

Abstract
The role of exosome-mediated mitophagy in the crosstalk between hepatocytes (HCs) and hepatic stellate cells (HSCs) in metabolic-associated fatty liver disease (MAFLD) remains unknown. Serum exosomal miR-27a levels were markedly increased and positively correlated with liver fibrosis in MAFLD patients and mice. Exosomal miR-27a was released from lipotoxic HCs and specifically transmitted to recipient-activated HSCs. PINK1, the key target of miR-27a, primarily mediates mitophagy. Overexpression of miR-27a or knockdown of PINK1 or lipotoxic HC-exosomal miR-27a impaired mitochondria (inhibiting mitophagy, respiration, membrane potential, and transcription while promoting reactive oxygen species production) in activated HSCs and stimulated HSC-derived fibroblasts (promoting activation and proliferation while inhibiting autophagy). High exosomal miR-27a serum levels and a lack of hepatic PINK1-mediated mitophagy were directly related to liver fibrosis in MAFLD mice. Lipotoxic HC exosome transplantation aggravated the degree of PINK1-mediated mitophagy suppression, steatohepatitis, lipidosis, and fibrosis in the livers of MAFLD mice with cirrhosis. Both in vitro and in vivo, exosomes derived from miR-27a-knockdown HCs could not facilitate the abovementioned deteriorating effects. In conclusion, lipotoxic HC-exosomal miR-27a plays a pivotal role in inhibiting mitophagy and in promoting MAFLD-related liver fibrosis by negatively regulating PINK1 expression.
Keywords: metabolic-associated fatty liver disease, MAFLD, mitophagy, exosomes, microRNA-27a, miR27a, PINK1, PTEN-induced putative protein kinase 1, hepatic stellate cell, HSC
Graphical abstract
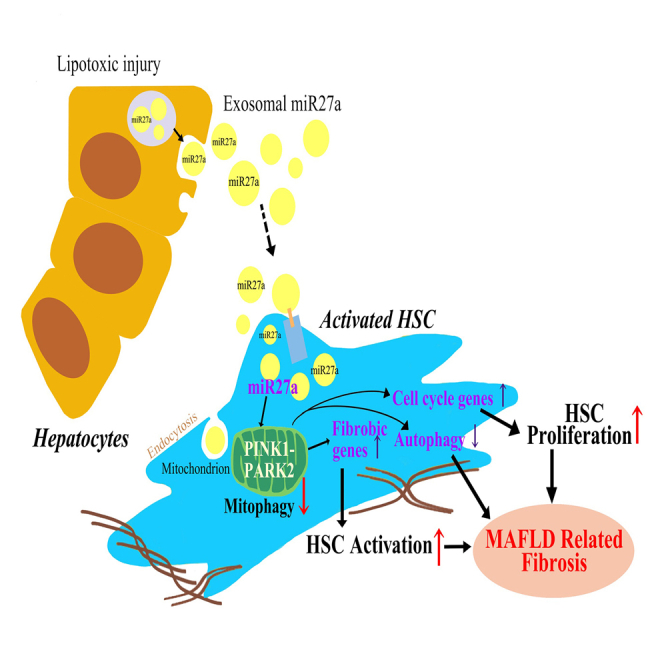
Lipotoxic hepatocytes derive rich exosomal miR-27a and transmit to the recipient hepatic stellate cells. Lipotoxic exosomal miR-27a played a pivotal role on inhibiting mitophagy and aggravating MAFLD-related liver fibrosis by negatively regulating PINK1. Serum exosomal miR-27a represents a good predictor for MAFLD-related liver fibrosis.
Introduction
Metabolic-associated fatty liver disease (MAFLD) has become the most common liver disease throughout the world. Advanced liver fibrosis (ALF) (fibrotic stage S ≥ 3) was identified as the major prognostic factor for MAFLD, and the fibrosis stage was independently associated with long-term overall mortality.1,2
Selective mitochondrial autophagy (mitophagy) is an important cell defense mechanism.3 Mitophagy failure has been suggested to occur in steatotic or fibrotic livers.4,5 Loss of phosphatase and tensin homolog (PTEN)-induced expression of putative protein kinase 1 (PINK1), the key regulator of the mitophagy pathway, might accelerate liver fibrosis.6
Exosomes are nanovesicles that epigenetically reprogram and alter the phenotype of their recipient cells.7 The majority of extracellular microRNAs (miRNAs) are secreted via exosomes.7 Hepatocytes (HCs) injured by lipotoxic fatty acids were found to produce exosomes enriched in miR-17 and miR-92 clusters, which were taken up by hepatic stellate cells (HSCs), ultimately leading to fibrogenic activation.8
However, the molecular mechanisms by which exosomal miRNAs are derived from HCs and transmitted to govern mitophagy in HSCs remain poorly understood.
Results
Lipotoxic fatty acids increase the release of exosomal miR27a from HCs and then affect recipient-activated HSCs
First, the exosomes isolated in our study were characterized. The exosomes derived from primary HCs (PHCs) exhibited a round morphology with a size of 50–200 nm (Figure S1A). PHC exosomes contained CD63 and TSG101 proteins (exosomal markers) but did not contain GAPDH (cellular marker) (Figure S1B). LO2 cells released the highest number of exosomes (Figure S1C). Based on this, LO2 exosomes were used for in vitro and in vivo transplantation studies. Interestingly, lipotoxic palmitic acid (PA) increased not only the size but also the number of PHC exosomes in a dose-dependent manner (Figure S1D). These findings indicate that HCs release additional exosomes into the environment after lipotoxic injury.
Second, we explored the majority of extracellular miRNAs secreted via exosomes from lipotoxic HCs. MAFLD and chronic hepatitis B patients in the ALF group had significantly higher serum miR27a levels than the moderate liver fibrosis (MLF) group; however, there was no difference in the livers of these two groups in the trend analysis of the miRNA microarray results (Figures 1A and S2). Serum miR27a levels could effectively distinguish ALF in MAFLD patients (area under the receiver operating characteristic [AUROC] = 0.730, Figure 1B). Therefore, we focused on miR27a for further study.
Figure 1.

PA promoted the release of exosomal miR-27a from HCs and affected activated HSCs
(A) Trend analysis of miR-27a expression in serum or liver tissues of MAFLD patients (MLF and ALF groups, each group n = 8). (B) AUROC of serum miR27a for ALF diagnosis is shown. (C) Exosomal miR-27a expression was examined in the serum of MAFLD patients (F0S0-2, F3+NASH, and F3+S3-4 groups, each group n = 3) and mice (LFD, HFD, and HFD + CCl4 groups, each group n = 3). (D) Exosomal and cellular miR-27a expression was examined in PHCs or LO2 cells (CN, PA200 μM, and PA400 μM groups). (E) Representative images of PKH26-labeled Exo-PA taken up by primary liver cells (1- or 7-day-cultured PHSCs and PKCs) are shown. The relative IF intensity in each type of cell was normalized to the cell number. Scale bar, 25 μm. (F) miR27a expression was assessed in activated LX2 cells incubated with 25–200 μg Exo-PA or 25 μg Exo-CN isolated from LO2 cells. (G) miR27a expression was detected in activated LX2 cells incubated with Exo-PA or Exo-CN isolated from LO2 cells transfected with miR-27a inhibitor or its negative control (LO2 in-NC or LO2 in-miR groups). All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test (A, B, D, E, and G) (for comparison between two groups) or one-way ANOVA followed by a Student-Newman-Keuls analysis (C and F) (for comparison of multiple groups). Statistical significance: ∗p < 0.05, compared with (A) MAFLD-MLF group, (C) F0S0-2 patients or LFD mice, (D) CN group, (E) PHC group, (F) CN group, (G) or Exo-CN of LO2 in-NC group; #p < 0.05, compared with (C) F3+NASH patients or HFD mice, (F) Exo-CN 25 μg-treated cells.
Third, we checked miR-27a levels in exosomes released from lipotoxic HCs. Importantly, the serum exosomes isolated from MAFLD patients with ALF (fibrosis stage S3-4) and HFD-fed mice treated with CCl4 contained the highest levels of miR-27a compared with their controls (Figure 1C). miR27a levels were dramatically increased in exosomes derived from lipotoxic HCs (including PHCs, LO2, AML12, HepG2, and Huh7 cells) compared with the untreated controls; however, miR27a levels showed no differences in the cells (Figures 1D and S3A). To further determine whether other types of hepatic cells also release exosomal miR27a under lipotoxic conditions, LX2, THP-1, and EGL-1 cells were treated with PA. PA did not affect exosomal or cellular miR-27a expression in these hepatic cells (Figure S3B). These data suggest that HCs are the primary source of exosomal miR27a under lipotoxic conditions.
Finally, we clarified the effect of lipotoxic HC exosomes on recipient cells (activated HSCs). To identify the major recipient cells of Exo-PA (exosomes derived from LO2 cells treated with PA), red immunofluorescence (IF) of the PKH26-labeled exosomes was examined in primary hepatic cells, including PHCs, quiescent primary HSCs (PHSCs) (1-day culture), activated PHSCs (7-day culture), and primary Kupffer cells (PKCs). After 6 h of incubation, the relative amount of PKH26-labeled Exo-PA was highest in activated PHSCs, followed by quiescent PHSCs, PKCs, and PHCs (red IF value for PKH26/4′6-diamidino-2-phenylindole (DAPI); PHSC-1d versus PHC: a 2.95 ± 0.25 increase, PHSC-7d versus PHC: a 6.81 ± 0.14 increase; Figure 1E). This indicates that lipotoxic HC exosomes were efficiently and rapidly taken up by activated PHSCs in a time-dependent manner. Given that miR-27a was primarily expressed in lipotoxic HC exosomes, these structures might play a critical role in transferring miR-27a from HCs to activated HSCs. Next, we examined whether Exo-PA could directly affect miR-27a expression in activated HSCs. miR27a expression was very low in control LX2 cells (CN group) and activated LX2 cells (treated with TGF-β1) incubated with exosomes from untreated LO2 cells (Exo-CN group, Figure 1F). Interestingly, Exo-PA dose dependently increased miR-27a levels in activated LX2 cells (Exo-PA 25–200-μg groups, p < 0.05; Figure 1F). However, Exo-PA derived from miR-27a-inhibited LO2 cells (LO2 in-miR group) could not elevate miR-27a levels in activated LX2 cells (Figure 1G). This suggests that lipotoxic HC exosomes were the source of miR-27a and that these structures transmitted exosomal miR-27a to recipient-activated HSCs.
Lipotoxic HC-exosomal miR27a was identified to negatively regulate PINK1 in HSCs
miR27a is known to participate in the pathogenesis of Parkinson's disease by impairing mitochondrial functions through targeting of PINK1.9 PINK1 has also been reported as an important regulator of MAFLD and liver fibrosis.6,10, 11, 12 Sequence alignment indicated the presence of a miR-27a binding site in the 3′ UTR of wild-type (WT) PINK1 (Figure S4B). As expected, PINK1 mRNA and protein levels were significantly suppressed in LX2 cells overexpressing miR27a (mimic miR27a and mi-miR27a) and clearly increased in LX2 cells with miR27a inhibition (in-miR27a), regardless of TGF-β1 treatment (Figures 2A and 2B). PINK1 mRNA and protein levels were also clearly reduced in LX2 cells (±TGF-β1) incubated with Exo-PA (Figure 2C). However, the levels were not different when activated LX2 cells were incubated with Exo-PA derived from in-miR27a LO2 cells (Figure 2D). Furthermore, mi-miR27a significantly reduced the mRNA stability of PINK1 in activated LX2 cells (Figure 2E). To determine the potential role of miR-27a in regulating PINK1 expression and activity, luciferase reporters containing the WT or mutant PINK1 3′ UTR were constructed and in combination with mi-miR27a or in-miR27a used to cotransfect activated LX2 cells. mi-miR27a significantly inhibited the luciferase activity of WT PINK1 (Figure 2F). These results indicate that PINK1 is the key target of lipotoxic HC-exosomal miR-27a in activated HSCs.
Figure 2.

Lipotoxic HC-exosomal miR27a negatively regulated PINK1 in HSC
PINK1 mRNA and protein levels were measured in LX2 cells transfected with mi-miR/in-miR ± TGF-β1 (A and B), LX2 cells incubated with Exo-CN/Exo-PA ± TGF-β1 (C), and activated LX2 cells incubated with Exo-CN/Exo-PA from LO2 cells transfected with in-NC/in-miR (D). (E) LO2 cells were transfected with mi-NC or mi-miR27a for 24 h before the addition of actinomycin D (5 μg/mL; time 0). Total cellular RNA was extracted at 0, 0.25, 0.5, 1, and 2 h after treatment with actinomycin D. Relative mRNA levels of PINK1 at different time points were determined by qPCR and compared with those at time 0. (F) LX2 cells were cotransfected with luciferase reporter constructs containing the wild-type or mutant 3′ UTR of PINK1 and miR-27a mimic or inhibitor for 24 h. The relative luciferase activity was normalized to Renilla luciferase luminescence. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with (A and B) CN group, (C) Exo-CN group, (D) Exo-CN of LO2 in-NC group, (E) mi-miR at 0 h group, or (F) mi-NC PINK1 wild-type group. #p < 0.05, compared with (F) the in-NC PINK1 wild-type group.
Negative regulation of PINK1 by miR-27a impairs mitophagy and stimulates fibroblasts in activated HSCs
Overexpressing miR27a (mi-miR27a) or knocking down PINK1 (siRNA-PINK1, siPINK1) distinctly decreased the protein levels of Parkin and microtubule-associated protein light chain 3B (LC3B) and increased the p62 level in activated LX2 cells (Figure 3A). Mitochondrial respiration is usually associated with transcriptional induction of several major regulators of mitochondrial biogenesis and energy metabolism (including nuclear respiratory factor [NRF]-2A/2B/1, transcription factor A/B1/B2 mitochondrial [TFAM, TFB1M/2M], and peroxisome proliferator-activated receptor [PPAR]-α/δ). mi-miR27a or siPINK1 significantly downregulated mitochondrial transcription-related genes, including PPAR-α/σ, TFB1M/2M, NRF-2A/B, and TFAM, in activated LX2 cells (Figure S5A). In addition, we measured the oxygen consumption rate (OCR) to assess mitochondrial respiration. A 30%–40% decrease in maximal mitochondrial respiration (assessed by treatment with carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone [FCCP]) was observed in activated LX2 cells in the mi-miR27a and siPINK1 groups versus their controls (Figures 3B and S5B) and resulted in a significant reduction in the oxidative phosphorylation reserve capacity and damage to respiration. The level of mitochondrial LC3B protein was significantly decreased, mitochondrial reactive oxygen species (ROS) production was dramatically increased and an extraordinary loss of mitochondrial membrane potential (MMP) was observed (Figure 3D) in activated LX2 cells in the mi-miR27a and siPINK1 groups. Based on these data, we conclude that the ability of miR-27a to negatively regulate PINK1 impairs mitochondrial functions, particularly mitophagy, in activated HSCs.
Figure 3.

mi-miR27a and si-PINK1 significantly impaired mitochondrial functions and promoted activation of activated HSCs
Activated LX2 cells were transfected with mi-miR27a and si-PINK1. (A) Parkin, p62, and LC3B protein levels were examined via western blotting. (B) Mitochondrial respiration was assessed using Seahorse. (C) α-SMA, cyclin D1, and PCNA proteins were detected in activated LX2 cells via western blotting. (D) LC3B (red IF) and COX4 (green IF) proteins and mtSOX red and mtCMXRos red were observed via IF staining. Scale bar, 25 μm. (E) Representative images and statistical analysis of cell morphology changes in PHSCs at day 7 are shown. Scale bar, 25 μm. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with the (B and D) mi-NC group, (E) mi-NC PHSC group at day 5 or 7; #p < 0.05, compared with the (B and D) si-NC group, (E) si-NC PHSC group at day 5 or 7.
We further assessed the effects of mi-miR27a or siPINK1 on activation, proliferation, and autophagy in HSCs. mi-miR27a or siPINK1 significantly increased the expression of profibrotic genes or proteins (including α-smooth muscle actin [α-SMA], fibronectin, and platelet-derived growth factor receptor [PDGFR]-β) in activated LX2 cells (Figures 3C and S5E). mi-miR27a or siPINK1 promoted activation of LX2 cells by significantly increasing the red IF values for α-SMA protein (a marker of fibrosis, Figure S5C). mi-miR27a or siPINK1 clearly promoted transdifferentiation of PHSCs because the percentage of activated cells obviously increased at days 5 and 7 (Figures 3E and S6). mi-miR27a or siPINK1 also distinctly promoted the expression of pro-proliferation genes and proteins (cyclin D1 and proliferating cell nuclear antigen [PCNA], Figures 3C and S5E) and notably increased cell proliferation, as measured by ethynyl-20-deoxyuridine (EdU) staining (Figure S5D), in activated LX2 cells. mi-miR27a or siPINK1 further markedly reduced levels of the autophagy-related protein lysosomal membrane protein (LAMP)-1/2 while increasing the levels of mammalian target of rapamycin (mTOR) and Rheb (Figure S5F). Based on this, we postulate that the negative regulation of PINK1 by miR-27a stimulates activation, promotes proliferation and inhibits autophagy in activated HSCs.
Lipotoxic HC-exosomal miR27a was the key player in inhibition of PINK1-mediated mitophagy and activation of HSCs
To determine whether lipotoxic HC-exosomal miR27a is a direct factor influencing HSC mitophagy and activation, HSCs were further incubated with Exo-PA derived from LO2 cells pretreated with miR27a inhibitor (in-miR27a) or miRNA-negative control (in-NC).
Exo-PA derived from in-NC-LO2 cells clearly reduced PINK1, Parkin, and LC3B levels and increased p62 protein levels; however, Exo-PA from in-miR27a-LO2 cells did not affect these proteins in activated LX2 cells (Figure 4A). Exo-PA from in-NC-LO2 cells also significantly downregulated the expression of mitochondrial transcription genes (including PPAR-α/σ, TFB1M/2M, NRF-2A/B, and TFAM), while Exo-PA from in-miR27a-LO2 cells did not affect these genes in activated LX2 cells (Figure S7A). Exo-PA from in-NC-LO2 cells led to a 40% decrease in maximal mitochondrial respiration, while Exo-PA from in-miR27a-LO2 cells did not affect mitochondrial respiration in activated LX2 cells (Figures 4B and S7B). Exo-PA from in-NC-LO2 cells dramatically reduced mitochondrial LC3B protein levels (Exo-PA group versus Exo-CN group from in-NC-LO2 cells: a 0.54-fold decrease in the red IF value), obviously increased mitochondrial ROS production (Exo-PA group versus Exo-CN group from in-NC-LO2 cells: a 2.39-fold increase in the red IF value), and markedly reduced MMP protein levels (Exo-PA group versus Exo-CN group from in-NC-LO2 cells: a 0.32-fold decrease in the red IF value) in activated LX2 cells (Figure 4C). Interestingly, Exo-PA from in-miR27a-LO2 cells did not produce these changes in activated LX2 cells (Figure 4C).
Figure 4.

The functions of lipotoxic HC-exosomal miR-27a in PINK1-related mitophagy and activation in HSCs
The functions of activated LX2 cells or PHSCs incubated with Exo-CN/Exo-PA derived from in-NC/in-miR27a LO2 cells were detected. (A) PINK1, Parkin, p62, and LC3B protein levels in activated LX2 cells were examined via western blotting. (B) Mitochondrial respiration of activated LX2 cells was assessed using Seahorse. (C) LC3B (red IF) and COX4 (green IF) proteins, mtSOX red, and mtCMXRos red in activated LX2 cells were observed via IF staining. Scale bar, 25 μm. (D) Representative images and statistical analysis of cell morphology changes in PHSCs at day 7 are shown. Scale bar, 25 μm. (E) Fibrosis-related (α-SMA), proliferation-related (cyclin D1 and PCNA) proteins were detected in activated LX2 cells via western blotting. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with the (B and C) Exo-CN group from in-NC LO2 cells, (D) Exo-CN group from in-NC LO2 cells among day 5 or 7 PHSCs.
On the other hand, Exo-PA from in-NC-LO2 cells distinctly increased the expression of pro-fibrosis genes or proteins (α-SMA, fibronectin, and PDGFR-β), increased pro-proliferation genes and proteins (cyclin D1 and PCNA), and reduced autophagy-related proteins (LAMP1/2, autophagy protein 7 [Atg7], Beclin1), but increased other autophagy-related proteins (mTOR and Rheb) in activated LX2 cells (Figures 4E, S7C, S8B, and S8C). Interestingly, these effects could be blocked by the miR-27a inhibitor (Figures 4E, S7C, S8B, and S8C). Exo-PA from in-NC-LO2 cells clearly promoted PHSC transdifferentiation (the percentage of activated cells increased on days 5 and 7), but the effect was restored by in-miR27a (Figures 4D and S8A). Exo-PA from in-NC-LO2 cells also strongly activated cells (as determined by red IF indicating α-SMA protein expression) and distinctly increased cell proliferation (assessed by EdU staining) in activated LX2 cells, and this effect was reversed by in-miR27a (Figures S7D and S7E).
These findings indicate that lipotoxic HC-exosomal miR27a can inhibit PINK1-related mitophagy and mitochondrial functions, promote activation and proliferation, and suppress autophagy in activated HSCs, and this can be reversed by a miR27a inhibitor.
High serum miR-27a and a lack of hepatic PINK1-mediated mitophagy were positively related to MAFLD progression in mice
To identify the role of miR-27a in negatively regulating PINK1 in MAFLD progression in vivo, WT mice were fed an LFD (low-fat diet) or HFD (high-fat diet) and then subjected to CCl4 (carbon tetrachloride) or olive oil (control group, CN) treatments. The liver index (liver/body weight) was significantly reduced; the serum alanine aminotransferase (ALT) level was elevated; and liver steatohepatitis, fibrosis, and lipid deposition were aggravated in HFD ± CCl4 mice compared with LFD ± CCl4 mice (Figures S9A–S9D).
A remarkable increase in serum miR-27a (Figure 5B) and decrease in hepatic PINK1 (Figure 5C) were observed in HFD ± CCl4 mice compared with LFD ± CCl4 mice. Dramatic inhibition of the PINK1-mediated mitophagy pathway (PINK1, Parkin, and LC3B protein levels were decreased, but the p62 level was increased) was shown in HFD ± CCl4 mice compared with LFD ± CCl4 mice (Figure 5A). Under an electronic microscope, the mitochondria were observed to be markedly swollen and abnormal in the livers of CCl4 mice. Compared with the LFD + CCl4 mice, mitochondria were obviously fused and lost their normal shape in the HFD + CCl4 mice (inside the red circle, Figure 5D). An HFD also resulted in a decrease in autophagosomes (shown by the yellow arrow) in CCl4-induced cirrhotic mice (Figure 5D). Furthermore, decreased mitochondrial respiration in response to HFD ± CCl4 was associated with alterations in major transcriptional regulators, such as NRF-2A/2B/1, TFAM, TFB1M/2M, and PPAR-α/δ, compared with levels observed in LFD ± CCl4 mice (Figure S9E). A significant reduction in mitochondrial LC3B protein expression was observed in HFD ± CCl4 mice compared with LFD ± CCl4 mice (orange IF; HFD versus LFD group: a 0.67-fold decrease; HFD + CCl4 versus LFD + CCl4 group: a 0.56-fold decrease; Figure 5E).
Figure 5.

Serum miR27a upregulation suppressed hepatic PINK1-induced mitophagy and aggravated liver fibrosis in MAFLD mice
WT mice were divided into four groups: LFD, HFD, LFD + CCl4, and HFD + CCl4 (each group: n = 10). (A) The protein levels of PINK1, Parkin, LC3B, and p62 were assessed via western blotting. (B and C) The expression of serum miR-27a and liver PINK1 mRNA was detected via PCR. (D) Mitochondria (red cycle) and autophagosomes (yellow arrow) were detected via TEM in LFD/HFD + CCl4 mice. Scale bar, 2 μm. (E) LC3B (red IF) and COX4 (green IF), α-SMA (red IF) and PINK1 (green IF), and α-SMA (red IF) and PCNA (green IF) protein expression in liver tissues was observed via IF staining. Scale bar, 25 μm. (F) The protein levels of α-SMA, cyclin D1, and PCNA were assessed via western blotting. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with mice in the LFD group; #p < 0.05, compared with mice in the LFD + CCl4 group.
Obvious upregulation of fibrosis-related genes or proteins (α-SMA, collagen I [ColI], fibronectin, or PDGFR-β) and proliferation-related genes or proteins (cyclin D1 or PCNA) was found in HFD + CCl4 mice compared with LFD + CCl4 mice (Figures 5F and S9F). The opposite presentation of PINK1 protein (showed green IF) and α-SMA protein (showed red IF) expression was seen in HFD ± CCl4 mice compared with the LFD ± CCl4 mice (the IF value of PINK1/α-SMA; HFD versus LFD group: a 0.65-fold decrease; HFD + CCl4 group versus LFD + CCl4 group: a 0.79-fold decrease; Figure 5E). A distinct increase in liver cells coexpressing α-SMA (shown by red IF) and PCNA (shown by green IF) was observed in HFD + CCl4 mice compared with LFD + CCl4 mice (the IF value of PCNA/α-SMA; HFD versus LFD group: a 1.21-fold increase; HFD + CCl4 versus LFD + CCl4 group: a 1.27-fold increase; Figure 5E). In addition, a noticeable decrease in pro-autophagy proteins (LAMP1/2, Atg7, Beclin1) and an increase in anti-autophagy proteins (mTOR) were observed in HFD + CCl4 mice compared with LFD + CCl4 mice (Figure S9G).
However, a high serum miR-27a level and loss of PINK1 consistently led to mitochondrial damage, fibrosis, and proliferation acceleration, and autophagy inhibition in the livers of HFD ± CCl4 mice compared with observations in LFD ± CCl4 mice.
Transplantation of lipotoxic HC-exosomal miR27a exerted an aggravating effect on hepatic mitochondrial damage and fibrosis in MAFLD mice
To elucidate the role of lipotoxic HC-exosomal miR27a in mitochondrial and fibrotic liver injury in vivo, MCD (methionine-choline-deficient diet)-fed WT mice were subjected to lipotoxic/control LO2 exosomes and CCl4/olive oil treatments (MCD, MCD + CCl4, and MCD + CCl4 + Exo groups). MCD + CCl4 + Exo mice exhibited an obviously reduced liver index and an elevated serum ALT level, followed by a more severe degree of steatohepatitis, fibrosis, and lipidosis than MCD ± CCl4 mice (Figures S10A–S10D).
Serum miR-27a levels gradually increased with progression of MAFLD injury in MCD + CCl4 mice during the 2- to 6-week treatment period (Figure 6A). MCD + CCl4 + Exo mice also exhibited a remarkable increase in the serum miR-27a level and a decrease in hepatic PINK1 expression compared with MCD ± CCl4 mice (Figures 6B and 6C). MCD + CCl4 + Exo treatment mostly inhibited PINK1-mediated mitophagy signaling (PINK1, Parkin, and LC3B protein levels decreased, but the p62 level increased; Figure 6D) and led to the most reduced levels of mitochondrial LC3B protein (orange IF; MCD + CCl4 + Exo group versus MCD + CCl4 group: a 0.48-fold decrease; MCD + CCl4 + Exo group versus MCD group: a 0.17-fold decrease; Figure 6F) compared with MCD ± CCl4 treatment in mice. Consistently, the mRNA levels of major mitochondrial transcriptional regulators (NRF-2A/2B/1, TFAM, TFB1M/2M, and PPAR-α/δ) were remarkably decreased in MCD + CCl4 + Exo mice compared with levels in MCD ± CCl4 mice (Figure S10E).
Figure 6.

Transplantation of lipotoxic HC-exosomal miR27a exerted an aggravating effect on mitochondrial damage and fibrosis in MAFLD mice
MCD-fed WT mice were established and divided into three groups (Ct, CCl4, and CCl4 + Exo, each group: n = 10). Relative serum miR-27a levels (A) in MCD + CCl4 mice of different ages (0, 2, 4, and 6 weeks) were detected via PCR. Serum miR-27a (B) and hepatic PINK1 (C) mRNA levels were detected via PCR. (D and E) The levels of proteins in the PINK1 pathway (PINK1, Parkin, LC3B, and p62), the fibrosis pathway (α-SMA), and the proliferation pathway (cyclin D1 and PCNA) were assessed via western blotting. (F) LC3B (red IF) and COX4 (green IF) proteins, α-SMA (red IF) and PINK1 (green IF) proteins, and α-SMA (red IF) and PCNA (green IF) proteins were determined by IF staining of liver tissues in MCD mice. Scale bar, 25 μm. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with (A) MCD + CCl4 mice (0 weeks), (B, C, and F) MCD mice in the Ct group; #p < 0.05, compared with MCD mice in the CCl4 group.
Furthermore, MCD + CCl4 + Exo treatment significantly activated fibrosis signaling (increased α-SMA protein level), stimulated proliferation signaling (increased cyclin D1 and PCNA protein levels), and suppressed autophagy signaling (decreased LAMP1/2, Atg7, and Beclin1 protein levels, but an increased mTOR level) compared with MCD ± CCl4 treatment in mice (Figures 6E and S10G). In addition, the mRNA levels of fibrosis signaling (α-SMA, ColⅠ, fibronectin, or PDGFR-β) and proliferation signaling (cyclin D1 or PCNA) factors were markedly elevated in MCD + CCl4 + Exo mice compared with levels in the MCD ± CCl4 mice (Figure S10F). The opposite trend was observed for PINK1 (green IF) and α-SMA (red IF) protein levels in MCD + CCl4 + Exo mice compared with MCD ± CCl4 mice (PINK1/α-SMA; MCD + CCl4 + Exo group versus MCD + CCl4 group: a 0.53-fold decrease; MCD + CCl4 + Exo group versus MCD group: a 0.39-fold decrease; Figure 6F). However, identical increases in PCNA (green IF) and α-SMA (red IF) protein levels were observed in MCD + CCl4 + Exo mice compared with MCD ± CCl4 mice (PCNA/α-SMA; MCD + CCl4 + Exo group versus MCD + CCl4 group: a 1.54-fold increase; MCD + CCl4 + Exo group versus MCD group: a 1.86-fold increase; Figure 6F).
These data demonstrate that transplantation of lipotoxic HC-exosomal miR27a damaged mitochondrial functions and aggravated MAFLD-related fibrosis in vivo.
Exosomal miR27a is also the key player in mitochondrial and fibrotic liver injury in MAFLD mice
To further determine the necessary role of lipotoxic exosomal miR27a in mitochondrial and fibrotic liver injury in vivo, purified exosomes were isolated from LO2 cells that were pretransfected with mi-miR27a or in-miR27a and then transplanted into HFD + CCl4 mice (divided into Exo-mi-CN, Exo-mi-miR27a, Exo-in-CN, and Exo-in-miR27a groups).
The liver index was apparently reduced and the serum ALT level was increased in mice in the Exo-mi-miR27a group compared with those values in mice in the Exo-mi-CN group; however, there was no difference between the Exo-in-CN and Exo-in-miR27a groups (Figure S11A). In addition, a more severe degree of steatohepatitis, fibrosis, and lipidosis was seen in the Exo-mi-miR27a group than in the Exo-mi-CN group but was relieved in the Exo-in-CN and Exo-in-miR27a groups (Figures S11B–S11D).
Exo-mi-miR27a (exosomes overexpressing miR27a) treatment resulted in an approximately 2-fold increase in miR27a in serum exosomes and a one-half decrease in PINK1 in the liver, and this effect was blocked by Exo-in-miR27a (exosomes with miR27a inhibition; Figure 7A). Exo-mi-miR27a caused a greater increase in the abnormal appearance of damaged mitochondria (Figures 7B and a decrease in autophagosome vesicles) that was accompanied by a consistently greater increase in CD63 and α-SMA coexpressing cells (shown by yellow IF; Figure 7E) in HFD + CCl4 mice. Simultaneously, Exo-mi-miR27a inhibited mitophagy-related proteins (PINK1/Parkin/LC3B decreased and p62 increased; Figure 7C) and resulted in missing mitochondrial LC3B proteins (orange IF; Exo-mi-miR group versus Exo-mi-CN group: a 0.65-fold decrease; Figure 7F), which could be blocked by Exo-in-miR27a. At the same time, the expression of mitochondrial transcriptional mRNAs (PPAR-α/σ, TFBM1/2, and TFAM) was significantly decreased in Exo-mi-miR27a mice compared with control mice, and this decrease was prevented by Exo-in-miR27a (Figure S11E).
Figure 7.

Lipotoxic HC-exosomal miR27a was the key player in mitochondrial and fibrotic liver injury in MAFLD mice
Different purified exosomes were transplanted into HFD + CCl4 mice, and the mice were divided into four groups (Exo-mi-CN, Exo-mi-miR, Exo-in-CN, and Exo-in-miR, each group: n = 10). (A) Serum exosomal miR-27a and hepatic PINK1 mRNA were detected via PCR. (B) Mitochondria (shown by red circles) and autophagosomes (shown by yellow arrows) were assessed via TEM. Scale bar, 2 μm (in the Exo-mi-CN and Exo-mi-miR groups). (C and D) The levels of proteins in the PINK1 pathway (PINK1, Parkin, LC3B, and p62), the fibrosis pathway (α-SMA), and the proliferation pathway (cyclin D1 and PCNA) in HFD + CCl4 mouse livers. (E) α-SMA (red IF) and CD63 (green IF) protein expression in HFD + CCl4 mice in the Exo-mi-CN and Exo-mi-miR groups was assessed via IF staining. Scale bar, 25 μm. (F) LC3B (red IF) and COX4 (green IF) proteins, α-SMA (red IF) and PINK1 (green IF) proteins, and α-SMA (red IF) and PCNA (green IF) proteins were detected via IF staining. Scale bar, 25 μm. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05, compared with HFD + CCl4 mice in the Exo-mi-CN group.
Moreover, Exo-mi-miR27a stimulated fibrosis- and proliferation-related proteins (increased α-SMA, cyclin D1, and PCNA; Figure 7D) and suppressed autophagy-related proteins (decreased LAMP1/2, Atg7, and Beclin1, and increased mTOR; Figure S11G), and this effect was also blocked by Exo-in-miR27a. The expression of pro-fibrotic mRNAs (α-SMA, ColⅠ, fibronectin, and PDGFR-β) and pro-proliferation mRNAs (cyclin D1 and PCNA) was markedly increased in Exo-mi-miR27a mice, and this effect was similarly prevented by Exo-in-miR27a (Figure S11F). The hepatic proteins α-SMA (red IF) and PINK1 (green IF) exhibited the opposite expression trend (PINK1/α-SMA; Exo-mi-miR27a group versus Exo-mi-CN group: a 0.60-fold decrease); however, α-SMA (red IF) and PCNA (green IF) exhibited a consistent expression trend (PCNA/α-SMA; Exo-mi-miR27a group versus Exo-mi-CN group: a 1.58-fold increase) in Exo-mi-miR27a mice compared with control mice, and this could be reversed by Exo-in-miR27a (Figure 7F).
Based on the above results, lipotoxic HC exosomes could not exert a promoting effect on mitochondrial and fibrotic liver injury without miR-27a in MAFLD mice.
Hepatic exosomal-miR27a inhibition of PINK1-mediated mitophagy was correlated with the severity of liver fibrosis in MAFLD patients
To elucidate the factors involved in exosome-induced liver fibrosis in MAFLD patients, we examined whether exosomal-miR27a-mediated inhibition of hepatic PINK1-induced mitophagy resulted in this response in MAFLD progression. Greater numbers of CD63 (green IF)- and α-SMA (red IF)-positive cells were indicative of increased levels of hepatic exosomes (CD63/α-SMA; ALF group versus MLF group: a 1.35-fold increase; Figure 8A) and were interrelated with the progression of MAFLD in patients with ALF (MAFLD-ALF). In MAFLD-ALF patients, a lack of mitochondrial LC3B proteins (orange IF; ALF group versus MLF group: a 0.35-fold decrease; Figure 8B) in livers was indicative of general inhibition of mitophagy. A negative correlation between hepatic α-SMA (red IF) and PINK1 (green IF) protein expression indicated that the PINK1-mitophagy pathway was inhibited in MAFLD-ALF patients (PINK1/α-SMA; ALF group versus MLF group: a 0.24-fold decrease; Figure 8C). Compared with MLF tissues, PINK1, Parkin, and LC3B proteins were significantly decreased and p62 was markedly increased in ALF tissues from three pairs of MAFLD patients (Figure 8D). Interestingly, both MAFLD-MLF and ALF patients exhibited higher serum miR27a levels than healthy control individuals (p < 0.05; Figure 8F). Linear regression analysis suggested that the expression of serum exosomal miR27a was positively correlated with the degree of liver fibrosis (based on the liver stiffness measurement (LSM) value determined by FibroScan, r2 = 0.418, p < 0.001; Figure 8E) and the fat content (based on the CAP value determined by FibroScan, r2 = 0.420, p < 0.001; Figure 8E). This phenomenon, in which exosomal miR-27a inhibits hepatic PINK1-induced mitophagy, was also demonstrated to be associated with the progression of MAFLD in patients with liver fibrosis.
Figure 8.

Hepatic exosomal-miR27a-mediated inhibition of PINK1-induced mitophagy was correlated with the severity of liver fibrosis in MAFLD patients
Liver tissues from MAFLD patients (MLF and ALF groups, each group n = 3) were examined, and serum exosomes were assessed in another MAFLD cohort (control, MLF, and ALF groups, each group n = 6–10). (A–C) α-SMA (red IF) and CD63 (green IF) proteins were assessed via IF staining of liver tissues. Scale bar, 50 μm. α-SMA (red IF) and PINK1 (green IF) proteins, and LC3B (red IF) and COX4 (green IF) proteins were assessed via IF staining of liver tissues. Scale bar, 25 μm. (D) The levels of protein in the PINK1-mediated mitophagy pathway (PINK1, Parkin, LC3B, and p62) were assessed via western blotting. (E) The relative serum exosomal miR27a level was plotted against the LSM and CAP levels. Linear regression analysis was performed. (F) Scatterplot of serum miR-27a expression levels assessed in MAFLD patients. All quantifications are presented as the mean ± SD, and p values were calculated using an unpaired Student's t test. Statistical significance: ∗p < 0.05 (A–D) compared with the MLF group, (F) compared with control group.
Discussion
MAFLD encompasses a spectrum of liver disorders that range from benign steatosis to steatohepatitis (NASH) or even cirrhosis.13 The incidence of NASH-related cirrhosis is rapidly increasing, as is the need for liver transplantation.14 A key event in the progression of NASH is differentiation of HSCs to myofibroblasts. However, whether and how HCs affect HSC transdifferentiation or activation in MAFLD remain unclear. Our pioneering study reveals that lipotoxic HC-derived exosomal miR27a plays a pivotal role in inhibiting PINK1-mediated mitophagy, which could result in activation of HSCs and promote MAFLD-related liver fibrosis.
Cellular interactions among all resident liver cell populations are important in the pathogenesis and progression of liver diseases, and exosomes are crucial in intercellular transduction.15 Exosomal miRNAs have particularly important epigenetic functions, and they can posttranscriptionally regulate gene expression.16 Increase and change in exosome contents were found in HCs due to lipid accumulation.17 Exploring the key exosomal miRNAs is meaningful to understanding MAFLD-related fibrosis. We found that serum exosomal miR-27a levels were significantly upregulated in MAFLD-ALF patients and in HFD-fed cirrhotic mice. In addition, lipotoxic HCs released additional exosomes carrying large amounts of miR27a, and these exosomes were the major source of miR27a in vitro. Therefore, for the first time, we demonstrated that serum exosomal miR-27a levels are positively correlated with the severity of liver fibrosis and could be used to effectively distinguish ALF in MAFLD patients. Recent findings have suggested that HC exosomes are essential players in regulating HSC proliferation and differentiation.18, 19, 20 In our study, through an in vitro primary cell culture experiment that mimicked proximal communication between HCs and HSCs, lipotoxic HC exosomes were demonstrated to be preferentially and rapidly taken up by activated HSCs. Then, we innovatively assumed that lipotoxic HC exosomes carried and specifically transmitted large amounts of miR27a to recipient-activated HSCs.
Previous studies have reported that miR-27a is overexpressed in cirrhotic patients and rats.21,22 miR27a can accelerate the proliferation and activation of HSCs and induce triglyceride accumulation in MAFLD mice.18,23 In Parkinson's disease,9 miR-27a was first demonstrated to regulate mitophagy by targeting PINK1 (the most important regulator of mitophagy). Thus, we focused on the relationship between miR-27a- and PINK1-mediated mitophagy, which was unexplored in MAFLD-related liver fibrosis. Mitophagy is an efficient means of clearing injured mitochondria to maintain homeostasis. However, few studies have investigated mitophagy in HSCs. Excessive mitophagy promotes cell death in HSCs;24 in contrast, inhibiting Parkin-mediated mitophagy suppresses apoptosis in HSCs.25 PINK1 recruits and activates the E3 ubiquitin ligase activity of Parkin, which in turn initiates mitophagy. Inhibition of PINK1-mediated mitophagy might activate the PDGFR/PI3K/AKT pathway and thus contribute to the development of pulmonary fibrosis.26, 27, 28 However, how lipotoxic HC-exosomal miR-27a regulates PINK1-mediated mitophagy and then influences recipient-activated HSCs is completely unknown.
Our data exhaustively revealed a new phenomenon in which mitophagy is significantly inhibited in parallel with exacerbated liver fibrosis in vitro and in vivo. In addition, we verified that the mechanism involved suppression of the PINK1-mediated mitophagy pathway in recipient HSCs by lipotoxic HC-exosomal miR27a. Negative regulation of PINK1 expression by miR27a impairs mitochondrial functions (leading to a substantially abnormal morphology; inhibition of mitophagy, transcription, respiration, and MMP; and an increase in ROS production), which stimulates HSC-derived fibroblasts (promoting activation and proliferation and suppressing autophagy), and finally accelerates liver fibrosis (aggravating inflammation, lipid deposition, and fibrosis) in vitro and in vivo.
To further confirm the novel mechanism of lipotoxic HC exosomes, we first reported that lipotoxic HC-exosomal miR27a can exercise identical abilities to promote recipient HSC activation by negatively targeting PINK1-induced mitophagy in vitro and in vivo. Both in vitro and in vivo, exosomes derived from HCs with knockdown of miR-27a could not negatively target PINK1-induced mitophagy and could not aggravate MAFLD-related liver fibrosis. Based on these results, it is clear that miR-27a carried in lipotoxic HC exosomes is the key effector that inhibits mitochondrial function and promotes fibrotic liver injury in MAFLD.
Our findings identified a link between mitophagy and hepatic fibrosis in MAFLD and provide a mechanism underlying the regulation of mitophagy-related apoptosis of HSCs. Simultaneously, our findings suggest for the first time that lipotoxic HCs could provide a rich source of exosomal-miR27a, which is the essential player in negatively targeting PINK1-induced mitophagy to subsequently aggravate MAFLD-related liver fibrosis.
However, the underlying mechanisms involved in miR-27a-PINK1 signaling-mediated HSC activation remain mostly unknown and require further study. Insufficient PINK1 gene function could activate TGF-β1-Smad2/3, PDGFR/mTOR, or Akt1 signaling in fibrotic disease.29, 30, 31 In contrast, we showed that miR-27a-PINK1 promoted HSC activation and proliferation by stimulating α-SMA, fibronectin, PDGFR-β, cyclin D1, and PCNA expression, and this is a critical event involved in the progression of MAFLD in patients with liver fibrosis.
In conclusion, our study indicates that lipotoxic HC-derived exosomes are preferentially taken up by activated HSCs. Exosomal miR27a negatively regulates hepatic PINK1-mediated mitophagy, which can enhance HSC activation and proliferation and thus accelerate liver fibrosis in MAFLD. These results suggest that inhibition of HC-exosomal miR-27a expression could be used as a potential diagnostic marker and therapeutic target for MAFLD-related liver fibrosis.
Materials and methods
Human study
A cohort of 46 individuals was enrolled. (1) Sixteen biopsy-confirmed MAFLD patients (MLF: S0-2; ALF: S3-4; each group n = 8) were enrolled, and the degrees of steatosis and fibrosis were based on the pathological steatosis scores (F0: <5% steatosis; ∼F3: >50% steatosis) and Scheuer's classification. (2) Thirty noninvasive diagnostic MAFLD patients (healthy control, MLF, and ALF, each group n = 10) were enrolled, and the steatosis and fibrosis degrees were based on CAP and LSM values determined by FibroScan. The control group consisted of ten matched healthy subjects with normal body mass index, liver enzyme levels, and FibroScan values. Details of these two cohorts are described in Tables S1 and S2. All enrolled patients provided written informed consent, and the study was approved by the ethics committee of Shanghai General Hospital.
Animal experiments
A total of 110 male C57BL/6 mice were randomized into 3 groups. (1) Forty mice were divided into 4 subgroups (LFD, HFD, LFD + CCl4, and HFD + CCl4; each group n = 10). (2) Thirty mice were divided into 3 subgroups (MCD, MCD + CCl4, and MCD + CCl4 + Exo; each group n = 10). Transplanted exosomes were derived from LO2 cells (incubated with 200 μM PA or 3% BSA as the vehicle control). (3) Forty mice were divided into 4 subgroups (Exo-mi-CN, Exo-mi-miR, Exo-in-CN, and Exo-in-miR; each group n = 10). Transplanted exosomes were derived from LO2 cells with different pretransfection treatments (miR27a mimic [mi-miR] and miR negative control mimic [mi-NC]; miR27a inhibitor [in-miR] and miR negative inhibitor [in-NC]). The details are described in the supplemental information and Table S3.
Exosome experiments
Exosomes were isolated from the serum of humans and mice and from cell culture medium via an ultracentrifugation method (Beckman Coulter, Miami, FL).32 To obtain lipotoxic exosomes, hepatic cells were treated with 200 μM PA for 24 h.
The morphology of exosomes was identified using transmission electron microscopy (TEM) (JEOL, Tokyo, Japan). The surface markers of exosomes, CD63 and TSG101 (1:1,000; Abcam, Cambridge, MA), were identified via western blotting. The size distribution and concentration of exosomes were measured using nanoparticle tracking analysis (Malvern Instruments, Worcestershire, UK).
For the exosome uptake experiment, exosomes were labeled with the red fluorescent dye PKH26 (Sigma-Aldrich) and added to the medium of cultured cells for 6 h. Then, the cells were fixed using 4% paraformaldehyde, followed by permeabilization with 0.5% Triton X-100 and staining with 1% DAPI (Sigma-Aldrich). The uptake of exosomes was observed under a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
MitoSOX and MitoTracker Red CMXRos
Mitochondrial ROS production was analyzed using a MitoSOX Red staining Kit (Invitrogen, Carlsbad, CA). The MMP was detected using a MitoTracker Red CMXRos Kit (Invitrogen). Representative images were captured using a TCS SP8 CARS confocal fluorescence microscope (Leica Microsystems). These experiments were repeated three times.
Mitochondrial respiration assay
The mitochondrial OCR was assessed using a Seahorse XF96 Analyzer (Seahorse Bioscience, North Billerica, MA). The cells were incubated with Seahorse medium (pH adjusted to 7.4) for 1 h in a 37°C air incubator. Basal OCR was measured. Then, different drugs, including 1.5 μM oligomycin (ATP uncoupler), 1.0 μM FCCP (electron transport chain accelerator), and 0.5 μM rotenone (complex I inhibitor)/antimycin A (complex III inhibitor), were added automatically during the mitochondrial stress test. The post-exposure OCR was measured, and the experiment was repeated three times.
Statistical analysis
Data are presented as the mean ± standard deviation. Differences between two groups were analyzed with an unpaired Student's t test. One-way analysis of variance followed by Student-Newman-Keuls analysis was used for comparisons of multiple groups. All statistical analyses were performed using SPSS 19.0 software (SPSS, Chicago). A p value < 0.05 was considered statistically significant.
Other details, including histology, TEM, serum ALT activity, IF, cell culture and transfection, EdU staining, luciferase reporter assays, quantitative real-time PCR, and western blotting, are described in the Materials and methods.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (nos. 81770597 and 82100607) and the National 13th Five-Year Major Project (no. 2017ZX10203202003005). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
M.-Y.X. conceived the original ideas, designed the study, and wrote the manuscript. X.L., Z.-X.X., J.-C.W., and S.-Z.L. carried out the experiments and data analysis.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.10.022.
Supplemental information
and Tables S1–S6
References
- 1.Angulo P., Kleiner D.E., Dam-Larsen S., Adams L.A., Bjornsson E.S., Charatcharoenwitthaya P., Mills P.R., Keach J.C., Lafferty H.D., Stahler A., et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149:389–397. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ekstedt M., Hagström H., Nasr P., Fredrikson M., Stål P., Kechagias S., Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–1554. doi: 10.1002/hep.27368. [DOI] [PubMed] [Google Scholar]
- 3.Archer S.L. Mitochondrial dynamics-mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 4.Kim C.S., Kwon Y., Choe S.Y., Hong S.M., Yoo H., Goto T., Kawada T., Choi H.S., Joe Y., Chung H.T., et al. Quercetin reduces obesity induced hepatosteatosis by enhancing mitochondrial oxidative metabolism via heme oxygenase-1. Nutr. Metab. (Lond). 2015;12:33. doi: 10.1186/s12986-015-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meira Martins L.A., Vieira M.Q., Ilha M., de Vasconcelos M., Biehl H.B., Lima D.B., Schein V., Barbé-Tuana F., Borojevic R., Guma F.C. The interplay between apoptosis, mitophagy and mitochondrial biogenesis induced by resveratrol can determine activated hepatic stellate cells death or survival. Cell Biochem. Biophys. 2015;71:657–672. doi: 10.1007/s12013-014-0245-5. [DOI] [PubMed] [Google Scholar]
- 6.Kang J.W., Hong J.M., Lee S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal. Res. 2016;60:383–393. doi: 10.1111/jpi.12319. [DOI] [PubMed] [Google Scholar]
- 7.Ying W., Riopel M., Bandyopadhyay G., Dong Y., Birmingham A., Seo J.B., Ofrecio J.M., Wollam J., Hernandez-Carretero A., Fu W., et al. Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell. 2017;171:372–384. doi: 10.1016/j.cell.2017.08.035. [DOI] [PubMed] [Google Scholar]
- 8.Povero D., Panera N., Eguchi A., Johnson C.D., Papouchado B.G., de Araujo Horcel L., Pinatel E.M., Alisi A., Nobili V., Feldstein A.E. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol. Gastroenterol. Hepatol. 2015;1:646–663.e4. doi: 10.1016/j.jcmgh.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J., Fiesel F.C., Belmonte K.C., Hudec R., Wang W.X., Kim C., Nelson P.T., Springer W., Kim J. MiR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1) Mol. Neurodegener. 2016;11:55. doi: 10.1186/s13024-016-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X., Hao M., Liu Y., Ma X., Lin W., Xu Q., Zhou H., Shao N., Kuang H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019;864:172715. doi: 10.1016/j.ejphar.2019.172715. [DOI] [PubMed] [Google Scholar]
- 11.Park J.S., Lee D.H., Lee Y.S., Oh E., Bae K.H., Oh K.J., Kim H., Bae S.H. Dual roles of ULK1 (unc-51 like autophagy activating kinase 1) in cytoprotection against lipotoxicity. Autophagy. 2019;25:1–20. doi: 10.1080/15548627.2019.1598751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang N.P., Liu X.J., Xie L., Shen X.Z., Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Invest. 2019;99:749–763. doi: 10.1038/s41374-018-0177-6. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim S.H., Hirsova P., Gores G.J. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. 2018;67:963–972. doi: 10.1136/gutjnl-2017-315691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandrakumaran A., Siddiqui M.S. Implications of nonalcoholic steatohepatitis as the cause of end-stage liver disease before and after liver transplant. Gastroenterol. Clin. North. Am. 2020;49:165–178. doi: 10.1016/j.gtc.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masyuk A.I., Masyuk T.V., Larusso N.F. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J. Hepatol. 2013;59:621–625. doi: 10.1016/j.jhep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemoinne S., Thabut D., Housset C., Moreau R., Valla D., Boulanger C.M., Rautou P.E. The emerging roles of microvesicles in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2014;11:350–361. doi: 10.1038/nrgastro.2014.7. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y.S., Kim S.Y., Ko E., Lee J.H., Yi H.S., Yoo Y.J., Je J., Suh S.J., Jung Y.K., Kim J.H., et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017;7:3710. doi: 10.1038/s41598-017-03389-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devhare P.B., Sasaki R., Shrivastava S., Di Bisceglie A.M., Ray R., Ray R.B. Exosome-mediated intercellular communication between hepatitis C virus-infected hepatocytes and hepatic stellate cells. J. Virol. 2017;91:e02225. doi: 10.1128/JVI.02225-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X., Chen R., Kemper S., Brigstock D.R. Extracellular vesicles from hepatocytes are therapeutic for toxin-mediated fibrosis and gene expression in the liver. Front. Cell Dev. Biol. 2020;7:368. doi: 10.3389/fcell.2019.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X.W., Zhou J.C., Peng D., Hua F., Li K., Yu J.J., Lv X.X., Cui B., Liu S.S., Yu J.M., et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy. 2020;16:782–796. doi: 10.1080/15548627.2019.1635383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H., Yan X.L., Guo X.X., Shi M.J., Lu Y.Y., Zhou Q.M., Chen Q.L., Hu Y.Y., Xu L.M., Huang S., et al. MiR-27a as a predictor for the activation of hepatic stellate cells and hepatitis B virus-induced liver cirrhosis. Oncotarget. 2017;9:1075–1090. doi: 10.18632/oncotarget.23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge S., Wang X., Xie J., Yi X., Liu F. Deep sequencing analysis of microRNA expression in porcine serum-induced hepatic fibrosis rats. Ann. Hepatol. 2014;13:439–449. [PubMed] [Google Scholar]
- 23.Teimouri M., Hosseini H., Shabani M., Koushki M., Noorbakhsh F., Meshkani R. Inhibiting miR-27a and miR-142-5p attenuate nonalcoholic fatty liver disease by regulating Nrf2 signaling pathway. IUBMB Life. 2020;72:361–372. doi: 10.1002/iub.2221. [DOI] [PubMed] [Google Scholar]
- 24.Denardin C.C., Martins L.A., Parisi M.M., Vieira M.Q., Terra S.R., Barbé-Tuana F.M., Borojevic R., Vizzotto M., Emanuelli T., Guma F.C., et al. Autophagy induced by purple pitanga (Eugenia uniflora L.) extract triggered a cooperative effect on inducing the hepatic stellate cell death. Cell Biol. Toxicol. 2017;33:197–206. doi: 10.1007/s10565-016-9366-5. [DOI] [PubMed] [Google Scholar]
- 25.Ding Q., Xie X.L., Wang M.M., Yin J., Tian J.M., Jiang X.Y., Zhang D., Han J., Bai Y., Cui Z.J., et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp. Mol. Med. 2019;51:1–13. doi: 10.1038/s12276-018-0199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bueno M., Lai Y.C., Romero Y., Brands J., St Croix C.M., Kamga C., Corey C., Herazo-Maya J.D., Sembrat J., Lee J.S., et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest. 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu G., Tzouvelekis A., Wang R., Herazo-Maya J.D., Ibarra G.H., Srivastava A., de Castro J.P.W., DeIuliis G., Ahangari F., Woolard T., et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018;24:39–49. doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi K., Araya J., Minagawa S., Hara H., Saito N., Kadota T., Sato N., Yoshida M., Tsubouchi K., Kurita Y., et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2016;197:504–516. doi: 10.4049/jimmunol.1600265. [DOI] [PubMed] [Google Scholar]
- 29.Li S., Lin Q., Shao X., Zhu X., Wu J., Wu B., Zhang M., Zhou W., Zhou Y., Jin H., et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic. Biol. Med. 2020;152:632–649. doi: 10.1016/j.freeradbiomed.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Tsubouchi K., Araya J., Kuwano K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018;38:18. doi: 10.1186/s41232-018-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H., Chen G., Wang J., Deng M., Yuan F., Gong J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/mitophagy signalling pathway. Cell Prolif. 2020;53:e12731. doi: 10.1111/cpr.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lobb R.J., Becker M., Wen S.W., Wong C.S., Wiegmans A.P., Leimgruber A., Möller A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and Tables S1–S6