Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Human Molecular Genetics, 10(9), 953–962. 10.1093/hmg/10.9.953 [DOI] [PubMed] [Google Scholar]
- Guazzarotti, L. , Tadini, G. , Mancini, G. E. , Giglio, S. , Willoughby, C. E. , Callea, M. , Sani, I. , Nannini, P. , Mameli, C. , Tenconi, A. A. , Mauri, S. , Bottero, A. , Caimi, A. , Morelli, M. , & Zuccotti, G. V. (2015). Phenotypic heterogeneity and mutational spectrum in a cohort of 45 Italian males subjects with X‐linked ectodermal dysplasia. Clinical Genetics, 87(4), 338–342. 10.1111/cge.12404 [DOI] [PubMed] [Google Scholar]
- Guazzarotti, L. , Tadini, G. , Mancini, G. E. , Sani, I. , Pisanelli, S. , Galderisi, F. , D’Auria, E. , Secondi, R. , Bottero, A. , & Zuccotti, G. V. (2018). WNT10A gene is the second molecular candidate in a cohort of young Italian subjects with ectodermal derivative impairment (EDI). Clinical Genetics, 93(3), 693–698. 10.1111/cge.13147 [DOI] [PubMed] [Google Scholar]
- Hammersen, J. , Wohlfart, S. , Goecke, T. W. , Köninger, A. , Stepan, H. , Gallinat, R. , Morris, S. , Bücher, K. , Clarke, A. , Wünsche, S. , Beckmann, M. W. , Schneider, H. , & Faschingbauer, F. (2019). Reliability of prenatal detection of X‐linked hypohidrotic ectodermal dysplasia by tooth germ sonography. Prenatal Diagnosis, 39(9), 796–805. 10.1002/pd.5384 [DOI] [PubMed] [Google Scholar]
- Headon, D. , Emmal, S. , Ferguson, B. , Tucker, A. , Justice, M. , Sharpe, P. , Zonana, J. , & Overbeek, P. (2001). Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature, 414(6866), 913–916. 10.1038/414913a [DOI] [PubMed] [Google Scholar]
- Kere, J. , Srivastava, A. K. , Montonen, O. , Zonana, J. , Thomas, N. , Ferguson, B. , Munoz, F. , Morgan, D. , Clarke, A. , Baybayan, P. , Chen, E. Y. , Ezer, S. , Saarialho‐Kere, U. , de la Chapelle, A. , & Schlessinger, D. (1996). X‐linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nature Genetics, 13(4), 409–416. 10.1038/ng0895-409 [DOI] [PubMed] [Google Scholar]
- Kowalczyk‐Quintas, C. , & Schneider, P. (2014). Ectodysplasin A (EDA) – EDA receptor signalling and its pharmacological modulation. Cytokine & Growth Factor Reviews, 25(2), 195–203. 10.1016/j.cytogfr.2014.01.004 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Yin, W. , & Bian, Z. (2017). Mutation detection and prenatal diagnosis of XLHED pedigree. PeerJ, 5, e3691. 10.7717/peerj.3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, G. , Wang, X. , Qin, M. , Sun, L. , & Zhu, J. (2019). A novel missense mutation p. S305R of EDA gene causes XLHED in a Chinese family. Archives of Oral Biology, 107, 104507. 10.1016/j.archoralbio.2019.104507 [DOI] [PubMed] [Google Scholar]
- Ma, X. , Lv, X. , Liu, H. Y. , Wu, X. , Wang, L. , Li, H. , & Chou, H. Y. (2018). Genetic diagnosis for X‐linked hypohidrotic ectodermal dysplasia family with a novel Ectodysplasin A gene mutation. Journal of Clinical Laboratory Analysis, 32(9), e22593. 10.1002/jcla.22593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola, M. L. (2009). Molecular aspects of hypohidrotic ectodermal dysplasia. American Journal of Medical Genetics. Part A, 149A(9), 2031–2036. 10.1002/ajmg.a.32855 [DOI] [PubMed] [Google Scholar]
- Mues, G. , Tardivel, A. , Willen, L. , Kapadia, H. , Seaman, R. , Frazier‐Bowers, S. , Schneider, P. , & D’Souza, R. N. (2010). Functional analysis of Ectodysplasin‐A mutations causing selective tooth agenesis. European Journal of Human Genetics, 18(1), 19–25. 10.1038/ejhg.2009.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen‐Nielsen, M. , Skovbo, S. , Svaneby, D. , Pedersen, L. , & Fryzek, J. (2013). The prevalence of X‐linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. European Journal of Medical Genetics, 56(5), 236–242. 10.1016/j.ejmg.2013.01.012 [DOI] [PubMed] [Google Scholar]
- Priolo, M. , & Laganà, C. (2001). Ectodermal dysplasias: A new clinical‐genetic classification. Journal of Medical Genetics, 38(9), 579–585. 10.1136/jmg.38.9.579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadier, A. , Viriot, L. , Pantalacci, S. , & Laudet, V. (2014). The ectodysplasin pathway: From diseases to adaptations. Trends in Genetics, 30(1), 24–31. 10.1016/j.tig.2013.08.006 [DOI] [PubMed] [Google Scholar]
- Schnabl, D. , Grunert, I. , Schmuth, M. , & Kapferer‐Seebacher, I. (2018). Prosthetic rehabilitation of patients with hypohidrotic ectodermal dysplasia: A systematic review. Journal of Oral Rehabilitation, 45(7), 555–570. 10.1111/joor.12638 [DOI] [PubMed] [Google Scholar]
- Schneider, H. , Faschingbauer, F. , Schuepbach‐Mallepell, S. , Körber, I. , Wohlfart, S. , Dick, A. , Wahlbuhl, M. , Kowalczyk‐Quintas, C. , Vigolo, M. , Kirby, N. , Tannert, C. , Rompel, O. , Rascher, W. , Beckmann, M. W. , & Schneider, P. (2018). Prenatal correction of X‐linked hypohidrotic ectodermal dysplasia. The New England Journal of Medicine, 378(17), 1604–1610. 10.1056/NEJMoa1714322 [DOI] [PubMed] [Google Scholar]
- Schneider, P. , Street, S. L. , Gaide, O. , Hertig, S. , Tardivel, A. , Tschopp, J. , Runkel, L. , Alevizopoulos, K. , Ferguson, B. M. , & Zonana, J. (2001). Mutations leading to X‐linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin‐A. Journal of Biological Chemistry, 276(22), 18819–18827. 10.1074/jbc.M101280200 [DOI] [PubMed] [Google Scholar]
- Swee, L. K. , Ingold‐Salamin, K. , Tardivel, A. , Willen, L. , Gaide, O. , Favre, M. , Demotz, S. , Mikkola, M. , & Schneider, P. (2009). Biological activity of ectodysplasin A is conditioned by its collagen and heparan sulfate proteoglycan‐binding domains. Journal of Biological Chemistry, 284(40), 27567–27576. 10.1074/jbc.M109.042259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak, W. H. , & Koczorowski, R. (2016). Molecular basis of hypohidrotic ectodermal dysplasia: An update. Journal of Applied Genetics, 57(1), 51–61. 10.1007/s13353-015-0307-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfart, S. , Söder, S. , Smahi, A. , & Schneider, H. (2016). A novel missense mutation in the gene EDARADD associated with an unusual phenotype of hypohidrotic ectodermal dysplasia. American Journal of Medical Genetics, Part A. 1, 249–253. 10.1002/ajmg.a.37412 [DOI] [PubMed] [Google Scholar]
- Wright, J. T. , Fete, M. , Schneider, H. , Zinser, M. , Koster, M. I. , Clarke, A. J. , Hadj‐Rabia, S. , Tadini, G. , Pagnan, N. , Visinoni, A. F. , Bergendal, B. , Abbott, B. , Fete, T. , Stanford, C. , Butcher, C. , D’Souza, R. N. , Sybert, V. P. , & Morasso, M. I. (2019). Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. American Journal of Medical Genetics, Part A, 179(3), 442–447. 10.1002/ajmg.a.61045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Han, D. , Song, S. , Wang, Y. , Zhao, H. , Pan, S. , Bai, B. , & Feng, H. (2011). Correlation between the phenotypes and genotypes of X‐linked hypohidrotic ectodermal dysplasia and non‐syndromic hypodontia caused by ectodysplasin‐A mutations. European Journal of Medical Genetics, 54(4), e377–382. 10.1016/j.ejmg.2011.03.005 [DOI] [PubMed] [Google Scholar]