

Abstract
α1-Antitrypsin deficiency (AATD) has been historically under-recognised and under-diagnosed; recently it has begun to receive greater interest in terms of attempts at deeper elucidation of pathology and treatment options. However, the concept of disease phenotypes within AATD (emphysema, chronic bronchitis, bronchiectasis or a combination of phenotypes) has not been proposed or studied. Of the three neutrophil serine proteases, neutrophil elastase was historically believed to be the sole contributor to disease pathology in AATD. Recently, Proteinase-3 has been increasingly studied as an equal, if not greater, contributor to the disease process. Cathepsin G, however, has not been extensively evaluated in this area. Matrix metalloproteinases have also been mentioned in the pathogenesis of AATD but have not been widely explored. This article considers the available evidence for differential protease activity in patients with AATD, including the contribution to distinct phenotypes of the disease. Owing to limited literature in this area, extrapolations from studies of other chronic lung diseases with similar phenotypes, including COPD and bronchiectasis, have been made. We consider a new framework of understanding defined by protease-driven endotypes of disease which may lead to new opportunities for precision medicine.
Short abstract
α1-antitrypsin deficiency is a heterogeneous disease driven by aberrant protease activity in the presence of low α1-AT levels. Understanding mechanisms driving pathological processes in α1-ATD phenotypes could identify novel therapeutic targets. https://bit.ly/2XeILCp
Introduction
α-1 antitrypsin deficiency (AATD) is an autosomal co-dominant condition initially described by Laurell and Eriksson in 1963 [1]. When manifested in the lungs, it is best described as an inherited form of COPD, mainly causing airflow obstruction and emphysema in susceptible individuals [2]. The α1-antitrypsin (AAT) protein is encoded by the SERPINA1 (serine protease inhibitor group A, member 1) gene [3] located on the long arm of chromosome 14q32.1, and codes for a 52-kDa protein consisting of 394 amino acids. This gene, also known as the protease inhibitor (PI) locus, has ∼123 single nucleotide polymorphisms listed so far. The PI phenotype is identified by the speed of migration of AAT protein variants on gel electrophoresis [4]. The different phenotypes of AATD are each denoted by a letter of the English alphabet: M allele, which is the normal allele, results in a protein with a medium rate of migration, the S allele results in a slower rate of migration, and the most severe form of the disease, resulting from the Z allele, has the slowest rate of migration. Null alleles result in no detectable protein and therefore no movement on gel electrophoresis. Consequently, individuals may have varying combinations of the above alleles or other rare alleles, with PiMM being the normal phenotype/genotype. The S mutation is caused by substitution of glutamic acid for valine at position 264. Substitution of glutamic acid for lysine at position 342 results in the Z mutation [5]. The ZZ homozygous state is the most common variant that results in disease manifestation. Direct DNA sequence analysis allows identification of variants not detected by protein electrophoresis. These techniques are able to identify >75 AAT gene alleles [6].
AATD is not uncommon in Europe and exhibits regional variance. It is believed that the ZZ allele probably arose in northern Europe [7], and is particularly prevalent in southern Scandinavia, Denmark, the Netherlands, northern France and the UK. The prevalence gradually declines towards south-east Europe, with the lowest prevalence in eastern Europe [8]. Overall, it is estimated that 3–4% of northern Europeans carry the Z allele, with a carrier frequency of 1 in 25 [9]. The prevalence of AATD ZZ homozygosity in the whole of Europe is estimated to be 1 in 2000 [10]. A population study of blood donors from the St Louis area of the US identified 1 in 5000 prevalence in this area [11]. The high prevalence and carrier frequency of AATD is noteworthy as the carrier frequency of cystic fibrosis (CF), a well-known chronic lung disease, is also 1 in 25 in the Caucasian population, with a disease prevalence of 1 in 2500 in Europe and the US. One important difference between the two conditions, however, is that unlike CF, AATD does not always exhibit full penetrance, which means that some patients with AATD develop no lung disease and remain well throughout life, whereas others develop markedly severe disease. Importantly, with the exception of smoking, factors that determine whether or not a patient with genetically inherited AATD will develop clinically evident lung disease is, as yet, largely unknown.
AAT is produced mainly in the liver and transported via the circulation and lymphatic system to the lungs, where it is involved in protection against inflammatory damage. AAT is also produced in small amounts in pulmonary alveolar cells, neutrophils and macrophages, and provides some local lung protection [10]. Common deficiency alleles such as S and Z mutations do not affect protein synthesis; rather they cause a large amount of protein to be degraded within hepatocytes, resulting in a reduction in protein secretion (∼15% of normal in the case of Z mutation). It is currently not known whether or not locally produced mutant AATD undergoes degradation similar to that in hepatocytes, but it is known that AATD has the potential to cause polymerisation of mutant AAT molecules within hepatocytes as well as in the lungs. AAT polymer accumulation in the liver has been established to be the cause of liver disease in AATD, which manifests as chronic hepatitis, cirrhosis, and increased risk of hepatocellular carcinoma [5, 12, 13]. Polymer accumulation in the lungs has also been demonstrated to be pro-inflammatory with chemotactic properties and resultant neutrophil recruitment, but whether this phenomenon directly contributes to lung disease has not been studied.
Pulmonary disease in AATD
The commonest clinical manifestation of AATD is emphysema, defined as enlargement of airspaces distal to terminal bronchioles and destruction of alveolar walls. AATD is the most widely recognised genetic cause of emphysema and characteristically results in a basal panlobular distribution of disease, unlike the upper lobe-predominant centrilobular emphysema associated with smoking-related lung disease. Inflammatory stimuli such as cigarette smoke, air pollution and recurrent bacterial colonisation are all factors known to accelerate disease progression. It has been demonstrated that cigarette smoke inactivates AAT through oxidisation of methionine residues on the active site of the AAT molecule and promotes its polymerisation [14–16], although it is important to note that these effects have only been demonstrated in vitro and that there are no clinical studies to support this concept. It has also been demonstrated that some never-smoker with AATD develop severe lung disease [17, 18]. Factors that determine whether a never-smoker will develop lung disease are not clearly understood.
Patients with AATD also show typical features of airflow obstruction characteristic of COPD. In usual COPD, airflow obstruction involves a complex interplay of inflammatory pathways including neutrophil and macrophage infiltration [19]. A study investigating factors involved in development of COPD in patients with AATD [20] found that cigarette smoking, chronic bronchitis and pneumonia were all risk factors for development of severe airflow obstruction. However, the exact cellular and molecular mechanisms involved in the development of airflow obstruction in AATD are incompletely understood.
Stockley et al. [21] showed correlation between microbial load and markers of inflammation in patients with AATD and usual COPD. In a study of AATD patients compared to usual COPD [22], total bacterial load, including Streptococcus pneumoniae and Moraxella catarrhalis, was reported to be lower in AATD patients. Interestingly, however, the relative proportion of isolation of Pseudomonas aeruginosa, Haemophilus influenzae and Staphylococcus aureus was not significantly different across the groups. As inflammation is directly related to progression of lung disease, it is probable that microbial load has a significant contribution to disease pathology and progression in patients with AATD. The role of the pulmonary microbiome in patients with AATD requires further exploration.
Bronchiectasis, defined as permanent dilatation and scarring of bronchial and bronchiolar walls, has been attributed to lung damage resulting from AATD [23]. Cuvelier et al. [6] reported no difference in distribution of PI alleles in patients with bronchiectasis compared to their control group; however the investigators found that the presence of emphysema in bronchiectatic patients was associated with an abnormal AAT allele distribution. Parr et al. [24] assessed 74 patients with AATD and observed clinically significant bronchiectasis in 27% of the analysed group. Interestingly, the investigators also observed that the distribution of bronchiectasis mirrored that of emphysema, with lower lobe-predominant disease in study subjects. It is plausible that the pathogenic processes involved in bronchiectasis and emphysema are inter-related, although this relationship is not yet clearly understood. One explanation offered by the above authors as to the presence of bronchiectasis in patients with AATD is that bronchiectasis may be a consequence of emphysema in patients with AATD, as Cuvelier et al. reported that patients with bronchiectasis without emphysema did not have an abnormal AAT distribution, as opposed to bronchiectatic patients with emphysema. However, isolated case reports of AATD have also documented bronchiectasis in the absence of emphysema [25, 26], suggesting that the pathways involved in the pathogenesis of bronchiectasis in AATD are not straightforward.
Based on the above evidence, it is becoming increasingly clear that AATD is not a single entity and that there are distinct phenotypes of disease, albeit with some overlap. Factors that determine what phenotype will be predominant in a particular patient are not currently understood, and research into chronic lung disease phenotypes such as COPD and bronchiectasis has historically excluded patients with AATD.
A curative therapy for AATD has not yet been identified, although a number of studies have evaluated AAT augmentation as a treatment option [27–29]. Whilst some benefit has been demonstrated, augmentation therapy does not appear to completely halt disease progression, although abrupt cessation of AAT in those already receiving it appeared to have consequences in terms of increased inflammatory markers, increased pulmonary exacerbations and hospitalisations [30]. Some authors have suggested that one of the reasons for this modest benefit is the possibility that intravenously administered AAT may not reach the required local physiological concentrations, although Stockley et al. [31] observed in their cohort that the standard dose of intravenous AAT was enough to reach protective threshold both in serum as well as in sputum. Another possibility is that in AATD there is inadequate local production of AAT, which may have an impact on local lung protection. In addition, augmentation therapy is administered periodically with peaks and troughs in drug concentrations, which may have an impact on lung protection. Furthermore, AAT augmentation does not address the issue of AAT polymerisation, including locally in the lungs, with resultant inflammation and tissue damage despite augmentation therapy. Augmentation therapy therefore may not be the ultimate solution for patients with AATD, and the disease pathology therefore requires deeper exploration.
Greater understanding of the roles and contributions of specific proteases involved in AATD may be key in defining new treatment strategies. Unfortunately, studies on the roles of specific proteases in phenotypes of AATD are lacking and we have therefore extrapolated some conclusions from studies on certain chronic lung disease phenotypes such as bronchiectasis, chronic bronchitis and emphysema.
Proteolytic activity in AATD
AATD-related lung pathology is thought to be driven mainly by neutrophils, which navigate via chemotaxis towards sources of inflammation. This chemotaxis may be in response to a variety of stimuli mentioned above such as cigarette smoke, air pollution and recurrent infection. Polymerisation of abnormal AAT, for example, from local production in alveolar cells, is also known to be pro-inflammatory, with resultant chemotaxis and neutrophil recruitment. This creates a vicious cycle of inflammation and extracellular matrix (ECM) destruction.
Neutrophils exert their protease-driven effects on the lungs via degranulation, with release of certain proteases. Neutrophils contain azurophilic granules, which consist of the main neutrophil serine proteases (NSPs): neutrophil elastase (NE), proteinase-3 (PR3) and cathepsin G (CG) [32]. NSPs are released upon neutrophil activation, and if unchecked, result in excessive proteolytic activity and tissue destruction via degradation of elastin. One of the main roles of AAT is to antagonise excessive NSP activity and protect lung tissue from damage.
AAT is the archetypal antiprotease and inhibits NSPs in a 1:1 ratio; however, the amount of NSPs far exceed the level of AAT in the vicinity of a degranulating neutrophil [33, 34]. This proteolytic effect is then diluted as NSPs diffuse into the extracellular environment. With a constantly high inflammatory state as that which exists in AATD, and with significantly reduced AAT levels, tissue damage is often inevitable.
In addition to AAT, two other main antiproteases have been described – secretory leukocyte proteinase inhibitor (SLPI) and elafin [35], which are both mainly produced locally by bronchial epithelial cells. AAT is thought to be the main inhibitor of the three main NSPs, while SLPI is known to inhibit NE and CG but not PR3, and elafin inhibits both NE and PR3 but not CG. Oxidation of antiproteases, for example, by cigarette smoke, is known to reduce their activity against proteolytic enzymes [36]. The summative roles of proteases and antiproteases in the lungs is depicted in figure 1.
FIGURE 1.

The role of serine proteases and antiproteases in α1-antitrypsin deficiency (AATD): protease release by neutrophils overcomes antiprotease production by innate immune cells and respiratory epithelial cells, with disruption of the protease/antiprotease balance and resultant lung damage. This process is accelerated by external drivers such as cigarette smoke and infection. Antiproteases involved in lung protection are highlighted in green, and destructive pathways involved in their inhibition are highlighted with red arrows. Created with BioRender.com. SLPI: secretory leukocyte proteinase inhibitor; LPS: lipopolysaccharide; AAT: α1-antitrypsin; MMP: matrix metalloproteinase.
Neutrophil serine proteases
NSPs are involved in a variety of physiological processes such as defence against infection, tissue remodelling, wound healing and fibrinolysis [37]. As part of their inflammatory activity, they also cause degradation of the ECM if their activities remain unchecked, and this is thought to be the main mechanism of lung damage in patients with AATD. Each neutrophil is estimated to store 1.1, 3.0 and 0.85 pg of NE, PR3 and CG, respectively [38]. The proportional activity of each NSP has not been clearly established; furthermore, their activity also depends on whether they are free or bound to the neutrophil cell membrane.
NE was historically thought to be the main elastase causing emphysema in AATD. More recently, other elastases such as PR3 and CG have been increasingly recognised as key players in lung damage. Guyot and colleagues [39] showed, in mice experiments, that all three proteases collectively caused more severe lung damage in emphysema than did NE alone, when exposed to cigarette smoke. They also demonstrated a positive feedback loop whereby these proteases activated a protease cascade which further intensified activities of other proteases such as matrix metalloproteinase-12 (MMP-12/ macrophage elastase) and matrix metalloproteinase-9 (MMP-9/ gelatinase B). In addition, NSPs have been demonstrated to inactivate tissue inhibitors of metalloproteinases (TIMPs), which are physiological inhibitors of MMPs [36]. These proteases therefore work in a synergistic manner to enhance tissue damage by creating a pro-inflammatory environment. Figure 2 summarises the interplay between the main proteases and antiproteases.
FIGURE 2.
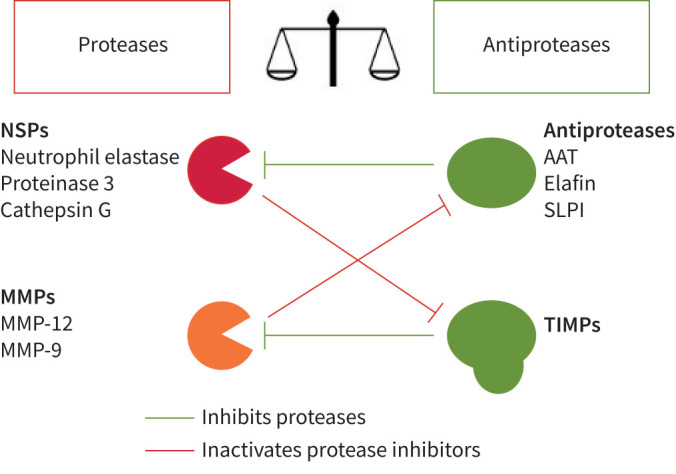
Interplay between proteases and antiproteases. NSP: neutrophil serine protease; MMP: matrix metalloproteinase; AAT: α1-antitrypsin; SLPI: secretory leukocyte proteinase inhibitor; TIMP: tissue inhibitor of metalloproteinases. Figure was formed with an image of scales, taken from The Noun Project. The Noun Project, 8800 Venice Blvd., Los Angeles, CA 90034. Work is licensed under the Attribution 3.0 Unported (CC BY 3.0). Image, titled “Scale” by Fahmihorizon, was downloaded from the justice scale collection from https://thenounproject.com on August 3, 2021.
Neutrophil elastase
NE is mainly involved in defence against bacteria but is also known to cause host tissue damage as a result of its inflammatory properties. This includes destruction of the ECM, damage to airway epithelial cells, mucous gland hyperplasia with increased mucus production and reduction in ciliary beat frequency [40]. It has also been suggested that under certain conditions, NE evades inactivation by AAT and retains its proteolytic activity, for example, when bound to the neutrophil membrane [41]. In bronchiectasis, it is hypothesised that NE avoids inactivation by combining with polyanions such as DNA and glycosaminoglycans as well as syndecan-1, which makes it inaccessible to inhibition [42].
NE has been demonstrated to inhibit ciliary function and muco-ciliary clearance [43]. Amitani et al. [40] showed that NE reduces ciliary beat frequency on bronchial epithelial cells, which may have a direct impact on development and progression of bronchiectasis in AATD. Interestingly, this study also showed that human AAT at a dose of 5 mg·mL−1 completely abolished slowing of ciliary beat frequency, although lower concentrations were less beneficial.
Excessive mucin production has also been associated with increased NE activity. Voynow et al. [44] showed that treatment with NE increased MUC5AC mucin gene expression, which is one of the major mucins expressed by the respiratory epithelium.
A recent study by Chalmers et al. [45], conducted over 3 years in patients with non-CF bronchiectasis, showed that greater NE activity was associated with increased exacerbation frequency, shorter time to next exacerbation and decline in lung function, as well as a higher bronchiectasis severity index.
Shapiro et al. [36] demonstrated that knockout of the NE gene in mice was associated with reduced airspace enlargement compared to wild-type mice. In this study, total lung neutrophils were similar in NE-knockout and wild-type mice following smoke exposure. However, in NE-knockout mice, the neutrophils were confined to the blood vessels, suggesting a defect in chemotaxis in the absence of NE. This group also showed that NE and MMP-12 complemented each other's activity by inactivating the inhibitor of the other, i.e. NE degraded TIMP while MMP-12 inhibited AAT.
A study evaluating the role of NE in patients with AAT-replete COPD [46] demonstrated that the NE-specific fibrinogen degradation product Aα-Val360 was associated with severity of COPD and was raised during acute exacerbations, and fell following resolution of exacerbations. The authors also found that the levels of Aα-Val360 were higher in subjects with radiological evidence of emphysema than those without, and that the baseline level of Aα-Val360 was related to subsequent progression in lower zone emphysema over 4 years. These results suggest that NE potentially plays a key role in exacerbations as well as in progression of emphysema, although these results have been extrapolated from patients with non-AATD COPD.
NE is also capable of disrupting the activity of SLPI. Weldon et al. [47] demonstrated that NE abrogates the activity of SLPI in bronchoalveolar lavage fluid (BALF) of patients with cystic fibrosis infected with P. aeruginosa. This phenomenon has direct clinical implications, as SLPI inhibits bacterial lipopolysaccharide (LPS) – induced inflammatory responses in monocytes and macrophages. These findings suggest that, in addition to causing direct elastolytic damage, NE causes indirect damage by increasing susceptibility of the lungs to bacterial infection. Interestingly, the same group showed that SLPI cleaved by NE still retained its antiprotease activity. This is because NE cleaves SLPI at a site different from its antiprotease active site.
NE has also been demonstrated to cleave elafin [48]. This is significant because, like SLPI, elafin and its precursor trappin-2 display antibacterial activities through inhibition of LPS, independent of their antiprotease activities. Like SLPI, elafin also interferes with NF-κB activation and associated downstream inflammatory pathways, suggesting that, in addition to its direct proteolytic effects, NE indirectly promotes inflammation and resultant ECM destruction.
In summary, NE overactivity has been associated with emphysema as well as bronchiectasis, but there are no studies exploring the role of NE activity particularly in patients with bronchiectasis-predominant AATD. Based on the above evidence, it is possible that NE is the dominant protease in patients with bronchiectasis-predominant AATD, although this hypothesis requires careful and extensive exploration.
Protease 3
Protease 3 (PR3), also known as myeloblastin, is mainly located in neutrophil granules and the neutrophil cell surface, but it is also on the surface of secretory vesicles and secondary granules [49]. PR3 has been demonstrated to invoke downstream inflammatory pathways such as activation of tumour necrosis factor-α (TNF-α) and interleukin (IL)-1β and has been shown to induce apoptosis.
Studies by Ying and Simon [50] reported that PR3 was not as potent as NE or CG in their experiments and was completely inhibited by AAT. This group suggested that PR3 was not as formidable a protease in elastin destruction as NE or CG, with the catalytic activity of PR3 being half that of CG, and one-eighth that of NE. It is important to note that this study used bovine neck ligament elastin, which is structurally different from human elastin fibres, and therefore may not be fully reflective of NSP activity on human lung elastin.
More recently, PR3 has been proposed as an equally, if not more, important protease than NE in the pathogenesis of chronic lung diseases including AATD [51]. Korkmaz et al. [52] studied the inhibitory capacity of NE, PR3 and CG in BALF of patients admitted with acute bacterial pneumonia and/or acute respiratory distress syndrome, and reported that AAT preferentially inhibited NE, and only inhibited PR3 and CG once all NE had been inactivated. This group also looked at the preferential inhibition of neutrophil membrane-bound proteases by BALF collected from patients and reported that NE was the first to be inhibited, whereas there was marked residual activity of the other two proteases.
Sinden et al. [53] analysed sputum from clinically stable patients with AATD and usual COPD, and reported that in their cohort, PR3 activity was greater in AATD patients compared to those with AAT-replete COPD. Newby and colleagues [54] identified Aα-Val541 as a specific fibrinogen cleavage product of PR3 and demonstrated that this marker correlated with airflow obstruction and gas transfer on pulmonary function tests in patients with AATD.
Membrane-bound PR3 has been demonstrated to be more resistant to inhibition by AAT and elafin than soluble PR3 [38]. It is noteworthy that, as mentioned earlier, while PR3 is susceptible to inhibition by AAT and elafin, SLPI has no effect on PR3. Similar to NE, PR3 may indirectly augment protease activity by degrading other antiproteases such as SLPI, thereby increasing availability of other proteases such as NE and CG. However, studies by Rao et al. [55] reported that while PR3 was not inhibited by SLPI and did indeed cleave SLPI, cleavage was at the non-reactive site and did not reduce its inhibitory activity against other NSPs. Nevertheless, it is plausible that cleavage of SLPI renders other anti-inflammatory properties of this protein redundant, leading to net effect of lung damage.
There are no studies assessing the role of PR3 in bronchiectasis or bronchiectasis-predominant AATD. However, the above studies lend proof to the concept that PR3 is a key player in the pathogenesis of AATD. It remains to be explored whether PR3 is dominant in a particular phenotype of AATD.
Cathepsin G
CG is also stored within the azurophilic granules of neutrophils. Although CG has been implicated in the pathogenesis of AATD, studies directly associating this protease with AATD are scarce.
CG is associated with activated neutrophils and macrophages and is therefore known to be involved in inflammatory lung diseases. As mentioned previously, studies by Guyot et al. [39] showed that elastin degradation was greater with all three NSPs combined than with each NSP in isolation, with a clear contribution demonstrated by CG.
Maryanoff et al. [56] showed that inhibition of CG in a cigarette smoke-exposed murine model resulted in a 66% reduction in the neutrophil burden in BALF compared to controls. This group also showed that there was a corresponding 55% reduction in cytokine-induced neutrophil-attracting chemokine (KC, murine equivalent of IL-8) following inhibition of CG. This is especially relevant as IL-8 is chemotactic to neutrophils, leading to a cycle of neutrophil degranulation and protease release.
CG has also been particularly implicated in the pathogenesis of bronchiectasis. Studies have demonstrated the role of CG in destruction of airway epithelium and dysfunction of ciliated cells [57]. Sepper et al. [58] demonstrated that, in patients with bronchiectasis, the activity of CG is significantly higher compared to that in controls, and that this activity increased depending on the severity of bronchiectasis. It is therefore plausible that, like NE, CG may have a role to play in patients with AATD with a bronchiectatic phenotype.
Other proteases implicated in pathogenesis of AATD: MMPs
Macrophages are known to comprise a large proportion of inflammatory cells that accumulate in the lungs of smokers and are the source of MMPs. MMPs are structurally similar proteolytic enzymes that have been implicated in inflammation, tissue destruction and lung tissue remodelling, especially in COPD [59]. Ostridge et al. [60] showed that MMPs in BALF are increased in patients with COPD, and that the levels of MMPs correlated with degree of small airways disease and emphysema measured on computed tomography. The authors noted that the correlation was greater for small airways disease than for emphysema, suggesting that MMPs may have a greater role to play in this phenotype of lung disease. While the implications of pathological activities of MMPs have been reported in usual COPD and emphysema, with the exception of MMP-12, their role in AATD has not been elucidated.
Churg and colleagues [61] reported, in their experiments, that MMP-12 is essential for development of emphysema. This group suggested that MMP-12 initiates release of TNF-α via alveolar macrophages, which in turn results in endothelial activation, neutrophil accumulation and subsequent NSP release resulting in matrix destruction.
Work by Sapey et al. [62] adds to the above hypothesis and shows that TNF polymorphism rs361525 is associated with more rapid decline in lung function in patients with AATD that carry this polymorphism. A study investigating MMP gene polymorphisms in a cohort of patients with COPD [63] demonstrated that they have significantly higher MMP-12 levels than controls.
AAT has been shown to suppress MMP-12 production by cigarette smoke-stimulated macrophages [64]. Interestingly, the investigators reported that this anti-inflammatory activity requires an intact AAT molecule, with oxidised and polymerised AAT molecules shown to be ineffective at preventing MMP-12 release. This phenomenon is relevant both because polymerisation of AAT is a feature of AATD and cigarette smoke oxidises AAT, rendering it ineffective as an antiprotease.
MMP-12 is also known to inactivate AAT [65] and therefore has the potential to extenuate lung damage through unopposed proteolytic activity of proteases other than MMP-12 that are usually inactivated by AAT.
Other MMPs, although not directly studied in AATD, have been implicated in the development of small airways disease, which is now widely believed to be the precursor to development of emphysema, as well as development of emphysema itself. These MMPs include MMPs −1, −2, −8, −9 and −14, which have been identified in lung samples from smokers and those with COPD. It has been suggested that activation of these MMPs via mechanisms other than NE (i.e. oxidant-related pathways) provides a direct as well as indirect mode of development of emphysema [51].
Once again, there are no clinical studies evaluating the role of MMPs in patients with AATD, and expansion of our understanding in this area will give greater insight into their role, which might pave the way to development of treatment options aimed at patients with this condition.
Current evidence for protease inhibitors in chronic lung disease
Protease inhibitors have been mentioned as potential options for treatment of chronic lung disease [66]; however, clinical trials evaluating efficacy of specific protease inhibitors remain scarce.
A study evaluating the role of an oral NE inhibitor (AZD9668) versus placebo for 28 days in patients with CF [67] showed no change in absolute and percentage sputum neutrophil counts between groups. However, urinary desmosine, a marker of elastin breakdown, was reduced by 30% in the treatment group compared to placebo, and this difference was statistically significant. Consideration of the mechanism of action of NE is paramount in analysis of these results, as NE, although pro-inflammatory with resultant release of chemo-attractants such as LTB4 and IL-8, is also responsible for direct elastin breakdown, and therefore a reduction in elastin breakdown may have been observed in the absence of a reduction in the pro-inflammatory activity of NE.
AZD9668 was also tested in patients with non-CF bronchiectasis over 28 days [68]. Once again, there was no significant difference in percentage sputum neutrophil count or NE activity between treatment group and placebo, although there was a trend for reduction in NE activity with active treatment. Importantly, the authors reported an improvement in forced expiratory volume in 1 s (FEV1) of 100 mL in the treatment group compared to placebo. Unlike the above study, however, there was no significant difference in serum or urinary desmosine levels between treatment and placebo groups.
Safety and tolerability of NE inhibitor BAY 85–8501 versus placebo [69] administered for 4 weeks in patients with non-CF bronchiectasis showed no change in pulmonary function between the two groups, but interestingly, the authors reported a significant reduction in NE activity compared to placebo in whole blood. However, this finding was not replicated in sputum NE activity, suggesting differences in systemic versus local delivery.
AZD9668 was also tested against placebo in subjects with non-AATD COPD for 12 weeks [70], and showed no difference in the primary end-point of pre-bronchodilator FEV1 between treatment and control groups.
Although the above studies showed a trend towards clinical/biochemical improvement, they did not always achieve their primary end-points. It is important to note the short duration of these trials, which may not have been long enough to discern beneficial effects. In the trial with COPD, the patients were not stratified by phenotype, and this may have been a key reason for lack of proven efficacy, an observation made by the authors themselves. We have suggested above that NE may be the dominant protease in patients with bronchiectasis-predominant AATD, and therefore NE inhibitors may prove useful in this subgroup of patients.
Different routes of administration of recombinant AATD have been explored, including intravenous and inhaled, but NE inhibitors so far trialled have used the oral route. It is possible that other routes of delivery such as intravenous and inhaled therapy might provide different results in terms of bioavailability, peak drug concentrations and patient outcomes.
Although the roles of PR3 and CG in lung disease have been discussed in the literature [71], and specific PR3 and CG inhibitors described [72, 73], there are no clinical trials exploring the roles of these specific inhibitors in lung disease.
The role of a dual MMP-9/MMP-12 inhibitor was explored in guinea pigs exposed to cigarette smoke for 6 months [74] and showed promising effects in terms of reduction in bronchoalveolar lavage (BAL) neutrophil, macrophage and desmosine levels as well as emphysema and small airways thickness in smoke-exposed models. Another study evaluating the effects of an MMP-12 inhibitor in mice exposed to cigarette smoke [75] showed a significant reduction in BAL inflammatory cells. Once again, these results have not yet been translated into clinical trials.
Most recently, a phase II study of bronchiectasis involving Brensocatib, an oral reversible inhibitor of dipeptidyl peptidase 1 (DPP-1, also known as cathepsin C), an enzyme responsible for activation of NSPs, demonstrated prolongation in the time to first exacerbation compared to placebo (primary end-point) [76]. The authors also reported a reduction in sputum neutrophil elastase activity in the treatment group compared to placebo. It is important to note that, unlike the above studies, the duration of this study was much longer, i.e. 24 weeks, thereby allowing time for beneficial effects to become evident. In addition, in approximately a quarter of patients in this study population, the sputum neutrophil elastase concentration was below the lower limit of quantification, making assessment of changes following treatment difficult. This may have been an issue in some of the above studies as well, highlighting the importance of sensitive and accurate methods of assessment of effects as well as adequate study duration.
Conclusion
Despite supposedly monogenic inheritance, AATD is being increasingly identified as a heterogeneous condition with disease phenotypes. Protease activity has been demonstrated to be integral to disease pathology in patients with AATD, but it is not yet clear which enzymes are involved and whether one predominates in a particular disease phenotype in AATD, including in contribution to irreversible lung damage. Several studies highlight the importance of NE in disease pathology in emphysema and bronchiectasis, and a clear role of PR3 in disease pathology in AATD has been demonstrated. The studies mentioned above have investigated the role of different proteases in inflammatory lung diseases not exclusive to AATD; in fact, all studies investigating COPD or bronchiectasis excluded patients with AATD. Studies directly investigating the role of specific protease activity in AATD, and particularly in various phenotypes of the disease, will enhance understanding of mechanistic pathways in disease development and progression, and will enable development of targeted therapies aimed at different phenotypes of disease. It is possible that, in future, depending on dominant protease activity, specific protease inhibitors will be appropriate for use in different phenotypes of disease including in AATD.
Acknowledgements
The authors thank Alastair Watson, University of Southampton, UK, for contributing figures to the article, and Karl Staples, University of Southampton, UK, for his suggestions and comments.
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: A. Fazleen has nothing to disclose.
Conflict of interest: T. Wilkinson reports grants and personal fees from AstraZeneca during the conduct of the study; and personal fees from and a directorship of MMH, grants and personal fees from GSK, AZ and Synairgen, and personal fees from BI, outside the submitted work.
References
- 1.Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. Scand J Clin Lab Invest 2009; 15: 132–140. doi: 10.1080/00365516309051324 [DOI] [Google Scholar]
- 2.Stockley RA. Turner AM. α-1-Antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol Med 2014; 20: 105–115. doi: 10.1016/j.molmed.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 3.Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis 2017; 12: 561–569. doi: 10.2147/COPD.S125389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly E, Greene CM, Carroll TP, et al. Alpha-1 antitrypsin deficiency. Respir Med CME 2011; 4: 1–8. doi: 10.1016/j.rmedc.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 5.Lomas DA, Evans DL, Finch JT, et al. The mechanism of Z alpha 1antitrypsin accumulation in the liver. Nature 1992; 357: 605–607. doi: 10.1038/357605a0 [DOI] [PubMed] [Google Scholar]
- 6.Cuvelier A, Muir JF, Hellot MF, et al. Distribution of α1-antitrypsin alleles in patients with bronchiectasis. Chest 2000; 117: 415–419. doi: 10.1378/chest.117.2.415 [DOI] [PubMed] [Google Scholar]
- 7.Cox DW, Woo SLC, Mansfield T. DNA restriction fragments associated with alpha 1antitrypsin indicate a single origin for deficiency allele PI Z. Nature 1985; 316: 79–81. doi: 10.1038/316079a0 [DOI] [PubMed] [Google Scholar]
- 8.Luisetti M, Seersholm N. α1-Antitrypsin deficiency. 1: epidemiology of α1-antitrypsin deficiency. Thorax 2004; 59: 164–169. doi: 10.1136/thorax.2003.006494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll TP, O'Connor CA, Floyd O, et al. The prevalence of alpha-1 antitrypsin deficiency in Ireland. Resp Res 2011; 12: 91. doi: 10.1186/1465-9921-12-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strnad P, McElvaney NG, Lomas DA. α1-antitrypsin deficiency. N Engl J Med 2020; 382: 1443–1455. doi: 10.1056/NEJMra1910234 [DOI] [PubMed] [Google Scholar]
- 11.Silverman EK, Miletich JP, Pierce JA, et al. Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis 1989; 140: 961–966. doi: 10.1164/ajrccm/140.4.961 [DOI] [PubMed] [Google Scholar]
- 12.Bouchecareilh M. Alpha-1 antitrypsin deficiency-mediated liver toxicity: why do some patients do poorly? What do we know so far? Chronic Obstr Pulm Dis 2020; 7: 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mela M, Smeeton W, Davies SE, et al. The alpha-1 antitrypsin polymer load correlates with hepatocyte senescence, fibrosis stage and liver-related mortality. Chronic Obstr Pulm Dis 2020; 7: 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubbard RC, Ogushi F, Fells GA, et al. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of alpha 1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J Clin Invest 1987; 80: 1289–1295. doi: 10.1172/JCI113204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taggart C, Cervantes-Laurean D, Kim G, et al. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem 2000; 275: 27258–27265. doi: 10.1016/S0021-9258(19)61505-X [DOI] [PubMed] [Google Scholar]
- 16.Alam S, Li Z, Janciauskiene S, et al. Oxidation of Z α1-antitrypsin by cigarette smoke induces polymerization: a novel mechanism of early-onset emphysema. Am J Respir Cell Mol Biol 2011; 45: 261–269. doi: 10.1165/rcmb.2010-0328OC [DOI] [PubMed] [Google Scholar]
- 17.Tanash HA, Nilsson PM, Nilsson JA, et al. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63: 1091–1095. doi: 10.1136/thx.2008.095497 [DOI] [PubMed] [Google Scholar]
- 18.Seersholm N, Kok-Jensen A. Clinical features and prognosis of life time non-smokers with severe alpha1-antitrypsin deficiency. Thorax 1998; 53: 265–268. doi: 10.1136/thx.53.4.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax 1998; 53: 129–136. doi: 10.1136/thx.53.2.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeMeo DL, Sandhaus RA, Barker AF, et al. Determinants of airflow obstruction in severe alpha-1-antitrypsin deficiency. Thorax 2007; 62: 806–813. doi: 10.1136/thx.2006.075846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stockley RA, Hill AT, Hill SL, et al. Bronchial inflammation: its relationship to colonizing microbial load and alpha(1)-antitrypsin deficiency. Chest 2000; 117: 291S–293S. doi: 10.1378/chest.117.5_suppl_1.291S [DOI] [PubMed] [Google Scholar]
- 22.Balbi B, Sangiorgi C, Gnemmi I, et al. Bacterial load and inflammatory response in sputum of alpha-1 antitrypsin deficiency patients with COPD. Int J Chron Obstruct Pulmon Dis 2019; 14: 1879–1893. doi: 10.2147/COPD.S207203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eden E, Choate R, Barker A, et al. The clinical features of bronchiectasis associated with Alpha-1 antitrypsin deficiency, common variable immunodeficiency and primary ciliary dyskinesia: results from the U.S. Bronchiectasis Research Registry. Chronic Obstr Pulm Dis 2019; 6: 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parr DG, Guest PG, Reynolds JH, et al. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2007; 176: 1215–1221. doi: 10.1164/rccm.200703-489OC [DOI] [PubMed] [Google Scholar]
- 25.Longstreth GF, Weitzman SA, Browning RJ, et al. Bronchiectasis and homozygous Alpha1-antitrypsin deficiency. Chest 1975; 67: 233–235. doi: 10.1378/chest.67.2.233 [DOI] [PubMed] [Google Scholar]
- 26.Scott JH, Anderson CL, Shankar PS, et al. Alpha1antitrypsin deficiency with diffuse bronchiectasis and cirrhosis of the liver. Chest 1977; 71: 535–538. doi: 10.1378/chest.71.4.535 [DOI] [PubMed] [Google Scholar]
- 27.Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in α1-antitrypsin deficiency. Eur Respir J 2009; 33: 1345–1353. doi: 10.1183/09031936.00159408 [DOI] [PubMed] [Google Scholar]
- 28.Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. The Lancet 2015; 386: 360–368. doi: 10.1016/S0140-6736(15)60860-1 [DOI] [PubMed] [Google Scholar]
- 29.Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of α1-antitrypsin augmentation therapy. Am J Resp Crit Care 1999; 160: 1468–1472. doi: 10.1164/ajrccm.160.5.9901055 [DOI] [PubMed] [Google Scholar]
- 30.McElvaney OJ, Carroll TP, Franciosi AN, et al. Consequences of abrupt cessation of α1-antitrypsin replacement therapy. N Engl J Med 2020; 382: 1478–1480. doi: 10.1056/NEJMc1915484 [DOI] [PubMed] [Google Scholar]
- 31.Stockley RA, Bayley DL, Unsal I, et al. The effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2002; 165: 1494–1498. doi: 10.1164/rccm.2109013 [DOI] [PubMed] [Google Scholar]
- 32.McCarthy C, Reeves EP, McElvaney NG. The role of neutrophils in alpha-1 antitrypsin deficiency. Ann Am Thorac Soc 2016; 13: Suppl 4, S297–S304. doi: 10.1513/AnnalsATS.201509-634KV [DOI] [PubMed] [Google Scholar]
- 33.Campbell EJ, Campbell MA, Boukedes SS, et al. Quantum proteolysis by neutrophils: implications for pulmonary emphysema in α1-antitrypsin deficiency. Chest 2000; 117: 303S. doi: 10.1378/chest.117.5_suppl_1.303S [DOI] [PubMed] [Google Scholar]
- 34.Sapey E. Neutrophil modulation in alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis 2020; 7: 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zani ML, Nobar SM, Lacour SA, et al. Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre-elafin (trappin-2) expressed in Pichia pastoris. Eur J Biochem 2004; 271: 2370–2378. doi: 10.1111/j.1432-1033.2004.04156.x [DOI] [PubMed] [Google Scholar]
- 36.Shapiro SD, Goldstein NM, Houghton AM, et al. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 2003; 163: 2329–2335. doi: 10.1016/S0002-9440(10)63589-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008; 90: 227–242. doi: 10.1016/j.biochi.2007.10.009 [DOI] [PubMed] [Google Scholar]
- 38.Campbell EJ, Campbell MA, Owen CA. Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J Immunol 2000; 165: 3366–3374. doi: 10.4049/jimmunol.165.6.3366 [DOI] [PubMed] [Google Scholar]
- 39.Guyot N, Wartelle J, Malleret L, et al. Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am J Pathol 2014; 184: 2197–2210. doi: 10.1016/j.ajpath.2014.04.015 [DOI] [PubMed] [Google Scholar]
- 40.Amitani R, Wilson R, Rutman A, et al. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am J Respir Cell Mol Biol 1991; 4: 26–32. doi: 10.1165/ajrcmb/4.1.26 [DOI] [PubMed] [Google Scholar]
- 41.Owen CA, Campbell MA, Sannes PL, et al. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, nonoxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 1995; 131: 775–789. doi: 10.1083/jcb.131.3.775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan SCH, Shum DKY, Ip MSM. Sputum sol neutrophil elastase activity in bronchiectasis: differential modulation by syndecan-1. Am J Resp Crit Care Med 2003; 168: 192–198. doi: 10.1164/rccm.200208-829OC [DOI] [PubMed] [Google Scholar]
- 43.Fischer B, Voynow J. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 2002; 26: 447–452. doi: 10.1165/ajrcmb.26.4.4473 [DOI] [PubMed] [Google Scholar]
- 44.Voynow JA, Young LR, Wang YQ, et al. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am J Physiol-Lung C 1999; 276: L835–LL43. doi: 10.1152/ajplung.1999.276.5.L835 [DOI] [PubMed] [Google Scholar]
- 45.Chalmers JD, Moffitt KL, Suarez-Cuartin G, et al. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Respir Crit Care Med 2017; 195: 1384–1393. doi: 10.1164/rccm.201605-1027OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carter RI, Ungurs MJ, Mumford RA, et al. Aalpha-Val360: a marker of neutrophil elastase and COPD disease activity. Eur Respir J 2013; 41: 31–38. doi: 10.1183/09031936.00197411 [DOI] [PubMed] [Google Scholar]
- 47.Weldon S, McNally P, McElvaney NG, et al. Decreased levels of secretory leucoprotease inhibitor in the Pseudomonas-infected cystic fibrosis lung are due to neutrophil elastase degradation. J Immunol 2009; 183: 8148–8156. doi: 10.4049/jimmunol.0901716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guyot N, Butler MW, McNally P, et al. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J Biol Chem 2008; 283: 32377–32385. doi: 10.1074/jbc.M803707200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Witko-Sarsat V, Cramer EM, Hieblot C, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 1999; 94: 2487–2496. doi: 10.1182/blood.V94.7.2487.419k07_2487_2496 [DOI] [PubMed] [Google Scholar]
- 50.Ying QL, Simon SR. Elastolysis by proteinase 3 and its inhibition by α1-proteinase inhibitor: a mechanism for the incomplete inhibition of ongoing elastolysis. Am J Resp Cell Mol 2002; 26: 356–361. doi: 10.1165/ajrcmb.26.3.4704 [DOI] [PubMed] [Google Scholar]
- 51.Stockley RA. Alpha-1 antitrypsin deficiency: have we got the right proteinase? Chronic Obstr Pulm Dis 2020; 7: 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korkmaz B, Poutrain P, Hazouard E, et al. Competition between elastase and related proteases from human neutrophil for binding to alpha1-protease inhibitor. Am J Respir Cell Mol Biol 2005; 32: 553–559. doi: 10.1165/rcmb.2004-0374OC [DOI] [PubMed] [Google Scholar]
- 53.Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur Respir J 2013; 41: 1042–1050. doi: 10.1183/09031936.00089712 [DOI] [PubMed] [Google Scholar]
- 54.Newby PR, Crossley D, Crisford H, et al. A specific proteinase 3 activity footprint in α1-antitrypsin deficiency. ERJ Open Res 2019; 5: 00095-2019. doi: 10.1183/23120541.00095-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao NV, Marshall BC, Gray BH, et al. Interaction of secretory leukocyte protease inhibitor with proteinase-3. Am J Resp Cell Mol 1993; 8: 612–616. doi: 10.1165/ajrcmb/8.6.612 [DOI] [PubMed] [Google Scholar]
- 56.Maryanoff BE, de Garavilla L, Greco MN, et al. Dual inhibition of cathepsin G and chymase is effective in animal models of pulmonary inflammation. Am J Respir Crit Care Med 2010; 181: 247–253. doi: 10.1164/rccm.200904-0627OC [DOI] [PubMed] [Google Scholar]
- 57.Rickard KA, Taylor J, Rennard SI. Observations of development of resistance to detachment of cultured bovine bronchial epithelial cells in response to protease treatment. Am J Respir Cell Mol Biol 1992; 6: 414–420. doi: 10.1165/ajrcmb/6.4.414 [DOI] [PubMed] [Google Scholar]
- 58.Sepper R, Konttinen YT, Ingman T, et al. Presence, activities, and molecular forms of cathepsin G, elastase, alpha 1-antitrypsin, and alpha 1-antichymotrypsin in bronchiectasis. J Clin Immunol 1995; 15: 27–34. doi: 10.1007/BF01489487 [DOI] [PubMed] [Google Scholar]
- 59.Churg A, Zhou S, Wright JL. Matrix metalloproteinases in COPD. Eur Respir J 2012; 39: 197–209. doi: 10.1183/09031936.00121611 [DOI] [PubMed] [Google Scholar]
- 60.Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax 2016; 71: 126–132. doi: 10.1136/thoraxjnl-2015-207428 [DOI] [PubMed] [Google Scholar]
- 61.Churg A, Wang RD, Xie C, et al. α-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med 2003; 168: 199–207. doi: 10.1164/rccm.200302-203OC [DOI] [PubMed] [Google Scholar]
- 62.Sapey E, Wood AM, Ahmad A, et al. Tumor necrosis factor-α rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 182: 192–199. doi: 10.1164/rccm.200912-1846OC [DOI] [PubMed] [Google Scholar]
- 63.Gilowska I, Majorczyk E, Kasper L, et al. The role of MMP-12 gene polymorphism – 82 A-to-G (rs2276109) in immunopathology of COPD in polish patients: a case control study. BMC Med Genet 2019; 20: 19. doi: 10.1186/s12881-019-0751-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Churg A, Wang X, Wang RD, et al. α1-antitrypsin suppresses TNF-α and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol 2007; 37: 144–151. doi: 10.1165/rcmb.2006-0345OC [DOI] [PubMed] [Google Scholar]
- 65.Desrochers PE, Jeffrey JJ, Weiss SJ. Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activity. J Clin Invest 1991; 87: 2258–2265. doi: 10.1172/JCI115262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Craciun I, Fenner AM, Kerns RJ. N-Arylacyl O-sulfonated aminoglycosides as novel inhibitors of human neutrophil elastase, cathepsin G and proteinase 3. Glycobiology 2016; 26: 701–709. doi: 10.1093/glycob/cww011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elborn JS, Perrett J, Forsman-Semb K, et al. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur Respir J 2012; 40: 969–976. doi: 10.1183/09031936.00194611 [DOI] [PubMed] [Google Scholar]
- 68.Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med 2013; 107: 524–533. doi: 10.1016/j.rmed.2012.12.009 [DOI] [PubMed] [Google Scholar]
- 69.Watz H, Nagelschmitz J, Kirsten A, et al. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85–8501 for the treatment of non-cystic fibrosis bronchiectasis: a randomized controlled trial. Pulm Pharmacol Ther 2019; 56: 86–93. doi: 10.1016/j.pupt.2019.03.009 [DOI] [PubMed] [Google Scholar]
- 70.Vogelmeier C, Aquino TO, O'Brien CD, et al. A randomised, placebo-controlled, dose-finding study Of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 2012; 9: 111–120. doi: 10.3109/15412555.2011.641803 [DOI] [PubMed] [Google Scholar]
- 71.Grzywa R, Lesner A, Korkmaz B, et al. Proteinase 3 phosphonic inhibitors. Biochimie 2019; 166: 142–149. doi: 10.1016/j.biochi.2019.03.005 [DOI] [PubMed] [Google Scholar]
- 72.Budnjo A, Narawane S, Grauffel C, et al. Reversible ketomethylene-based inhibitors of human neutrophil proteinase 3. J Med Chem 2014; 57: 9396–9408. doi: 10.1021/jm500782s [DOI] [PubMed] [Google Scholar]
- 73.Swedberg JE, Li CY, de Veer SJ, et al. Design of potent and selective cathepsin G inhibitors based on the sunflower trypsin inhibitor-1 scaffold. J Med Chem 2017; 60: 658–667. doi: 10.1021/acs.jmedchem.6b01509 [DOI] [PubMed] [Google Scholar]
- 74.Churg A, Wang R, Wang X, et al. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 2007; 62: 706–713. doi: 10.1136/thx.2006.068353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Quement C, Guenon I, Gillon JY, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Br J Pharmacol 2008; 154: 1206–1215. doi: 10.1038/bjp.2008.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chalmers JD, Haworth CS, Metersky ML, et al. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med 2020; 383: 2127–2137. doi: 10.1056/NEJMoa2021713 [DOI] [PubMed] [Google Scholar]