

Abstract
Variants in the LMNA gene, which encodes for Lamin A/C, are associated with cardiac conduction disease (CCD). We previously reported that Lamin A/C variants p.R545H and p.A287Lfs*193, which were identified in CCD patients, decreased peak I Na in HEK‐293 cells expressing Nav1.5. Decreased peak I Na in the cardiac conduction system could account for patients’ atrioventricular block. We found that serine 22 (Ser 22) phosphorylation of Lamin A/C was decreased in the p.R545H variant and hypothesized that lamin phosphorylation modulated Nav1.5 activity. To test this hypothesis, we assessed Nav1.5 function in HEK‐293 cells co‐transfected with LMNA variants or treated with the small molecule LBL1 (lamin‐binding ligand 1). LBL1 decreased Ser 22 phosphorylation by 65% but did not affect Nav1.5 function. To test the complete loss of phosphorylation, we generated a version of LMNA with serine 22 converted to alanine 22 (S22A‐LMNA); and a version of mutant R545H‐LMNA that mimics phosphorylation via serine 22 to aspartic acid 22 substitution (S22D‐R545H‐LMNA). We found that S22A‐LMNA inhibited Lamin‐mediated activation of peak I Na by 63% and shifted voltage‐dependency of steady‐state inactivation of Nav1.5. Conversely, S22D‐R545H‐LMNA abolished the effects of mutant R545H‐LMNA on voltage‐dependency but not peak I Na. We conclude that Lamin A/C Ser 22 phosphorylation can modulate Nav1.5 function and contributes to the mechanism by which R545H‐LMNA alters Nav1.5 function. The differential impact of complete versus partial loss of Ser 22 phosphorylation suggests a threshold of phosphorylation that is required for full Nav1.5 modulation. This is the first study to link Lamin A/C phosphorylation to Nav1.5 function.
Keywords: cardiac conduction disease, Lamin phosphorylation, Nav1.5
Lamin A/C Ser 22 phosphorylation can modulate Nav1.5 function and contributes to the mechanism by which R545H‐LMNA alters Nav1.5 function in cardiac conduction disease. Abolishing phosphorylation with S22A‐LMNA reduces channel function, but decreasing phosphorylation with the small molecule LBL1 does not, suggesting a threshold of Ser 22 phosphorylation required for Nav1.5 modulation. Mimicking phosphorylation with S22D‐R545H‐LMNA abolished the effects of mutant R545H‐LMNA on voltage‐dependency but not peak I Na.

1. INTRODUCTION
Laminopathies are a broad spectrum of diseases involving cardiac and skeletal anomalies attributed to mutations in the LMNA gene, which encodes for Lamin A/C. These disorders comprise over 12 clinically heterogeneous syndromes (Bertrand et al., 2011). Lamin A/C is a type‐V intermediate filament that forms the nuclear lamina underlying the inner membrane of the nucleus (Houben et al., 2013). LMNA mutations are highly prevalent in patients with cardiac conduction disease (CCD), often associated with eventual dilated cardiomyopathy (DCM; Anselme et al., 2013; Barra et al., 2012; Hermida‐Prieto et al., 2004; Keller et al., 2012; Malek et al., 2011; Mounkes et al., 2005; Olaopa et al., 2018; Saj et al., 2010; Shaw et al., 2002; Zaragoza et al., 2016). A clinical study of familial autosomal DCM found that approximately 33% of patients were positive for LMNA mutations (Arbustini et al., 2002). LMNA patients require implantable cardioverter‐defibrillator or pacemaker therapy to prevent cardiac arrest and sudden death (Anselme et al., 2013; Kumar et al., 2016; Olaopa et al., 2018). The clinical presentations of these patients, including atrioventricular (AV) block and progressive CCD, are similar to those observed in disorders caused by mutations in the SCN5A gene (Olaopa et al., 2018; Wang et al., 2002), which encodes for the cardiac sodium channel (Nav1.5).
Nav1.5 is primarily localized within the sarcolemma, in contrast to the nuclear localization of Lamin A/C. Mutations in SCN5A are a major cause of AV block, sick sinus syndrome, progressive CCD, and eventual DCM leading to sudden death (Amin et al., 2010; Chockalingam et al., 2012; Holst et al., 2010; Lee et al., 2016; Makita, 2009; Samani, Ai, et al., 2009; Samani, Wu, et al., 2009; Shuraih et al., 2007). Due to the similarities between clinical presentations of disorders associated with both LMNA and SCN5A mutations, studies by our group and others have sought to functionally link variants in Lamin A/C to Nav1.5 activity (Liu et al., 2016; Markandeya et al., 2016; Olaopa et al., 2018). Our group reported that two Lamin A/C variants (p.R545H and p.A287Lfs*193), found in patients with CCD, significantly decrease peak sodium current (I Na) and shift the voltage‐dependency of steady‐state inactivation of Nav1.5 (Olaopa et al., 2018). The p.R545H variant is due to a missense point mutation (c.1634G>A); while the p.A287Lfs*193 variant is caused by a single nucleotide deletion (c.859delG) and subsequent frame shift, leading to a premature termination codon (Olaopa et al., 2018).
Although the transcriptional and post‐translational regulation of Lamin A/C within the heart is not fully understood, its phosphorylation has functional importance (Buxboim et al., 2014; Haas & Jost, 1993; Kochin et al., 2014; Mitsuhashi et al., 2010; Swift & Discher, 2014; Torvaldson et al., 2015; Wu et al., 2011). Phosphorylation at serine 22 (Ser 22) plays an important role in cell cycle regulation, nuclear stability, and signaling between the nucleoskeleton and cytoskeletal structures––the LINC (Linker of Nucleoskeleton and Cytoskeleton) complex (Buxboim et al., 2014; Kochin et al., 2014; Osmanagic‐Myers et al., 2015; Swift & Discher, 2014; Torvaldson et al., 2015). Additionally, a p.S22L Lamin A/C variant was identified in a genetic screen of cardiac transplant patients with DCM (Pethig et al., 2005), indicating a clinically relevant role for Ser 22 in cardiac disease. Thus, we sought to determine if Ser 22 is involved in the mechanism by which the p.R545H variant affects Nav1.5 function, with the aim of identifying a potential therapeutic target for CCD patients with similar LMNA mutations. We used a novel small molecule (lamin‐binding ligand, LBL1) that binds Lamin A/C in the N‐terminal region encompassing Ser 22 (Chao et al., 2017; Li et al., 2018), as well as genetic (Ser 22 modification) approaches to modulate Ser 22 phosphorylation and determine its role in Nav1.5 function.
2. MATERIALS AND METHODS
2.1. Cloning and generation of plasmids
The cDNA of wild‐type LMNA (ORF NM_005572) was purchased (OriGene) in a pCMV6‐AC‐GFP (C‐terminus fused) vector backbone. Mutant R545H‐LMNA and phosphorylation LMNA constructs (S22A‐LMNA and S22D‐R545H‐LMNA) were generated using site‐directed mutagenesis kit (Qiagen). We substituted alanine (GCG) for serine (TCG) at position 22 (p.S22A) in wild‐type LMNA to prevent phosphorylation; to mimic phosphorylation in the R545H‐LMNA mutant we substituted aspartic acid (GAC) for serine (TCG) at position 22 (p.S22D) in the R545H‐LMNA plasmid. DNA sequencing and western blot analyses confirmed successful mutagenesis (Figure 1).
FIGURE 1.
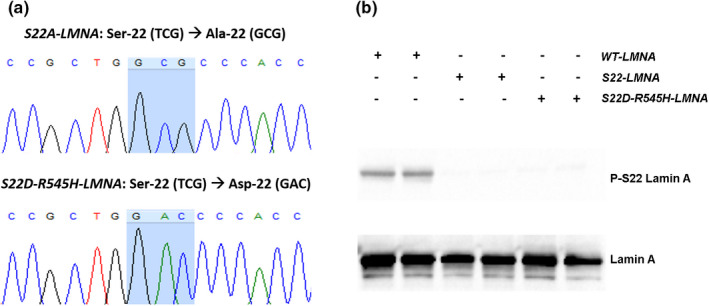
Confirmation of LMNA phosphorylation plasmids. (a) Electropherographs of sequencing results confirming mutagenesis of targeted codon (blue highlight) for each respective LMNA phosphorylation plasmid. (b) Western blot on protein lysates extracted from HEK‐293 cells transfected with each respective LMNA plasmid
2.2. Cell culture and transfection
HEK‐293 cells were grown and maintained in DMEM media (Thermo Fisher), supplemented with 10% fetal bovine serum (ATCC). Cells were serum‐starved overnight and transiently transfected with identical amounts of plasmid DNA using Effectene (Qiagen). Cells were treated 24 hrs post‐transfection with vehicle dimethyl sulfoxide (1% DMSO), 5 or 10 µM LBL1. Cells were harvested 24 hrs post‐treatment or 48 hrs post‐transfection.
2.3. Protein isolation and western blot
Protein was isolated in lysis buffer (in mM): Tris 20, etheylenediaminetetraacetic acid (EDTA) 10, NaCl 100, 1% SDS, 1× complete protease inhibitor (Roche), and 1× phosphatase cocktail inhibitor (Sigma Aldrich). SDS‐PAGE used 4%–15% gradient Tris–HCl polyacrylamide precast gels in 1× Tris/Glycine/SDS running buffer (Bio‐Rad). For high molecular weight proteins, overnight wet transfer was performed (20 V, 16 h); for medium and low molecular weight proteins, fast semi‐dry transfer was performed (25 V, 40 min) using a Novex Semi‐Dry Blotter apparatus (Invitrogen). Transfers were onto nitrocellulose membranes (GE Life Sciences). Primary antibodies : mouse monoclonal anti‐pan‐sodium channel (1:500) and anti‐beta‐tubulin (1:10,000; Sigma‐Aldrich); rabbit anti‐total‐lamin (1:2000), anti‐phospho‐lamin (Ser 22, 1:2000), and anti‐alpha‐actinin 2 (1:2000) (Cell Signaling). Secondary antibodies (Thermo Fisher): goat anti‐mouse‐HRP (1:5000) and goat anti‐rabbit‐HRP (1:5000). Primary antibodies were incubated overnight at 4°C. Secondary antibodies were incubated at room temperature for 1 h, developed using SuperSignal West Pico or Femto (Thermo Fisher), and imaged using ChemiDoc XRS+ (Bio‐Rad). Membranes were stripped with Restore Stripping buffer (Thermo Fisher) at 55°C for 15 min followed by shaking at room temperature for 30 min. Antibody and blocking solutions were prepared in 0.1% Tween‐20 in Tris‐buffered saline solution (in mM): NaCl 150, Tris 10 (pH 7.4). Bands were quantified with ImageJ (NIH).
2.4. Patch clamp experiments
Cells were harvested using trypsin‐EDTA (Thermo Fisher) for 2 min and transferred to the patch chamber for whole‐cell recording of GFP‐labeled cells, which was performed as previously described (Olaopa et al., 2018; Yu et al., 2014). Whole‐cell configuration was made in bath solution (in mM): NaCl 140, KCl 5, CaCl2 1.8, MgCl2 1, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) 5, and Glucose 10 (pH 7.4 adjusted with NaOH). Pipette resistances were 1.5–3 MΩ; solution (in mM): NaF 10, CsF 110, CsCl 20, ethylene glycol tetraacetic acid (EGTA) 10, and HEPES 10 (pH 7.35 adjusted with CsOH). After achieving a giga‐seal, the test pulse current was nulled by adjusting the pipette capacitance compensator with both fast and slow components. After break‐in, the whole‐cell charging transient was nulled by adjusting whole‐cell capacitance and series resistance. Voltage control protocols were generated with Axopatch 200B amplifier/Digidata 1440A or MultiClamp 700A acquisition system using pCLAMP‐10 software (Molecular Devices). Whole‐cell recording was analyzed using Clampfit 10.2 (Molecular Devices). All experiments were carried out at room temperature. Conductance G (V) was calculated by the equation:
where I is the peak current, E rev is the measured reversal potential, and V m is the membrane potential. The normalized peak conductance was plotted as a function of membrane potentials. Steady‐state inactivation was estimated by pre‐pulse protocols (500 ms) from a holding potential of −140 mV. Steady‐state activation and inactivation were fitted with the Boltzmann equation: y = [1 + exp ((V m − V h)/k)] − 1, where y represents variables; V h, midpoint; k, slope factor; and V m, membrane potential.
2.5. Statistical analyses and data availability
The Mann–Whitney–Wilcoxon rank test was performed for patch clamp analyses. One‐way ANOVA test was performed for western blot densitometry. For statistical significance p < 0.05 was used. Data are presented as mean ± SE. The data associated with this manuscript will be made available.
3. RESULTS
3.1. Mutagenesis of Ser 22 residue
We generated LMNA phosphorylation plasmids that either mimic loss of phosphorylation in wild type (S22A‐LMNA) or constitutive phosphorylation in the R545H‐LMNA mutant (S22D‐R545H‐LMNA). We selected the R545H‐LMNA mutant for our study due to the previously described decrease in Ser 22 phosphorylation. To mimic loss of phosphorylation, we genetically substituted alanine (GCG codon) for serine (TCG codon) residue at position 22 (p.S22A); conversely, to mimic phosphorylation, we genetically substituted aspartic acid (GAC codon) for serine (TCG codon) residue at position 22 (p.S22D). We confirmed successful mutagenesis by sequencing and western blot (Figure 1). Cells that expressed either the S22A‐LMNA or S22D‐R545H‐LMNA plasmids appeared negative for Ser 22 phosphorylation, indicating the serine residue had been successfully mutated.
3.2. Ser 22 Lamin phosphorylation is reduced in R545H‐LMNA mutant
We found that Ser 22 Lamin A/C phosphorylation was reduced by 60% in cells expressing R545H‐LMNA compared to wild type (Figure 2a,c). The A287Lfs‐LMNA mutation decreased total Lamin A/C levels but did not impact Ser 22 phosphorylation (Figure 2a,d) and was therefore not a subject of this study. R545H‐LMNA did not alter Nav1.5 whole‐cell protein content (Figure 2b), indicating that the decrease in peak I Na was not due to a change in total channel expression. We did not assess localization of Nav1.5 to the membrane, and cannot rule out a role for decreased cell surface expression of sodium channels contributing to the decrease in peak I Na. Alpha‐actinin 2, which has been shown to modulate Nav1.5 function via direct interaction (Ziane et al., 2010), was likewise unchanged by R545H‐LMNA (Figure 2b).
FIGURE 2.
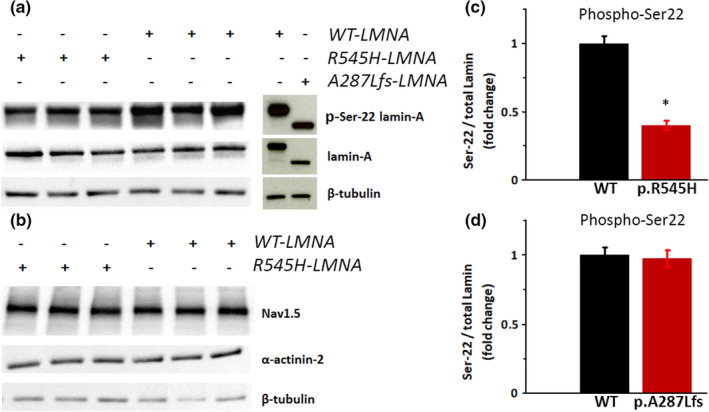
Serine 22 phosphorylation in LMNA‐transfected cells. (a, b) Representative western blots of HEK‐293 cells transfected with the combination of plasmids noted. (c, d) Densitometry showing fold change of Ser 22 phosphorylation in: (c) WT‐LMNA (N = 6) and R545H‐LMNA (N = 6); *p < 0.05. (d) WT‐LMNA (N = 6) and A287Lfs‐LMNA (N = 4); p = N.S. Data are mean ± SE
3.3. Ablation of Ser 22 phosphorylation recapitulates R545H‐LMNA effect on I Na
R545H‐LMNA, which decreased Ser 22 phosphorylation, also decreased peak I Na and shifted voltage‐dependency of steady‐state inactivation rightward, with no effect on steady‐state activation (Olaopa et al., 2018). To determine if Ser 22 Lamin A/C phosphorylation contributed to the regulation of Nav1.5, we assessed peak I Na in cells transfected with wild‐type LMNA or S22A‐LMNA, which cannot be phosphorylated on Ser 22. S22A‐LMNA significantly decreased peak I Na by 60% compared to wild‐type LMNA (S22A: −125 ± 12 pA/pF, N = 10 vs. wild type: −339 ± 45 pA/pF, N = 10, p < 0.0005; Figure 3a,b). Similar to the R545H‐LMNA mutant (Olaopa et al., 2018), S22A‐LMNA also led to a rightward shift of steady‐state of inactivation (S22A: V h = −88.10 ± 0.51 mV, N = 7 vs. wild type: V h = −93.20 ± 0.89 mV, N = 10, p < 0.05) while leaving steady‐state activation unaltered (Figure 4a; Table 1).
FIGURE 3.
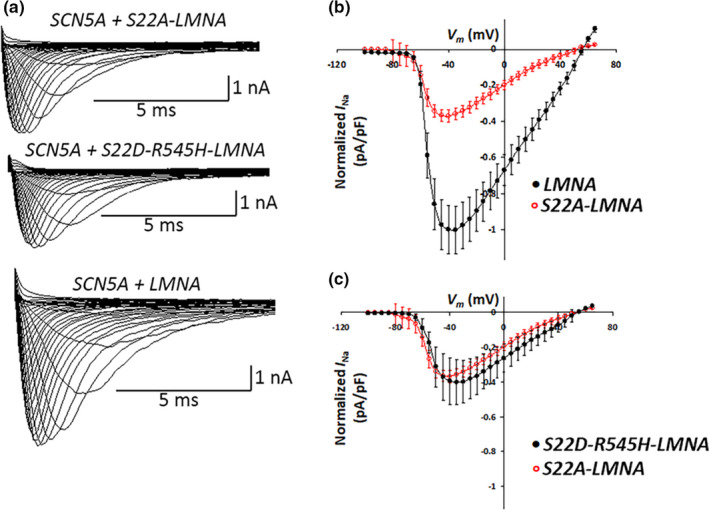
S22A‐LMNA phosphorylation mutant alters peak I Na. (a) Representative superimposed current traces drawn to scale obtained from cells co‐transfected with SCN5A and LMNA variants; step‐pulse protocol (inset). (b) I–V relationship comparing cells co‐transfected with SCN5A + WT‐LMNA (black), N = 10 versus SCN5A + S22A‐LMNA (red), N = 10; p < 0.0005. Peak current for SCN5A + WT‐LMNA = 1. (c) I–V relationship comparing cells co‐transfected with SCN5A + S22A‐LMNA (red), N = 10 versus SCN5A + S22D‐R545H‐LMNA (black), N = 7; p = N.S. Peak current for SCN5A + WT‐LMNA is set at 1
FIGURE 4.
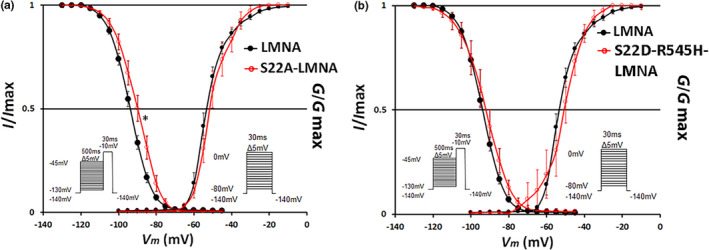
4 S22A‐LMNA phosphorylation mutant alters steady‐state inactivation. Voltage‐dependency of the steady‐state inactivation and activation of cells co‐transfected with SCN5A + WT‐LMNA (black) versus SCN5A + S22A‐LMNA (red, a) or S22D‐R545H‐LMNA (red, b). Inactivation: SCN5A + WT‐LMNA versus SCN5A + S22A‐LMNA p < 0.05. No significant differences in other inactivation or activation curves. Insets show pre‐pulse inactivation (left) and step‐pulse activation (right) protocols. Data are mean ± SE
TABLE 1.
Summary of I Na parameters
| WT | LBL1 (10 µM) | p.S22A | p.S22D‐R545H | |
|---|---|---|---|---|
| I–V relationship | N = 10 | N = 7 | N = 10 | N = 7 |
| Peak I Na (pA/pF) | −339 ± 45 | −319 ± 26 | −125 ± 12*** | −137 ± 44*** |
| Steady‐state inactivation | N = 10 | N = 8 | N = 7 | N = 6 |
| V h (mV) | −93.20 ± 0.89 | −95.91 ± 1.72 | −88.10 ± 0.51* | −92.60 ± 1.41 |
| K | 5.62 ± 0.07 | 7.42 ± 0.17 | 6.84 ± 0.16 | 7.23 ± 0.30 |
| Steady‐state activation | N = 10 | N = 7 | N = 10 | N = 7 |
| V h (mV) | −52.38 ± 0.41 | −55.92 ± 1.23 | −50.21 ± 1.48 | −51.70 ± 1.39 |
| K | 5.21 ± 0.36 | 5.37 ± 0.17 | 5.28 ± 0.22 | 6.62 ± 0.34 |
p < 0.05,
p < 0.0005 (vs. WT).
3.4. Mimicking Ser 22 phosphorylation in R545H‐LMNA partially restores Nav1.5 function
To test if Ser 22 played a role in the mechanism by which R545H‐LMNA decreased peak I Na (Olaopa et al., 2018), we generated a version of R545H‐LMNA that substituted aspartic acid for serine at position 22 (S22D‐R545H‐LMNA) to mimic phosphorylation at that site. We then measured peak I Na in cells transfected with S22D‐R545H‐LMNA. We anticipated that S22D‐R545H‐LMNA would restore peak I Na to the levels seen with wild‐type LMNA. However, a similar loss of peak I Na occurred in cells expressing S22D‐R545H‐LMNA, indicating that it was unable to rescue the R545H‐LMNA effect on peak I Na (S22D‐R545H: −137 ± 44 pA/pF, N = 7 vs. S22A: −125 ± 12 pA/pF, N = 10, p = N.S.; Figure 3a,c). In contrast, S22D‐R545H‐LMNA prevented the shift in the steady‐state inactivation of the voltage‐dependency. Cells transfected with S22D‐R545H‐LMNA were indistinguishable from wild‐type LMNA (Figure 4b; S22D‐R545H: V h = −92.60 ± 1.41 mV, N = 6 vs. wild type: V h = −93.20 ± 0.89 mV, N = 10, p = N.S.). Thus, mimicking Ser 22 phosphorylation partially rescued Nav1.5 function disrupted by the R545H‐LMNA mutation, as normal voltage‐dependency was restored but peak I Na levels were not (Table 1).
3.5. LBL1 inhibits Ser 22 phosphorylation but does not affect Nav1.5 function
LBL1, which binds Lamin A/C in the N‐terminal region encompassing Ser 22, was identified based on its ability to inhibit the growth of cancer cells (Chao et al., 2017; Li et al., 2018). LBL1 inhibits the proliferation of cancer cells at micromolar concentrations, so we treated LMNA‐transfected HEK‐293 cells with 5 and 10 µM LBL1 to determine if that was sufficient to prevent the phosphorylation of Ser 22. Treating LMNA‐transfected cells with 10 µM LBL1 reduced Ser 22 phosphorylation by 65% while 5 µM LBL1 had no effect (Figure 5a–c). To determine the effect of LBL1‐suppressed Lamin A/C phosphorylation on Nav1.5 function, we assessed peak I Na and voltage‐dependency in cells treated with 10 µM LBL1 compared to vehicle. We anticipated that inhibition of Lamin Ser 22 phosphorylation would lead to changes in Nav1.5 function similar to those seen with the R545H‐LMNA or S22A‐LMNA mutations. However, decreasing Ser 22 phosphorylation by LBL1 in wild‐type Lamin did not change peak I Na or steady‐state inactivation or activation (Figure 5d–f; Table 1).
FIGURE 5.
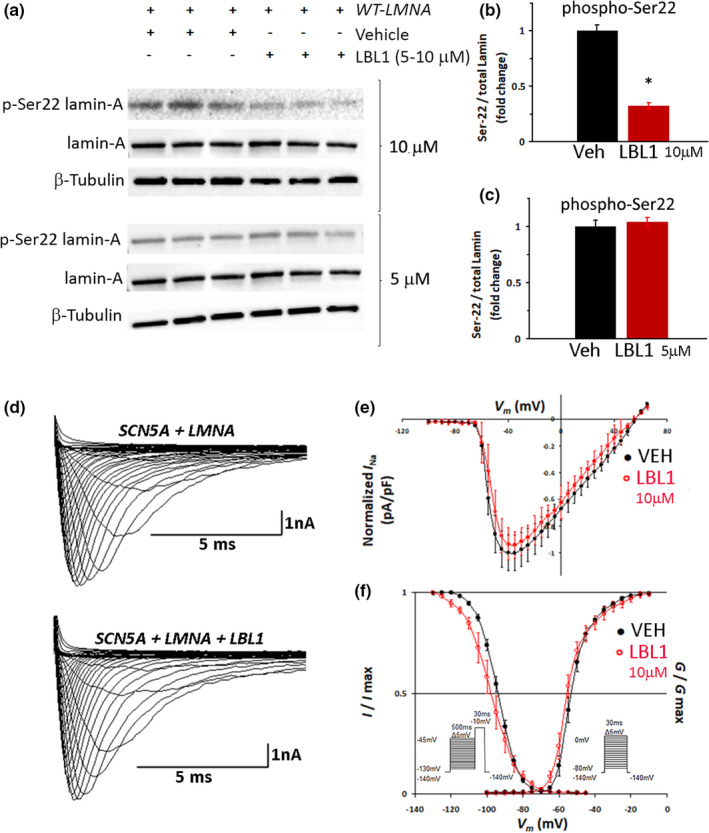
LBL1 decreases Ser 22 Phosphorylation but does not alter I Na . (a) Representative western blot of HEK‐293 cells transfected with WT‐LMNA and treated with 10 µM LBL1 (top) or 5 µM LBL1 (bottom). (b, c) Quantification of Ser 22 phosphorylation in cells treated with vehicle, 10 µM (b) or 5 µM (c) LBL1 (red bars). N = 6; *p < 0.05. Ser 22 phosphorylation in vehicle‐treated cells was set at 1. (d) Representative superimposed current traces obtained from respective cells. (e) I–V relationship of cells co‐transfected with SCN5A + WT‐LMNA; vehicle (black), N = 10 versus 10 µM LBL1 (red), N = 7. Peak current for vehicle is set at 1. (f) Voltage‐dependency of the steady‐state inactivation and activation of cells co‐transfected with SCN5A + WT‐LMNA. Inactivation: vehicle (black), N = 10 versus 10 µM LBL1 (red), N = 8; p = N.S. Activation: vehicle (black), N = 10 versus 10 µM LBL1 (red), N = 7; p = N.S. Insets show pre‐pulse inactivation (left) and step‐pulse activation (right) protocols. Data are mean ± SE
4. DISCUSSION
The goal of this study was to understand the effect of Lamin A/C Ser 22 phosphorylation on Nav1.5 function, with the aim of identifying a novel therapeutic target for CCD patients with LMNA mutations. Our findings indicate that Ser 22 phosphorylation plays a role in Lamin‐mediated Nav1.5 modulation and can alter peak I Na and voltage‐dependency of the steady‐state inactivation. However, there appears to be a threshold level of phosphorylation necessary in wild‐type Lamin A/C. Partial loss of Ser 22 phosphorylation via the lamin‐binding small molecule LBL1 did not affect Nav1.5 function, but complete loss of Ser 22 phosphorylation via genetic substitution had significant effects on channel function that were consistent with the R545H‐LMNA variant (Table 1).
Another possible explanation for the discrepancy between the effect of partial and complete loss of Ser 22 phosphorylation is that other Lamin A/C phosphorylation sites, in addition to Ser 22, may play a cumulative role in modulating Nav1.5. This alternative interpretation is supported by our findings, which show that mimicking constitutive Ser 22 phosphorylation in the R545H‐LMNA mutant only partially restored Nav1.5 function. This suggests that there are other factors and/or phosphorylation sites that also play a role in governing the effect of R545H‐LMNA or other LMNA mutations on sodium channel function or cell surface expression. To explore this possibility, future studies will be focused on assessing the role of additional Lamin A/C phosphorylation sites and developing chemical and genetic tools that can specifically identify and modulate these sites.
Lamin A/C is an intermediate filament organized into a tripartite structure: a non‐helical N‐terminal head domain encompassing the Ser 22 phosphorylation site; an α‐helical coiled‐coil rod domain encompassing the 287 residue; and an immunoglobulin fold (Ig‐fold) domain at the C‐terminal end encompassing the 545 residue (Ho & Lammerding, 2012; Osmanagic‐Myers et al., 2015). The Ig‐fold domain, which spans residues 430–545, is believed to be involved in Lamin A/C’s protein–protein functional interactions (Dhe‐Paganon et al., 2002; Krimm et al., 2002; Shumaker et al., 2008), including with cytoskeletal linkers that provide the machinery by which sarcolemma proteins like Nav1.5 might be modulated (Markandeya et al., 2016; Olaopa et al., 2018; Stroud et al., 2014). Consistent with this, both the R545H and A287Lfs mutations alter the Ig‐fold domain. R545H is a point mutation at the end of the Ig‐fold, a location that has been implicated in other laminopathies including DCM and CCD with loss of peak INa (Chan et al., 2016; Liu et al., 2016; Malek et al., 2011; Saj et al., 2010). The A287Lfs frame shift mutation alters the sequence from A287 in the rod domain to the premature stop codon in the Ig‐fold. In contrast, LBI1 binds Lamin A/C in the N‐terminal region encompassing Ser 22 (Chao et al., 2017; Li et al., 2018).
Dimerization of Lamin A/C is driven by coiled‐coil formation of its central rod domains (Ho & Lammerding, 2012) (Dhe‐Paganon et al., 2002; Krimm et al., 2002; Stuurman et al., 1998). Lamin A/C dimers assemble head‐to‐tail into polar polymers, which require an overlapping interaction between the head and tail domains (Heitlinger et al., 1992; Sasse et al., 1998). These polymers then laterally assemble in an anti‐parallel fashion into nonpolar filaments (Ben‐Harush et al., 2009). These reports, in addition to our findings, lead us to propose a structural paradigm by which the R545H mutation could affect Ser 22 phosphorylation via antiparallel head‐to‐tail interaction with neighboring dimers (graphical abstract). In the case of the A287Lfs mutant, which does not result in loss of Ser 22 phosphorylation levels, this head‐to‐tail interaction is likely abolished due to the frame shift and subsequent truncation of this region of the protein. This truncation could explain why modulation of Ser 22 phosphorylation level does not occur in the A287Lfs mutant. The complex structure of lamin polymers may also explain why a twofold increase in LBL1 concentration induced a significant loss of Ser 22 phosphorylation. Experimental approaches aimed at exploring these possibilities, while important, are outside the scope and focus of this study.
From a clinical perspective, our results suggest that Lamin A/C phosphorylation may be a potential therapeutic target for patients with specific LMNA mutations and CCD. Small molecules that enhance, rather than block, Ser 22 phosphorylation might partially restore Nav1.5 function in disease caused by R545H and similar LMNA mutations. This would mimic the partial rescue of Nav1.5 function we observed in cells expressing the S22D‐R545H‐LMNA variant. It could also help to improve diagnosis and prognosis in patients with similar LMNA mutations by developing tools that could assess their Lamin A/C phosphorylation state.
In summary, our data indicate that Lamin A/C Ser 22 phosphorylation modulates Nav1.5 function. This phosphorylation appears to be part of the mechanism by which the R545H‐LMNA mutation affects Nav1.5 function. To our knowledge, this is the first study to link Lamin A/C phosphorylation and Nav1.5 function.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Michael A. Olaopa performed the research. Michael A. Olaopa and Tomohiko Ai performed the analyses. Michael A. Olaopa, Tomohiko Ai, Matteo Vatta, and Beth A. Habecker contributed to conceptualization and design of the study. Bo Chao, Xiangshu Xiao, and Beth A. Habecker contributed analytic tools and resources. Michael A. Olaopa wrote the manuscript with significant contributions from all co‐authors. All authors reviewed and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Peng‐Sheng Chen, MD (Indiana University) for his logistical support; and Dr. Mark Doyle, PhD (Oregon Health & Science University) for his technical assistance. This study was supported, in part, by NIH HL093056 (B.A.H); the Methodist Research Institute, Showalter Cardiovascular Research Fund (T.A.); the IU Health–IU School of Medicine Strategic Research Initiative (T.A. and M.V.); and by the Dr. Charles Fisch Cardiovascular Research Award endowed by Dr. Suzanne B. Knoebel (M.A.O.); and NIH CA211866 (X.X.).
Olaopa, M. A. , Ai, T. , Chao, B. , Xiao, X. , Vatta, M. , & Habecker, B. A. (2021). Phosphorylation of Lamin‐A/C at serine‐22 modulates Nav1.5 function. Physiological Reports, 9, e15121. 10.14814/phy2.15121
REFERENCES
- Amin, A. S. , Asghari‐Roodsari, A. , & Tan, H. L. (2010). Cardiac sodium channelopathies. Pflugers Archiv. European Journal of Physiology, 460, 223–237. 10.1007/s00424-009-0761-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselme, F. , Moubarak, G. , Savoure, A. , Godin, B. , Borz, B. , Drouin‐Garraud, V. , & Gay, A. (2013). Implantable cardioverter‐defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 10, 1492–1498. 10.1016/j.hrthm.2013.06.020 [DOI] [PubMed] [Google Scholar]
- Arbustini, E. , Pilotto, A. , Repetto, A. , Grasso, M. , Negri, A. , Diegoli, M. , Campana, C. , Scelsi, L. , Baldini, E. , Gavazzi, A. , & Tavazzi, L. (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect‐related disease. Journal of the American College of Cardiology, 39, 981–990. 10.1016/S0735-1097(02)01724-2 [DOI] [PubMed] [Google Scholar]
- Barra, S. N. , Providencia, R. , Paiva, L. , Nascimento, J. , & Marques, A. L. (2012). A review on advanced atrioventricular block in young or middle‐aged adults. Pacing and Clinical Electrophysiology, 35, 1395–1405. 10.1111/j.1540-8159.2012.03489.x [DOI] [PubMed] [Google Scholar]
- Ben‐Harush, K. , Wiesel, N. , Frenkiel‐Krispin, D. , Moeller, D. , Soreq, E. , Aebi, U. , Herrmann, H. , Gruenbaum, Y. , & Medalia, O. (2009). The supramolecular organization of the C. elegans nuclear lamin filament. Journal of Molecular Biology, 386(5), 1392–1402. 10.1016/j.jmb.2008.12.024 [DOI] [PubMed] [Google Scholar]
- Bertrand, A. T. , Chikhaoui, K. , Yaou, R. B. , & Bonne, G. (2011). Clinical and genetic heterogeneity in laminopathies. Biochemical Society Transactions, 39, 1687–1692. 10.1042/BST20110670 [DOI] [PubMed] [Google Scholar]
- Buxboim, A. , Swift, J. , Irianto, J. , Spinler, K. R. , Dingal, P. C. , Athirasala, A. , Kao, Y. R. , Cho, S. , Harada, T. , Shin, J. W. , & Discher, D. E. (2014). Matrix elasticity regulates lamin‐A, C phosphorylation and turnover with feedback to actomyosin. Current Biology, 24, 1909–1917. 10.1016/j.cub.2014.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, D. , McIntyre, A. D. , Hegele, R. A. , & Don‐Wauchope, A. C. (2016). Familial partial lipodystrophy presenting as metabolic syndrome. Journal of Clinical Lipidology, 10, 1488–1491. 10.1016/j.jacl.2016.08.012 [DOI] [PubMed] [Google Scholar]
- Chao, B. , Li, B. X. , & Xiao, X. (2017). Design, synthesis and evaluation of antitumor acylated monoaminopyrroloquinazolines. Bioorganic & Medicinal Chemistry Letters, 27, 3107–3110. 10.1016/j.bmcl.2017.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chockalingam, P. , Clur, S. A. , Breur, J. M. , Kriebel, T. , Paul, T. , Rammeloo, L. A. , Wilde, A. A. , & Blom, N. A. (2012). The diagnostic and therapeutic aspects of loss‐of‐function cardiac sodium channelopathies in children. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 9, 1986–1992. 10.1016/j.hrthm.2012.08.011 [DOI] [PubMed] [Google Scholar]
- Dhe‐Paganon, S. , Werner, E. D. , Chi, Y. I. , & Shoelson, S. E. (2002). Structure of the globular tail of nuclear lamin. Journal of Biological Chemistry, 277, 17381–17384. 10.1074/jbc.C200038200 [DOI] [PubMed] [Google Scholar]
- Haas, M. , & Jost, E. (1993). Functional analysis of phosphorylation sites in human lamin A controlling lamin disassembly, nuclear transport and assembly. European Journal of Cell Biology, 62, 237–247. [PubMed] [Google Scholar]
- Heitlinger, E. , Peter, M. , Lustig, A. , Villiger, W. , Nigg, E. A. , & Aebi, U. (1992). The role of the head and tail domain in lamin structure and assembly: Analysis of bacterially expressed chicken lamin A and truncated B2 lamins. Journal of Structural Biology, 108, 74–89. [DOI] [PubMed] [Google Scholar]
- Hermida‐Prieto, M. , Monserrat, L. , Castro‐Beiras, A. , Laredo, R. , Soler, R. , Peteiro, J. , Rodriguez, E. , Bouzas, B. , Alvarez, N. , Muniz, J. , & Crespo‐Leiro, M. (2004). Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. The American Journal of Cardiology, 94, 50–54. 10.1016/j.amjcard.2004.03.029 [DOI] [PubMed] [Google Scholar]
- Ho, C. Y. , & Lammerding, J. (2012). Lamins at a glance. Journal of Cell Science, 125, 2087–2093. 10.1242/jcs.087288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst, A. G. , Liang, B. , Jespersen, T. , Bundgaard, H. , Haunso, S. , Svendsen, J. H. , & Tfelt‐Hansen, J. (2010). Sick sinus syndrome, progressive cardiac conduction disease, atrial flutter and ventricular tachycardia caused by a novel SCN5A mutation. Cardiology, 115, 311–316. 10.1159/000312747 [DOI] [PubMed] [Google Scholar]
- Houben, F. , De Vos, W. H. , Krapels, I. P. , Coorens, M. , Kierkels, G. J. , Kamps, M. A. , Verstraeten, V. L. , Marcelis, C. L. , van den Wijngaard, A. , Ramaekers, F. C. , & Broers, J. L. (2013). Cytoplasmic localization of PML particles in laminopathies. Histochemistry and Cell Biology, 139, 119–134. 10.1007/s00418-012-1005-5 [DOI] [PubMed] [Google Scholar]
- Keller, H. , Finsterer, J. , Steger, C. , Wexberg, P. , Gatterer, E. , Khazen, C. , Stix, G. , Gerull, B. , Hoftberger, R. , & Weidinger, F. (2012). Novel c.367_369del LMNA mutation manifesting as severe arrhythmias, dilated cardiomyopathy, and myopathy. Heart and Lung, 41, 382–386. 10.1016/j.hrtlng.2011.07.007 [DOI] [PubMed] [Google Scholar]
- Kochin, V. , Shimi, T. , Torvaldson, E. , Adam, S. A. , Goldman, A. , Pack, C. G. , Melo‐Cardenas, J. , Imanishi, S. Y. , Goldman, R. D. , & Eriksson, J. E. (2014). Interphase phosphorylation of lamin A. Journal of Cell Science, 127, 2683–2696. 10.1242/jcs.141820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimm, I. , Ostlund, C. , Gilquin, B. , Couprie, J. , Hossenlopp, P. , Mornon, J. P. , Bonne, G. , Courvalin, J. C. , Worman, H. J. , & Zinn‐Justin, S. (2002). The Ig‐like structure of the C‐terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure, 10, 811–823. 10.1016/S0969-2126(02)00777-3 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Baldinger, S. H. , Gandjbakhch, E. , Maury, P. , Sellal, J. M. , Androulakis, A. F. , Waintraub, X. , Charron, P. , Rollin, A. , Richard, P. , Stevenson, W. G. , Macintyre, C. J. , Ho, C. Y. , Thompson, T. , Vohra, J. K. , Kalman, J. M. , Zeppenfeld, K. , Sacher, F. , Tedrow, U. B. , & Lakdawala, N. K. (2016). Long‐term arrhythmic and nonarrhythmic outcomes of Lamin A/C mutation carriers. Journal of the American College of Cardiology, 68, 2299–2307. 10.1016/j.jacc.2016.08.058 [DOI] [PubMed] [Google Scholar]
- Lee, Y. S. , Olaopa, M. A. , Jung, B. C. , Lee, S. H. , Shin, D. G. , Park, H. S. , Cho, Y. , Han, S. M. , Lee, M. H. , & Kim, Y. N. (2016). Genetic variation of SCN5A in Korean patients with sick sinus syndrome. Korean Circulation Journal, 46, 63–71. 10.4070/kcj.2016.46.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. X. , Chen, J. , Chao, B. , David, L. L. , & Xiao, X. (2018). Anticancer pyrroloquinazoline LBL1 targets nuclear lamins. ACS Chemical Biology. 10.1021/acschembio.8b00266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Shan, H. , Huang, J. , Li, N. , Hou, C. , & Pu, J. (2016). A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin‐like fold associated with left ventricular non‐compaction. Europace, 18, 617–622. 10.1093/europace/euv044 [DOI] [PubMed] [Google Scholar]
- Makita, N. (2009). Phenotypic overlap of cardiac sodium channelopathies: Individual‐specific or mutation‐specific? Circulation Journal, 73, 810–817. [DOI] [PubMed] [Google Scholar]
- Malek, L. A. , Labib, S. , Mazurkiewicz, L. , Saj, M. , Ploski, R. , Tesson, F. , & Bilinska, Z. T. (2011). A new c.1621 C > G, p. R541G lamin A/C mutation in a family with DCM and regional wall motion abnormalities (akinesis/dyskinesis): Genotype‐phenotype correlation. Journal of Human Genetics, 56, 83–86. 10.1038/jhg.2010.137 [DOI] [PubMed] [Google Scholar]
- Markandeya, Y. S. , Tsubouchi, T. , Hacker, T. A. , Wolff, M. R. , Belardinelli, L. , & Balijepalli, R. C. (2016). Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA‐N195K mutation. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 13, 2228–2236. 10.1016/j.hrthm.2016.08.007 [DOI] [PubMed] [Google Scholar]
- Mitsuhashi, H. , Hayashi, Y. K. , Matsuda, C. , Noguchi, S. , Wakatsuki, S. , Araki, T. , & Nishino, I. (2010). Specific phosphorylation of Ser458 of A‐type lamins in LMNA‐associated myopathy patients. Journal of Cell Science, 123, 3893–3900. 10.1242/jcs.072157 [DOI] [PubMed] [Google Scholar]
- Mounkes, L. C. , Kozlov, S. V. , Rottman, J. N. , & Stewart, C. L. (2005). Expression of an LMNA‐N195K variant of A‐type lamins results in cardiac conduction defects and death in mice. Human Molecular Genetics, 14, 2167–2180. 10.1093/hmg/ddi221 [DOI] [PubMed] [Google Scholar]
- Olaopa, M. A. , Spoonamore, K. G. , Bhakta, D. , Chen, Z. , Celestino‐Soper, P. B. S. , Chen, P. S. , Ai, T. , & Vatta, M. (2018). Lamin A/C variants found in patients with cardiac conduction disease reduce sodium currents. Cardiogenetics, 8, 4–8. 10.4081/cardiogenetics.2018.7127 [DOI] [Google Scholar]
- Osmanagic‐Myers, S. , Dechat, T. , & Foisner, R. (2015). Lamins at the crossroads of mechanosignaling. Genes & Development, 29, 225–237. 10.1101/gad.255968.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethig, K. , Genschel, J. , Peters, T. , Wilhelmi, M. , Flemming, P. , Lochs, H. , Haverich, A. , & Schmidt, H. H. (2005). LMNA mutations in cardiac transplant recipients. Cardiology, 103, 57–62. 10.1159/000082048 [DOI] [PubMed] [Google Scholar]
- Saj, M. , Jankowska, A. , Lewandowski, M. , Szwed, H. , Szperl, M. , Ploski, R. , & Bilinska, Z. T. (2010). Dilated cardiomyopathy with profound segmental wall motion abnormalities and ventricular arrhythmia caused by the R541C mutation in the LMNA gene. International Journal of Cardiology, 144, e51–53. 10.1016/j.ijcard.2008.12.083 [DOI] [PubMed] [Google Scholar]
- Samani, K. , Ai, T. , Towbin, J. A. , Brugada, R. , Shuraih, M. , Xi, Y. , Wu, G. , Cheng, J. , & Vatta, M. (2009). A nonsense SCN5A mutation associated with Brugada‐type electrocardiogram and intraventricular conduction defects. Pacing and Clinical Electrophysiology, 32, 1231–1236. 10.1111/j.1540-8159.2009.02470.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samani, K. , Wu, G. , Ai, T. , Shuraih, M. , Mathuria, N. S. , Li, Z. , Sohma, Y. , Purevjav, E. , Xi, Y. , Towbin, J. A. , Cheng, J. , & Vatta, M. (2009). A novel SCN5A mutation V1340I in Brugada syndrome augmenting arrhythmias during febrile illness. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 6, 1318–1326. 10.1016/j.hrthm.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse, B. , Aebi, U. , & Stuurman, N. (1998). A tailless Drosophila lamin Dm0 fragment reveals lateral associations of dimers. Journal of Structural Biology, 123, 56–66. 10.1006/jsbi.1998.4006 [DOI] [PubMed] [Google Scholar]
- Shaw, T. , Elliott, P. , & McKenna, W. J. (2002). Dilated cardiomyopathy: A genetically heterogeneous disease. Lancet, 360, 654–655. 10.1016/S0140-6736(02)09879-3 [DOI] [PubMed] [Google Scholar]
- Shumaker, D. K. , Solimando, L. , Sengupta, K. , Shimi, T. , Adam, S. A. , Grunwald, A. , Strelkov, S. V. , Aebi, U. , Cardoso, M. C. , & Goldman, R. D. (2008). The highly conserved nuclear lamin Ig‐fold binds to PCNA: Its role in DNA replication. Journal of Cell Biology, 181, 269–280. 10.1083/jcb.200708155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuraih, M. , Ai, T. , Vatta, M. , Sohma, Y. , Merkle, E. M. , Taylor, E. , Li, Z. , Xi, Y. , Razavi, M. , Towbin, J. A. , & Cheng, J. (2007). A common SCN5A variant alters the responsiveness of human sodium channels to class I antiarrhythmic agents. Journal of Cardiovascular Electrophysiology, 18, 434–440. 10.1111/j.1540-8167.2007.00777.x [DOI] [PubMed] [Google Scholar]
- Stroud, M. J. , Banerjee, I. , Veevers, J. , & Chen, J. (2014). Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circulation Research, 114, 538–548. 10.1161/CIRCRESAHA.114.301236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuurman, N. , Heins, S. , & Aebi, U. (1998). Nuclear lamins: Their structure, assembly, and interactions. Journal of Structural Biology, 122, 42–66. 10.1006/jsbi.1998.3987 [DOI] [PubMed] [Google Scholar]
- Swift, J. , & Discher, D. E. (2014). The nuclear lamina is mechano‐responsive to ECM elasticity in mature tissue. Journal of Cell Science, 127, 3005–3015. 10.1242/jcs.149203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torvaldson, E. , Kochin, V. , & Eriksson, J. E. (2015). Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus, 6, 166–171. 10.1080/19491034.2015.1017167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. W. , Viswanathan, P. C. , Balser, J. R. , George Jr., A. L. , & Benson, D. W. (2002). Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation, 105, 341–346. [DOI] [PubMed] [Google Scholar]
- Wu, W. , Muchir, A. , Shan, J. , Bonne, G. , & Worman, H. J. (2011). Mitogen‐activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation, 123, 53–61. 10.1161/CIRCULATIONAHA.110.970673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, C. C. , Ai, T. , Weiss, J. N. , & Chen, P. S. (2014). Apamin does not inhibit human cardiac Na+ current, L‐type Ca2+ current or other major K+ currents. PLoS One, 9, e96691. 10.1371/journal.pone.0096691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza, M. V. , Fung, L. , Jensen, E. , Oh, F. , Cung, K. , McCarthy, L. A. , Tran, C. K. , Hoang, V. , Hakim, S. A. , & Grosberg, A. (2016). Exome sequencing identifies a novel LMNA splice‐site mutation and multigenic heterozygosity of potential modifiers in a family with sick sinus syndrome, dilated cardiomyopathy, and sudden cardiac death. PLoS One, 11, e0155421. 10.1371/journal.pone.0155421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziane, R. , Huang, H. , Moghadaszadeh, B. , Beggs, A. H. , Levesque, G. , & Chahine, M. (2010). Cell membrane expression of cardiac sodium channel Na(v)1.5 is modulated by alpha‐actinin‐2 interaction. Biochemistry, 49, 166–178. 10.1021/bi901086v [DOI] [PMC free article] [PubMed] [Google Scholar]