

ABSTRACT
Background
Patient‐reported adverse events (AEs) may be a useful adjunct to clinician‐assessed AEs for assessing tolerability in early phase, dose‐finding oncology trials (DFOTs). We reviewed DFOTs on ClinicalTrials.gov to describe trends in patient‐reported outcome (PRO) use.
Methods
DFOTs commencing 01 January 2007 – 20 January 2020 with ‘PROs’ or ‘quality of life’ as an outcome were extracted and inclusion criteria confirmed. Study and PRO characteristics were extracted. Completed trials that reported PRO outcomes and published manuscripts on ClinicalTrials.gov were identified, and PRO reporting details were extracted.
Results
5.3% (548/10 372) DFOTs included PROs as an outcome. 231 (42.2%) were eligible: adult (224, 97%), solid tumour (175, 75.8%), and seamless phase 1/2 (108, 46.8%). PRO endpoints were identified in more trials (2.3 increase/year, 95% CI: 1.6–2.9) from an increasing variety of countries (0.7/year) (95% CI: 0.4–0.9) over time. PROs were typically secondary endpoints (207, 89.6%). 15/77 (19.5%) completed trials reported results on the ClinicalTrials.gov results database, and of those eight included their PRO results. Eighteen trials had published manuscripts available on ClinicalTrials.gov. Three (16.7%) used PROs to confirm the maximum tolerated dose. No trials identified who completed the PROs or how PROs were collected.
Conclusions
PRO use in DFOT has increased but remains limited. Future work should explore the role of PROs in DFOT and determine what guidelines are needed to standardise PRO use.
Keywords: clinical trials, drug development, oncology, patient‐reported outcomes, phase 1, quality of life
PRO use in DFOT has increased but remains limited. Future work should explore the role of PROs in DFOT and determine what guidelines are needed to standardise PRO use.

1. BACKGROUND
The primary purpose of early phase (phase 1 and phase 1/2) oncology trials is to establish the safety and tolerability of novel anti‐cancer agents. Clinician‐assessed adverse event (AE) reporting using the Common Terminology Criteria for Adverse Events (CTCAE) is critical for describing a drug's safety profile. CTCAE data, in conjunction with data on dose modifications, discontinuations and hospitalisations, is then used to select tolerable doses and regimens, including the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D).
However, clinician‐assessed AE reporting has limitations. Firstly, it may miss up to half of symptomatic AEs compared with patient self‐reports. 1 , 2 , 3 , 4 , 5 Some AEs may be difficult to observe (e.g., fatigue) and not adequately characterised. This may under‐estimate toxicities and result in suboptimal toxicity management. Secondly, the patient's perspective on AEs is noticeably absent in the selection of tolerable doses and regimens.
Changes in the types of therapies in early phase oncology trials can also produce challenges in defining tolerable doses. Immunotherapy and targeted therapy may not produce toxicities in a dose‐dependent manner. Toxicities may be longer in duration or occur later than the traditional dose‐limiting toxicity (DLT) period, which is often defined as the first cycle of treatment. 6 , 7 Low‐grade toxicities experienced over the medium term to long term could reduce a drug's tolerability but are not taken into consideration. This can make defining the MTD, and consequently RP2D, challenging. Alternative methods of determining tolerable doses and regimens are, therefore, urgently needed.
There is increasing interest from clinicians, industry, and regulators, including the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), to enhance the accuracy of toxicity reporting and incorporate the patient's experience in the drug development process through the use of patient‐reported outcomes (PROs). 8 , 9 , 10
PROs are defined as measurements of a patient's health status that come directly from the patient. 11 PROs have been extensively studied in the clinical trial and routine care settings and are reliable, feasible and valued by clinicians and patients. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 PROs, in conjunction to CTCAE data, could provide different yet complementary data to inform a more complete understanding of a drug's tolerability and support the selection of tolerable doses and schedules. 21 PRO collection in early phase trials may become more crucial as drugs developed for specific molecular subtypes with small patient populations may receive regulatory approval on the basis of early phase trials resulting in PROs not being collected in later‐phase trials. A recent example is the FDA approval of the neutrotrophic tyrosine receptor kinase (NTRK) inhibitor entrectinib in patients with NTRK gene fusion solid tumours or ROS1 positive metastatic non‐small‐cell‐lung cancer. 22
The FDA has taken steps towards the inclusion of the patient perspective in all stages of the drug development process, including issuing guidance on the use of PROs in drug development, and collaborating with industry to form the PRO consortium with the aim of developing robust patient‐reported symptom measurement tools. 11
Despite the desire to increase PRO use in drug development, there is minimal literature regarding the use of PROs in early phase oncology trials. A review of PRO use across all clinical trials (including non‐oncological trials) from 2007 to 2013 showed phase 1 trials were less likely to include a PRO measure (PROM) compared with phase 2 trials. 23 A systematic review from 2012 to 2016 identified 15 phase 1 oncology trials with health‐related quality of life (HRQOL) as an endpoint, none of which used HRQOL to inform the RP2D. 24 However, this study only included published trials, placing it at risk of publication bias. PRO inclusion is also a relatively recent phenomena, so examining published trials alone may reflect choices made by trial investigators from several years prior. Trials in progress may have higher rates of PRO inclusion due to increasing awareness of PROs among early phase trialists.
The feasibility and importance of assessing PROs was highlighted in a single‐centre study in which 80 PRO‐CTCAE items were assessed in phase 1 oncology patients at baseline, mid‐cycle 1 and 2. 25 , 26 , 27 Completion rates were high (98.7%). Significant under‐reporting of certain AEs by clinicians was noted, including fatigue, anxiety, and pain. There was also poor patient‐clinician agreement on sexual health, cognition, and urination. This emphasised the potential role of PROs in complementing clinician‐assessed CTCAE gradings to formulate a more comprehensive understanding of a drug's tolerability. PRO‐CTCAE responses, particularly interference, have clinically relevant consequences and correspond with DLTs, dose interruption, and drug discontinuation. This provides further evidence of their usefulness in assessing drug tolerability. 28
We conducted a database search of ClinicalTrials.gov to describe characteristics and trends of PRO use and reporting in early phase oncology trials. ClinicalTrials.gov is the largest global trial registry and a valuable resource for assessing the trends and characteristics of registered clinical trials. 29 By including trials that are completed and in progress, this will provide a more current and complete assessment of PRO use.
2. METHOD
2.1. Study design and search strategy
This cross‐sectional study included early phase dose‐finding oncology trials (DFOT) with start dates 01/01/2007–20/01/2020 registered on ClinicalTrials.gov. Ethics approval was not required for this database review of ClinicalTrials.gov. Data were extracted in XML format on 1 February 2020. DFOT were identified using the following search strategy in the advanced search function: condition or disease – cancer; study type – interventional (clinical trial); study results – all trials; recruitment status – not yet recruiting, recruiting, enrolling by invitation, active not recruiting, suspended and completed; expanded access – available, no longer available, temporarily not available, approved for marketing; age: child, adult, older adult; sex – all; study phase – early phase 1, phase 1; funder type – NIH, other US federal agency, industry, all others; date restriction (01/01/2007–20/01/2020). The search was then repeated to identify DFOT with a PRO endpoint by adding outcome measures – ‘patient‐reported outcome’ OR ‘Health‐related quality of life’ OR ‘quality of life’ OR ‘QOL’ OR ‘PRO’ OR ‘PROMS’ OR ‘HRQOL’.
The ClinicalTrials.gov database entry for each search result was reviewed to confirm inclusion criteria were met. Eligible trials were DFOT (phase 1 or phase 1/2 oncology trial with a dose finding component) assessing an intervention (drug or radiotherapy), which included at least one PRO as an endpoint. Haematology and paediatric trials were included. Trials that did not meet all eligibility criteria were excluded.
2.2. Data extraction
Data was extracted using a pre‐defined data abstraction form in Microsoft Excel (Text S1). Study characteristics were extracted: title, study period, sponsor's country of origin, sponsor type, number of participating centres, number of patients enrolled, study phase, dose escalation study design, tumour type, type of therapy undergoing dose escalation, primary endpoint for phase 1, and current study activity status. PRO characteristics were then extracted: number of PROs included, type of PROs, PRO endpoint (primary, secondary, exploratory), phase in trial in which PROs were collected, method of collection, person completing the PRO, frequency of PRO assessment and duration of PRO follow‐up.
2.2.1. Patient‐reported outcome reporting
Data were extracted regarding PRO outcomes and the statistical methods used for PRO analysis from two sources:
Completed trials with PRO results available on the ClinicalTrials.gov database
Trials with published manuscripts listed on the ClinicalTrials.gov database.
There are no guidelines regarding how PRO data should be reported in non‐randomised trials. The following details were extracted based on the CONSORT‐PRO checklist for randomised trials 30 : whether PRO results were published, whether PRO results were described in the abstract, whether a PRO hypothesis was stated, whether relevant PRO domains were identified, the content validity of the PRO, timing of PRO assessments, statistical methods for PRO analysis, presence of missing data and reasons for missing data, handling of missing data, whether a minimal clinically important difference (MCID) was described and whether PRO data informed the MTD or RP2D.
All data were checked for internal consistency and disagreements resolved by discussion among the investigators.
2.3. Data analysis
Data were analysed using R version 3.6.1. For continuous variables, summary statistics of median and range were displayed. For categorical variables, frequencies (number) and percentages were displayed. Linear regression models with ‘year of study initiation’ as the independent variable were fitted to assess the trend of the number and percentage of trials, and number of sponsoring countries with PRO endpoints over time.
3. RESULTS
A total of 10 372 DFOTs were identified on ClinicalTrials.gov. In total, 548 (5.3%) DFOTs included a PRO endpoint, and 231 (42.2%) met eligibility criteria (Figure 1): adult (224, 97%), paediatric (7, 3%), solid tumour (175, 75.8%) and haematology (56, 24.2%) (Table 1). Phase 1/2 (108, 46.8%) trials were most common, followed by phase 1 dose escalation (101, 43.7%) and phase 1 dose escalation and expansion trials (22, 9.5%).
FIGURE 1.
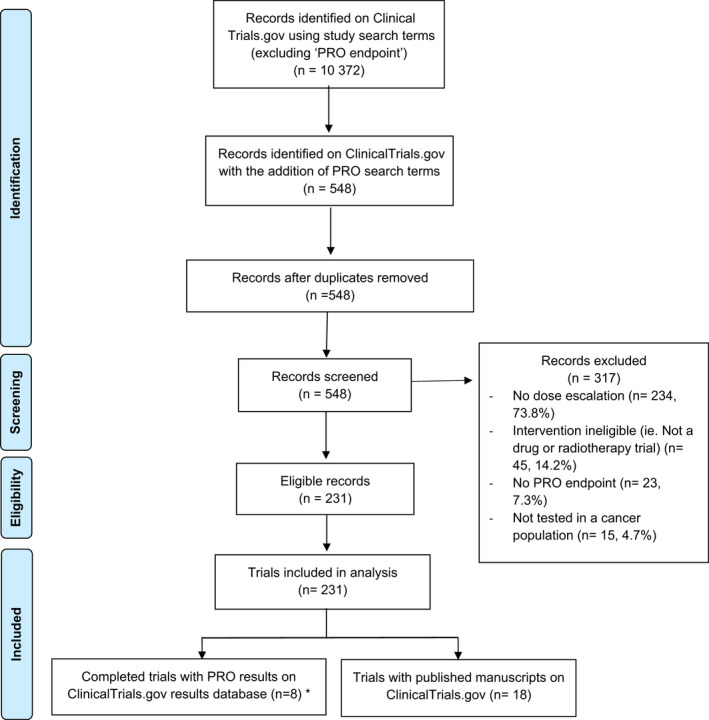
PRISMA diagram. * 3 trials had published manuscripts on ClinicalTrials.gov
TABLE 1.
All eligible trials – study characteristics (n = 231)
| Number (%) | |
|---|---|
| Study population | |
| Adult | 224 (97.0%) |
| Paediatric | 7 (3.0%) |
| Cancer | |
| Solid tumour | 175 (75.8%) |
| Haematology | 56 (24.2%) |
| Phase | |
| Phase 1 dose esc | 101 (43.7%) |
| Phase 1 dose esc and exp | 22 (9.5%) |
| Phase 1 and 2 | 108 (46.8%) |
| Anti‐cancer agent | |
| Drug combinations | 119 (51.5%) |
| Single agents | 110 (47.6%) |
| Not described | 2 (0.9%) |
| Escalating treatment in dose escalation | |
| Targeted therapy | 94 (40.7%) |
| Immunotherapy | 33 (14.3%) |
| Radiotherapy | 33 (14.3%) |
| Chemotherapy | 32 (13.9%) |
| Complementary/alternative | 20 (8.7%) |
| Vaccine | 6 (2.6%) |
| Radionuclide | 5 (2.2%) |
| Other | 5 (2.2%) |
| Hormonal | 2 (0.9%) |
| Antibiotic | 1 (0.4%) |
| Primary endpoint | |
| MTD | 107 (34.9%) |
| Safety | 95 (30.9%) |
| DLT | 57 (18.6%) |
| RP2D | 26 (8.5%) |
| Response rate | 16 (5.2%) |
| Feasibility | 2 (0.7%) |
| HRQOL | 2 (0.7%) |
| Recurrence‐free survival | 1 (0.3%) |
| Missing | 1 (0.3%) |
| Sponsor type | |
| All others (individuals, universities, organisations) | 135 (58.4%) |
| Industry | 83 (35.9%) |
| US NIH | 13 (5.6%) |
| Number of centres | |
| 1 | 108 (46.8%) |
| 2–5 | 56 (24.2%) |
| 6–10 | 20 (8.7%) |
| >10 | 46 (19.9%) |
| Not specified | 1 (0.4%) |
| Study design | |
| Other | 151 (65.4%) |
| 3+3 dose escalation | 64 (27.7%) |
| Continual reassessment method | 12 (5.2%) |
| Accelerated titration | 3 (1.3%) |
| Rolling 6 | 1 (0.4%) |
| Study activity | |
| Recruiting | 114 (49.4%) |
| Completed | 77 (33.3%) |
| Active, not recruiting | 33 (14.3%) |
| Not yet recruiting | 6 (2.6%) |
| Suspended | 1 (0.4%) |
| PRO endpoint | |
| Primary | 4 (1.7%) |
| Secondary | 207 (89.6%) |
| Exploratory | 18 (7.8%) |
| Primary and secondary | 2 (0.9%) |
| PRO phase | |
| Dose escalation only | 115 (49.8%) |
| Phase 1 and 2 | 54 (23.4%) |
| Dose escalation and expansion | 31 (13.4%) |
| Phase 2 | 28 (12.1%) |
| Dose expansion only | 3 (1.3%) |
| Number of PRO measures included | |
| 1 | 137 (59.3%) |
| 2 |
67 (29.0%) |
| 3 | 17 (7.4%) |
| 4 | 5 (2.2%) |
| 5 | 4 (1.7%) |
| 7 | 1 (0.4%) |
| PRO measure (N = 119, top 10 listed) | |
| EORTC QLQ C30 | 78 (21.1%) |
| Not specified | 49 (13.3%) |
| EQ 5D−5L | 19 (5.1%) |
| Brief Pain Inventory | 10 (2.7%) |
| PRO‐CTCAE | 10 (2.7%) |
| FACT‐prostate | 7 (1.9%) |
| FACT‐General | 7 (1.9%) |
| EORTC QLQ LC13 | 7 (1.9%) |
| FACT‐lymphoma | 6 (1.6%) |
| EORTC QLQ Brain‐20 | 6 (1.6%) |
| PRO suites | |
| Others | 124 (33.6%) |
| EORTC | 119 (32.2%) |
| FACT | 52 (14.1%) |
| Not specified | 49 (13.3%) |
| PROMIS | 11 (3.0%) |
| MDASI | 8 (2.2%) |
| Peds QOL | 6 (1.6%) |
| Frequency assessment | |
| Unknown | 140 (60.6%) |
| Other | 87 (37.7%) |
| Monthly | 2 (0.9%) |
| Weekly | 2 (0.9%) |
| Method collection | |
| Unknown | 230 (99.6%) |
| Electronic | 1 (0.4%) |
| Person completing PRO | |
| Not stated | 218 (94.4%) |
| Patient | 12 (5.2%) |
| Patient or carer | 1 (0.4%) |
Abbreviations: MTD, maximum tolerated dose; DLT, dose‐limiting toxicity; RP2D, Phase 2 recommended dose; PRO, patient‐reported outcome; QoL, quality of life; HRQOL, health‐related quality of life.
3.1. All eligible trials
3.1.1. Study characteristics
The number and percentage of DFOT with PRO endpoints increased by 2.3 trials per year (95% confidence interval (CI): 1.6–2.9) (Figure 2A) and 0.3% per year (95% CI: 0.2–0.4) (Figure 2B) respectively. There was also an increasing number of countries with institutions sponsoring DFOT with PRO endpoints, from three countries in 2007 to 11 in 2019 (Figure 3A), at a rate of 0.7 countries per year (95% CI: 0.4–0.9) (Figure 3B).
FIGURE 2.
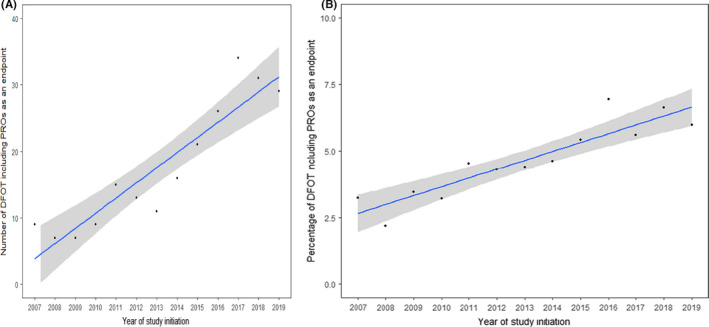
(A) Trends in the number of DFOT using PROs as an endpoint on ClinicalTrials.gov (2007–2019). (B) Trends in the percentage of DFOT using PROs as an endpoint on ClinicalTrials.gov (2007–2019)
FIGURE 3.

(A) Trends in PRO usage by sponsor country of origin on ClinicalTrials.gov (2007–2019). (B) Trends in the number of sponsoring countries using PROs as an endpoint on ClinicalTrials.gov (2007–2019)
Trials were predominantly conducted in adults (224, 97.0%) with solid tumours (175, 75.8%), sponsored by individuals, universities or other organisations (135, 58.4%). A similar number involved drug combinations (119, 51.5%) or single agents (110, 47.6%). The most common therapies in escalation were targeted therapy (94, 40.7%), immunotherapy (33, 14.3%) and radiotherapy (33, 14.3%). Most trials did not specify their study design (151, 65.4%). 64 (27.7%) used a ‘3+3’ dose escalation design and 12 (5.2%) used a continual reassessment method. MTD (107, 34.9%) and safety (95, 30.9%) were the most common primary endpoints. A total of 114 (49.4%) were still recruiting, and 77 (33.3%) were completed.
3.1.2. Patient‐reported outcome characteristics
PROs were typically secondary endpoints (207, 89.6%). Most trials (137, 59.3%) used 1 PROM (range: 1–7). PROs were most frequently collected in the dose escalation (115, 49.8%) and phase 1 and 2 (54, 23.4%). A total of 119 unique PROMs were used. The most common were the EORTC QLQ C30 (78, 21.1%), EQ‐5D‐5L (19, 5.1%), the Brief Pain Inventory (10, 2.7%) and PRO‐CTCAE (10, 2.7%). Information on the frequency of PRO assessment was missing in 140 (60.6%) trials, and the method of PRO collection was missing in 230 (99.6%) trials. Only 13 trials (5.6%) specified the person completing the PRO.
3.2. Eligible trials with reported results on ClinicalTrials.gov
Fifteen completed trials had results available on ClinicalTrials.gov database, of which eight reported their PRO results specifically (Figure 1). The study characteristics and PRO reporting characteristics are presented in Table S1. Seven trials presented descriptive statistics, and one presented least‐square means of treatment effects from a linear mixed effects (LME) model.
3.3. Eligible trials with published manuscripts listed on ClinicalTrials.gov
3.3.1. Study characteristics
Eighteen trials had published manuscripts listed on the ClinicalTrials.gov database (Table 2). Trials were primarily sponsored by individuals, universities or other organisations (9, 50.0%). Most used a ‘3+3’ dose escalation design (10, 55.6%). The treatments most common in dose escalation were targeted therapy (7, 38.9%) and chemotherapy (6, 33.3%). All trials except one had safety and/or MTD as their primary endpoint. 31
TABLE 2.
Published trials with manuscripts available on ClinicalTrials.gov – study characteristics (n = 18)
| Study | Sponsor country of origin | Sponsor type | Study design | Population | Treatment in dose escalation | Dose escalation design | Primary endpoint |
|---|---|---|---|---|---|---|---|
| United States | Individuals, universities or other organisations | Phase 1 dose escalation | Adults, recurrent ovarian/ primary peritoneal/ fallopian tube cancer | Chemotherapy – intraperitoneal oxaliplatin 32 or intraperitoneal docetaxel 33 | 3+3 | MTD, DLT | |
|
Wyatt et al. 34 |
United States | Individuals, universities or other organisations | Phase 1/2 | Adults, prostate cancer | Complementary therapy – Saw Palmetto | Continual reassessment method | MTD, feasibility, efficacy |
|
Van Zandwijk 35 |
Australia | Industry | Phase 1 dose escalation | Adults, mesothelioma or metastatic non–small cell lung cancer |
Targeted therapy – Epidermal Growth Factor Receptor –targeted, EnGeneIC Delivery vehicle packaged, miR‐16 mimic (TargomiRs) |
3+3 | MTD, DLT, efficacy |
|
Haddad et al. 36 |
United States | US National Institute of Health | Phase 1 dose escalation | Adult, advanced solid tumours |
Targeted therapy – Cilengitide |
3+3 | MTD, safety |
|
Sampath et al. 37 |
United States | Individuals, universities or other organisations | Phase 1 dose escalation | Adults, prostate cancer | Radiotherapy – Stereotactic radiotherapy | Modified rolling 6 | MTD |
|
Reiss et al. 38 |
United States | US National Institute of Health | Phase 1 dose escalation | Adults, peritoneal carcinomatosis | Targeted therapy – veliparib | 3+3 | MTD |
|
Dinkic et al. 39 |
Germany | Individuals, universities or other organisations | Phase 1 dose escalation | Adults, recurrent ovarian cancer | Targeted therapy – pazopanib | 6 patients to be treated at each dose level or less if 2 dose limiting toxicities (DLTs) developed at 1 dose level | MTD |
|
Laetsch et al. 31 |
Germany | Industry | Phase 1 dose escalation | Paediatric, central nervous system tumours | Targeted therapy‐ LOXO‐101 | Modified rolling 6 | Safety |
|
Aguilar et al. 40 |
United States | Industry | Phase 1 dose escalation | Adults, pancreas |
Immunotherapy‐ Gene‐mediated cytotoxic immunotherapy |
Not specified | Safety |
|
Subbiah et al. 41 |
United States | Industry | Phase 1/2 | Adult, advanced solid tumours | Chemotherapy‐ Nanoparticle cisplatin | Continual reassessment method | MTD, RP2D |
|
Crew et al. 42 |
United States | US National Institute of Health | Phase 1 dose escalation | Adult, breast cancer | Complementary therapy‐ Polyphenon E | Time to event continual reassessment method | MTD |
|
Watanabe et al. 49 |
United States | Individuals, universities or other organisations | Phase 1/2 | Adult, squamous non‐small cell lung cancer | Chemotherapy‐ gemcitabine | 3+3 | MTD |
|
Kumar et al. 43 |
United States | Individuals, universities or other organisations | Phase 1/2 | Adult, multiple myeloma/ light chain amyloidosis | Chemotherapy‐ cyclophosphamide | 3+3 | MTD |
|
Verstovsek et al. 44 |
United States | Industry | Phase 1/2 | Adult, primary myelofibrosis and polycythaemia vera/ essential thrombocythemia | Targeted therapy‐ JAK2 inhibitor INCB018424 | 3+3 | Safety, MTD |
|
Boulin et al. 46 |
France | Individuals, universities or other organisations | Phase 1 dose escalation | Adult, hepatocellular carcinoma | Chemotherapy‐ idarubicin loaded microspheres | Modified continual assessment method | MTD, safety |
|
Verstovsek et al. 45 |
Japan | Individuals, universities or other organisations | Phase 1 dose escalation | Adult, primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythemia myelofibrosis | Targeted therapy‐ NS‐018 (JAK 2 inhibitor) | 3+3 | Safety, MTD |
|
Siegel et al. 47 |
United States | Individuals, universities or other organisations | Phase 1 dose escalation | Adult, hepatocellular carcinoma | Complementary therapy – Siliphos (milk thistle) | 3+3 | MTD |
|
Martin‐Broto et al. 48 |
Italy | Individuals, universities or other organisations | Phase 1/2 | Adult, sarcoma | Chemotherapy – trabectidin | 3+3 | DLT, RP2D |
Abbreviations: DLT: dose‐limiting toxicity; MTD: maximum tolerated dose; RP2D: Phase 2 recommended dose.
Three trials also listed their PRO results on the ClinicalTrials.gov database. As expected, more detailed descriptive and inferential analyses were provided in the manuscripts compared with the entries on the ClinicalTrials.gov database.
3.3.2. PRO reporting
One trial (NCT00692900) conducted the dose escalation in two parallel arms and reported each arm separately in two publications. 32 , 33 This resulted in 19 publications from 18 trials (Table 3). 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 Only one trial included PROs as a co‐primary endpoint. 34 No trials stated a PRO hypothesis. All trials used a valid PRO instrument. The most commonly used PROMs were the EORTC QLQ C30 (7, 26.9%), FACT‐hepatobiliary (2, 7.7%), International Prostate Symptom Score (2, 7.7%) and the Myelofibrosis Symptom Assessment Form (2, 7.7%). The majority collected PROs during the Phase I dose escalation (11, 61.1%). Most (16, 88.9%) collected PROs at baseline and subsequent time points. No trials identified who was responsible for completing the PROs or how PROs were collected.
TABLE 3.
Published trials with manuscripts available on ClinicalTrials.gov – PRO characteristics (n = 18)
| Study | PRO result in abstract | PRO instrument | Phase | Frequency of assessment | Statistical methods | How were the PRO outcomes analysed? | MCID described? | Missing PRO data present? | Number of patients with missing PRO data described? | Reasons for missing PRO data described? | Method for managing missing PRO data described? | PRO considered for MTD/RP2D? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yes | MDASI with ovarian cancer specific items | Phase 1 dose escalation | Weekly | Descriptive analysis |
Over dose Over time (baseline to each time period) |
No | Yes | Yes | Partial | No | Used to confirm the MTD | |
|
Wyatt et al. 34 |
Yes | IPSS, FACT‐Prostate | Phase 1/2 | IPSS: baseline, weeks 2–10, 12, 14 and 22. FACT‐P: baseline, week 12, 14 and 22 | Descriptive analysis and inferential statistic |
Baseline differences between treatment groups [two‐sample t‐test] Over time and between treatment groups [LME] |
No | Not described | No | No | Yes | No |
|
Van Zandwijk 35 |
No | EORTC QLQ C30 | Phase 1 dose escalation | Weekly | Descriptive analysis | Over time(from baseline to 8 treatment weeks) | No | Yes | Yes | No | No | No |
|
Haddad et al. 36 |
No | PRO‐CTCAE | Phase 1 dose escalation | Weekly | Descriptive analysis | Not described | No | Yes | Yes | Yes | No | No |
|
Sampath et al. 37 |
Yes | IPSS, SHIM, Merrick rectal function scale | Phase 1 dose escalation | Baseline and at each follow‐up appointment for 3 years | Descriptive analysis and Inferential statistic | Over dose and over time, and between patient subgroups [statistical test not stated] | No | Yes | No | No | No | No |
|
Reiss et al. 38 |
Yes | EORTC QLQ C30 | Phase 1 dose escalation | Baseline, every 2 cycles, during follow‐up | Inferential statistic |
Over time [paired t‐test]; Over different patient subgroups [two‐sample t‐test] |
Yes | Yes | Yes | Yes | Yes | Used to confirm the MTD |
|
Dinkic et al. 39 |
No | EORTC QLQ C30, EORTC Ovar‐28 | Phase 1 dose escalation | Baseline, every 3 cycles, during follow‐up | Unclear | Over time [statistical test not stated] | No | Not described | No | No | No | No |
|
Laetsch et al. 31 |
No | Peds QL Infant, Peds QL Core module, Wong Baker Pain Faces | Phase 1 dose escalation | Every cycle | Descriptive analysis | Over time (baseline to each time period) | Yes | Yes | Yes | No | No | No |
|
Aguilar et al. 40 |
Yes | FACT‐hepatobiliary | Phase 1 dose escalation | Baseline, follow up | Descriptive analysis | Over time (baseline to each time period) and over different treatment arms | No | Yes | Yes | No | No | No |
|
Subbiah et al. 41 |
Yes | EORTC QLQ C30 | Phase 1/2 | Not stated | Descriptive analysis | Over time (baseline to last assessment) | No | Yes | Yes | No | No | No |
|
Crew et al. 42 |
No | SF‐36 | Phase 1 dose escalation | Baseline, 6 months | Unclear | Over time [statistical test not stated] | No | Not described | No | No | No | No |
|
Watanabe et al. 49 |
No | LCSS, ASBI | Phase 1/2 | Not stated | Descriptive analysis and Inferential analysis | Time to worsening via Kaplan–Meier estimates and over treatment groups [cox regression] | Yes | Not described | No | No | No | No |
|
Kumar et al. 43 |
No | FACT/GOG neurotoxicity questionnaire | Phase 1/2 | Baseline, after each cycle for 4 cycles, then every 3 cycles | Descriptive analysis | Over time (for each cycle) | No | Yes | Yes | No | No | No |
|
Verstovsek et al. 44 |
No | Myelofibrosis Symptom Assessment Form, EORTC QLQ C30 | Phase 1/2 | Baseline, 1 and 6 months | Descriptive analysis and Inferential statistic | Over dose, over time and over treatment groups [statistical test not stated] | No | No | No | No | No | No |
|
Boulin et al. 46 |
No | EORTC QLQ C30 | Phase 1 dose escalation | Baseline, D15, D30 and D60 post TACE | Descriptive analysis | Over dose and over time (baseline to 1 month at each dose) [Mean difference and 95% CI stated] | Yes | Yes | Yes | No | No | Used to confirm the MTD |
|
Verstovsek et al. 45 |
Yes | Myelofibrosis Symptom Assessment Form | Phase 1/2 | Baseline, Day 1 of Cycles 2 and 4, Day 1 of every 3 cycles thereafter | Descriptive analysis | Over time (after 4 and 12 weeks of treatment) | No | Yes | No | No | No | No |
|
Siegel et al. 47 |
No | FACT‐hepatobiliary | Phase 1 dose escalation | Week 1, 6, and 12 | Unclear | Only baseline data was reported | No | Not applicable | No | No | No | No |
|
Martin‐Broto et al. 48 |
No | EORTC QLQ C30 | Phase 1/2 | Every 3 months for 24 months | Descriptive analysis and Inferential statistic | Over time (baseline to cycle3) [Mann‐Whitney or Kruskal‐Wallis] | No | Not described | No | No | No | No |
Abbreviations: ASBI, Average Symptom Burden Index; IPSS, International Prostate Symptom Scale; LCSS, Lung Cancer Symptom Scale; LME, linear mixed effects; MCID, minimal clinically important difference; MDASI, MD Anderson Symptom Inventory; MTD, maximum tolerated dose; RP2D, phase 2 recommended dose; SF‐36, 36‐item Short Form Survey; SHIM, Sexual Health Inventory for Men; TACE, trans‐arterial chemoembolization.
The statistical methods for PRO analysis were variably described. Analysis approaches were classified into three categories: descriptive statistics, inferential statistics (using the observed data to draw inferences, generalise and make judgements about the larger population, usually via hypothesis testing with p‐values) or a combination. Nine (50.0%) used descriptive statistics, 1 (5.6%) used inferential statistics, 5 (27.8%) used both. Three (16.7%) did not provide sufficient details on their statistical methods to enable classification. Trials using descriptive analysis described changes in PRO scores over time using different categories, such as those whose PRO scores improved, remained stable or declined. 35 , 41 , 44 , 45 Among those trials using inferential statistics, most conducted simple statistical tests such as the t‐test to assess whether there were significant differences in PRO scores between baseline and after start of treatment, or among different treatment arms. One trial used a Cox regression model to compare time of worsening of symptom scores between two randomised treatment groups in the phase 2 component and presented the estimated hazard ratio and CI. Another trial used an LME model to account for repeated measures over time and allowed for missing data at random. Least‐square means of the treatment effects were obtained from an LME model (with adjustment for baseline PRO scores) to assess whether the experimental treatment had a significant effect on PROs over time compared with placebo in its exploratory randomised controlled component. 34 Sixteen (88.9%) analysed PRO data over time, four (22.2%) analysed PRO data over different doses. Four (22.2%) trials described an MCID. Nine (50.5%) reported the number of patients with missing data, and two (11.1%) reported the reasons for missing data. Two (11.1%) described their methods for dealing with missing data.
With regards to using PROs to define tolerable doses and regimens, 3 (16.7%) used PROs to confirm the MTD. However, these do not state whether PROs were reviewed prior to determining the MTD or whether the MTD was retrospectively deemed tolerable after PRO data were reviewed.
Seven (39.8%) trials included PRO results in their abstract. All trials reported their PRO results in their primary trial manuscript rather than in a separate manuscript.
4. DISCUSSION
This is the first study to examine current trends in PRO use and reporting in DFOT. PRO use in DFOT increased over time and in a wider variety of settings. The trial characteristics are representative of the current drug development landscape, with a trend towards combinations of therapies in seamless phase 1/2 trials with novel dose‐finding statistical designs such as the Continual Reassessment Method. 50 , 51 , 52 , 53 Around half (58.4%) were sponsored by individuals, universities or other organisations, primarily representing academic institutions. This is nearly double the rate of academic sponsorship of DFOT on ClinicalTrials.gov for the same time period (2628/10372, 25.3%), indicating the key role that academic‐sponsored trials play in driving the inclusion of patient‐centred endpoints.
Nevertheless, overall use remains limited. DFOT including PROs remain a small proportion of all DFOT (Figure 2B), and this only increased by 2.3 trials per year (Figure 2A). PROs were predominantly used as a secondary or exploratory endpoint, indicating that researchers are not using PROs for the primary endpoint of dose determination. Generic cancer or disease‐specific PROMs were mainly used. However, these may not adequately capture the breadth of toxicities a patient may experience on DFOT and may not be fit for the purpose of assessing tolerability. Some trials used item libraries such as the PRO‐CTCAE (10, 2.7%) and EORTC item library (1, 0.3%), which may be useful for examining specific toxicities from novel agents not covered by legacy measures. Further work is needed to determine how best to select these items. A balance will need to be struck between thoroughness (such as including all 78 symptomatic toxicities in the PRO‐CTCAE to ensure unexpected toxicities are not missed) and feasibility (whether patients will complete lengthy PROMs on a weekly basis). 54 A possible compromise 55 is to collect a core set of items from an item library representing common and clinically relevant treatment‐related symptoms 56 and anticipated toxicities from pre‐clinical trials, 57 with a free text item to ensure unexpected toxicities are not missed. 58
The number of trials reporting PRO results was also limited. Only 8 completed trials had PRO results available on the ClinicalTrials.gov database, and only 18 trials published their PRO results in a manuscript listed on ClinicalTrials.gov, indicating a significant amount of research waste. This is also an issue in later‐phase trials with around 20% reporting their PRO results. 59 , 60
Although there are no guidelines to inform how PRO results should be reported for non‐randomised trials, the CONSORT‐PRO extension provides a useful checklist of items that should be reported to enable interpretation of PRO results. Key information such as a PRO objective, the person completing the PRO, and the method of PRO collection was not described in any publications. Furthermore, the statistical approach to the PRO data, MCID, the presence of missing data (and the reasons for missing data) were only described in some publications. This limits the interpretation and usefulness of the PRO data for making decisions regarding tolerability. It also limits the capacity to develop hypotheses and inform power calculations for later‐phase trials. This is again consistent with the later‐phase trial setting, where studies have shown that PRO reporting quality is suboptimal. 61 , 62 , 63 , 64
Although most trials collected PRO data over different doses and over several time points, only 22.2% analysed PRO data over different doses and time. More than 60% reported PRO data over time only. This fails to use the richness of the data to further inform tolerability at different doses. In addition to descriptive summary measures, several regression approaches can be used to analyse PRO data over time and different doses such as LME model or generalised estimating equation framework, which can account for the correlation of PRO measurements at different time points within an individual. 65 , 66 Only a small proportion (16.7%) used PROs to confirm the MTD. It is unclear how the PRO information was incorporated to inform the MTD, and whether it was reviewed prior or after the determination of MTD.
The strengths of this study are that by searching ClinicalTrials.gov rather than published trials we generated a current and complete assessment of PRO use. Many DFOT are never published or there can be a significant lag time between completion and publication. 67 , 68 Therefore, searching for published trials only may provide a limited picture of PRO use. We included all forms of DFOT, including haematology, paediatric and radiotherapy trials, which have been excluded from prior reviews. 24 Our search strategy included all trials commencing from 2007 when registration of US‐based trials to ClinicalTrials.gov became compulsory, ensuring this review is as current as possible. However, not all trials sponsored outside the United States are registered on ClinicalTrials.gov, which may result in those trials being under‐represented in our study.
Other limitations include the use of the ClinicalTrials.gov advanced search function, which can return slight variation in the number of search results day‐to‐day. This was mitigated by creating a copy of the database prior to data extraction. Our search strategy did not include specific PROMs, which may have resulted in some DFOT being missed. However, given that 119 unique PROMs were identified using our search strategy, it is likely that the vast majority of DFOT using PROs were captured by using generic QOL‐ and PRO‐related terms. There was also significant variability in the quality of data provided for each trial. We cannot be certain these data are accurate as we did not review the individual trial protocols or published manuscripts. However, the Clinical Trials Transformation Initiative suggests that information is more likely to be complete if the trial is evaluating drugs/devices that are sponsored by US or multinational organisations which the majority of these trials were. 69 Although inaccuracies and missing data may impact on the quality and characteristics of the trials, the purpose of this study was to examine trends and characteristics in PRO use. This is more likely to introduce random error rather than bias as the issues are likely to be distributed similarly across time.
The evaluation of PRO reporting quality was not performed as a systematic review. There may be trials which have published their results but have not updated their records on ClinicalTrials.gov. However, there were several similarities noted with our study and the systematic review by Fiteni et al., 24 including the type of PROMs used and inadequacies in PRO result reporting, reinforcing that our findings are a fair representation of the characteristics and quality of PRO reporting in DFOT.
Further work is necessary to determine the degree of acceptability of PROs to key stakeholders, potential benefits and barriers and the role of PRO data in defining tolerable doses and regimens. A qualitative study examined patient and clinician attitudes towards the collection of electronic PROs in early phase oncology trials. 70 While most agreed PROs could provide a more comprehensive understanding of a drug's toxicity, clinicians expressed concerns about monitoring PRO data and the need for careful decision‐making regarding data flow and symptom attribution. Our group is currently conducting a global survey exploring these questions in clinicians, statisticians, trial managers, funders and regulators.
Adaptation of existing methodological guidelines for the inclusion of PROs in randomised trials (e.g. SPIRIT‐PRO extension, CONSORT‐PRO extension, SISAQOL) 30 , 71 , 72 will be needed to standardise protocol content, reporting and statistical analysis in early phase trials. Guidance from regulators such as the EMA and FDA will also enhance uptake. 11 Simultaneous publication of PRO results alongside the main trial manuscript should be encouraged to minimise research waste. 73
5. CONCLUSION
PRO use in DFOT has increased over time, and across a wider variety of settings. However, overall use remains limited. Few trials reported their PRO results and the quality of the PRO reporting was highly variable. Further work is needed to understand key stakeholder attitudes towards PROs in DFOT, and the potential benefits and barriers to their inclusion. Consensus is needed as how to best integrate PRO data into dose‐finding trials. If PROs are to be included in a meaningful way, adaptation of methodological guidelines for protocol content, statistical analysis and reporting will be required to standardise and improve the quality of PRO data in dose‐finding oncology trials.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONFLICTS OF INTEREST
AM has served on advisory boards and received fees from Merck, FARON, Novartis, Bayer and Janssen, which are all unrelated to this work. CY has received fees from FARON and honorarium from Celgene, which are all unrelated to this work. JLK and ZY declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
JLK, CY – study design, review of ClinicalTrials.gov, analysis of results, manuscript preparation, approval of final manuscript; ZY– analysis of results, manuscript preparation, approval of final manuscript; AM – study design, analysis of results, manuscript preparation, approval of final manuscript.
Supporting information
Table S1
Text S1
Lai‐Kwon J, Yin Z, Minchom A, Yap C. Trends in patient‐reported outcome use in early phase dose‐finding oncology trials – an analysis of ClinicalTrials.gov. Cancer Med. 2021;10:7943–7957. doi: 10.1002/cam4.4307
Funding information
This study represents independent research supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at the Royal Marsden NHS Foundation Trust and the Institute of Cancer Research. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Data Availability Statement
Data are available on request from the corresponding author.
REFERENCES
- 1. Atkinson TM, Li Y, Coffey CW, et al. Reliability of adverse symptom event reporting by clinicians. Qual Life Res. 2012;21(7):1159‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Basch E, Jia X, Heller G, et al. Adverse symptom event reporting by patients vs clinicians: relationships with clinical outcomes. J Natl Cancer Inst. 2009;101(23):1624‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fromme EK, Eilers KM, Mori M, Hsieh YC, Beer TM. How accurate is clinician reporting of chemotherapy adverse effects? A comparison with patient‐reported symptoms from the Quality‐of‐Life Questionnaire C30. J Clin Oncol. 2004;22(17):3485‐3490. [DOI] [PubMed] [Google Scholar]
- 4. Basch E, Iasonos A, McDonough T, et al. Patient versus clinician symptom reporting using the National Cancer Institute Common Terminology Criteria for Adverse Events: results of a questionnaire‐based study. Lancet Oncol. 2006;7(11):903‐909. [DOI] [PubMed] [Google Scholar]
- 5. Di Maio M, Gallo C, Leighl NB, et al. Symptomatic toxicities experienced during anticancer treatment: agreement between patient and physician reporting in three randomized trials. J Clin Oncol. 2015;33(8):910‐915. [DOI] [PubMed] [Google Scholar]
- 6. Harrington JA, Hernandez‐Guerrero TC, Basu B. Early phase clinical trial designs: state of play and adapting for the future. Clin Oncol. 2017;29(12):770‐777. [DOI] [PubMed] [Google Scholar]
- 7. Postel‐Vinay S, Gomez‐Roca C, Molife LR, et al. Phase I trials of molecularly targeted agents: should we pay more attention to late toxicities? J Clin Oncol. 2011;29(13):1728‐1735. [DOI] [PubMed] [Google Scholar]
- 8. Basch E. Toward patient‐centered drug development in oncology. N Engl J Med. 2013;369(5):397‐400. [DOI] [PubMed] [Google Scholar]
- 9. Calvert MJ, O'Connor DJ, Basch EM. Harnessing the patient voice in real‐world evidence: the essential role of patient‐reported outcomes. Nat Rev Drug Discov. 2019;18(10):731‐732. [DOI] [PubMed] [Google Scholar]
- 10. Kluetz PG, O'Connor DJ, Soltys K. Incorporating the patient experience into regulatory decision making in the USA, Europe, and Canada. Lancet Oncol. 2018;19(5):e267‐e274. [DOI] [PubMed] [Google Scholar]
- 11. Food and Drug Administration . Guidance for industry — patient‐reported outcomes measures: use in medical product development to support labeling claims. Silver Spring; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cleeland CS, Wang XS, Shi Q, et al. Automated symptom alerts reduce postoperative symptom severity after cancer surgery: a randomized controlled clinical trial. J Clin Oncol. 2011;29(8):994‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gilbert JE, Howell D, King S, et al. Quality improvement in cancer symptom assessment and control: the Provincial Palliative Care Integration Project (PPCIP). J Pain Symptom Manage. 2012;43(4):663‐678. [DOI] [PubMed] [Google Scholar]
- 14. Valderas JM, Kotzeva A, Espallargues M, et al. The impact of measuring patient‐reported outcomes in clinical practice: a systematic review of the literature. Qual Life Res. 2008;17(2):179‐193. [DOI] [PubMed] [Google Scholar]
- 15. Chen J, Ou L, Hollis SJ. A systematic review of the impact of routine collection of patient reported outcome measures on patients, providers and health organisations in an oncologic setting. BMC Health Serv Res. 2013;13:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kotronoulas G, Kearney N, Maguire R, et al. What is the value of the routine use of patient‐reported outcome measures toward improvement of patient outcomes, processes of care, and health service outcomes in cancer care? A systematic review of controlled trials. J Clin Oncol. 2014;32(14):1480‐1501. [DOI] [PubMed] [Google Scholar]
- 17. Detmar SB, Muller MJ, Schornagel JH, Wever LD, Aaronson NK. Health‐related quality‐of‐life assessments and patient‐physician communication: a randomized controlled trial. JAMA. 2002;288(23):3027‐3034. [DOI] [PubMed] [Google Scholar]
- 18. Basch E, Artz D, Dulko D, et al. Patient online self‐reporting of toxicity symptoms during chemotherapy. J Clin Oncol. 2005;23(15):3552‐3561. [DOI] [PubMed] [Google Scholar]
- 19. Velikova G, Booth L, Smith AB, et al. Measuring quality of life in routine oncology practice improves communication and patient well‐being: a randomized controlled trial. J Clin Oncol. 2004;22(4):714‐724. [DOI] [PubMed] [Google Scholar]
- 20. Berry DL, Blumenstein BA, Halpenny B, et al. Enhancing patient‐provider communication with the electronic self‐report assessment for cancer: a randomized trial. J Clin Oncol. 2011;29(8):1029‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nipp RD, Temel JS. Harnessing the power of patient‐reported outcomes in oncology. Clin Cancer Res. 2018;24:1777‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marcus L, Donoghue M, Aungst S, et al. FDA approval summary: entrectinib for the treatment of NTRK gene fusion solid tumors. Clin Cancer Res. 2021;27:928‐932. [DOI] [PubMed] [Google Scholar]
- 23. Scoggins JF, Patrick DL. The use of patient‐reported outcomes instruments in registered clinical trials: evidence from ClinicalTrials.gov. Contemp Clin Trials. 2009;30(4):289‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fiteni F, Ray IL, Ousmen A, Isambert N, Anota A, Bonnetain F. Health‐related quality of life as an endpoint in oncology phase I trials: a systematic review. BMC Cancer. 2019;19(1):361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Veitch ZW, Shepshelovich D, Gallagher C, et al. Implementation of PRO‐CTCAE in phase I clinical trials identifies under reporting of adverse events. Ann Oncol. 2019;30:v718‐v746. [Google Scholar]
- 26. Shepshelovich D, McDonald K, Spreafico A, et al. Feasibility assessment of using the complete patient‐reported outcomes version of the common terminology criteria for adverse events (PRO‐CTCAE) item library. Oncologist. 2019;24(4):e146‐e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Veitch ZW, Shepshelovich D, Gallagher C, et al. Underreporting of symptomatic adverse events in phase I clinical trials. J Natl Cancer Inst. 2021;113:980‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watson GA, Veitch ZWN, Shepshelovich D, et al. Mapping PRO‐CTCAE responses to clinician‐graded adverse events, dose reductions, interruptions, and discontinuations in phase I cancer trials. J Clin Oncol. 2020;38(15 Suppl):2014.32243222 [Google Scholar]
- 29. Gresham G, Meinert JL, Gresham AG, Meinert CL. Assessment of Trends in the Design, Accrual, and Completion of Trials Registered in ClinicalTrials.gov by Sponsor Type, 2000‐2019. JAMA Network Open. 2020;3(8):e2014682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Calvert M, Kyte D, Mercieca‐Bebber R, et al. Guidelines for inclusion of patient‐reported outcomes in clinical trial protocols: the SPIRIT‐PRO extension. JAMA. 2018;319(5):483‐494. [DOI] [PubMed] [Google Scholar]
- 31. Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open‐label, phase 1/2 study. Lancet Oncol. 2018;19(5):705‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor SE, Li R, Petschauer JS, et al. Phase I study of intravenous (IV) docetaxel and intraperitoneal (IP) oxaliplatin in recurrent ovarian and fallopian tube cancer. Gynecol Oncol. 2015;138(3):548‐553. [DOI] [PubMed] [Google Scholar]
- 33. Taylor SE, Petschauer JS, Donovan H, et al. Phase I study of intravenous oxaliplatin and intraperitoneal docetaxel in recurrent ovarian cancer. Int Jo Gynecol Cancer. 2019;29(1):147‐152. [DOI] [PubMed] [Google Scholar]
- 34. Wyatt GK, Sikorskii A, Safikhani A, McVary KT, Herman J. Saw palmetto for symptom management during radiation therapy for prostate cancer. J Pain Symptom Manage. 2016;51(6):1046‐1054. [DOI] [PubMed] [Google Scholar]
- 35. van Zandwijk N, Pavlakis N, Kao SC, et al. Safety and activity of microRNA‐loaded minicells in patients with recurrent malignant pleural mesothelioma: a first‐in‐man, phase 1, open‐label, dose‐escalation study. Lancet Oncol. 2017;18(10):1386‐1396. [DOI] [PubMed] [Google Scholar]
- 36. Haddad T, Qin R, Lupu R, et al. A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(6):1221‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sampath S, Frankel P, Vecchio BD, et al. Stereotactic body radiation therapy to the prostate bed: results of a phase 1 dose‐escalation trial. Int J Radiat Oncol Biol Phys. 2020;106(3):537‐545. [DOI] [PubMed] [Google Scholar]
- 38. Reiss KA, Herman JM, Zahurak M, et al. A Phase I study of veliparib (ABT‐888) in combination with low‐dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin Cancer Res. 2015;21(1):68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dinkic C, Eichbaum M, Schmidt M, et al. Pazopanib (GW786034) and cyclophosphamide in patients with platinum‐resistant, recurrent, pre‐treated ovarian cancer ‐ Results of the PACOVAR‐trial. Gynecol Oncol. 2017;146(2):279‐284. [DOI] [PubMed] [Google Scholar]
- 40. Aguilar LK, Shirley LA, Chung VM, et al. Gene‐mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol Immunother. 2015;64(6):727‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Subbiah V, Grilley‐Olson JE, Combest AJ, et al. Phase Ib/II trial of NC‐6004 (Nanoparticle Cisplatin) plus gemcitabine in patients with advanced solid tumors. Clin Cancer Res. 2018;24(1):43‐51. [DOI] [PubMed] [Google Scholar]
- 42. Crew KD, Brown P, Greenlee H, et al. Phase IB randomized, double‐blinded, placebo‐controlled, dose escalation study of polyphenon E in women with hormone receptor‐negative breast cancer. Cancer Prev Res. 2012;5(9):1144‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar SK, Buadi FK, LaPlant B, et al. Phase 1/2 trial of ixazomib, cyclophosphamide and dexamethasone in patients with previously untreated symptomatic multiple myeloma. Blood Cancer J. 2018;8(8):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, myelofibrosis. N Engl J Med. 2010;363(12):1117‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verstovsek S, Talpaz M, Ritchie E, et al. A phase I, open‐label, dose‐escalation, multicenter study of the JAK2 inhibitor NS‐018 in patients with myelofibrosis. Leukemia. 2017;31(2):393‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boulin M, Hillon P, Cercueil JP, et al. Idarubicin‐loaded beads for chemoembolisation of hepatocellular carcinoma: results of the IDASPHERE phase I trial. Aliment Pharmacol Ther. 2014;39(11):1301‐1313. [DOI] [PubMed] [Google Scholar]
- 47. Siegel AB, Narayan R, Rodriguez R, et al. A phase I dose‐finding study of silybin phosphatidylcholine (milk thistle) in patients with advanced hepatocellular carcinoma. Integr Cancer Ther. 2014;13(1):46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin‐Broto J, Hindi N, Lopez‐Pousa A, et al. Assessment of safety and efficacy of combined trabectedin and low‐dose radiotherapy for patients with metastatic soft‐tissue sarcomas: a nonrandomized phase 1/2 clinical trial. JAMA Oncol. 2020;6(4):535‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe S, Yoshioka H, Sakai H, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first‐line treatment for stage IV squamous non‐small cell lung cancer: A phase 1b and randomized, open‐label, multicenter, phase 2 trial in Japan. Lung Cancer. 2019;129:55‐62. [DOI] [PubMed] [Google Scholar]
- 50. Wong KM, Capasso A, Eckhardt SG. The changing landscape of phase I trials in oncology. Nat Rev Clin Oncol. 2016;13(2):106‐117. [DOI] [PubMed] [Google Scholar]
- 51. Le Tourneau C, Gan HK, Razak AR, Paoletti X. Efficiency of new dose escalation designs in dose‐finding phase I trials of molecularly targeted agents. PLoS One. 2012;7(12):e51039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manji A, Brana I, Amir E, et al. Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single‐agent phase I cancer trials. J Clin Oncol. 2013;31(33):4260‐4267. [DOI] [PubMed] [Google Scholar]
- 53. Riviere MK, Le Tourneau C, Paoletti X, Dubois F, Zohar S. Designs of drug‐combination phase I trials in oncology: a systematic review of the literature. Ann Oncol. 2015;26(4):669‐674. [DOI] [PubMed] [Google Scholar]
- 54. Basch E, Yap C. Patient‐reported outcomes for tolerability assessment in phase I cancer clinical trials. J Natl Cancer Inst. 2021;113(8):943‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Trask PC, Dueck AC, Piault E, Campbell A. Patient‐reported outcomes version of the common terminology criteria for adverse events: methods for item selection in industry‐sponsored oncology clinical trials. Clin Trials. 2018;15(6):616‐623. [DOI] [PubMed] [Google Scholar]
- 56. Reeve BB, Mitchell SA, Dueck AC, et al. Recommended patient‐reported core set of symptoms to measure in adult cancer treatment trials. J Natl Cancer Inst. 2014;106(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sodergren SC, Wheelwright SJ, Fitzsimmons D, et al. Developing symptom lists for people with cancer treated with targeted therapies. Target. Oncol. 2021;16(1):95‐107. [DOI] [PubMed] [Google Scholar]
- 58. Chung AE, Shoenbill K, Mitchell SA, et al. Patient free text reporting of symptomatic adverse events in cancer clinical research using the National Cancer Institute’s Patient‐Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO‐CTCAE). J Am Med Inform Assoc. 2019;26(4):276‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schandelmaier S, Conen K, von Elm E, et al. Planning and reporting of quality‐of‐life outcomes in cancer trials. Ann Oncol. 2015;26(9):1966‐1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weingärtner V, Dargatz N, Weber C, et al. Patient reported outcomes in randomized controlled cancer trials in advanced disease: a structured literature review. Expert Rev Clin Pharmacol. 2016;9(6):821‐829. [DOI] [PubMed] [Google Scholar]
- 61. Kyte D, Retzer A, Ahmed K, et al. Systematic evaluation of patient‐reported outcome protocol content and reporting in cancer trials. J Natl Cancer Inst. 2019;111(11):1170‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bylicki O, Gan HK, Joly F, Maillet D, You B, Péron J. Poor patient‐reported outcomes reporting according to CONSORT guidelines in randomized clinical trials evaluating systemic cancer therapy. Ann Oncol. 2015;26(1):231‐237. [DOI] [PubMed] [Google Scholar]
- 63. Smith AB, Cocks K, Parry D, Taylor M. Reporting of health‐related quality of life (HRQOL) data in oncology trials: a comparison of the European Organization for Research and Treatment of Cancer Quality of Life (EORTC QLQ‐C30) and the Functional Assessment of Cancer Therapy‐General (FACT‐G). Qual Life Res. 2014;23(3):971‐976. [DOI] [PubMed] [Google Scholar]
- 64. Efficace F, Fayers P, Pusic A, et al. Quality of patient‐reported outcome reporting across cancer randomized controlled trials according to the CONSORT patient‐reported outcome extension: a pooled analysis of 557 trials. Cancer. 2015;121(18):3335‐3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Laird NM, Ware JH. Random‐effects models for longitudinal data. Biometrics. 1982;38(4):963‐974. [PubMed] [Google Scholar]
- 66. Liang K‐Y, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73(1):13‐22. [Google Scholar]
- 67. DeVito NJ, Goldacre B. Evaluation of compliance with legal requirements under the FDA amendments act of 2007 for timely registration of clinical trials, data verification, delayed reporting, and trial document submission. JAMA Intern Med. 2021;181(8):1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shepshelovich D, Goldvaser H, Wang L, Abdul Razak AR. Comparison of published and unpublished phase I clinical cancer trials: an analysis of the CliniclTrials.gov database. Invest New Drugs. 2018;36(5):933‐938. [DOI] [PubMed] [Google Scholar]
- 69. Clinical Trials Transformation Initiative . Points to consider for statistical analysis using the database for aggregate analysis of ClinicalTrials.gov. 2013. https://www.ctti‐clinicaltrials.org/file/aact2013statisticalpointstoconsider122713docx [Accessed 13 May, 2020].
- 70. Kennedy F, Shearsmith L, Ayres M, et al. Online monitoring of patient self‐reported adverse events in early phase clinical trials: Views from patients, clinicians, and trial staff. Clin Trials. 2021;18(2):168‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Calvert M, Blazeby J, Altman DG, et al. Reporting of patient‐reported outcomes in randomized trials: the CONSORT PRO extension. JAMA. 2013;309(8):814‐822. [DOI] [PubMed] [Google Scholar]
- 72. Coens C, Pe M, Dueck AC, et al. International standards for the analysis of quality‐of‐life and patient‐reported outcome endpoints in cancer randomised controlled trials: recommendations of the SISAQOL Consortium. Lancet Oncol. 2020;21(2):e83‐e96. [DOI] [PubMed] [Google Scholar]
- 73. Cella D. In our patient‐centered era, it is time we gave patient‐reported outcomes their due. Cancer. 2020;126(11):2592‐2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Text S1
Data Availability Statement
Data are available on request from the corresponding author.