

Abstract
Objectives
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular disorder characterized by recurrent epistaxis, telangiectasia, and visceral arteriovenous malformations (AVMs). Activin A receptor-like type 1 (ACVRL1/ALK1) and endoglin (ENG) are the principal genes whose mutations cause HHT. No multicenter study has yet investigated correlations between genetic variations and clinical outcomes in Korean HHT patients.
Methods
Seventy-two members from 40 families suspected to have HHT based on symptoms were genetically screened for pathogenic variants of ACVRL1 and ENG. Patients with genetically diagnosed HHT were also evaluated.
Results
In the HHT genetic screening, 42 patients from 24 of the 40 families had genetic variants that met the pathogenic criteria (pathogenic very strong, pathogenic strong, pathogenic moderate, or pathogenic supporting) based on the American College of Medical Genetics and Genomics Standards and Guidelines for either ENG or ACVRL1: 26 from 12 families (50%) for ENG, and 16 from 12 families (50%) for ACVRL1. Diagnostic screening of 42 genetically positive HHT patients based on the Curaçao criteria revealed that 24 patients (57%) were classified as having definite HHT, 17 (41%) as having probable HHT, and 1 (2%) as unlikely to have HHT. Epistaxis was the most common clinical presentation (38/42, 90%), followed by visceral AVMs (24/42, 57%) and telangiectasia (21/42, 50%). Five patients (12%) did not have a family history of HHT clinical symptoms.
Conclusion
Only approximately half of patients with ACVRL1 or ENG genetic variants could be clinically diagnosed as having definite HHT, suggesting that genetic screening is important to confirm the diagnosis.
Keywords: Hereditary Hemorrhagic Telangiectasia, ACVRL1, ENG, Genetic Screening
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber disease, is a rare vascular disease with a worldwide prevalence of 1 in 5,000–8,000 [1]. HHT is clinically diagnosed by the Curaçao diagnostic criteria: (1) recurrent nosebleeds (epistaxis); (2) cutaneous or mucosal telangiectasia; (3) visceral arteriovenous malformations (AVMs); and (4) a family history of HHT. The HHT diagnosis is probable or definite if two or three criteria are present, respectively [2].
The clinical presentation of HHT is highly variable even among affected members within a family. The most common clinical symptom is epistaxis; more than 90% of HHT patients experience epistaxis and the average age of onset is 12 years [3]. Severe gastrointestinal bleeding occurs in 25% of patients who are over 60 years old [4]. Another common symptom is AVMs in internal organs, including the brain, lung, and liver. Cerebral AVMs (CAVMs) can cause life-threatening complications, such as intracranial hemorrhage [1]. Pulmonary AVMs (PAVMs) increase the risk of developing cerebral abscess and embolic stroke due to the failure to filter pathogens and thrombus particles [5,6].
Three genes encoding proteins involved in the transforming growth factor-β signaling pathway have been reported to be associated with HHT: endoglin (ENG), activin A receptor-like type 1 (ACVRL1/ALK1), and SMAD family member 4 (SMAD4) [1,7,8]. Among these, ENG (HHT1) and ACVRL1 (HHT2) account for up to 96% of HHT cases [7,9]. More than 1,000 variants have been identified in ENG and ACVRL1 loci [10]. SMAD4 mutations cause a combination of HHT and juvenile polyposis and are found in fewer than 2% of HHT patients [9]. Genetic characterizations of Korean HHT patients have been reported [11-13]. However, these studies have included only a few HHT patients. In this study, we screened genetic variants in ENG and ACVRL1 in 72 individuals enrolled at multiple centers and investigated correlations between genetic variants and clinical outcomes in Korean HHT patients.
MATERIALS AND METHODS
Genetic screening
A total of 72 members from 40 families at six participating hospitals were included in this study. Serum samples from the patients were transferred to Gachon HHT center (Incheon, Korea) for genetic screening of ENG and ACVRL1. The study group included four previously described families (family No. 1, 2, 5, 6) including a total of 27 individuals with known pathogenic variants and clinical symptoms [12,13]. The genetic variant analyses were performed after obtaining informed consent from patients and their family members. All experimental procedures were reviewed and approved by the Institutional Review Board of the Lee Gil Ya Cancer and Diabetes Institute, Gachon University (Incheon, Korea; IRB No. GU1102-001).
Genomic DNA isolation
Blood samples were collected into vacuum blood collection tubes containing ethylenediaminetetraacetic acid (EDTA). Genomic DNA was isolated from 200 μL of the collected whole blood samples using the Exgene Blood SV mini kit (GeneAll, Seoul, Korea) according to the manufacturer’s protocol.
Polymerase chain reaction amplification and sequence analysis of ENG and ACVRL1
All exons of ENG and ACVRL1 and their flanking sequences were amplified by polymerase chain reaction (PCR) using PrimeSTAR HS DNA Polymerase (Takara Bio, Shiga, Japan) using appropriate primer sets as previously described [12]. The PCR products, which were separated on a 1%–2% agarose gel, were purified using the QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany) or the Expin PCR SV Kit (GeneAll). Then, the purified PCR products were subjected to direct sequencing. The GenBank accession numbers for the reference sequences of ENG and ACVRL1 are followings: NC_000009.12 (ENG genomic DNA), NM_000118.3 (ENG cDNA), NP_000109.1 (ENG protein), NC_000012.12 (ACVRL1 genomic DNA), NM_000020.3 (ACVRL1 cDNA), and NP_000011.2 (ACVRL1 protein). Missense variants were analyzed using PolyPhen-2 (http://genetics. bwh.harvard.edu/pph2/) [14], SIFT (https://sift.bii.a-star.edu.sg/) [15], and PROVEAN (http://provean.jcvi.org/index.php) [16], which are in silico predictive programs for predicting the possible impact of an amino acid substitution on a human protein. PolyPhen-2 predicts the substitution impact using straightforward physical and comparative considerations [14]; SIFT uses sequence homology and the physical properties of amino acids [15]; PROVEAN uses an alignment-based score approach [16]. The genome aggregation database (v3.1) (https://gnomad.broadinstitute.org/) was used to obtain the frequencies of variants in large populations. Variants were classified as pathogenic very strong (PVS), pathogenic strong (PS), pathogenic moderate (PM), pathogenic supporting (PP), benign stand-alone (BA), benign strong (BS), and benign supporting (BP), and then the selected criteria were combined according to the scoring rules to identify a pathogenic classification from five categories (pathogenic, likely pathogenic, benign, likely benign, or uncertain significance) based on American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines [17].
Clinical analysis
We retrospectively reviewed the clinical symptoms, physical examinations, family histories, and clinical histories of 42 patients from 24 families who were positive in the HHT genetic screening (ENG1 or ACVRL1). Patients meeting more than three of the Curaçao criteria were classified as definite HHT, patients meeting two criteria were classified as probable HHT, and patients meeting less than two criteria were classified as unlikely. We analyzed the clinical manifestations according to the Curaçao criteria and the genetic variants in the 42 patients. The clinical manifestations of 30 patients from 16 families who were negative for genetic screening were also reviewed (Fig. 1).
Fig. 1.
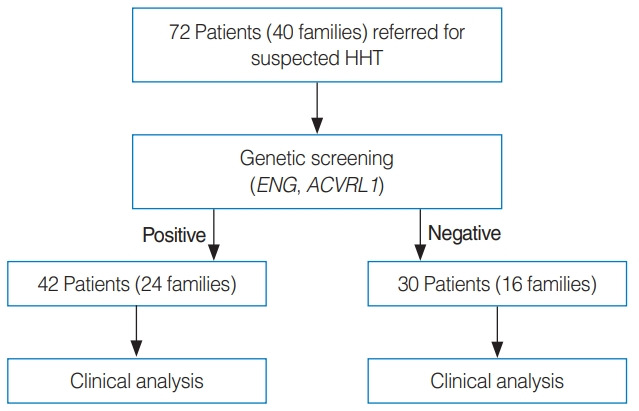
Seventy-two family members of patients with definite or probable hereditary hemorrhagic telangiectasia (HHT) were genetically screened for pathogenic variants in known HHT genes (ENG and ACVRL1). Clinical and genetic analyses were performed of the genetic variant-positive and negative patients.
Statistics
Statistical analyses were carried out using GraphPad Prism ver. 7.03 (GraphPad, San Diego, CA, USA). The clinical presentations of patients between the ENG and ACVRL1 pathogenic variant groups were compared using Fisher’s exact test.
RESULTS
Gene variant analyses of Korean patients with suspected HHT
We performed genetic analyses for all family members who agreed to undergo testing. The analysis showed that all symptomatic members in each family had the same genetic variant. Among 72 individuals in 40 families who were referred for genetic screening, a total of 42 had genetic variants that met the pathogenic criteria (PVS, PS, PM, or PP) based on the ACMG Standards and Guidelines [17]. Twenty-six patients in 12 families (50%) had genetic variants in ENG, and 16 patients in 12 families (50%) had genetic variants in ACVRL1 (Tables 1-3). Among the 12 ENG genetic variants, 11 were classified as pathogenic or likely pathogenic, and one was classified as of uncertain significance according to the ACMG Standards and Guidelines [17]. These variants included splicing defects by substitution (3/11), frameshifts by deletion (3/11), frameshifts by duplication (2/11), a frameshift by deletion-insertion (1/11), a nonsense variant by deletion-insertion (1/11), and a translation defect by substitution (1/11) (Table 2). One missense variant by substitution (family 21 c.821C>T; p.Thr274Ile) was found in a definite HHT patient and her daughter who exhibited epistaxis. This variant was also reported in two members of a Danish family who suffered from epistaxis, telangiectasia, and CAVMs [18]. In silico prediction results of this variant were inconsistent. The PolyPhen-2 [14] and PROVEAN [16] programs predicted the variant as being provably damaging/deleterious, while the SIFT [15] program predicted it as being tolerated (Supplementary Table 1). Although the variant was classified as being of uncertain significance according to ACMG Standards and Guidelines [17], we suspect that this variant was pathogenic because (1) at least four definite HHT patients had this variant (PP4), (2) the variant was not detected in the genome aggregation database (PM2), (3) two out of three in silico programs predicted this variant as to be deleterious, and (4) the p.Thr274Ile variant was not detected in ENG homologs. It is of interest to note that the same splicing defect variant (c.360+1G>A) was detected in three unrelated families (1, 20, and 26) (Table 2). There were 12 families with ACVRL1 genetic variants, which were classified as pathogenic (5/12), likely pathogenic (3/12), or of uncertain significance (4/12) [17], including frameshifts by deletion (2/12), frameshifts by duplication (2/12), missense variants by substitution (6/12), a splicing defect by substitution (1/12), and a nonsense variant by substitution (1/12) (Table 3). Among six missense variants, two variants (family 6c.199C>T; p.Arg67Trp and family 10 c.925G>A; p.Gly309Ser) were previously reported in several HHT families in more than five different studies [10] and classified as likely pathogenic [17]. One missense variant (family 32 c.1124A>G; p.Tyr375Cys) that was previously reported in a Chinese patient [19] and three novel missense variants (family 12 c.781G>C; p.Ala261Pro, family 30 c.605T>G; p.Val202Gly, and family 38 c.1005T>G; p.Asn335Lys) were classified as being of uncertain significance [17]. We suspected that the four missense variants were pathogenic because (1) these variants were not detected in the genome aggregation database (PM 2) and ENG homologs and (2) three in silico programs (PolyPhen-2 [14], SIFT [15], and PROVEAN [16]) predicted these variants to be deleterious (PM3) (Supplementary Table 1). A substitution variant (c.1048+5G>A) found in family 40 was reported to cause a splicing defect based on a functional test [20].
Table 1.
Results of HHT genetic screening in 24 families
| Gene | No. of families | No. of tested family members | No. of members with genetic variants |
|---|---|---|---|
| ENG | 12 | 35 | 26 |
| ACVRL1 | 12 | 19 | 16 |
| Total | 24 | 54 | 42 |
HHT, hereditary hemorrhagic telangiectasia.
Table 2.
ENG genetic variants in HHT patients and their family members (12 families)
| Family no. | Variant location | Variant type | Nucleotide changea) | Protein changea) | Classificationb) | Reference |
|---|---|---|---|---|---|---|
| 1 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 2 | 5′-UTR | Substitution/translation defect | c.1-127C>T | New upstream translation start codon | Likely pathogenic (PS3, PM2, PP1, PP4) | [12] |
| 8 | Exon 13 | Deletion/frameshift | c.1687del | p.Glu563Lysfs*10 | Pathogenic (PVS1, PM2, PP4, PP5) | [10] |
| 18 | Exon 13 | Deletion/frameshift | c.1687del | p.Glu563Lysfs*10 | Pathogenic (PVS1, PM2, PP4, PP5) | [10] |
| 20 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 21 | Exon 7 | Substitution/missense | c.821C>T | p.Thr274Ile | Uncertain significance (PM2, PP4) | [10] |
| 22 | Exon 1 | Deletion/frameshift | c.63del | p.Thr22Glnfs*21 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 24 | Exon 3 | Deletion-insertion/nonsense | c.276_277delinsAT | p.Arg93* | Pathogenic (PVS1, PM2, PP4) | This study |
| 26 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 31 | Exon 13 | Duplication/frameshift | c.1690_1693dup | p.His565Argfs*3 | Pathogenic (PVS1, PM2, PP4) | This study |
| 33 | Exon 7 | Deletion-insertion/frameshift | c.857_858delinsG | p.Asn286Argfs*73 | Pathogenic (PVS1, PM2, PP4) | This study |
| 34 | Exon 11 | Duplication/frameshift | c.1342dup | p.Leu338Profs*53 | Pathogenic (PVS1, PM2, PP4) | This study |
HHT, hereditary hemorrhagic telangiectasia; p.?, an effect on the protein level is expected, but it is not possible to give a reliable prediction of the consequences; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; *, termination by nonsense mutation.
The reference sequences to describe variants were NC_000009.12 (genomic DNA), NM_000118.3 (coding DNA), and NP_000109.1 (protein).
The classification was based on the Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [17].
Table 3.
ACVRL1 genetic variants in HHT patients and their family members (12 families)
| Family No. | Variant location | Variant type | Nucleotide changea) | Protein changea) | Classificationb) | Reference |
|---|---|---|---|---|---|---|
| 5 | Exon 3 | Duplication/frameshift | c.252dup | p.Val85Argfs*84 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 6 | Exon 3 | Substitution/missense | c.199C>T | p.Arg67Trp | Likely pathogenic (PM2c), PP1, PP3, PP4, PP5) | [10] |
| 10 | Exon 7 | Substitution/missense | c.925G>A | p.Gly309Ser | Likely pathogenic (PM2, PP1, PP3, PP4, PP5) | [10] |
| 12 | Exon 7 | Substitution/missense | c.781G>C | p.Ala261Pro | Uncertain significance (PM2, PP3, PM4) | This study |
| 16 | Exon 10 | Substitution/nonsense | c.1435C>T | p.Arg479* | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 17 | Exon 8 | Deletion/frameshift | c.1118del | p.Lys373Serfs*42 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 28 | Exon 3 | Duplication/frameshift | c.121dup | p.Cys41Leufs*128 | Pathogenic (PVS1, PM2, PP4) | This study |
| 30 | Exon 5 | Substitution/missense | c.605T>G | p.Val202Gly | Uncertain significance (PM2, PP3, PP4) | This study |
| 32 | Exon 8 | Substitution/missense | c.1124A>G | p.Tyr375Cys | Uncertain significance (PM2, PP3, PP4) | [10] |
| 35 | Exon 9 | Deletion/frameshift | c.1311_1315del | p.Asp437Glufs*15 | Pathogenic (PVS1, PM2, PP4) | This study |
| 38 | Exon 7 | Substitution/missense | c.1005T>G | p.Asn335Lys | Uncertain significance (PM2, PP3, PP4) | This study |
| 40 | Intron 7 | Substitution/splicing defect | c.1048+5G>A | p.? | Likely pathogenic (PS3, PM2, PP4) | [9,20] |
HHT, hereditary hemorrhagic telangiectasia; *, termination by nonsense mutation; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting; p.?, an effect on the protein level is expected, but it is not possible to give a reliable prediction of the consequences; PS, pathogenic strong.
The reference sequences to describe variants are NC_000012.12 (genomic DNA), NM_000020.3 (coding DNA), and NP_000011.2 (protein).
The classification was based on the Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [17].
One c.199C>T variant indicated as pathogenic is reported in the genome aggregation database.
Clinical characteristics of Korean patients with HHT
Of the 42 patients with HHT genetic variants, 55% were male and their mean age was 53.9 years. Twenty-six (62%) and 16 (38%) patients had ENG and ACVRL1 genetic variants, respectively. Epistaxis was the most common clinical presentation (38/42, 90%), followed by visceral AVMs (24/42, 57%) and telangiectasia (21/42, 50%). Thirty-seven patients (88%) had a family history of HHT. Among the patients with AVMs, 12 patients (29%) had PAVMs, six patients (14%) had CAVMs, four patients (10%) had hepatic AVMs (HAVMs), and two patients (5%) had gastrointestinal tract lesions. Based on the Curaçao criteria, 24 patients (57%) had definite HHT, 17 (41%) had probable HHT, and 1 (2%) were unlikely to have HHT (Table 4).
Table 4.
Clinical characteristics of the 42 patients who had HHT genetic variants
| Characteristics | Value |
|---|---|
| Sex | |
| Male | 23 (55) |
| Female | 19 (45) |
| Age (yr) | 53.9 |
| Genotype | |
| ENG | 26 (62) |
| ACVRL1 | 16 (38) |
| Clinical presentation | |
| Epistaxis | 38 (90) |
| Telangiectasia | 21 (50) |
| Family history | 37 (88) |
| AVM | 24 (57) |
| Pulmonary | 12 (29) |
| Cerebral | 6 (14) |
| Hepatic | 4 (10) |
| Gastrointestinal | 2 (5) |
| Curaçao criteriaa) | |
| Definite | 24 (57) |
| Probable | 17 (41) |
| Unlikely | 1 (2) |
Values are presented as number (%).
HHT, hereditary hemorrhagic telangiectasia; AVM, arteriovenous malformation.
Classification according to the Curaçao criteria: definite, three or more criteria; probable, two criteria; unlikely; less than two criteria.
A comparison between the clinical presentations of the ENG and ACVRL1 genetic variant groups revealed that more patients with ENG genetic variants (65%) met the definite HHT Curaçao criteria than patients with ACVRL1 genetic variants (44%) (Table 5). PAVMs were more frequent in ENG patients than in ACVRL1 patients. Among the 30 patients who were negative in the genetic variant screening, six showed definite clinical manifestations of HHT (Table 6).
Table 5.
Comparison of the clinical presentations of patients in the ENG and ACVRL1 genetic variant groups
| Clinical characteristics | ENG (n=26) | ACVRL1 (n=16) | P-value |
|---|---|---|---|
| Mean age (yr) | 55.3 | 53.4 | - |
| Curaçao criteriaa) | - | ||
| Definite | 17 (65) | 7 (44) | |
| Probable | 9 (35) | 8 (50) | |
| Unlikely | 0 | 1 (6) | |
| Clinical presentation | |||
| Epistaxis | 24 (92) | 14 (88) | 0.628 |
| Telangiectasia | 12 (46) | 9 (56) | 0.751 |
| Family history | 25 (96) | 12 (75) | 0.061 |
| AVM | 18 (42) | 9 (56) | 0.511 |
| Pulmonary | 12 (46) | 0 | <0.001 |
| Cerebral | 3 (12) | 4 (25) | 0.397 |
| Hepatic | 1 (4) | 3 (19) | 0.146 |
| Gastrointestinal | 2 (8) | 2 (13) | 0.628 |
Values are presented as number (%).
AVM, arteriovenous malformation.
Classification according to the Curaçao criteria: definite, three or more criteria; probable, two criteria; unlikely; less than two criteria.
Table 6.
Clinical analysis of family members who were negative in the genetic screening
| Curaçao criteriaa) | No. of members (%) |
|---|---|
| Definite | 6 (20) |
| Probable | 0 |
| Unlikely | 24 (80) |
Classification according to the Curaçao criteria: definite, three or more criteria; probable, two criteria; unlikely; less than two criteria.
Case presentation
A proband was a 49-year-old man (family 16), diagnosed with HHT, who presented with recurrent epistaxis, PAVM, and telangiectasia lesions; he also had a family history of epistaxis and telangiectasia. Telangiectasia lesions were detected on the tongue, lips, and nasal mucosa. He underwent surgery for a brain abscess that was suspected to be a complication associated with PAVM (Fig. 2). His two sons also had epistaxis and telangiectasia lesions and had a history of syncope.
Fig. 2.
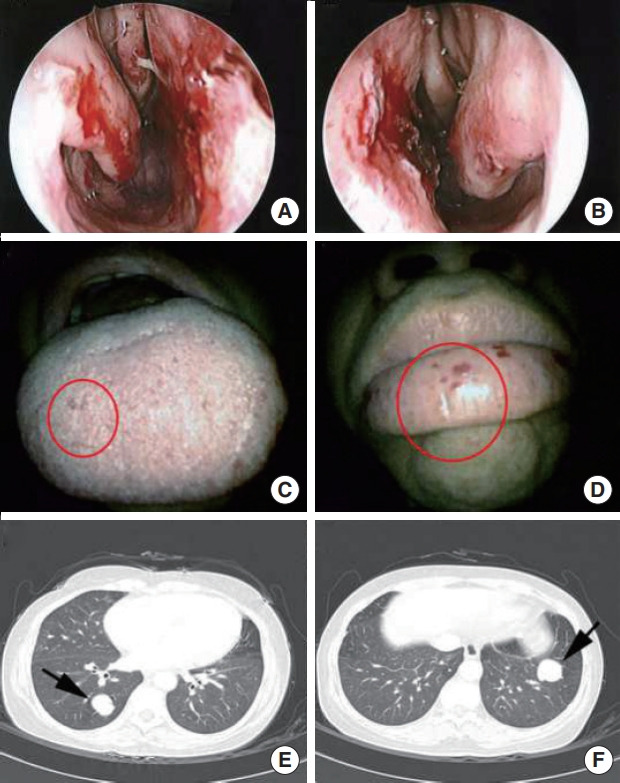
Telangiectasia lesions (circles) were detected on the nasal mucosa (A, B), tongue (C), and lip (D) in an hereditary hemorrhagic telangiectasia (HHT) patient (family 16). Pulmonary arteriovenous malformation lesions (arrows) in lung computed tomography images in the same HHT patient (E, F).
DISCUSSION
To our knowledge, this study is the first to present the results of large-scale HHT genetic testing in Korea. In this study, we performed genetic analyses of ENG and ACVRL1, but not SMAD4 because the majority (up to 96%) of patients with HHT have pathogenic variants of ENG or ACVRL1 [7,9]. We found an equal number of families with ENG or AVCRL1 genetic variants. In a Japanese HHT study, 27 families had ENG pathogenic variants and 17 families had AVCRL1 pathogenic variants [21]. A study on Danish patients with HHT showed that ENG pathogenic variants were found in 47 families (44%), AVCRL1 pathogenic variants in 45 families (42%), SMAD4 pathogenic variants in three families (3%), and unknown mutations in 12 families (11%) among 107 unrelated families [18]. In an Austrian patient cohort, pathogenic variants in ENG (37.5%) and ACVRL1 (62.5%) were identified as the cause of HHT [22].
In total, 341 ENG and 260 ACVRL1 pathogenic or suspected pathogenic variants have been reported in the HHT Mutation Database [10]. In our study, the same splicing defect in the ENG gene (c.360+1G>A) was found in three unrelated patients in families 1, 20, and 26. According to the HHT Mutation Database [10], this splicing variant is one of the most frequent pathogenic variants in HHT patients and has been reported in 13 different studies [10]. The pathogenicity of genetic variants classified as being of uncertain significance based on ACMG Standards and Guidelines [17] requires further evaluation.
Several genetic studies investigating the correlations between genetic variants and clinical phenotypes in HHT patients have yielded variable results. The case records of 21 Swedish HHT patients indicated that pathogenic variants in ENG, ACVRL1, and SMAD4 resulted in different HHT phenotypes [23]. For example, the prevalence of PAVMs was reported to be higher in patients with ENG variants than in patients with ACVRL1 variants (60% vs. 8%), whereas HAVMs were more common in HHT patients with ACVRL1 variants [23]. In a study of 78 patients in Japan, which included 53 HHT patients with ENG variants and 25 HHT patients with ACVRL1 variants, PAVMs and CAVMs were predominantly observed in patients with ENG variants, while HAVMs were more commonly detected in patients with ACVRL1 variants [21]. In our study, the most common symptoms were epistaxis (90%), a family history of HHT (88%), and AVMs (57%). The incidence of PAVMs was significantly higher in patients with ENG variants (46%) than that in patients with ACVRL1 variant (0%), but the incidence of HAVMs was higher in patients with ACVRL1 variants. Unlike the Japanese study, CAVMs were predominantly observed in patients with ACVRL1 variants.
HHT can only be diagnosed by clinical symptoms according to the Curaçao criteria. However, genetic testing is considered to be the gold standard. Of the genetic variant-positive patients in our study, 57% were classified as having definite HHT, while 41% had probable HHT. A previous study showed that 9.1% of patients with ENG or ACVRL1 variants and 45.5% of patients without those variants were classified as having probable HHT [24].
The penetration of HHT is age-dependent. Therefore, genetic screening of HHT-related genes should be performed in young individuals to prevent critical complications later in life [1]. The average age at diagnosis was 48.6 years for HHT2 and 35.1 years for HHT1, although typically the first symptom was epistaxis, which started at juvenile ages in Japanese patients [21]. If there is an HHT patient with a pathogenic variant in a family, other at-risk family members should undergo genetic analysis, clinical examination, and a presymptomatic screening. Because of the risks associated with HHT, early genetic screening and prompt treatment of PAVM in affected family members are important. In patients who are positive for an HHT gene variant, even those without severe symptoms, annual examinations should be performed.
In a previous study, members of 12 out of 107 families (11%) had no pathogenic variants in the genes responsible for HHT, even though they met the Curaçao criteria [18]. In our study, five families (six patients) who were negative in the genetic analysis were classified as having definite HHT by the Curaçao criteria, suggesting that they suffered from HHT nonetheless. In these families, the pathogenic variants might reside outside of the coding sequence, such as the CT-rich hotspot in ACVRL1 intron 9 [25] or other genetic loci including SMAD4 in these families. In one family, a patient had CAVMs and another patient had PAVMs. However, even after exhaustive exome analyses, the genetic cause remained unknown. Because we only conducted exon sequencing of the ENG and ACVRL1 genes, exome or whole genome sequencing would be useful to identify genetic variants in these variant-negative families. Although these patients were negative in the genetic study, they should be closely monitored for clinical progression.
The diagnosis of HHT based on clinical symptoms according to the Curaçao criteria is insufficient. Genetic analysis is required for a definitive diagnosis of HHT, which can increase the accuracy of the HHT diagnosis. Therefore, genetic testing should be performed during presymptomatic screenings of at-risk family members, as affected family members should be diagnosed as early as possible to prevent severe complications.
HIGHLIGHTS
▪ Among the enrolled Korean hereditary hemorrhagic telangiectasia (HHT) families, 50% had ENG and 50% had ACVRL1 genetic variants.
▪ Epistaxis was the most common symptom, followed by visceral arteriovenous malformations and telangiectasia in Korean HHT patients.
▪ Genetic screening is important to confirm the HHT diagnosis.
Acknowledgments
This research was supported by Korea Mouse Phenotyping Project (NRF-2014M3A9D5A01073528) of the Ministry of Science, ICT and Future Planning through the National Research Foundation and by a research grant of the Life Insurance Philanthropy Foundation (1-16) to YJL.
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant no. HI16C2319) to STK.
We thank Dr. S. Paul Oh for the useful discussion and advice.
Footnotes
Dae Woo Kim is an editor-in-chief of the journal but was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
AUTHOR CONTRIBUTIONS
Conceptualization: STK, YJL. Methodology: STK, YJL, BGK, JHJ, MJK, EHM. Visualization: STK, YJL, BGK, JHJ. Formal analysis: STK, YJL. Investigation: BGK, JHJ, MJK, EHM, JHO, JWP, HEC, JHK, YJK, JWC, KBH, HRJ, YJJ, SWK, SKC, DWK. Data curation: STK, YJL, BGK, JHJ, MJK, EHM. Funding acquisition: YJL, STK. Project administration: BGK, JHJ. Resources: STK, EHM, JHO, JWP, HEC, JHK, YJK, JWC, KBH, HRJ, YJJ, SWK, SKC, DWK, JHJ. Writing–original draft: STK, YJL, BGK, JHJ. Writing–review & editing: MJK, EHM, JHO, JWP, HEC, JHK, YJK, JWC, KBH, HRJ, YJJ, SWK, SKC, DWK.
Supplementary Materials
Supplementary materials can be found via https://doi.org/10.21053/ceo.2020.02124.
In silico prediction of missense variants
REFERENCES
- 1.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010 Nov;24(6):203–19. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000 Mar;91(1):66–7. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.AAssar OS, Friedman CM, White RI., Jr The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 1991 Sep;101(9):977–80. doi: 10.1288/00005537-199109000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Kjeldsen AD, Kjeldsen J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2000 Feb;95(2):415–8. doi: 10.1111/j.1572-0241.2000.01792.x. [DOI] [PubMed] [Google Scholar]
- 5.Kjeldsen AD, Oxhoj H, Andersen PE, Green A, Vase P. Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (HHT) J Intern Med. 2000 Sep;248(3):255–62. doi: 10.1046/j.1365-2796.2000.00725.x. [DOI] [PubMed] [Google Scholar]
- 6.Larsen L, Marker CR, Kjeldsen AD, Poulsen FR. Prevalence of hereditary hemorrhagic telangiectasia in patients operated for cerebral abscess: a retrospective cohort analysis. Eur J Clin Microbiol Infect Dis. 2017 Oct;36(10):1975–80. doi: 10.1007/s10096-017-3023-7. [DOI] [PubMed] [Google Scholar]
- 7.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994 Dec;8(4):345–51. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 8.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006 Feb;43(2):97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald J, Wooderchak-Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015 Jan;6:1. doi: 10.3389/fgene.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.University of Utah . ARUP scientific resource for research and education: HHT disease databases. Salt Lake City (UT): ARUP Laboratories; 2011. [Google Scholar]
- 11.Lee ST, Kim JA, Jang SY, Kim DK, Do YS, Suh GY, et al. Clinical features and mutations in the ENG, ACVRL1, and SMAD4 genes in Korean patients with hereditary hemorrhagic telangiectasia. J Korean Med Sci. 2009 Feb;24(1):69–76. doi: 10.3346/jkms.2009.24.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MJ, Kim ST, Lee HD, Lee KY, Seo J, Lee JB, et al. Clinical and genetic analyses of three Korean families with hereditary hemorrhagic telangiectasia. BMC Med Genet. 2011 Oct;12:130. doi: 10.1186/1471-2350-12-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha M, Kim YJ, Kwon KA, Hahm KB, Kim MJ, Kim DK, et al. Gastric angiodysplasia in a hereditary hemorrhagic telangiectasia type 2 patient. World J Gastroenterol. 2012 Apr;18(15):1840–4. doi: 10.3748/wjg.v18.i15.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012 Jul;40:W452–7. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015 Aug;31(16):2745–7. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torring PM, Brusgaard K, Ousager LB, Andersen PE, Kjeldsen AD. National mutation study among Danish patients with hereditary haemorrhagic telangiectasia. Clin Genet. 2014 Aug;86(2):123–33. doi: 10.1111/cge.12269. [DOI] [PubMed] [Google Scholar]
- 19.Chen YJ, Yang QH, Liu D, Liu QQ, Eyries M, Wen L, et al. Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia-associated pulmonary hypertension. Eur J Clin Invest. 2013 Oct;43(10):1016–24. doi: 10.1111/eci.12138. [DOI] [PubMed] [Google Scholar]
- 20.Alaa El Din F, Patri S, Thoreau V, Rodriguez-Ballesteros M, Hamade E, Bailly S, et al. Functional and splicing defect analysis of 23 ACVRL1 mutations in a cohort of patients affected by hereditary hemorrhagic telangiectasia. PLoS One. 2015 Jul;10(7):e0132111. doi: 10.1371/journal.pone.0132111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komiyama M, Ishiguro T, Yamada O, Morisaki H, Morisaki T. Hereditary hemorrhagic telangiectasia in Japanese patients. J Hum Genet. 2014 Jan;59(1):37–41. doi: 10.1038/jhg.2013.113. [DOI] [PubMed] [Google Scholar]
- 22.Koenighofer M, Parzefall T, Frohne A, Allen M, Unterberger U, Laccone F, et al. Spectrum of novel hereditary hemorrhagic telangiectasia variants in an Austrian patient cohort. Clin Exp Otorhinolaryngol. 2019 Nov;12(4):405–11. doi: 10.21053/ceo.2019.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlsson T, Cherif H. Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: experience from the Center for Osler’s Disease, Uppsala University Hospital. Ups J Med Sci. 2018 Sep;123(3):153–7. doi: 10.1080/03009734.2018.1483452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Gent MW, Velthuis S, Post MC, Snijder RJ, Westermann CJ, Letteboer TG, et al. Hereditary hemorrhagic telangiectasia: how accurate are the clinical criteria. Am J Med Genet A. 2013 Mar;161(3):461–6. doi: 10.1002/ajmg.a.35715. [DOI] [PubMed] [Google Scholar]
- 25.Wooderchak-Donahue WL, McDonald J, Farrell A, Akay G, Velinder M, Johnson P, et al. Genome sequencing reveals a deep intronic splicing ACVRL1 mutation hotspot in hereditary haemorrhagic telangiectasia. J Med Genet. 2018 Dec;55(12):824–30. doi: 10.1136/jmedgenet-2018-105561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In silico prediction of missense variants