

Abstract
Transitional cell carcinoma (TCC), also known as urothelial carcinoma, is the most common bladder cancer in humans and dogs. Approximately one-quarter of human TCCs are muscle-invasive and associated with a high risk of death from metastasis. Canine TCC (cTCC) tumours are typically high-grade and muscle-invasive. Shared similarities in risk factors, histopathology, and clinical presentation suggest that cTCC may serve as a model for the assessment of novel therapeutics that may inform therapies for human muscle-invasive TCC. The goal of this study was to characterize cTCC at the molecular level to identify drivers of oncogenesis and druggable targets. We performed whole exome sequencing (WES) of 11 cTCC tumours and three matched normal samples, identifying 583 variants in protein-coding genes. The most common variant was a V-to-E missense mutation in BRAF, identified in 4 out of 11 samples (36%) via WES. Sanger sequencing identified BRAF variants in 8 out of the same 11 cTCC samples, as well as in 22 out of 32 formalin-fixed paraffin embedded (FFPE) cTCC samples, suggesting an overall prevalence of 70%. RNA-Seq was performed to compare the gene expression profiles of cTCC tumours to normal bladder tissue. cTCC tumours exhibited up-regulation of genes involved in the cell cycle, DNA repair, and antiviral immunity. We also analysed the immune landscape of cTCC using immune gene signatures and immunohistochemical analysis. A subset of tumours had characteristics of a hot tumour microenvironment and exhibited high expression of signatures associated with complete response to PD-1/PD-L1 blockade in human bladder cancer.
Keywords: bladder cancer, BRAF, canine, immunotherapy, PD-L1, transitional cell carcinoma, urothelial carcinoma
1 |. INTRODUCTION
Bladder cancer comprises over 4% of diagnosed human malignancies in the United States, with an estimated 80 000 new cases diagnosed in 2020.1 TCC, the most common bladder cancer in humans, is broken into superficial, non-muscle-invasive TCC (~70%) and muscle-invasive TCC (~30%).2 Non-muscle-invasive TCC has a five-year survival rate of 90% but also has a high rate of recurrence.2 Muscle-invasive TCC has a poorer prognosis, with a five-year survival rate of 50% for all patients and only 5% for those with distant metastasis.2 Increased focus on cancer as a molecular disease has emphasized the relevance of comparative oncology, providing a translational opportunity to identify mechanisms underlying cancer progression and to evaluate novel therapeutics in spontaneous tumours in companion animals that may inform studies in human cancer patients.3,4
Most bladder tumours in dogs are TCCs, accounting for 2% of diagnosed canine cancers.5 The majority of cTCCs are muscle-invasive tumours of intermediate- to high-grade at diagnosis.6 Treatment of cTCC typically consists of cyclooxygenase inhibitors and/or chemotherapeutic agents; however, cTCC has a poor prognosis with a median survival time of less than a year for most treatments.5,7 Risk factors common to humans and dogs include living in urban areas, environmental exposure to benzene and polycyclic aromatic hydrocarbons, and prior treatment with cyclophosphamide.8 Both species also have race- or breed-associated subpopulations that exhibit increased risk.6,8 Additionally, human and canine muscle-invasive TCCs share similarities in histopathology, clinical presentation, and sites of metastasis.6
The Cancer Genome Atlas performed a genomic analysis of 412 chemotherapy-naïve, muscle-invasive human bladder tumours, identifying molecular alterations that may aid in future diagnosis and treatment of the disease.9 Inactivation of the p53/cell cycle pathway occurred in ~90% of tumours, mainly via mutations in TP53 and RB1, copy number loss of CDKN2A, and amplification/overexpression of MDM2.9 Activating alterations in the RTK/Ras/PI(3)K pathway were common, including mutations in FGFR3, PIK3CA, and the RAS gene family.9 Human muscle-invasive TCCs also frequently harbour mutations in chromatin modifiers such as histone demethylases and methyltransferases.9,10 The molecular landscape of canine TCC is less defined relative to that of human TCC. One of the most striking features of cTCC described so far is the disease’s high prevalence of BRAF mutations occurring in approximately three-quarters of tumours.11,12 Overexpression and activation of ERBB2 are also common occurrences in cTCC, as well as in human TCC.13–15 A more recent advancement in our understanding of human muscle-invasive TCC was the identification of basal and luminal molecular subtypes that may respond differently to chemotherapeutics.9,16 These basal and luminal molecular subtypes were subsequently identified in cTCC.17
In this study, we characterize the molecular features of 11 cTCC tumours by integrating WES and RNA-Seq to identify alterations contributing to pathogenesis as well as putative druggable targets. A major advantage of comparative oncology is the ability to evaluate novel immunotherapies in naturally occurring tumours under normal immunosurveillance. Thus, we examined the immune landscape of cTCC using both gene expression and immunohistochemical (IHC). Herein, we identify an immunologically hot subset of cTCC tumours exhibiting high expression of gene signatures associated with complete response to PD-1/PD-L1 blockade in human bladder cancer.
2 |. METHODS
2.1 |. Patients and samples
TCC tumour and matched normal tissue samples were collected through our institution’s tumour biorepository program with IACUC approval and informed owner consent. Pathological review identified 11 TCC samples containing at least 70% tumour by mass. Nine out of 11 patients were male and the average age of diagnosis was 10.9 years (Table 1). Patient-matched normal samples were obtained for three tumours. Normal bladder tissue samples were obtained from healthy research hounds.
TABLE 1.
Patient and sample characteristics
| Sample Name | Breed | Sex | Age at Diagnosis (yrs) | Estimated DFI (days) | Analysis Platform |
|---|---|---|---|---|---|
| Tumour | |||||
| T-1025 | Airedale Terrier | MC | 8.5 | 300 | WES/RNA-Seq |
| T-113 | Mix | MC | 13.7 | 102 | WES/RNA-Seq |
| T-22 | Dachshund - Long Haired Standard | MI | 11.1 | 480 | WES/RNA-Seq |
| T-353 | Mix | MC | 11.8 | 240 | WES/RNA-Seq |
| T-36 | Mix | MC | 7.3 | 95 | WES/RNA-Seq |
| T-400 | Mix | MC | 10.4 | 120 | WES/RNA-Seq |
| T-500 | Shetland sheepdog | FS | 8 | 90 | WES/RNA-Seq |
| T-522 | Dalmatian | FS | 13.2 | 690 | WES/RNA-Seq |
| T-730 | Mix | MC | 14.7 | 35 | WES/RNA-Seq |
| T-735 | Mix | MC | 12.3 | 65 | WES/RNA-Seq |
| T-868 | Mix | MC | 9.3 | 150 | WES/RNA-Seq |
| Patient-matched normal | |||||
| N-353 | Mix | MC | WES | ||
| N-400 | Mix | MC | WES | ||
| N-500 | Shetland sheepdog | FS | WES | ||
| Normal | |||||
| N-6970 | Research hound | FI | RNA-Seq | ||
| N-6994 | Research hound | FI | RNA-Seq | ||
| N-6998 | Research hound | FI | RNA-Seq | ||
2.2 |. Genomic DNA and RNA isolation
Tumour and normal tissues were freeze-fractured and homogenized in TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA). RNA and DNA were isolated according to the manufacturer’s recommendations. RNA was purified using the RNeasy Cleanup Kit (Qiagen, Hilden, Germany). Genomic DNA was purified using either DNeasy Blood and Tissue or QIAamp DNA Micro Kits (Qiagen).
2.3 |. Whole exome sequencing and analysis pipeline
Three μg of genomic DNA from 11 cTCC tumours and three matched normal samples was fragmented by sonication for a mean fragment size of 300 bp. Fragments were prepared for sequencing and captured using the Canine SureSelect V1 capture kit (53.59 Mb, #5190–5452, Agilent, Santa Clara, CA) based on the CanFam2.0 genome assembly. Sequencing was performed on an Illumina HiSeq 2500 (Illumina, San Diego, CA) generating 100 bp, paired-end reads.
FASTQ files were trimmed using Trimmomatic (v0.36).18 Reads were mapped to CanFam3.1 using BWA-MEM (v0.7.15).19 Duplicate reads were identified using the Picard (v1.119) tool MarkDuplicates. Base Quality Score Recalibration was performed on alignment files using the GATK (v4) tools BaseRecalibrator and ApplyBQSR in accordance with GATK best practices.20
Tumour and matched normal variants were called using Freebayes, with a min-alternate-count of two and a min-alternate-fraction of 0.05. Raw variants for tumour and matched normal samples were filtered for a minimum depth (DP) of 10. Raw tumour variants were further filtered for a minimum quality (QUAL) of one using SnpEff (v4.3t).21 Tumour variants were screened against both variants from an in-house panel of normals created from 43 canine normal samples, including the three matched normals in this study (Table S1) and against previously identified canine germline variants from four external studies.22–25 A list of 3340 somatic variants was obtained after removing variants with genotype 0/0. Annotation of these somatic variants was performed using the Ensembl Variant Effect Predictor (v99).26 Mutational burden was determined by dividing the number of somatic variants in a sample by the size of the Canine SureSelect capture in megabases (53.59 Mb).
2.4 |. RNA-Seq analysis
RNA sequencing was performed on 11 cTCC tumour samples and three normal bladder samples obtained from healthy dogs. A poly(A) selected library was prepared using a Universal mRNA-Seq Library Preparation Kit (NuGEN Technologies, Inc., San Carlos, CA) and sequenced on an Illumina NovaSEQ 6000 generating 150 bp, pair-end reads.
Raw FASTQ reads were trimmed using Trimmomatic (v0.36)18 and mapped against the CanFam3.1 genome with Tophat (v2.1.0).27 Count data was determined using HTseq-Count (v0.11.4) and relative FPKM expression values were extracted using Cufflinks (v2.2.1).28,29 Genes that are unexpressed or expressed at very low levels were removed by filtering for a minimum count of 10 in at least three samples, resulting in 17 225 expressed genes with Ensembl identifiers. Differentially expressed genes (DEGs) were identified using the ‘DESeq2’ R package,30 requiring a Benjamini and Hochberg adjusted p-value cutoff of 0.05 and a minimum log2 fold change of 2 (when comparing tumours to normal bladder samples) or 1 (when comparing between tumours). Heatmaps were generated in R using the ‘ComplexHeatmap’ package.31
2.5 |. Data availability
Raw FASTQ sequences from the WES and RNA-Seq analysis were submitted to NCBI’s SRA database.32 Sequence data for tumour and normal samples can be found under BioProject PRJNA616374 and PRJNA503860, respectively.
2.6 |. Additional materials and methods
Additional methodology for Sanger sequencing, functional analyses using DAVID, gene set enrichment analysis (GSEA), gene set variation analysis (GSVA), and IHC can be found in the online Supporting Materials and Methods.
3 |. RESULTS
3.1 |. Mutation profile of cTCC
WES yielded an average of 58.3 million reads per tumour, ranging from 31.7 million (T-22) to 82.1 million (T-730) reads. The mean target coverage was 57X, which ranged from 33X (T-22) to 87X (T-730) (Table S2). A total of 3340 somatic variants were identified among 11 cTCC tumours (Table S3). Mutational burden was less than 10 mutations per captured Mb for all samples, ranging from 1.9 to 7.9 mutations/Mb, with a mean of 5.7 mutations/Mb (Figure 1(A)). C > T transitions were the most prevalent base alteration, comprising 45% of single base substitutions, followed by T > C transitions (18%) and C > A transversions (15%) (Figure 1(B)). Eighty percent of somatic variants were single nucleotide polymorphisms (SNPs), while insertions and deletions made up 6% and 7% of somatic variants, respectively.
FIGURE 1.
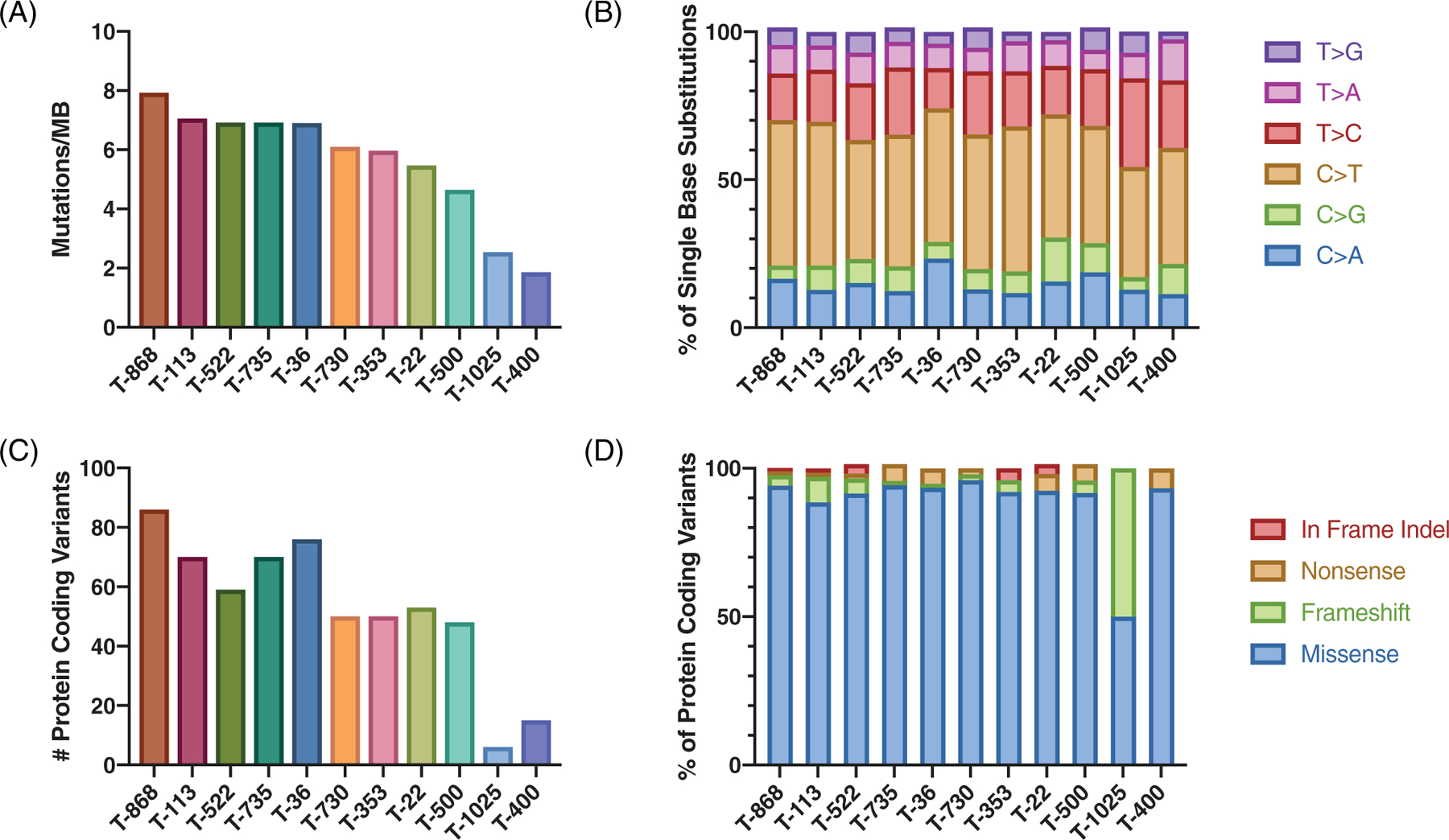
Mutation profile of canine transitional cell carcinoma (cTCC). (A) Mutational burden shown as somatic mutations per captured megabase. (B) Relative abundance of single base substitutions for somatic variants. (C) Number of protein-coding variants per sample. (D) Relative abundance of protein coding variants
Somatic variants were filtered for protein-coding variants, including in-frame indels and missense, frameshift, and nonsense variants. A total of 583 protein-altering variants were identified, ranging from 6 (T-1025) to 86 (T-868) per sample (Figure 1(C), Figure S1, Table S4). Missense mutations comprised 92% of protein-coding variants (Figure 1(D)). DAVID functional annotation tool was used to identify enriched pathways and Gene Ontology (GO) terms among the list of protein-coding variants. The top enriched pathways include Axon guidance, Focal adhesion, and MAPK signalling. The top enriched GO Molecular Function and Biological Process terms were “ATP binding” and “negative regulation of neuron apoptosis”, respectively (Table S5).
3.2 |. BRAF V596E variant identified in 70% of tumours
To identify potential drivers of oncogenesis, protein-coding variants were screened against the COSMIC Cancer Gene Census, identifying 42 variants in 32 cancer-related genes (Figure 2). BRAF, a kinase that plays a regulatory role in the MAPK signalling pathway, was mutated in 4 out of 11 samples (T-353, T-36, T-522, and T-22). BRAF variants were T > A substitutions resulting in V-to-E missense mutations at amino acid 588 (ENSCAFT00000006305). This variant is homologous to the BRAFV600E oncogenic mutation identified in human melanoma, thyroid, and colorectal cancers.33,34 We previously used rapid amplification of cDNA ends to determine the entire sequence of canine BRAF, identifying the V-to-E mutation in cTCC cell lines at amino acid 596 (MN581672).35 Sanger sequencing of the 11 cTCC tumours identified BRAF mutations in eight samples including four tumours whose BRAF variants were not detected via WES (T-500, T-113, T-400, and T-730). Further examination of raw WES results confirmed the presence of BRAF variants in these samples at low frequencies. Sanger sequencing of a larger cohort of 32 FFPE cTCC samples identified the BRAFV596E mutation in 22 out of 32 samples (Table S6). Thus, the BRAFV596E variant was detected in 30 out of 43 cTCCs, indicating an overall prevalence of 70%.
FIGURE 2.
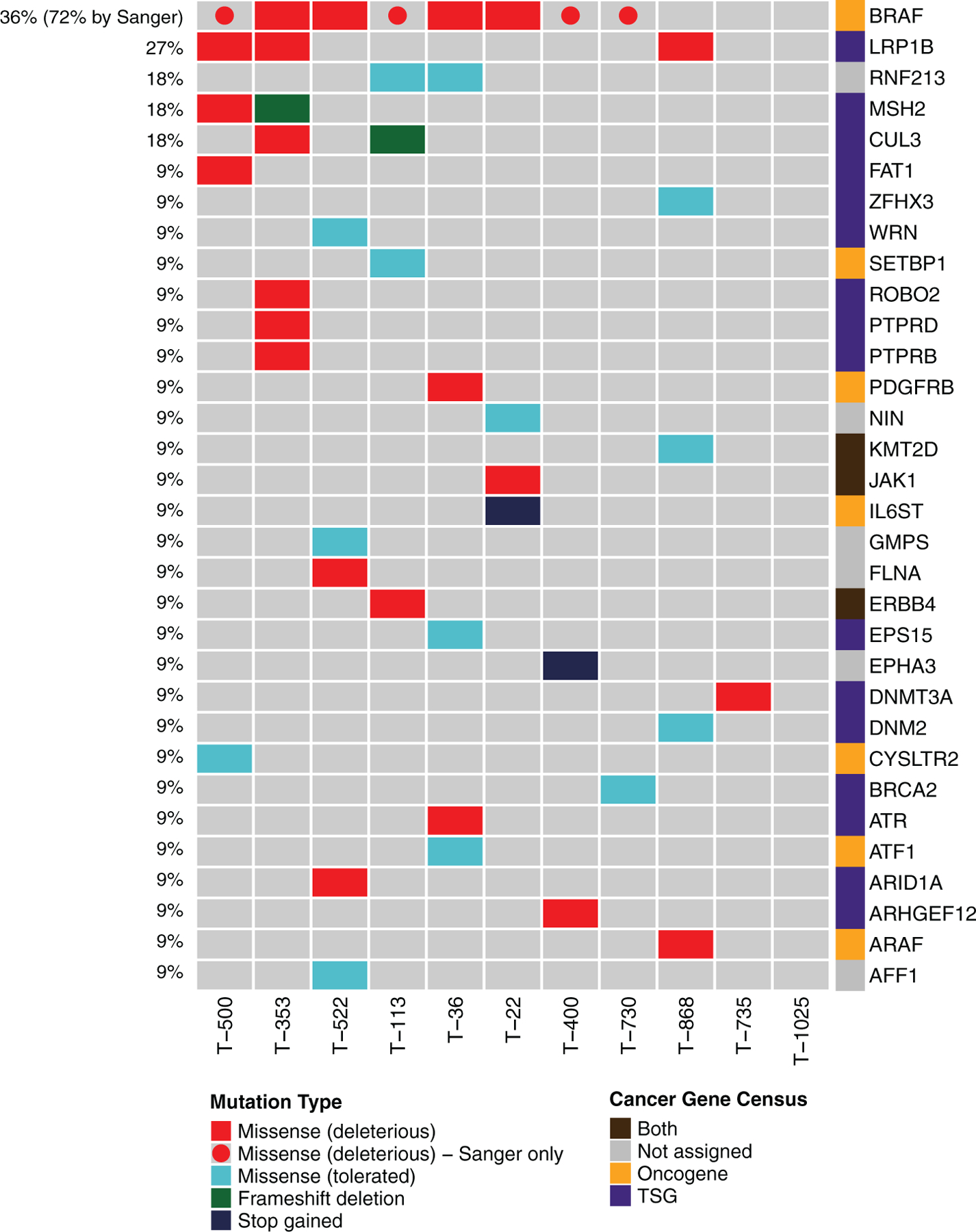
Oncoplot of cancer variants. Oncoplot depicting protein-coding variants in genes present in COSMIC’s Cancer Gene Census (v90). Colours on the oncoplot represent the type of variant. Missense variants with SIFT <0.05 are considered deleterious. BRAF variants that were detected via Sanger sequencing, but at allelic frequencies lower than WES detection limits, are shown as circles. Colours next to gene names represent a gene’s designation in the Cancer Gene Census
Deleterious variants in the low-density lipoprotein receptor LRP1B were detected in three samples (T-353, T-500, T-868). Other cancer-related genes that were mutated in more than one sample encode the E3 ubiquitin ligases CUL3 (deleterious missense and frameshift) and RNF213 (tolerated missense) as well as the mismatch repair protein MSH2 (deleterious missense and frameshift). Mutations occurred more frequently in tumour suppressor genes than oncogenes. No cancer gene variants were detected in T-1025. No mutations were detected in TP53, MDM2, CDKN2A, FGFR3, PIK3CA, or HRAS, which are frequently altered genes in human muscle-invasive TCC.9
3.3 |. Transcriptomic alterations in cTCC relative to normal bladder
RNA-Seq analysis was performed to assess the transcriptomes of 11 cTCC tumours and three normal bladder samples (Table 1). An average of 102 million reads were sequenced per sample, with a range of 80 million (T-22) to 123 million (T-730) reads per sample (Table S7). Principal component analysis resulted in a clear separation between tumours and normal bladder samples (Figure 3(A)). BRAF mutant tumours clustered separately from BRAF wild type tumours. Differential gene expression analysis identified 1219 and 1347 genes up- and down-regulated in tumour relative to normal tissue, respectively (Figure 3(B), Table S8). Functional analysis of up-regulated DEGs revealed enrichment of cell cycle-related processes as well as DNA repair and immune system processes (Table 2). Many of the immune terms were related to antiviral immunity. Genes down-regulated in tumours relative to normal bladder samples were associated with extracellular matrix organization, cell adhesion molecules, muscle-related terms, and second messenger signalling (Table 2).
FIGURE 3.
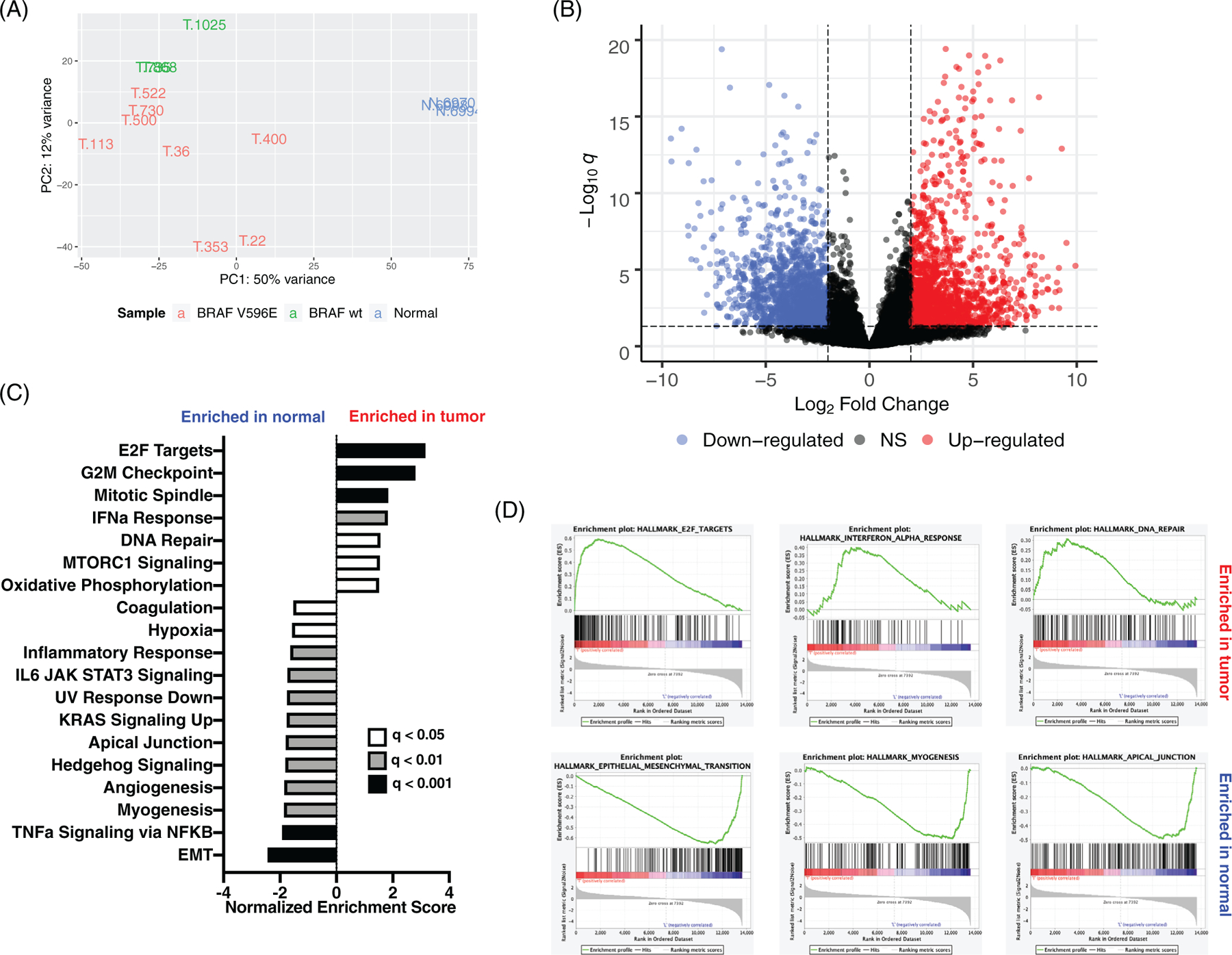
RNA-Seq analysis of canine transitional cell carcinoma (cTCC) tumours and normal bladder. (A) Principal component analysis of normalized gene counts. (B) 1219 and 1347 genes up-(red) and down-regulated (blue) in TCC tumour relative to normal bladder (logFC >2 and adj. p < 0.05). (C) Gene sets significantly enriched in tumour and normal (FDR q-val < 0.05). (D) Gene set enrichment analysis (GSEA) enrichment plots for significantly enriched gene sets
TABLE 2.
Functional annotation of DEGs between cTCC tumours and normal bladder tissue
| Process | Terms/Pathways | Genes |
|---|---|---|
| Up-regulated in tumour | ||
| Immune | T cell receptor signalling pathway | CSF2, LAT, IL5, CD3G, MAPK12, CD3D, CD3E, CD40LG, PAK5, CTLA4, PDCD1 |
| Immune response | CSF2, IL5, VPREB3, ENPP3, OAS3, CTLA4, OAS1, OAS2, CX3CL1, TNFRSF4, CCL28, LAT, CCL13, OASL, TNFSF11, RELT, AIRE, DLA-DMA, IL12B, BLNK | |
| 2’−5’-oligoadenylate synthetase activity | OAS3, OAS1, OAS2 | |
| Negative regulation of viral genome replication | RNASEL, OASL, ISG15, OAS3, RSAD2, SLPI, OAS1, CCL5, ISG20 | |
| Response to virus | LCN2, GATA3, CDK6, STMN1, MST1R, CCL5, DHX58 | |
| Defence response to virus | IFIT2, RNASEL, OASL, UNC13D, ISG15, DDX60, F2RL1, OAS3, RSAD2, OAS1, OAS2, IL12B, ISG20 | |
| Cell cycle | Cell cycle | CDK1, CDC6, E2F2, PKMYT1, TTK, CHEK1, ESPL1, CDC20, CDK6, MCM2, CDC25C, MCM5, CCNE2, CDC45, CCNB3, CCNB2, PLK1, BUB1, BUB1B, ORC6, ORC1, CCNA2 |
| DNA replication initiation | CCNE2, CDC6, CDC45, GINS4, ORC6, POLA2, MCM2, MCM10, MCM5 | |
| Mitotic sister chromatid segregation | CDCA8, PLK1, DSN1, CENPA, NEK2, ZWINT, KIF18A, KIF18B, ESPL1, KNSTRN | |
| Mitotic cytokinesis | KIF23, PLK1, NUSAP1, ANLN, MITD1, STMN1, RACGAP1, KIF20A | |
| DNA damage | Fanconi anaemia pathway | FANCM, RAD51C, FANCI, FANCD2, BRIP1, RMI2, BRCA1, UBE2T, RAD51 |
| Regulation of double-strand break repair via homologous recombination | TEX15, RAD51AP1, CHEK1, RAD51 | |
| Base-excision repair | RECQL4, DNA2, NEIL3, NEIL1, FEN1 | |
| G2 DNA damage checkpoint | CLSPN, PLK1, DTL, CHEK1, BRCA1 | |
| Down-regulated in tumour | ||
| Cell adhesion | ECM-receptor interaction | TNC, COL3A1, ITGA11, COL2A1, VTN, CHAD, COL6A6, TNR, COL6A3, COL6A1, SV2A, THBS1, COL11A1, THBS3, COL4A4, COL4A3, TNXB, HSPG2, COL4A6, COL4A5, LAMA2, ITGA9, LAMA4, CD36, LAMC3, ITGA5, ITGA8, RELN |
| Positive regulation of cell-substrate adhesion | EGFL6, SPOCK2, NPNT, DMP1, CCDC80, VTN, NID1, ECM2, VIT, ABI3BP, EMILIN1, SMOC2, ALOX15, FBLN2 | |
| Heterophilic cell-cell adhesion via plasma membrane cell adhesion molecules | VCAM1, TENM4, SELP, CADM3, TENM1, FAT4, ITGA5, TENM2, NLGN1, CDH2, SELE, DCHS1, SCARF2 | |
| Myogenesis | Vascular smooth muscle contraction | KCNMA1, ADCY3, ADCY2, ACTA2, ADCY5, PPP1R12B, CALD1, MRVI1, NPR2, PRKG1, KCNMB1, MYL9, KCNMB2, AGTR1, PTGIR, CACNA1C, MYLK, PPP1R14A |
| Dilated cardiomyopathy | ADCY3, CACNA2D1, ADCY2, ADCY5, CACNG7, ITGA11, CACNB2, IGF1, CACNA2D3, TPM2, CACNA2D2, TPM1, TNNT2, ITGA9, DES, ITGA5, DMD, ITGA8, SGCD, CACNA1C, SGCA | |
| Smooth muscle contraction | PDE4D, BDKRB2, HTR2B, MYLK, HTR2A | |
| Second messenger signalling | cGMP-PKG signalling pathway | ADCY3, SLC8A3, ADCY2, GNAI1, ATP1B2, ADCY5, MRVI1, BDKRB2, PRKG1, ADORA1, KCNMB1, KCNMB2, MYL9, ATP2B2, AGTR1, ADRA2A, NOS3, AKT3, KCNMA1, SLC8A1, ATP1A3, CREB5, NPR2, PDE2A, KCNJ8, PDE5A, CACNA1C, MYLK |
| cAMP signalling pathway | ADCY3, FXYD1, ADCY2, DRD2, ATP1B2, GNAI1, ADCY5, FFAR2, ADORA1, VIPR2, MYL9, ATP2B2, GRIN2D, PDE4B, CAMK2A, AKT3, PTGER2, ATP1A3, GRIA3, PDE4D, CREB5, GRIA4, GRIA1, CACNA1C, TSHR | |
| Calcium signalling pathway | SLC8A3, ADCY3, SLC8A1, ADCY2, PHKG1, BDKRB2, AGTR1, ATP2B2, P2RX1, HRH2, P2RX3, GRIN2D, PDE1A, CACNA1G, CACNA1H, PLCD4, NOS3, HTR2B, CACNA1C, CAMK2A, MYLK, HTR2A | |
Gene expression data was further analysed using GSEA to identify enrichment of MSigDB hallmark gene sets in cTCC tumours relative to normal bladder samples. GSEA identified significant enrichment of gene sets related to cell growth and proliferation in cTCC tumours: E2F Targets, G2M Checkpoint, Mitotic Spindle, and MTORC1 Signalling (Figure 3(C),(D)). Additionally, tumour samples exhibited enrichment of Interferon Alpha (IFN-α) Response, Oxidative Phosphorylation, and DNA Repair gene sets. Twelve hallmark gene sets were enriched in normal samples, including Epithelial Mesenchymal Transition (EMT), TNF-α signalling via NFKB, myogenesis, and angiogenesis (Figure 3(C),(D)). We also specifically focused on genes whose copy number or mRNA expression is frequently altered in human muscle-invasive TCC (TP53, MDM2, RB1, CDKN2A, E2F3, FGFR3, and PPARG).9 Of these genes, only FGFR3 was identified as a DEG between cTCC and normal bladder (5.4-fold upregulated). However, one tumour (T-868) exhibited dramatically increased expression of MDM2 relative to normal bladder (19-fold), suggesting loss of p53 activity in this tumour.
GSEA was also used to compare the gene expression profiles of BRAF mutant and wild type tumours. BRAF mutant tumours showed significant enrichment of the hallmark gene sets E2F Targets and Myogenesis; whereas, wild type tumours showed significant enrichment of IFN-α and -γ Response gene sets as well as Hedgehog Signalling (Figure S2). We also analysed MAPK pathway activity among the tumours since 8 out of 11 harbour BRAF mutations. The MAPK Pathway Activation Score (MPAS) is a gene signature quantifying relative MAPK activity based on expression levels of 10 downstream targets of the MAPK pathway and correlates with sensitivity to MAPK inhibitors in both cell lines and patients.36 The MPAS has previously been applied to a panel of canine cancer cell lines and TCC cell lines exhibited the highest MPAS values relative to other cancer types.35 In this study, all three normal samples and the three BRAF wild type samples had negative MPAS values (Figure S3). Only three of the BRAF mutant tumours had positive MPAS values, possibly indicating sensitivity to MAPK pathway inhibitors. T-353, a BRAF mutant tumour, exhibited extremely high MAPK pathway activation relative to the other tumour samples.
Molecular subtypes have been identified in both human and canine muscle-invasive TCC.16,17,37 cTCC tumours were clustered using a gene signature described by Dhawan et al. that is reported to separate cTCC into basal and luminal subtypes, which yielded two distinct clusters (Figure S4). One cluster, comprised of two BRAF mutant tumours, exhibited high expression of basal gene markers such as CAV1, MMP9, and VIM. The remaining nine tumours exhibited increased expression of luminal markers including PPARG, UPK3A, and ERBB2.
3.4 |. Identification of an immunologically hot subset of cTCC
Several immune-related, antiviral genes were up-regulated in cTCC tumours relative to normal bladder samples. Additionally, the IFN-α Response gene set was significantly enriched in cTCC tumours relative to normal bladder. We performed hierarchical clustering of tumour and normal samples based on expression of genes within this gene set and a subset of five tumours with high expression of IFN-α response genes clustered separately from the other tumours and normal bladder samples (Figure 4(A), Figure S5). To determine whether this subset of tumours exhibits an immunologically hot tumour microenvironment, we used GSVA to analyse the relative enrichment of gene sets representing specific immune cell types and processes described by Rooney et al (Table S9).38 We chose to employ GSVA since this method determines relative enrichment scores for each sample in an unsupervised manner. Clustering of tumours based on their GSVA scores resulted in two distinct clusters representing immunologically hot (T-1025, T-36, T-500, T-730) and cold tumours, termed “TME-Hot” and “TME-Cold”, respectively (Figure 4(B)). T-868, which has high expression of IFN-α response genes, clustered with the TME-Cold tumours. TME-Hot tumours exhibited significant enrichment of gene sets representing cytolytic activity, CD8+ T cells, plasmacytoid dendritic cells (pDCs), co-stimulatory and co-inhibitory receptor expression by antigen presenting cells (APCs) and T cells, major histocompatibility complex (MHC) class I expression, CD4+ regulatory T cells (Tregs), and type I IFN response (Figure 4(B)).
FIGURE 4.
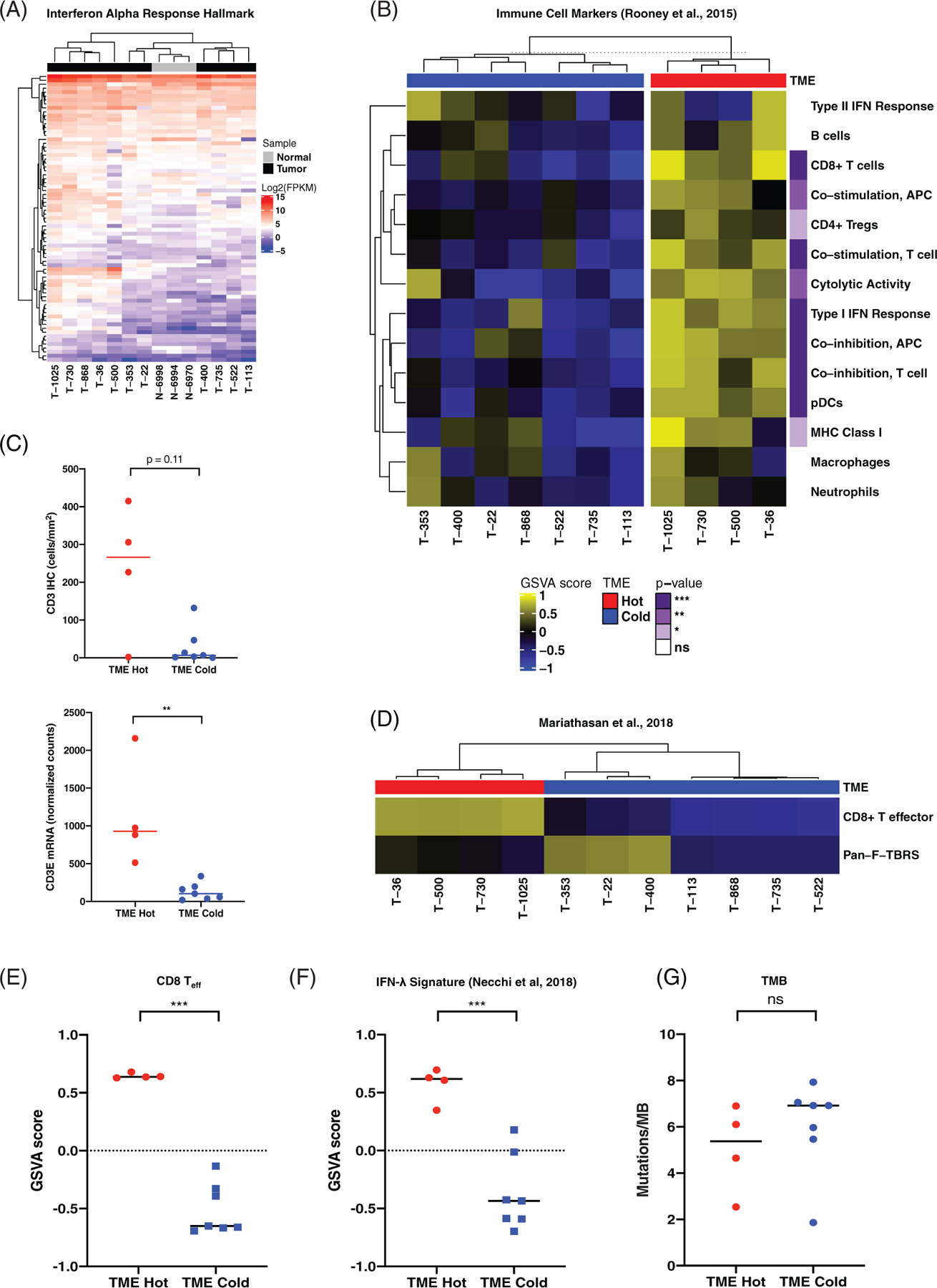
Identification of an immunologically hot subset of canine transitional cell carcinoma (cTCC) tumours associated with genomic indicators of response to PD-1/PD-L1 blockade in human bladder cancer. (A) Hierarchical clustering of cTCC tumours and normal bladder based on expression of genes present in the IFN-α Response Hallmark gene set. Colours on the heat map represent log2-FPKM expression values. See Figure S5 for the complete heatmap with gene names. (B) Heatmap of GSVA scores for gene sets representing immune cell types and processes described by Rooney et al. Significantly enriched gene sets were determined using linear models and moderated t tests implemented in the ‘limma’ R package. p values are represented in purple on the right. (C) CD3 immunohistochemical (IHC) (top) and CD3E mRNA expression (bottom) in tumour microenvironment (TME)-Hot versus TME-Cold tumours compared using the Mann Whitney test. (D) Heatmap representing cTCC GSVA scores for CD8 Teff and F-TBRS gene signatures associated with good and bad responses to PD-L1 blockade in human bladder cancer, respectively. (E) CD8 Teff (left) and (F) IFN-λ (right) GSVA scores in HME-Hot versus TME-Cold tumours. Statistical significance was determined same as in panel B. (G) Tumour mutation burden in TME-Hot versus TME-Cold tumours compared using the Mann Whitney test
IHC analysis was performed to quantify T cell abundance in cTCC tumours. CD3+ T cell counts were variable, ranging from 1 to 415 cells/mm2 (Figure S6). Though not statistically different, several TME-Hot tumours had higher T cell infiltration than any of the TME-Low tumours (Figure 4(C), top). Despite exhibiting an immunologically hot gene expression signature, T-730 had very little T cell infiltration. TME-Hot tumours did, however, exhibit significantly higher CD3E mRNA expression (Figure 4(C), bottom), which was highly correlated with CD3 IHC staining (p < 0.001, R2 = 0.86). Tumours with high CD3 staining showed enrichment of the immune-related hallmarks gene sets IFN-α Response, IFN-γ Response, and Allograft Rejection, as well as the cell cycle-related gene sets E2F Targets and G2M Checkpoint (Figure S7). Tumours with low CD3+ staining exhibited enrichment of hallmark gene sets for TNF-α Signalling via NFKB, TGF-β Signalling, and Hypoxia.
We also stained for MAC387 expression as a means of quantifying myeloid cells (Figure S6). MAC387+ cell counts ranged from 3 to 614 cell/mm2 and did not correlate with CD3+ T cell counts (p = 0.24, R2 = 0.15) or with S100A9 mRNA expression (p = 0.44, R2 = 0.07). This discrepancy between the IHC and gene expression data could be explained by tumour heterogeneity, lack of specificity of the antigen as a cell marker, or subsequent regulatory processes.
3.5 |. TME-Hot cTCCs exhibit high expression of genomic biomarkers of response to PD-1/PD-L1 inhibition in human bladder cancer
Immune checkpoint inhibitors targeting PD-1 and PD-L1 have shown efficacy in a subset of human bladder cancer patients.39,40 A CD8+ T-effector gene signature (CD8 Teff) has been identified as a genomic indicator of response to the PD-L1 inhibitor atezolizumab in bladder cancer patients enrolled in the phase 2 IMvigor210 clinical trial.41,42 Additionally, Mariathasan et al. generated a pan-fibroblast TGF-β response signature (F-TBRS) that was associated with lack of response.41 We applied these human-derived gene signatures to the canine TCC dataset in this study, generating relative CD8 Teff and F-TBRS scores for each sample using GSVA (Table S9). Clustering of tumours based on their CD8+ Teff and F-TBRS scores resulted in a clear separation between TME-Hot and TME-Cold tumours (Figure 4 (D)). TME-Hot tumours exhibited significantly higher CD8+ Teff scores relative to TME-Cold tumours (Figure 4(E)). Three tumours, all of which are TME-Cold and harbour BRAF mutations, exhibited high F-TBRS scores (T-353, T-22, T-400), suggestive of TGF-β signalling by cancer-associated fibroblasts.41 Two out of these three tumours (T-353 and T-22) were the basal tumours identified in Supporting Figure S4.
We next evaluated an IFN-γ-related gene signature associated with complete response to the PD-1 inhibitor pembrolizumab in the neoadjuvant setting.43,44 Indeed, TME-Hot tumours exhibited significantly higher IFN-γ scores relative to TME-Cold tumours (Figure 4(F)). High tumour mutation burden (TMB) has also been identified as a correlate of response to PD-1/PD-L1 blockade in several human cancers, including bladder.42 Notably, we did not detect a difference in TMB between TME-Hot and TME-Cold tumours (Figure 4(F)). Overall, these results suggest that the TME-Hot cTCC tumours identified in this study represent a subset of canine bladder cancer patients that may achieve clinical benefit from checkpoint inhibitors targeting the PD-1/PD-L1 axis.
4 |. DISCUSSION
Canine cancers have become increasingly established as valuable models for human cancer. Oncology clinical trials in dogs serve to evaluate novel anticancer strategies with the goal of advancing treatment for both canines and humans.3,4 Our genomic characterization of cTCC contributes to the growing body of evidence supporting the comparative relevancy of the disease. We identify novel and previously described drivers of oncogenesis as well as putative therapeutic targets. Additionally, a subset of cTCC tumours in this study exhibited an inflamed tumour microenvironment with high expression of genomic biomarkers of response to checkpoint inhibition in human bladder cancer.
The mutation profile of the cTCC tumours analysed in this study was similar to that of human muscle-invasive TCC. cTCC tumours exhibited a mean somatic mutation rate of 5.7 mutations/Mb, similar to 7.7 mutations/Mb reported in human bladder cancer.45 C > T transitions were the most common single base substitution, consistent with frequencies observed in human cancers, including TCC.9,46 The most common protein-coding variant identified in this study was a BRAF V-to-E missense mutation homologous to the V600E driving variant frequently observed in human cancer.33,34 BRAF variants were detected in this study at an overall prevalence of 70%, consistent with other studies of canine bladder cancer.11,12 Several small molecule inhibitors targeting mutant BRAF, as well as BRAF’s downstream target MEK1/2, have been approved for BRAF mutant human cancers; however, intrinsic and acquired drug resistance pose major roadblocks to treatment.47 We previously found that BRAF mutant cTCC cell lines are sensitive to the “paradox-breaking” BRAF inhibitor PLX7904 and the MEK inhibitor trametinib.35 However, MAPK pathway reactivation was observed is most cell lines following treatment, suggesting cTCC may serve as a model for interrogating combination therapies that circumvent resistance to MAPK pathway inhibition.35 Parker et al. recently determined genes and pathways differentially enriched in BRAF mutant versus BRAF wild type cTCC, identifying enrichment of cell cycle/cell death pathways in BRAF mutant cTCC and enrichment of immune-related gene sets in BRAF wild type cTCC.48 Similarly, we observed enrichment of E2F target genes in BRAF mutant tumours and enrichment of IFN-α and IFN-γ response gene sets in BRAF wild type tumours.
In total, 36 mutated genes identified in this study were previously identified in other genomic analyses of cTCC. Nine of these genes are present in the COSMIC Cancer Gene Census (BRAF, LRP1B, BRCA2, FAT1, PDGFRB, KMT2D, FLNA, ARHGEF12, and AFF1), potentially validating the importance of these alterations in cTCC pathogenesis.11,49 Deleterious missense mutations in LRP1B were detected in three tumours analysed in this study as well as one tumour analysed by Ramsey et al.49 LRP1B is a member of the LDL receptor family, and its inactivation has been observed in a number of human cancers, including bladder, where it is predicted to act as a tumour suppressor.50,51 Shapiro et al. identified deletions in a region on chromosome 19 containing LRP1B (corresponding to chromosome 13 in humans) as a common copy number aberration in canine bladder cancer that is conserved in human bladder cancer.52 Together, these results suggest that inactivation of LRP1B may be an important feature of cTCC oncogenesis.
Our transcriptomic analysis identified 2566 differentially expressed genes between cTCC and normal bladder tissue, of which 1561 (61%) were identified in at least one prior RNA-Seq analysis of canine bladder cancer.14,48,49 Functional analysis of up-regulated DEGs identified in this study revealed enrichment of cell cycle, DNA repair, and immune-related genes in cTCC tumours. Whereas, genes down-regulated in cTCC were related to focal adhesion, myogenesis, and second messenger signalling. Ramsey et al. performed a comparative analysis of the transcriptomes of canine and human bladder cancer, finding a high level of similarity between the two; whereby, both species exhibit up-regulation of cell cycle and DNA repair machinery and down-regulation of cytoskeletal, cell adhesion, and muscle-related genes.49
Activation of ErbB signalling has been previously reported in cTCC.13,14,48 Here, we identified up-regulation of ERBB2 and ERBB3 by 6- and 7-fold, respectively, in cTCC tumours relative to normal bladder (Table S8). Up-regulation of these genes was also observed in the transcriptomic analyses by Parker et al. and Ramsey et al. ERBB2’s gene product, HER2, is overexpressed in more than half of cTCC tumours.13 Additionally, EGFR and ERBB2 are predicted upstream regulators of overexpressed genes in canine bladder cancer.14 We previously identified up-regulation of EGFR and ERBB2 expression in cTCC cell lines relative to other canine cancer cell lines and demonstrated synergism between combined ErbB receptor inhibition and BRAF or MEK inhibition.35 Activation of the ErbB family of receptors is also common in human muscle-invasive bladder cancer, specifically via mutations in ERBB2/ERBB3 and amplification of EGFR/ERBB2.45
Immune checkpoint inhibitors targeting PD-1 and PD-L1 have been effective in approximately one-quarter of human bladder cancer patients.39,40 One of the major challenges to cancer immunotherapy has been the lack of pre-clinical models that adequately recapitulate the natural process of immunoediting that occurs during tumour development. Canine clinical trials provide the opportunity to evaluate novel immunotherapies in spontaneous tumours in the presence of an intact immune system. A recent study analysed PD-L1 expression across various canine malignant cancers using an antibody with high sensitivity for canine PD-L1 and observed high PD-L1 expression (tumour proportion score of 50% or greater) in 20 out of 20 cTCC samples.53
Four out of 11 cTCC tumours analysed in this study exhibited characteristics of a hot tumour microenvironment (TME-Hot) with high expression of gene markers for CD8+ T cells, pDCs, co-inhibitory/co-stimulatory receptor expression by T cells and APCs, cytolytic activity, MHC class I expression, and type I IFN response. Mariathasan et al. analysed pre-treatment tumour samples from human metastatic bladder cancer patients treated with the PD-L1 inhibitor atezolizumab, identifying PD-L1 expression on immune cells, high TMB, and a CD8 Teff signature as biomarkers of complete response.41 This group also derived a signature representing TGF-β signalling by fibroblasts (F-TBRS), which was associated with lack of response. Powles et al. evaluated similar biomarkers of response to atezolizumab as a neoadjuvant therapy for human bladder cancer, identifying CD8 Teff and F-TBRS signatures as indicators of response and resistance, respectively.54 Neither PD-L1 expression nor TMB correlated with response in the neoadjuvent setting. The TME-Hot cTCC tumours in this study exhibited strong expression of the CD8 Teff gene signature, as well as a similar IFN-γ gene signature associated with complete response to the PD-1 inhibitor pembrolizumab.43,44 These results suggest that TME-Hot tumours exhibit pre-existing CD8+ T cell immunity and may represent a subset of cTCC that would be responsive to checkpoint inhibitor therapy. Three TME-Cold tumours exhibited high expression of the F-TBRS signature, consistent with immunosuppressive TGF-β signalling.41
Additionally, three out of four TME-Hot tumours harbour BRAF mutations. BRAF inhibition in human melanoma increases antigen expression, promotes CD8+ lymphocyte infiltration and function, and decreases expression of immunosuppressive cytokines.55–57 Acquired resistance to BRAF inhibition is often accompanied by loss of antigen expression and CD8+ T cell infiltration, but can be partially reversed with the addition of a MEK inhibitor.56,57 Combining BRAF and/or MEK inhibitors with checkpoint inhibitors has been explored in BRAF mutant melanoma; however, toxicity remains a challenge. A triple combination of BRAF, MEK, and PD-1 inhibition causes increased toxicity relative to either BRAF/MEK combined inhibition or PD-1 blockade alone.58,59 However, this triple combination resulted in increased progression free survival and duration of response relative to combined BRAF/MEK inhibition in a phase II study.59 Genomic analysis of pre- and post-treatment biopsies of patients receiving the triple combination revealed increased expression of CD8 and MHC Class I and II molecules following treatment, as well as increased expression of the same IFN-γ gene signature used in this study, suggesting an enhanced antitumor immune response.44,58 The prevalence of BRAF mutations in cTCC provides the unique opportunity to evaluate novel strategies combining checkpoint inhibition with BRAF and/or MEK inhibition. Overall, this study emphasizes the importance of conducting clinical trials for dogs with TCC, where the evaluation of immunotherapies alone and in combination with MAPK-targeted therapies may benefit canines as well as humans.
Supplementary Material
Funding information
Anschutz Foundation; Colorado State University CVMBS College Research Council; Merial Veterinary Scholars Program; Morris Animal Foundation, Grant/Award Number: D16CA-071; National Institutes of Health, Grant/Award Numbers: K01OD022982, L30TR002126, T35OD015130; Shipley University Chair in Comparative Oncology; University of Colorado Comprehensive Cancer Center Support Grant, Grant/Award Number: P30-CA046934
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
DATA AVAILABILITY STATEMENT
Raw FASTQ sequences from the WES and RNA-Seq analysis were submitted to NCBI’s SRA database. Sequence data for tumor and normal samples can be found under BioProject PRJNA616374 and PRJNA503860, respectively.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015; 15(1):25–41. [DOI] [PubMed] [Google Scholar]
- 3.Gordon I, Paoloni M, Mazcko C, Khanna C. The comparative oncology trials consortium: using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 2009;6(10): e1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeBlanc AK, Mazcko CN. Improving human cancer therapy through the evaluation of pet dogs. Nat Rev Cancer. 2020;20:727–742. [DOI] [PubMed] [Google Scholar]
- 5.Knapp DW, McMillan SK. Tumors of the urinary system. In: Withrow SJ, Page RL, Vail DM, eds. Withrow and MacEwen’s Small Animal Clinical Oncology. 5th ed. St. Louis, MO: Elsevier Saunders; 2013: 572–582. [Google Scholar]
- 6.Knapp DW, Ramos-Vara JA, Moore GE, Dhawan D, Bonney PL, Young KE. Urinary bladder cancer in dogs, a naturally occurring model for cancer biology and drug development. ILAR J. 2014;55(1): 100–118. [DOI] [PubMed] [Google Scholar]
- 7.Fulkerson CM, Knapp DW. Management of transitional cell carcinoma of the urinary bladder in dogs: a review. Veterinary J. 2015;205(2): 217–225. [DOI] [PubMed] [Google Scholar]
- 8.Mutsaers AJ, Widmer WR, Knapp DW. Canine transitional cell carcinoma. J Vet Intern Med. 2003;17(2):136–144. [DOI] [PubMed] [Google Scholar]
- 9.Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171(3): 540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker B, Parker HG, Dhawan D, et al. Homologous mutation to human BRAF V600E is common in naturally occurring canine bladder cancer–evidence for a relevant model system and urine-based diagnostic test. Mol Cancer Res. 2015;13(6):993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mochizuki H, Kennedy K, Shapiro SG, Breen M. BRAF mutations in canine cancers. PLoS One. 2015;10(6):e0129534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millanta F, Impellizeri J, McSherry L, Rocchigiani G, Aurisicchio L, Lubas G. Overexpression of HER-2 via immunohistochemistry in canine urinary bladder transitional cell carcinoma - a marker of malignancy and possible therapeutic target. Vet Comp Oncol. 2018;16 (2):297–300. [DOI] [PubMed] [Google Scholar]
- 14.Maeda S, Tomiyasu H, Tsuboi M, et al. Comprehensive gene expression analysis of canine invasive urothelial bladder carcinoma by RNA-Seq. BMC Cancer. 2018;18(1):472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jalali Nadoushan MR, Taheri T, Jouian N, Zaeri F. Overexpression of HER-2/neu oncogene and transitional cell carcinoma of bladder. Urol J. 2007;4(3):151–154. [PubMed] [Google Scholar]
- 16.Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2): 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhawan D, Hahn NM, Ramos-Vara JA, Knapp DW. Naturally-occurring canine invasive urothelial carcinoma harbors luminal and basal transcriptional subtypes found in human muscle invasive bladder cancer. PLoS Genet. 2018;14(8):e1007571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinfo. 2013;43:11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elvers I, Turner-Maier J, Swofford R, et al. Exome sequencing of lymphomas from three dog breeds reveals somatic mutation patterns reflecting genetic background. Genome Res. 2015;25(11):1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai B, Zhao WM, Tang BX, et al. DoGSD: the dog and wolf genome SNP database. Nucleic Acids Res. 2015;43(Database issue):D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plassais J, Kim J, Davis BW, et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat Commun. 2019;10(1):1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decker B, Davis BW, Rimbault M, et al. Comparison against 186 canid whole-genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Res. 2015;25(11):1646–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2): 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5): 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 (12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32 (18):2847–2849. [DOI] [PubMed] [Google Scholar]
- 32.Sayers EW, Agarwala R, Bolton EE, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2019;47(D1):D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turski ML, Vidwans SJ, Janku F, et al. Genomically driven tumors and Actionability across Histologies: BRAF-mutant cancers as a paradigm. Mol Cancer Ther. 2016;15(4):533–547. [DOI] [PubMed] [Google Scholar]
- 34.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37(24):3183–3199. [DOI] [PubMed] [Google Scholar]
- 35.Cronise KE, Hernandez BG, Gustafson DL, Duval DL. Identifying the ErbB/MAPK signaling Cascade as a therapeutic target in canine bladder cancer. Mol Pharmacol. 2019;96(1):36–46. [DOI] [PubMed] [Google Scholar]
- 36.Wagle MC, Kirouac D, Klijn C, et al. A transcriptional MAPK pathway activity score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis Oncol. 2018;2(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Damrauer JS, Hoadley KA, Chism DD, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014;111(8):3110–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376 (11):1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariathasan S, Turley SJ, Nickles D, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387 (10031):1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm. Phase II Study J Clin Oncol. 2018;36(34):3353–3360. [DOI] [PubMed] [Google Scholar]
- 44.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507 (7492):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;174(4): 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 2019;9(3):329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parker HG, Dhawan D, Harris AC, et al. RNAseq expression patterns of canine invasive urothelial carcinoma reveal two distinct tumor clusters and shared regions of dysregulation with human bladder tumors. BMC Cancer. 2020;20(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramsey SA, Xu T, Goodall C, et al. Cross-species analysis of the canine and human bladder cancer transcriptome and exome. Genes Chromosomes Cancer. 2017;56(4):328–343. [DOI] [PubMed] [Google Scholar]
- 50.Langbein S, Szakacs O, Wilhelm M, et al. Alteration of the LRP1B gene region is associated with high grade of urothelial cancer. Lab Investigation J Tech Method Pathol. 2002;82(5):639–643. [DOI] [PubMed] [Google Scholar]
- 51.Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapiro SG, Raghunath S, Williams C, et al. Canine urothelial carcinoma: genomically aberrant and comparatively relevant. Chromosome Res Int J Mol Supramol Evolution Aspects Chromosome Biol. 2015;23(2): 311–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maekawa N, Konnai S, Nishimura M, et al. PD-L1 immunohistochemistry for canine cancers and clinical benefit of anti-PD-L1 antibody in dogs with pulmonary metastatic oral malignant melanoma. NPJ Precis Oncol. 2021;5(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Powles T, Kockx M, Rodriguez-Vida A, et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat Med. 2019;25(11):1706–1714. [DOI] [PubMed] [Google Scholar]
- 55.Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. [DOI] [PubMed] [Google Scholar]
- 56.Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hugo W, Shi H, Sun L, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162(6):1271–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ribas A, Lawrence D, Atkinson V, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med. 2019;25(6):936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ascierto PA, Ferrucci PF, Fisher R, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019;25(6):941–946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw FASTQ sequences from the WES and RNA-Seq analysis were submitted to NCBI’s SRA database.32 Sequence data for tumour and normal samples can be found under BioProject PRJNA616374 and PRJNA503860, respectively.
Raw FASTQ sequences from the WES and RNA-Seq analysis were submitted to NCBI’s SRA database. Sequence data for tumor and normal samples can be found under BioProject PRJNA616374 and PRJNA503860, respectively.