

Abstract
Macrophages are the principal innate immune cells that populate all major organs and provide the first line of cellular defense against infections and/or injuries. The immediate and early-responding macrophages must mount a robust pro-inflammatory response to protect the host by eliminating deleterious agents. The effective pro-inflammatory macrophage response requires the activation of complex transcriptional programs that modulate the dynamic regulation of inflammatory and metabolic gene expression. Therefore, transcription factors that govern pro-inflammatory and metabolic gene expression play an essential role in shaping the macrophage inflammatory response. Herein, we identify the basic helix-loop-helix family member e40 (BHLHE40), as a critical transcription factor that promotes broad pro-inflammatory and glycolytic gene expression by elevating HIF1α levels in macrophages. Our in vivo studies revealed that myeloid-BHLHE40 deficiency significantly attenuates macrophage and neutrophil recruitment to the site of inflammation. Our integrated transcriptomics and gene set enrichment analysis (GSEA) studies show that BHLHE40 deficiency broadly curtails inflammatory signaling pathways, hypoxia response, and glycolytic gene expression in macrophages. Utilizing complementary gain- and loss-of-function studies, our analyses uncovered that BHLHE40 promotes LPS-induced HIF1α mRNA and protein expression in macrophages. More importantly, forced overexpression of oxygen stable form of HIF1α completely reversed attenuated pro-inflammatory and glycolytic gene expression in BHLHE40-deficient macrophages. Collectively, these results demonstrate that BHLHE40 promotes macrophage pro-inflammatory gene expression and functions by elevating HIF1α expression in macrophages.
Keywords: BHLHE40, macrophages, inflammation, HIF1α, hypoxia, glycolysis
Introduction
Experimental and clinical studies support an important role for macrophages in the development and progression of inflammatory disease conditions (1). Macrophages are the unique cell types that are ubiquitously distributed in all organs and tissue types. They serve as sentinels of infection and play an essential role in tissue repair following injury by modulating both innate and adaptive immune responses (1). Interestingly, macrophages are also the major producers of inflammatory mediators that help to recruit additional inflammatory cells to the site of injury or infection (2). Macrophages recognize pathogens, foreign agents, and tissue damages by expressing an unmatched repertoire of pattern recognition receptors, including pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (2). Macrophages exploit PAMPs/DAMPs to relay extracellular cues to the nucleus to facilitate the expression of genes that helps to adapt and tailor an effective inflammatory response (2). These adaptive responses are precisely orchestrated by activation of key transcription factors that facilitate swift target gene expression and effective macrophage activation (3). The tissue microenvironment at the sites of inflammation are characterized by low levels of oxygen and nutrients supply in tandem with a high concentration of lactate, reactive free oxygen radicals, and reductive metabolites (4). Under these hypoxic environments, macrophages predominantly utilize the process of glycolysis to maintain the energy supply and perform effective biological functions (5). Previous studies have shown that the expression of key enzymes involved in the glycolytic pathway is exclusively governed by hypoxia-inducible factor 1 (HIF1) (6). HIF1 is a heterodimeric member of the basic helix-loop-helix family of transcription factors that consists of oxygen-sensitive HIF1alpha (HIF1α) and constitutively expressed HIF1beta (HIF1β) subunits (7). The expression of HIF1α is tightly controlled at the mRNA and protein levels (8). In addition to hypoxia, several cytokines, bacterial endotoxins, and inflammatory agents are also known to elevate HIF1α mRNA expression and protein accumulation under normoxic conditions (9). Macrophages that are deficient in HIF1α displayed a significant reduction in glycolysis, ATP production, cellular motility, and invasiveness (10). Further, HIF1α deficiency broadly attenuated bacterial endotoxin-induced pro-inflammatory gene expression in macrophages (10). More importantly, myeloid-HIF1α-deficient mice are protected from chronic and acute inflammatory disease conditions such as inflammatory bowel disease (11), arterial stenosis (12), arthritis, asthma, adipose tissue inflammation, obesity, type-2 diabetes, atherosclerosis, and endotoxin-induced sepsis (9). At the molecular level, a few transcription factors are known to regulate HIF1α mRNA expression in macrophages(12–14). In this study, we identify the basic helix-loop-helix family member e40 (BHLHE40) as a novel regulator of HIF1α expression and functions in macrophages.
BHLHE40 is a member of basic helix-loop-helix family transcription factors and is highly conserved across mammalian species (15). Structurally, BHLHE40 consists of an N-terminal basic DNA binding domain, a helix-loop-helix dimerization domain, and a protein interacting orange domain (15). BHLHE40 is known to regulate a wide variety of essential cellular processes, including cell cycle, cellular proliferation, programmed cell death, cellular development and differentiation, and circadian rhythm (15). In addition, BHLHE40 has been implicated in chondrocyte differentiation, memory CD8+ T cell development, transplant rejection, adipogenesis, skeletal muscle regeneration, and initiation of DNA damage response (15). BHLHE40 expression and functions are dysregulated in Burkett’s lymphoma, osteosarcoma, non-small cell lung cancer, breast cancer, and pancreatic carcinoma, gastric/colorectal cancers (16). Previous studies have shown that transcription factor BHLHE40 promotes TH1 and TH17 cells mediated neuroinflammation in experimental autoimmune encephalomyelitis (17, 18). In colorectal cancer patients, BHLHE40 is predominantly expressed in CD4+ T cells (19). Likewise, CD4+ T cell deficiency of Bhlhe40 significantly attenuated the development of colitis as well as curtailed IFNγ production by CD4+ T cells (20). Concordantly, mice that are deficient in CD4+ T cell- Bhlhe40 were also highly susceptible to T. gondii or M. tuberculosis infection (20, 21). It is also plausible that BHLHE40 is required for the normal functioning of dendritic cells, as mice deficient in dendritic cell-Bhlhe40 were highly prone to lethal infection of M. tuberculosis (21). Recently, studies have shown that Bhlhe40+GM-CSF+CD4+ T-cells promote pathogenic alloantigen presentation acute graft-versus-host disease and pathological damages associated with inflammatory cytokine production (22). In addition, BHLHE40 regulates cytokine production in human/murine CD4+ T cells as well as modulate proliferation and metabolism in murine B-cells and tissue-resident CD8+ T cells (23). Further, BHLHE40 has been implicated in antihelminth immunity and repression of IL10 production (20, 21, 24). Interestingly, BHLHE40 has also been shown to modulate alveolar macrophage proliferation, self-renewal, and type-II immunity (25, 26). Similarly, BHLHE40 promotes IFNγ production in iNKT cells (27). However, given the importance of macrophages in the pathogenesis of numerous chronic and acute inflammatory disease conditions, whether BHLHE40 regulates macrophage inflammatory and metabolic gene programs has not yet been investigated. In this study, we provide evidence that BHLHE40 promotes broad pro-inflammatory response gene programming and supports the progression of inflammatory disease pathogenesis in vivo.
Materials and methods
Generation of myeloid-BHLHE40 deficient mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and conformed to guidelines established by the American Association for Accreditation of Laboratory Animal Care. All mice were bred and maintained under pathogen-free conditions, fed standard laboratory chow (2916, ENVIGO, Indianapolis, IN), and kept on a 12-h light/dark cycle. The mouse line expressing lysozyme-M promoter-driven Cre recombinase (Lyz2 cre/cre) was obtained from The Jackson Laboratory (Bar Harbor, ME). Bhlhe40 floxed (Bhlhe40fl) mice were created by flanking exon 4 with loxP site, and neomycin selection cassette was excised by Flp-mediated deletion (Ozgene Pty Ltd, Perth, Australia). The Bhlhe40 floxed allele genotyping was performed using site-specific primers (forward primer, 5′- TTGCTTCCTTCTCTTTGGATGTGG-3′, and reverse primer, 5′-GACATACTGGCTTGGTTTGGTTTC-3′). For the generation of myeloid Bhlhe40-specific null mice, Bhlhe40fl/fl (pure C57BL/6J background) mice were crossed with Lyz2 cre/cre mice (pure C57BL/6J background) to generate a mouse line harboring the Bhlhe40 floxed and Lyz2cre alleles. These mice were further cross-bred to generate male and female offspring expressing two Lyz2cre and Bhlhe40 floxed alleles (Bhlhe40fl/fl:Lyz2cre/cre). Mice with two Lyz2cre alleles (Lyz2cre/cre) were used as the control group.
Lung inflammation studies
The lung inflammation model was established by intratracheal delivery of 15 mg/kg body weight zymosan solution to the 8–10 weeks old Bhlhe40fl/fl:Lyz2cre/cre and Lyz2cre/cre mice. The control groups that received saline were served as vehicle controls. These mice were euthanized on day-3 and intubated with one ml of saline to collect bronchoalveolar lavage. The lavages were centrifuged to separate the cellular and liquid components for further analyses. In parallel studies, lung tissues were obtained from Bhlhe40fl/fl:Lyz2cre/cre and Lyz2cre/cre mice on day-3 after the zymosan challenge. These lung tissues were fixed in 10% buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin. To determining the macrophage and neutrophil infiltration, paraffin-sections of lung tissues were deparaffinized in xylene and rehydrated in graded ethanol series. The tissue sections were subjected to antigen retrieval steps with antigen unmasking solution. The tissue sections were further treated with 0.3% H2O2 for 30 min at room temperature, and the nonspecific binding was blocked with blocking buffer. Samples were incubated with rabbit anti-F4/80 or rat anti-Ly6G antibody overnight at 4°C and were subsequently incubated with biotin-conjugated goat anti-rabbit IgG or goat anti-rat IgG respectively for 30 min at room temperature. Samples were further incubated in ABC reagent, and the immunostaining was visualized using a DAB reagent. Images were acquired utilizing a microscope, and macrophage or neutrophil areas were quantified by Image J software version 1.53e (NIH, Bethesda, MD; http://imagej.nih.gov/ij).
Cell Culture and treatment conditions
RAW264.7 and J774.1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 10 μg/ml streptomycin, and 2 mM glutamine in a humidified incubator at 5% CO2 and 37°C. Mouse thioglycollate-elicited peritoneal macrophages (PMs) were obtained by inducing peritonitis with 3% thioglycollate broth in 8- to 12-week-old mice. These mice were euthanized on day-3, and peritoneal lavages were obtained to purify primary macrophages. Peritoneal lavage non-target cells were magnetically labeled with a cocktail of biotin-conjugated monoclonal antibodies using a macrophage isolation kit (Miltenyi Biotec). The magnetically labeled non-target cells were depleted by capturing them within a magnetic separator, while the unlabeled macrophages pass through the column. These purified macrophages were examined for contaminating cells by fluorescence-activated cell sorting (FACS) analysis using fluorescently labeled anti-CD11b and anti-SiglecF antibodies as described in our previous publications (28, 29). Macrophage populations that exhibited above ninety-eight percent purity were maintained in complete DMEM and utilized for indicated experiments. Similarly, lung lavages were obtained from 8–10 weeks old Bhlhe40fl/fl:Lyz2cre/cre and Lyz2cre/cre mice after three days of intratracheal delivery of zymosan or saline. Macrophages were purified by using a macrophage isolation kit (Miltenyi Biotec) as described above. These purified macrophages were evaluated for contaminating cells by FACS analysis using fluorescently labeled anti-CD11b and anti-SiglecF antibodies (28, 29). Macrophage populations that exhibited above ninety-eight percent purity were utilized in our studies. Bone marrow-derived macrophages (BMDMs) were generated by ex vivo differentiation of bone marrow cells. Briefly, bone marrow cells from 8-week-old wild-type, Bhlhe40fl/fl:Lyz2cre/cre and Lyz2cre/cre mice were harvested from the femur and tibia. These bone marrow cells were cultured in complete DMEM supplemented with recombinant mouse macrophage colony-stimulating factor (M-CSF) for 7 days. These BMDMs were collected and utilized for the indicated experiments. For hypoxia treatment, the cell culture plates were incubated in a modular incubator chamber that was subsequently infused with a mixture of 1% O2, 5% CO2, 94% N2 gases and placed at 37°C for the indicated period.
RNAseq analysis
Total RNA samples from primary macrophages were obtained using the High Pure RNA Isolation Kit. Quality control of total RNA samples was assessed using Qubit (Thermo Fisher Scientific) for quantification and Agilent 2100 BioAnalyzer (Santa Clara, CA) to assess the quality using a cut-off of RIN > 7.0 to select specimens for further analysis. cDNA library for RNAseq was generated from 150 ng of total RNA using the Illumina TruSeq Stranded Total RNA kit with Ribo Zero Gold for rRNA removal according to the manufacturer’s protocol. The resulting purified mRNA was used as input for the Illumina TruSeq kit in which libraries are tagged with unique adapter-indexes. Final libraries were validated on the Agilent 2100 BioAnalyzer, quantified via qPCR, and pooled at equimolar ratios. Pooled libraries were diluted, denatured, and loaded onto the Illumina NextSeq 550 System using a high output flowcell. STAR Aligner was used for mapping the sequencing reads to the mm10 mouse reference genome. The aligned reads were then analyzed with Cuffdiff to obtain gene-level expression data using the GENCODE gene annotation for mm10 and reported as fragments per kilobase per million reads mapped (FPKM). Differential expression analysis was also performed using the Cuffdiff package and significantly differentially expressed genes were defined using an adjusted p-value < 0.05 (FDR corrected). Gene expression tables for relevant pairwise comparisons were assessed by gene set enrichment analysis (GSEA) using GenePattern (Broad Institute) (30). We specifically utilized Hallmark pathways datasets for current studies. A gene set was considered to be significantly enriched using an FWER cutoff < 0.05. Heatmaps were generated using ClustVis (31). The sequencing data reported in this manuscript has been deposited in Gene Expression Omnibus (GEO:GSE176174).
RNA extraction, Real-time quantitative PCR, and western blot
Total RNA samples were isolated from indicated samples using the High Pure RNA Isolation Kit. One microgram of total RNA was reverse-transcribed into cDNA using M-MuLV reverse transcriptase in the presence of random hexamers and oligo (dT) primers. Real-time quantitative PCR was performed using Universal SYBR Green PCR Master Mix or TaqMan Universal Master Mix on Applied Biosystems (Foster City, CA) Step One Plus real-time PCR machine in the presence of gene-specific primers. The list of primers and materials utilized in this study are provided in the supplementary tables. The indicated primary cells and cell lines were lysed in ice-cold RIPA buffer containing protease and phosphatase inhibitors. Protein concentrations were quantified by BCA protein assay kit. An equal amount of protein samples were electrophoresed using 8% or 4–15% Mini-PROTEAN TGXTM precast gels (Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk or 5% bovine serum albumin in TBS-T for 1 h at room temperature. These nitrocellulose membranes were further incubated with primary antibodies diluted in 5% BSA in TBS-T. After overnight incubation, primary antibodies were removed by washing with TBS-T. These nitrocellulose membranes were incubated for one hour at room temperature in horseradish peroxidase-conjugated secondary antibodies. The target proteins were visualized using enhanced chemiluminescence Western Blotting Substrate. The primary antibodies were used at the following dilutions: HIF1α and BHLHE40 (1:1000); and β-actin (1:5000).
Transient transfection and Luciferase assay
Transfection of RAW264.7 cells was performed using Lipofectamine transfection reagents (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. These transfected cells were stimulated with either LPS (100 ng/ml), or PBS (control), and used for indicated experiments. RAW264.7 cells were transfected with luciferase reporter plasmids driven by the HIF1α (HRE-Luc) or were co-transfected with pcDNA3-BHLHE40 plasmid or Bhlhe40 specific siRNA using Lipofectamine® transfection reagent (Invitrogen). These cells were exposed to 100 ng/ml LPS for 18 h. Luciferase reporter activity was measured and normalized according to the manufacturer’s instructions. Results are presented as relative luciferase activity over the control group.
Quantification and statistical analyses
All data, unless indicated, are presented as mean ± standard deviation (SD). The statistical significance of differences between the two groups was analyzed by Student’s t-test or two-way ANOVA with Bonferroni multiple comparison tests. p< 0.05 was considered statistically significant.
Supporting information
Supporting information includes two tables.
Results
BHLHE40 expression and impact of myeloid-Bhlhe40 deficiency on blood cells
Previous reports have shown that transcription factor BHLHE40 promotes TH1 and TH17 cells mediated neuroinflammation (17, 18). Recent studies demonstrated that Bhlhe40+GM-CSF+CD4+ T-cells promote pathogenic alloantigen presentation in acute graft-versus-host disease and pathological damages associated with inflammatory cytokine production (22). However, the expression level of BHLHE40 in fully differentiated primary macrophages and macrophage cell lines has not been examined. Analyses of bone marrow-derived macrophages (BMDMs) and thioglycollate-elicited peritoneal macrophages (PMs) revealed that Bhlhe40 mRNA expression is approximately 8-fold higher in PMs compared to BMDMs (Figure 1A). Similarly, Bhlhe40 mRNA expression is ~5-fold higher in J774.1 cells compared to RAW264.7 cells (Figure 1B). Further, our survey of Bhlhe40 expression across multiple tissue types shows that Bhlhe40 is most abundantly expressed in primary macrophages (Figure 1C). Next, we examined whether pro-inflammatory agents such as lipopolysaccharides (LPS) exposure alter BHLHE40 mRNA and protein expression in macrophages. As shown in Figures 1D and E, LPS challenge modestly elevated BHLHE40 mRNA and protein expression at one hour. However, prolonged exposure of LPS markedly attenuated BHLHE40 expression in wild-type mice BMDMs (Figure 1D and E). To investigate the functional role of BHLHE40 in macrophage inflammatory response, we generated myeloid-specific BHLHE40-deficient mice by crossing Bhlhe40fl/fl mouse with Lyz2cre/cre mouse line. Our analyses show that greater than 95% reduction of Bhlhe40 at mRNA levels in Bhlhe40fl/fl:Lyz2cre/cre mice PMs and BMDMs compared to Lyz2cre/cre mice (Figure 1F). Concordantly, western blot analysis corroborated complete loss of BHLHE40 protein in Bhlhe40fl/fl: Lyz2cre/cre mice PMs and BMDMs (Figure 1G). Since BHLHE40 is known to play an essential role in immune cell functions (23), we examined whether myeloid-BHLHE40 deficiency alters adult mice hematopoietic cell compartment. Our analyses show that no significant difference between Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice lymphocyte, neutrophil, monocyte, eosinophil, and basophil populations (Figure 1H). Further analysis of additional hematological components revealed a modest decrease in mean corpuscular hemoglobin content and volume (MCH and MCV) are noticed in Bhlhe40fl/fl:Lyz2cre/cre mice (Figure 1H). However, we did not observe any significant difference in other hematological components between Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice (Figure 1H). Collectively, our analyses reveal that BHLHE40 is abundantly expressed in macrophages, and myeloid-BHLHE40 deficiency did not substantially reshape the hematopoietic cell components in adult mice.
Figure 1. BHLHE40 expression and impact of myeloid-Bhlhe40 deficiency on blood cells.
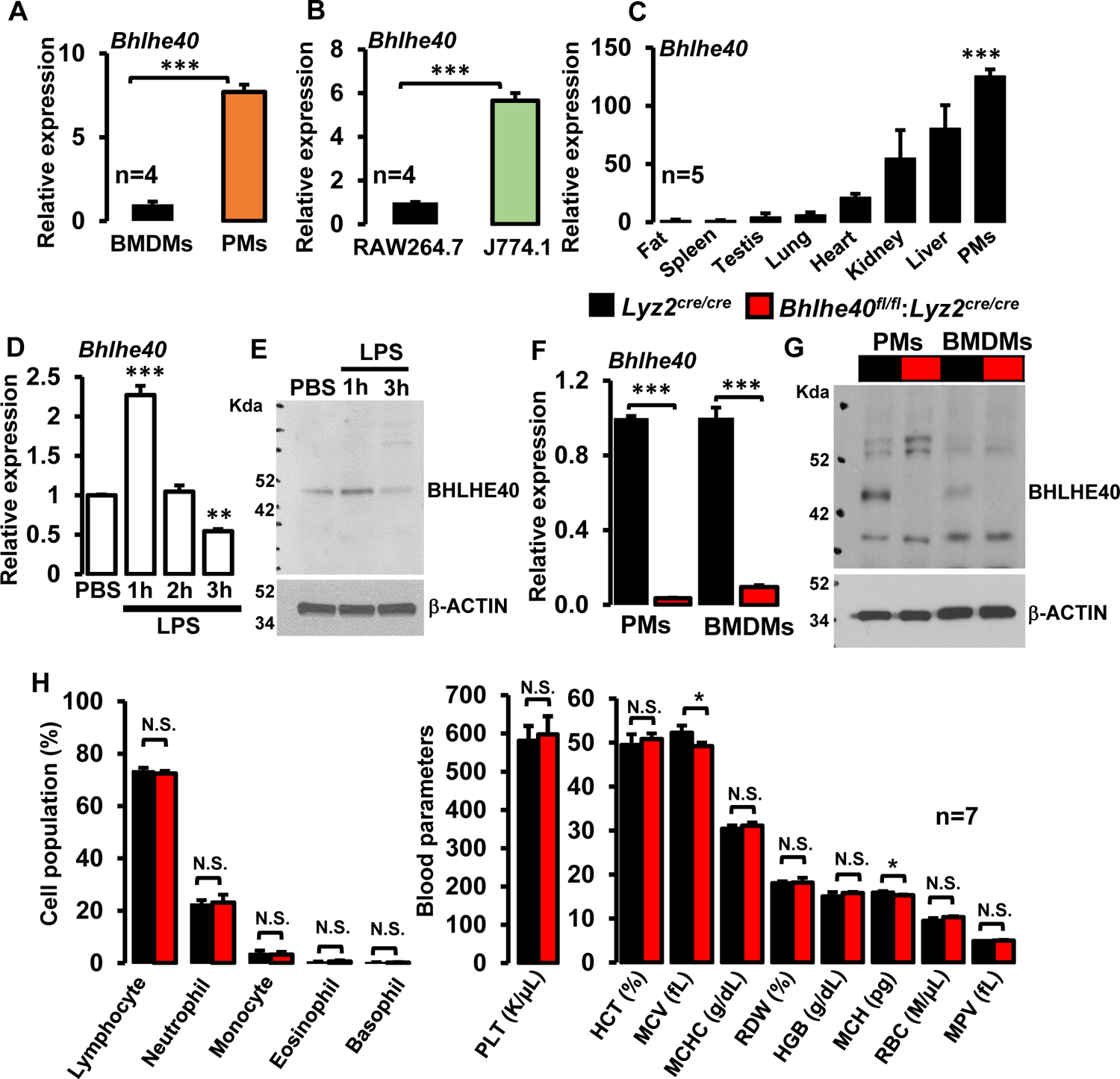
(A and B) Bhlhe40 mRNA expression in bone marrow-derived macrophages (BMDMs), thioglycollate-elicited peritoneal macrophages (PMs), RAW264.7, and J774.1 cells were analyzed by RT-qPCR (n=4). (C) Bhlhe40 mRNA expression levels were analyzed in mouse adipose tissue, spleen, testis, lung, heart, kidney, liver, and PMs by RT-qPCR (n=5). (D and E) Wild-type murine BMDMs were stimulated with 100 ng/ml of LPS for 0–3 hours. BHLHE40 mRNA (D) and protein (E) expression were evaluated by quantitative PCR, and Western blot analyses, respectively (n=4). (F and G) PMs and BMDMs from Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were analyzed for BHLHE40 mRNA and protein expression by RT-qPCR (F) and Western blot (G) analyses respectively (n=5). Actin and 36B4 were used as housekeeping genes for western blot, and RT-qPCR analyses, respectively. (H) Age and sex-matched Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice blood samples were analyzed by Hemavet 950™ hematology profiling unit. Bar graphs represent the mean cellular population with standard deviation (n=7). Data were analyzed either by Student’s t-test (A, B, F, H) or 2-way ANOVA (C and D). N.S. not significant. *P<0.05; **P<0.001, and ***P<0.001. All values are reported as mean ± S.D.
Myeloid-BHLHE40 deficiency is protective against zymosan-induced lung inflammation
Our analyses above (Figure 1D and E) show that BHLHE40 is an immediate-early response gene in macrophages. Therefore, we hypothesized that myeloid-BHLHE40 is required for the establishment of inflammatory response in vivo. To test this hypothesis, we utilized the zymosan‐induced acute lung inflammation and injury model. Accordingly, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were challenged by delivering zymosan through the intratracheal route. As expected, zymosan exposure robustly heightened immune cell recruitment to Lyz2cre/cre mice lungs (Figure 2A and D). Surprisingly, zymosan-induced immune cells recruitment was significantly attenuated in Bhlhe40fl/fl:Lyz2cre/cre mice compared to Lyz2cre/cre mice group challenged with zymosan (Figure 2A and D). Previous studies have shown that macrophages and neutrophils are the major innate immune cells recruited to lungs in the face of zymosan challenge (32, 33). Thus, we evaluated whether myeloid-BHLHE40 deficiency amends macrophage or neutrophil recruitment to lungs following zymosan challenge. Our histological analyses show that the zymosan challenge significantly elevated macrophage recruitment in Lyz2cre/cre mice lungs (Figure 2B and E). Remarkably, zymosan-induced macrophage accumulation was significantly attenuated in Bhlhe40fl/fl:Lyz2cre/cre mice lungs compared to Lyz2cre/cre mice (Figure 2B and E). Concurrently, we evaluated whether myeloid-BHLHE40 deficiency modulates neutrophil recruitment to lungs following zymosan exposure. Interestingly, zymosan-induced neutrophil accumulation was also significantly attenuated in myeloid-BHLHE40 deficient mice compared to Lyz2cre/cre mice group (Figure 2C and F). Our analyses thus far show that myeloid-BHLHE40 deficient mice are protected against pathological inflammatory response in vivo (Figure 2A–F). Thus, we intend to examine whether BHLHE40 deficiency affects pro-inflammatory gene expression in vivo. Accordingly, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were challenged with zymosan, and myeloid cells from bronchoalveolar lavages were utilized for inflammatory gene expression analyses. As shown in Figure 2G, zymosan challenge significantly heightened expression of Nos2, Ifnγ, Tnf, Ccl7, Cxcl9, and Cfb in Lyz2cre/cre mice bronchoalveolar myeloid cells. Strikingly, BHLHE40 deficiency significantly attenuated zymosan-induced expression of these pro-inflammatory cytokines and chemokines in bronchoalveolar myeloid cells (Figure 2G). Taken together, our analyses show that BHLHE40 deficiency significantly attenuates pro-inflammatory cytokine/chemokine expression and alleviates inflammatory innate immune cell recruitment to the site of inflammation.
Figure 2. Myeloid-BHLHE40 deficiency attenuates zymosan-induced lung inflammation.
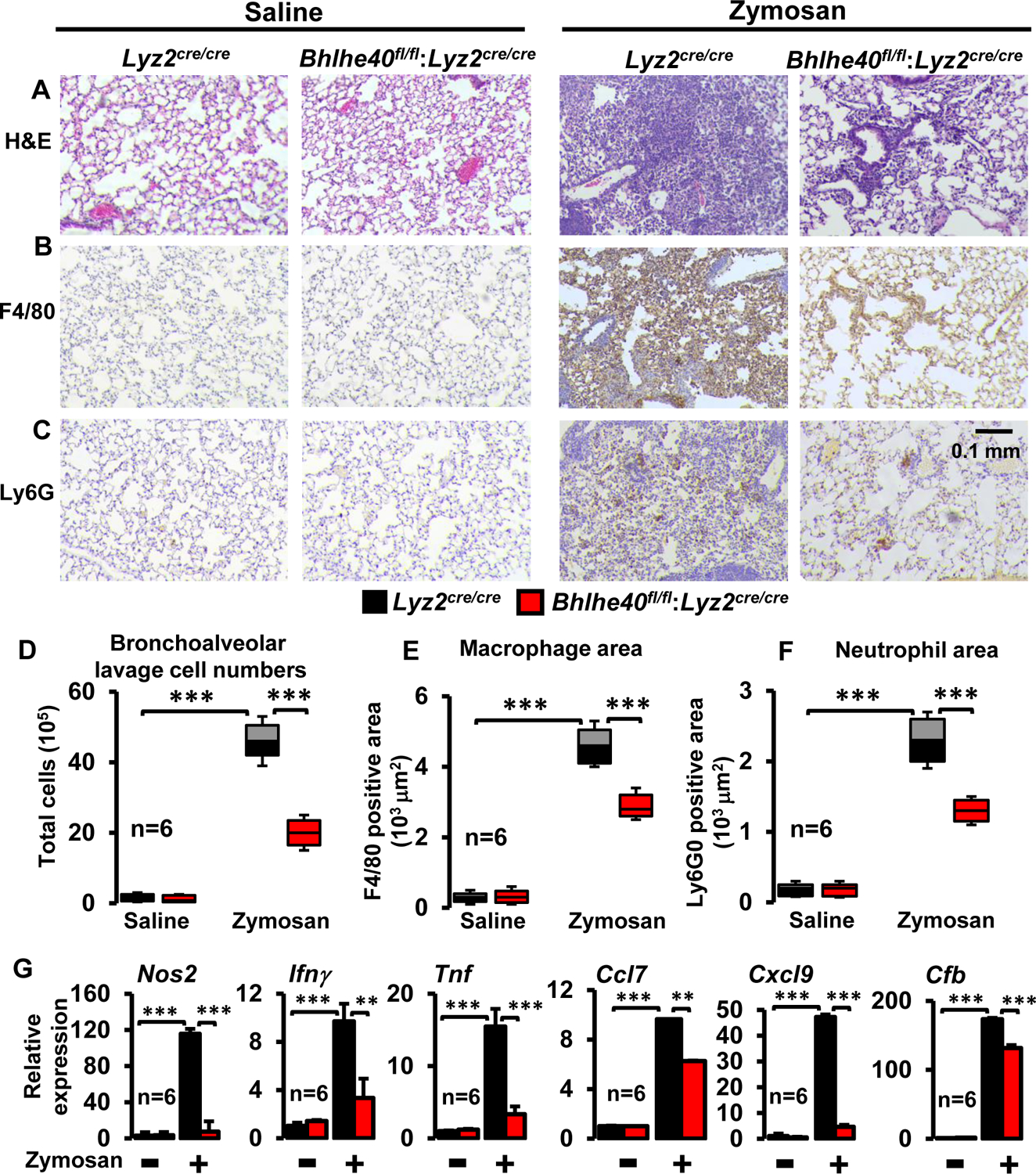
(A-C) The Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were challenged with intratracheal delivery of saline or zymosan. The lungs were collected after three days of zymosan challenge, and lung sections were stained for hematoxylin-eosin (A). The macrophages and neutrophils localization within the lung tissue were determined by anti-F4/80 (B) and anti-Ly6G (C) antibodies staining, respectively (n=6/group). (D) Bronchoalveolar lavage from Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were collected 3-days after intratracheal delivery of zymosan or saline (n=6/group). Total cell numbers are enumerated. (E and F) Area of F4/80 (E) and Ly6G (F) positive cells were quantified using Image J software (n=6/group). (G) The total RNA samples derived from bronchoalveolar lavage cells were evaluated for expression of Nos2, Ifnγ, Tnf, Ccl7, Cxcl9, and Cfb by RT-qPCR (n=6/group). The 36B4 gene was used as a housekeeping gene for RT-qPCR analyses. The box plot represents the median with first and third quartiles, and whiskers represent minimum/maximum. All other values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. * p < 0.05; ** p < 0.01; *** p < 0.001.
BHLHE40 deficiency curbs broad pro-inflammatory gene programs
Our in vivo studies demonstrate that myeloid-BHLHE40 deficiency significantly attenuates pro-inflammatory gene expression and curtails pathological inflammatory response (Figure 2A–G). Therefore, we postulated that BHLHE40 deficiency might alleviate expansive pro-inflammatory gene expression in macrophages. To test this notion, PMs derived from Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were stimulated with LPS, and total RNA samples were obtained to perform unbiased RNAseq analyses. To identify BHLHE40 regulated gene targets and signaling pathways, RNAseq data were subjected to Gene Set Enrichment Analysis (GSEA) (30). Remarkably, our GSEA studies show that BHLHE40 deficiency significantly curtails allograft rejection, IFNγ response, inflammatory response, IFNα response, complement response, activation of reactive oxygen species pathway, hypoxia response, MTORC1 signaling, xenobiotic metabolism, IL6-JAK-STAT3 signaling, and glycolysis pathway (Figure 3A–G). Next, we carefully examined the expression of pro-inflammatory and pathogenic gene expression in our GSEA studies. As anticipated, LPS challenge robustly elevated a large number of pro-inflammatory genes involved in the allograft rejection pathway in Lyz2cre/cre mice PMs (Figure 4A). However, the induction of these pro-inflammatory gene targets (Nos2, Ifnγ, Irf8, Il6, Cxcl9, Il12a, Ccl7, Nlrp3, Tnf…etc) were significantly attenuated in Bhlhe40fl/fl:Lyz2cre/cre mice PMs stimulated with LPS (Figure 4A). Similarly, LPS exposure significantly elevated a substantial number of IFNγ and IFNα response gene targets in Lyz2cre/cre mice PMs (Figure 4B). Remarkably, LPS-induced IFNγ and IFNα response gene expression (Btg1, Cd69, Socs3, Cd274, Irf5, Cd38, Ptgs2, Casp3…etc.) were significantly dampened in BHLHE40 deficient macrophages (Figure 4B). Concordantly, LPS treatment vigorously elevated a substantial number of inflammatory and IL6-JAK-STAT3 response gene targets in Lyz2cre/cre mice PMs (Figure 4C). Strikingly, expression of these gene targets (Cd14, Cxcl3, Cd82, Irak2, Mmp14, Itga4, Ccr7…etc) were significantly diminished in Bhlhe40fl/fl:Lyz2cre/cre mice PMs challenged with LPS (Figure 4C). Heat maps for allograft rejection, IFNγ/IFNα response, and inflammatory response represent the top fifty differentially expressed genes in the presence and absence of LPS (Figure 4A–C). Further, LPS challenge also robustly elevated a large number of complement response genes in Lyz2cre/cre mice PMs (Figure 4D). Surprisingly, BHLHE40 deficiency significantly diminished LPS-induced complement response gene (Ctsb, Cebpb, Adm9, C3, Serpinb2, Mmp13…etc.) expression in macrophages (Figure 4D). Collectively, our analyses show that BHLHE40 deficiency broadly attenuates pro-inflammatory signaling pathways and gene expression in macrophages.
Figure 3. BHLHE40 deficiency curbs broad pro-inflammatory gene programs.
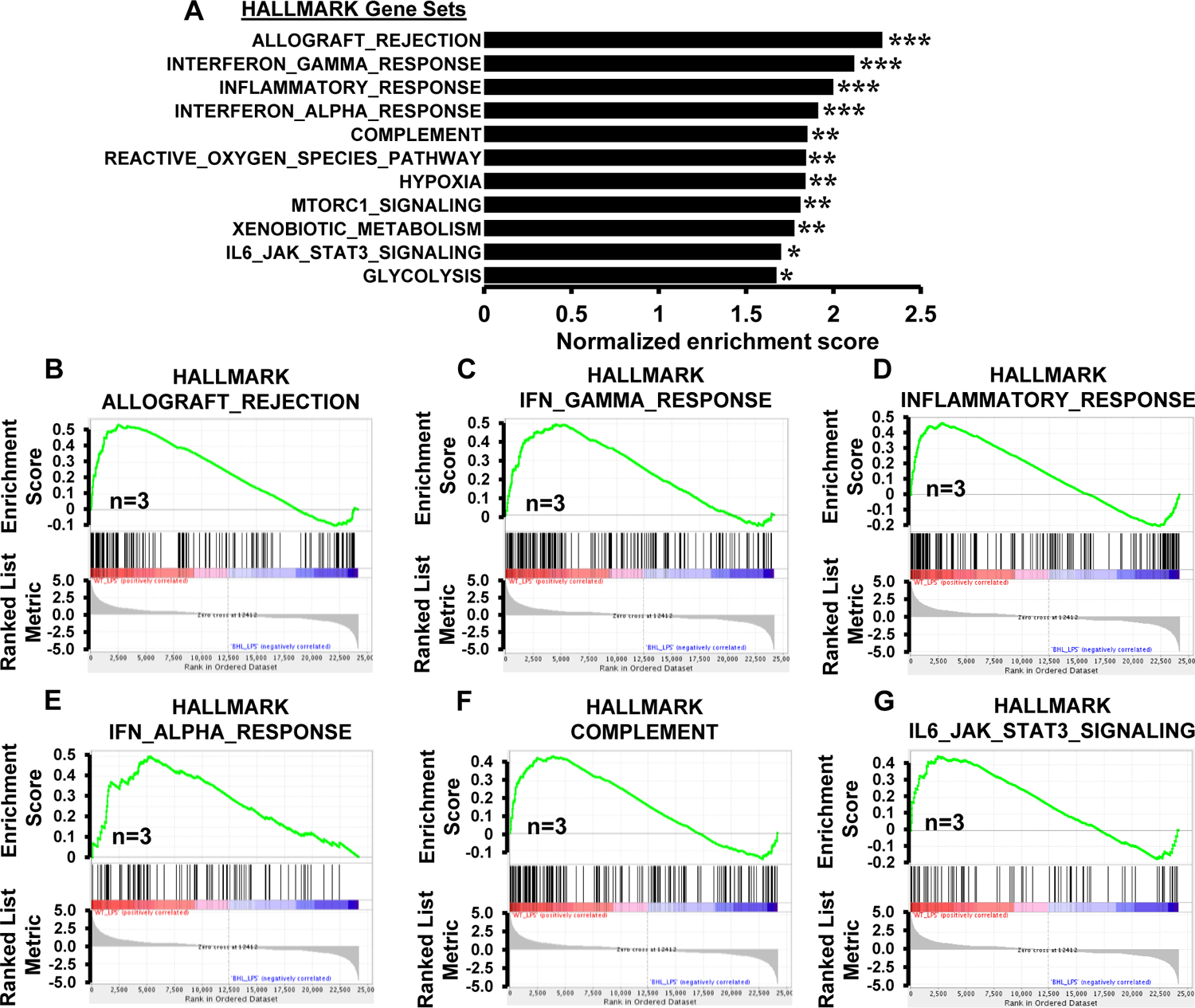
(A) GSEA of RNAseq data that are altered in Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs following 4 hours of 100 ng/ml LPS treatment. FWER p-value less than 0.05 was considered significant (n=3). (B-G) Enrichment plots of indicated gene set obtained by GSEA comparing Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs RNAseq data following LPS treatment (n=3).
FWER p-value * p < 0.05; ** p < 0.01; *** p < 0.001.
Figure 4. BHLHE40 deficiency dampens pro-inflammatory gene expression.
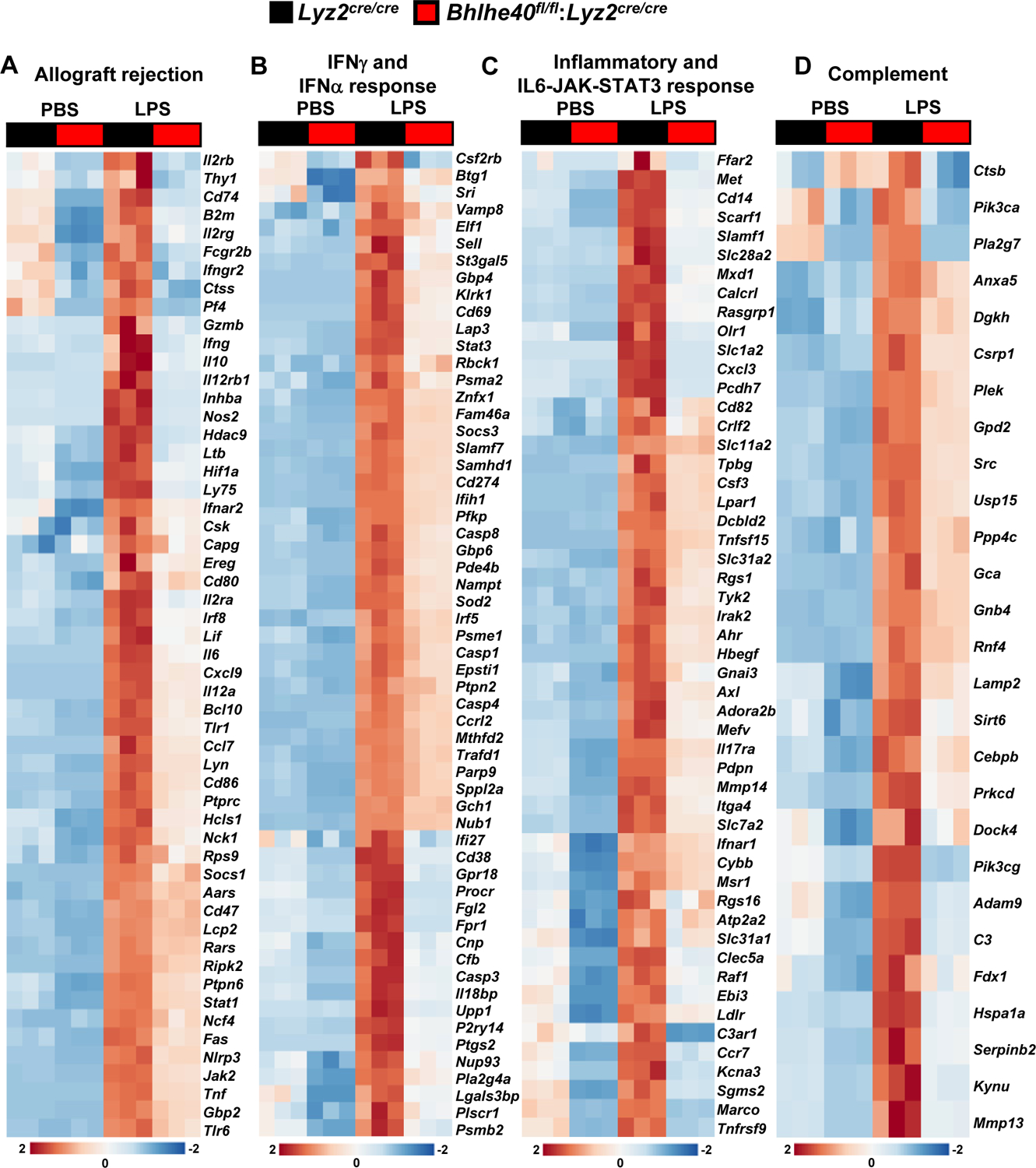
(A-D) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100 ng/ml LPS for 4 hours, and RNAseq data were subjected to GSEA studies. The LPS challenge significantly elevated gene targets involved in allograft rejection (A), IFNγ and INFα response (B), inflammatory and IL6-JAK-STAT3 response (C), and complement response (D) in Lyz2cre/cre mice macrophages. These inflammatory signaling responses were significantly dampened in Bhlhe40fl/fl:Lyz2cre/cre mice macrophages (n=3). Heat maps for allograft rejection, IFNγ/IFNα response, and inflammatory response represent the top fifty differentially expressed genes in the presence and absence of LPS. GSEA results from IFNγ and IFNα response data set were combined to generate panel B. Similarly, Inflammatory and IL6-JAK-STAT3 were combined to generate panel C.
BHLHE40 deficiency diminishes inducible pro-inflammatory gene expression
Our studies thus far show that BHLHE40 deficiency broadly attenuates pro-inflammatory gene programs (Figures 3 and 4) and is protective against pathological inflammatory response in vivo (Figure 2). Therefore, we posited that BHLHE40 is required for inducible pro-inflammatory gene expression. To test this concept, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were challenged with LPS, and total RNA samples were evaluated for classical pro-inflammatory gene expression. As shown in Figure 5A, LPS exposure significantly elevated pro-inflammatory genes involved in allograft rejection signaling including, Nos2, Ifnγ, Il12a, Tnf, Nlrp3, and Cxcl9 expression in Lyz2cre/cre macrophages. However, BHLHE40 deficiency significantly curtailed induction of these pro-inflammatory genes following LPS challenge (Figure 5A). Likewise, LPS exposure also heightened the expression of classical IFNγ and IFNα response genes including, Cd38, Ptgs2, Fpr1, Socs3, Cd274, and Irf8 in Lyz2cre/cre macrophages (Figure 5B). Remarkably, LPS-induced upregulation of these pro-inflammatory genes were significantly attenuated in Bhlhe40fl/fl:Lyz2cre/cre mice PMs (Figure 5B). Similarly, treatment of Lyz2cre/cre mice PMs with LPS robustly enhanced IL6-JAK-STAT3 signaling target gene expression (Cd14, Ccr7, Il6, Slc7a2, Itga4, and Ccl7) (Figure 5C). Nevertheless, the expression of these pro-inflammatory gene targets were significantly extenuated in LPS-induced Bhlhe40fl/fl:Lyz2cre/cre mice PMs (Figure 5C). Further, LPS treatment also augmented the expression genes involved in complement response such as Mmp14, Kynu, C3, Mmp13, Serpinb2, Cfb, Il1β, and Icam1 in Lyz2cre/cre mice PMs (Figure 5D). However, BHLHE40 deficiency significantly curtailed upregulation of these pro-inflammatory genes following LPS treatment (Figure 5D). Next, we assessed whether this shift in the inflammatory gene expression were observed at the protein level. Accordingly, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were challenged with LPS, and cell culture supernatants were evaluated for TNF, IFNγ, IL6, and IL1β levels by ELISA. As shown in Figure 6A, the LPS challenge significantly elevated TNF, IFNγ, IL6, and IL1β secretion in Lyz2cre/cre mice PMs. However, secretion of these pro-inflammatory cytokines were greatly diminished in BHLHE40 deficient macrophages (Figure 6A). Taken together, our analyses show that BHLHE40 deficiency significantly attenuates inducible pro-inflammatory gene expression in macrophages.
Figure 5. Impact of BHLHE40 deficiency on LPS-induced pro-inflammatory gene expression.
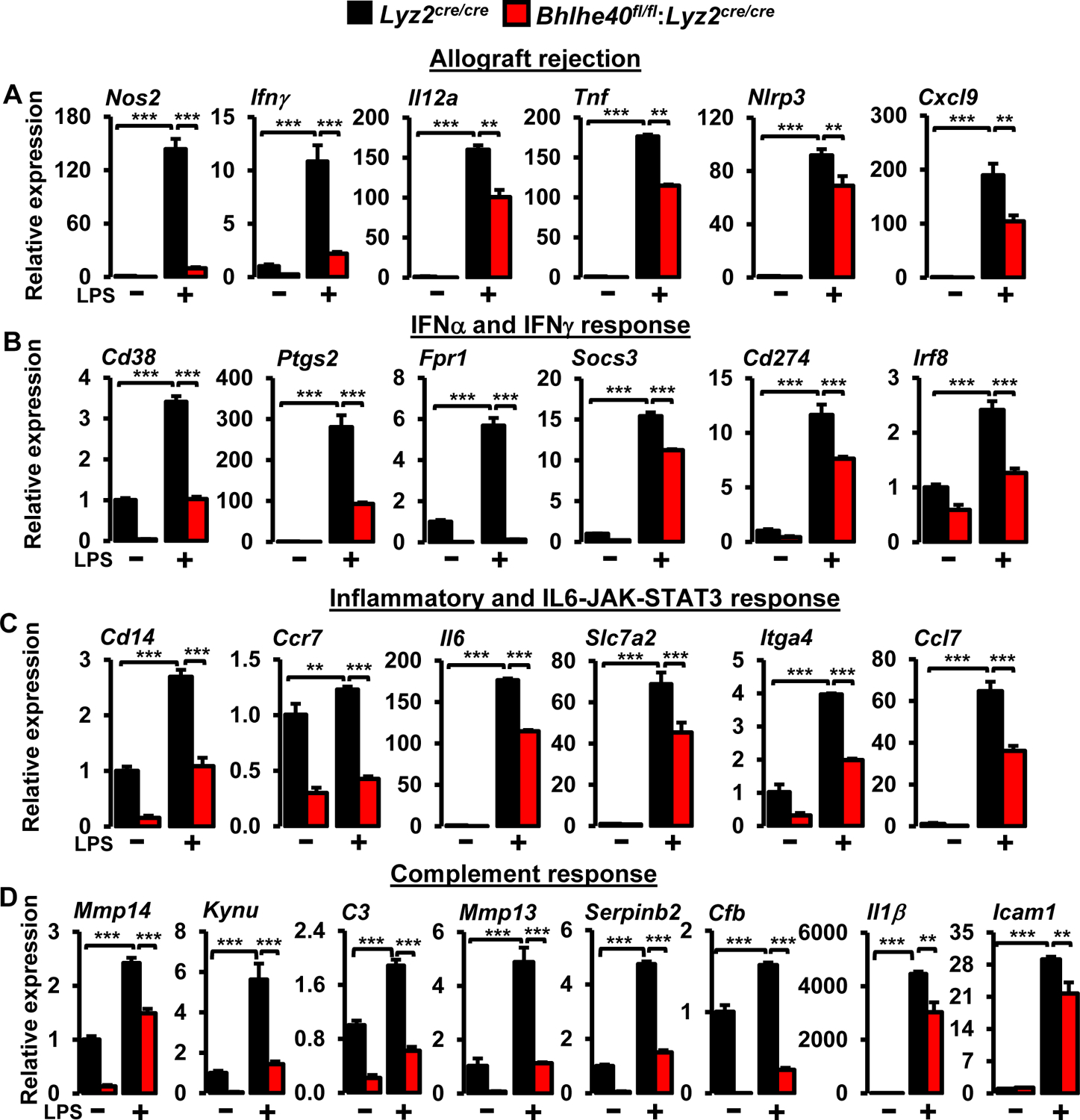
(A-D) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100 ng/ml of LPS for 4 hours. Total RNA samples were analyzed for expression of (A) allograft rejection (Nos2, Ifnγ, Il12a, Tnf, Nlrp3, Cxcl9), (B) IFNα/IFNγ response (Cd38, Ptgs2, Fpr1, Socs3, Cd274, Irf8), (C) inflammatory and IL6-JAK-STAT3 response (Cd14, Ccr7, Il6, Slc7a2, Itga4, Ccl7), and (D) complement (Mmp14, Kynu, C3, Mmp13, Serpinb2, Cfb, Il1β, Icam1) target genes were analyzed by RT-qPCR. 36B4 was used as a housekeeping gene (n=3). Values are reported as mean ± SD. Data were analyzed by Student’s t-test. *p < 0.05, ** p < 0.01 and ***p < 0.001.
Figure 6. BHLHE40 promotes LPS-induced HIF1α expression in macrophages.
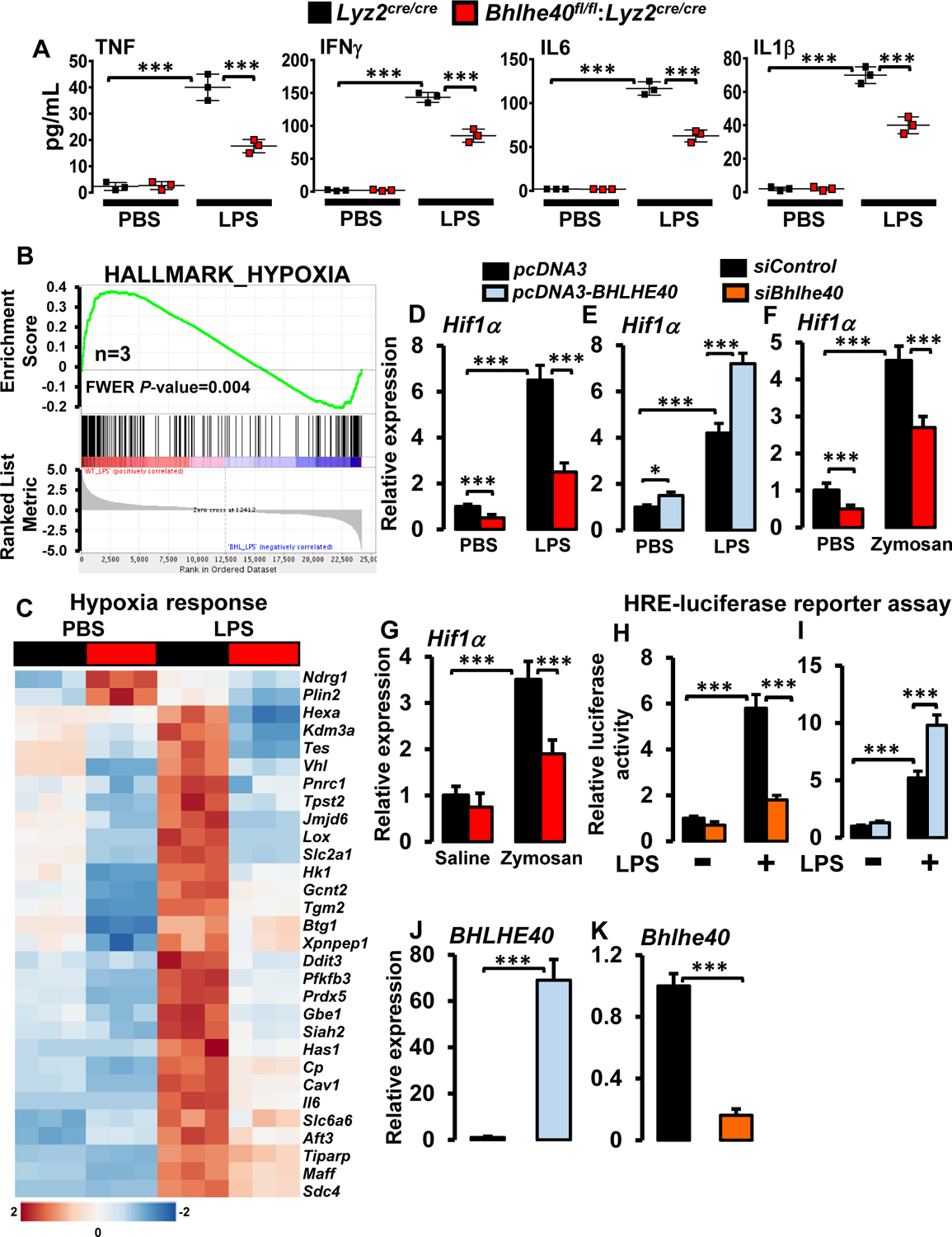
(A) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100 ng/ml of LPS for 12 hours. Cell culture supernatant TNF, IFNγ, IL6, and IL1β levels were quantified by ELISA and normalized to total cell numbers (n=3). (B and C) Enrichment plot of hypoxia gene set obtained by GSEA (B) and heatmap showing clustering of hypoxia response genes (C) generated by comparing RNAseq data from Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs following LPS treatment (n=3). (D and E) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs (D) or RAW264.7 cells transfected with pcDNA3-BHLHE40 (E) were stimulated with 100 ng/ml LPS for 4 hours. Total RNA samples were analyzed for Hif1α expression by RT-qPCR (n=4). (F) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated 0.2μg/ml zymosan for 4 hours. Total RNA samples were analyzed for Hif1α expression by RT-qPCR (n=3). (G) The Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were challenged with intratracheal delivery of saline or zymosan. Macrophages derived from bronchoalveolar lavages were evaluated for Hif1α expression by RT-qPCR (n=5). (H and I) RAW264.7 cells were transfected with an HRE-luciferase reporter construct in the presence of Bhlhe40-specific siRNA (H) or pcDNA3-BHLHE40 plasmid (I). These cells were stimulated with 100 ng/ml LPS for 18 hours, and cell lysates were analyzed for luciferase activity (n=3). (J and K) RAW264.7 cells transfected with pcDNA3-BHLHE40 (J) or BHLHE40 specific siRNA (K). Total RNA samples were obtained and evaluated for BHLHE40 expression by RT-qPCR (n=3). 36B4 or SHDA were used as housekeeping genes for RT-qPCR analyses. Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. ***p < 0.001.
BHLHE40 promotes LPS-induced HIF1α expression in macrophages
Previous studies have established that HIF1α is a central regulator of macrophage inflammatory response and pathogenesis of inflammation (7, 10). Interestingly, myeloid-HIF1α deficient mice are protected from inflammatory disease conditions (10) and exhibited attenuated pro-inflammatory gene expression (10, 34). In this context, our analyses show that BHLHE40 deficiency significantly attenuates inflammatory gene expression (Figures 3–5) as well as inflammatory disease pathogenesis in vivo (Figure 2). Thus, we hypothesized that BHLHE40 might promote inflammatory gene response by supporting HIF1α signaling in macrophages.
To evaluate this concept, we carefully reviewed GSEA of RNAseq data for a shift in hypoxia response signaling. Our analyses show that LPS challenge significantly enriched hypoxia response gene expression in Lyz2cre/cre mice PMs (Figures 3A and 6B). Concordantly, LPS exposure robustly elevated a large number of hypoxia response gene expressions in Lyz2cre/cre mice PMs (Figure 6C). However, LPS-induced upregulation of these classical hypoxia response genes were significantly attenuated in BHLHE40 deficient macrophages (Figure 6C). Interestingly, our RNAseq analyses have identified that LPS-induced Hif1α expression is significantly curtailed in BHLHE40 deficient macrophages (Figure 4A). Thus, we evaluated whether altering BHLHE40 levels affect LPS-induced HIF1α expression in macrophages. Accordingly, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were exposed to LPS, and Hif1α mRNA levels were evaluated by RT-qPCR analysis. As shown in Figure 6D, LPS-induced Hif1α mRNA levels are significantly attenuated in BHLHE40 deficient macrophages compared to Lyz2cre/cre mice PMs. Concordantly, overexpression of BHLHE40 significantly elevated basal as well as LPS-induced Hif1α mRNA expression in RAW264.7 macrophage cell line (Figure 6E).
Our in vivo studies have shown that myeloid-BHLHE40 deficient mice are protected from zymosan-induced lung inflammation (Figures 2A–G). Therefore, we examined whether BHLHE40 deficiency altered zymosan-induced Hif1α expression in macrophages. As shown in Figure 6F, zymosan-induced Hif1α expression was significantly attenuated in BHLHE40 deficient PMs. Further, our analysis of lung lavage macrophages also shows that BHLHE40 deficiency significantly curtails Hif1α expression following zymosan challenge in vivo (Figure 6G). Next, we assessed whether altering the BHLHE40 expression affects HIF1α transcriptional activity in macrophages. Accordingly, RAW264.7 cells were cotransfected with hypoxia response element (HRE)-driven luciferase reporter plasmid in the presence of siBhlhe40 or pcDNA3-Bhlhe40. These cells were challenged with LPS, and luciferase activities were recorded. As shown in Figures 6H and I, BHLHE40 knockdown attenuated, and overexpression of BHLHE40 enhanced LPS-induced HIF1α driven luciferase reporter activity in macrophages. The forced overexpression and knockdown of BHLHE40 in RAW264.7 cells were confirmed by RT-qPCR analyses (Figures 6J and K). Concurrently, we examined whether these observations were recapitulated at the HIF1α protein level. As shown in Figures 7A and B, zymosan challenge significantly elevated HIF1α protein expression in Lyz2cre/cre mice lung macrophages in vivo. However, zymosan exposure induced HIF1α protein expression was significantly attenuated in BHLHE40 deficient mice lung macrophages in vivo (Figures 7A and B). Furthermore, BHLHE40 deficiency substantially attenuated LPS (Figures 7C and D) or hypoxia-induced (Figures 7E and F) HIF1α protein levels in primary macrophages. Next, we examined whether experimentally overexpressed BHLHE40 utilizes HIF1α signaling to elevate pro-inflammatory gene expression in macrophages. Accordingly, RAW264.7 cells were overexpressed with BHLHE40 in the presence or absence of HIF1α specific siRNA. These cells were stimulated with LPS, and total RNA samples were evaluated for expression of BHLHE40 targets by RT-qPCR analyses. As shown in Figure 7G, BHLHE40 overexpression significantly elevated basal as well as LPS-induced Ptgs2, Fpr1, Mmp13, and Cd14 expression. Interestingly, HIF1α knockdown in BHLHE40 overexpressing cells significantly attenuated LPS-induced Ptgs2, Fpr1, Mmp13, and Cd14 expression in macrophages (Figure 7G). Collectively. our analyses show that BHLHE40 supports inducible HIF1α expression in macrophages and promotes pro-inflammatory gene expression in HIF1α dependent manner.
Figure 7. BHLHE40 boosts HIF1α and pro-inflammatory gene expression in macrophages.
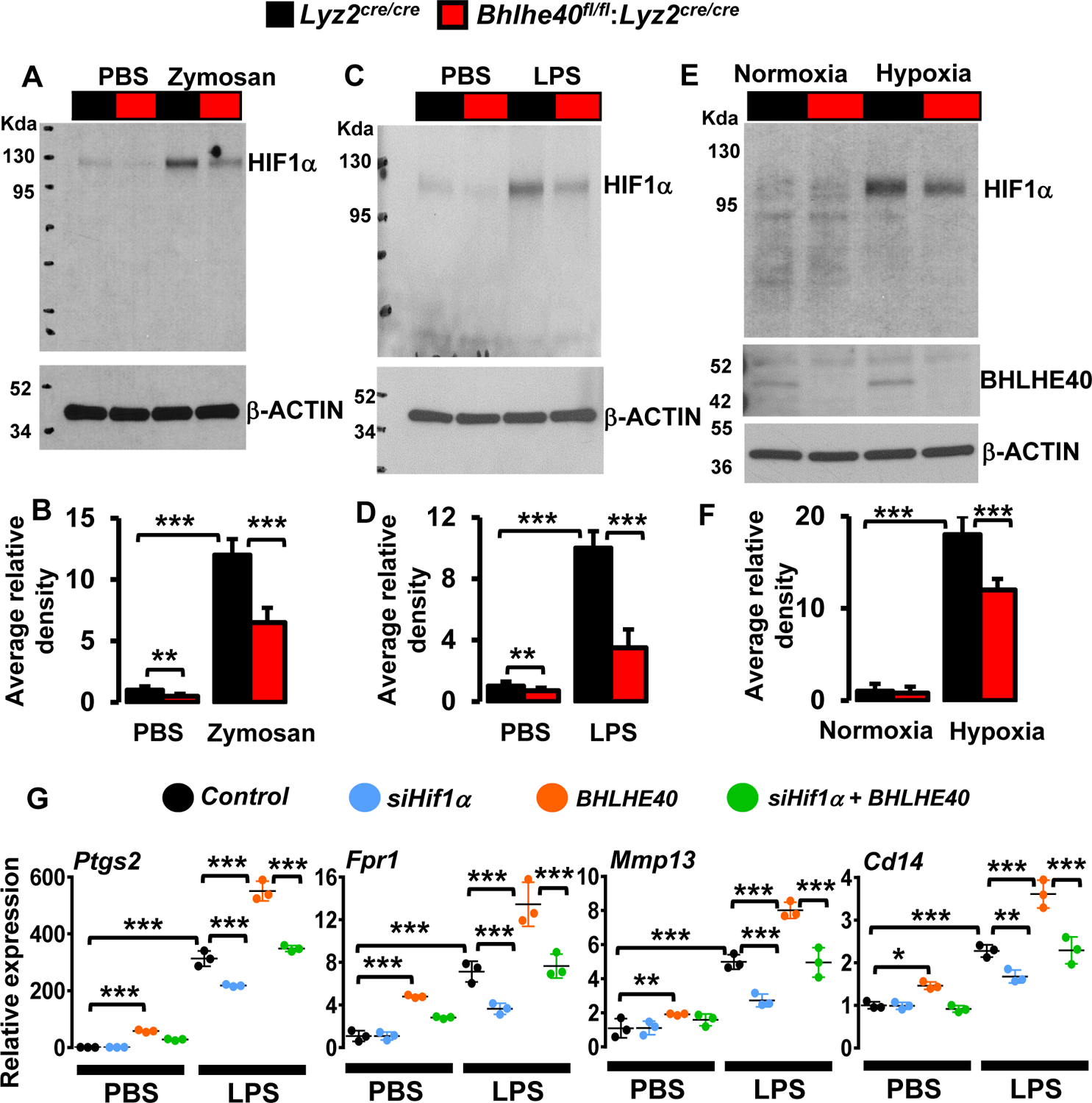
(A and B) The Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice were challenged with intratracheal delivery of saline or zymosan (n=3). Macrophages derived from bronchoalveolar lavages were evaluated for HIF1α protein levels by western blot (A). The total HIF1α levels were quantified by densitometry analysis (B). (C-F) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100 ng/ml LPS (C) or exposed to hypoxic conditions (E) for 4 hours. Total protein extracts were evaluated for the expression of HIF1α by western blot (n=3). The total HIF1α levels were quantified by densitometry analysis (D and F). (G) RAW264.7 cells were transfected with Hif1α-specific siRNA or/and pcDNA3-BHLHE40 plasmid. These cells were stimulated with 100 ng/ml LPS for 4 hours. Total RNA from these experiments were evaluated for expression of Ptgs2, Fpr1, Mmp13, and Cd14 by RT-qPCR (n=4). Actin and 36B4 were used as housekeeping genes for western blot and RT-qPCR analyses, respectively. Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. *p < 0.05, ** p< 0.01 and ***p < 0.001.
BHLHE40 deficiency attenuates processes of glycolysis in macrophages
Macrophages derive most of the energy through the process of glycolysis. Previous studies have shown that glycolysis is indispensable for macrophage activation and pro-inflammatory gene expression (35). More importantly, HIF1α governs key enzymes involved in the process of glycolysis (6). In this context, our analyses show that BHLHE40 promotes HIF1α expression (Figures 6–7), and BHLHE40 deficiency attenuates pro-inflammatory gene expression in macrophages (Figures 4 and 5). Therefore, we postulate that BHLHE40 deficiency may limit glycolytic gene expression in macrophages. Indeed, GSEA of our RNAseq data shows that LPS challenge significantly enriched glycolysis gene expression in Lyz2cre/cre mice PMs (Figures 3A and 8A). Consistently, LPS challenge robustly heightened the substantial number of glycolytic gene expressions in Lyz2cre/cre mice PMs (Figure 8B). Remarkably, BHLHE40 deficiency significantly attenuated LPS-induced escalation of these glycolytic gene expressions in primary macrophages (Figure 8B). Next, these differentially expressed glycolytic genes were used as a lead to probe the key glycolytic gene targets that are dysregulated in the absence of BHLHE40. Accordingly, Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with LPS, and total RNA samples were analyzed for glycolytic gene expression by RT-qPCR. As shown in Figures 8C and D, LPS challenge robustly heightened critical glycolytic gene expression (Glut1, Hk2, Pfkp, Pfkfb3, Pgk1, Eno2, Ldha, and Mct4) expression in Lyz2cre/cre mice PMs. Concordant with our RNAseq results, BHLHE40 deficiency significantly abrogated LPS-induced upregulation of key glycolytic gene expression in primary macrophages (Figures 8 C and D). Next, we examined whether BHLHE40 deficiency alters LPS-induced glycolytic process in macrophages. Our analyses show that LPS challenge significantly elevated cellular glucose uptake (Figure 8E), ATP generation (Figure 8F), and lactate production (Figure 8G) in Lyz2cre/cre mice PMs. Interestingly, BHLHE40 deficiency significantly curtailed LPS-induced glucose uptake, ATP generation, and lactate production in macrophages (Figure 8E–G). Taken together, our analyses demonstrate that BHLHE40 deficiency broadly attenuates inflammatory agent-induced glycolytic gene expression and the process of glycolysis in macrophages.
Figure 8. BHLHE40 deficiency attenuates the process of glycolysis in macrophages.
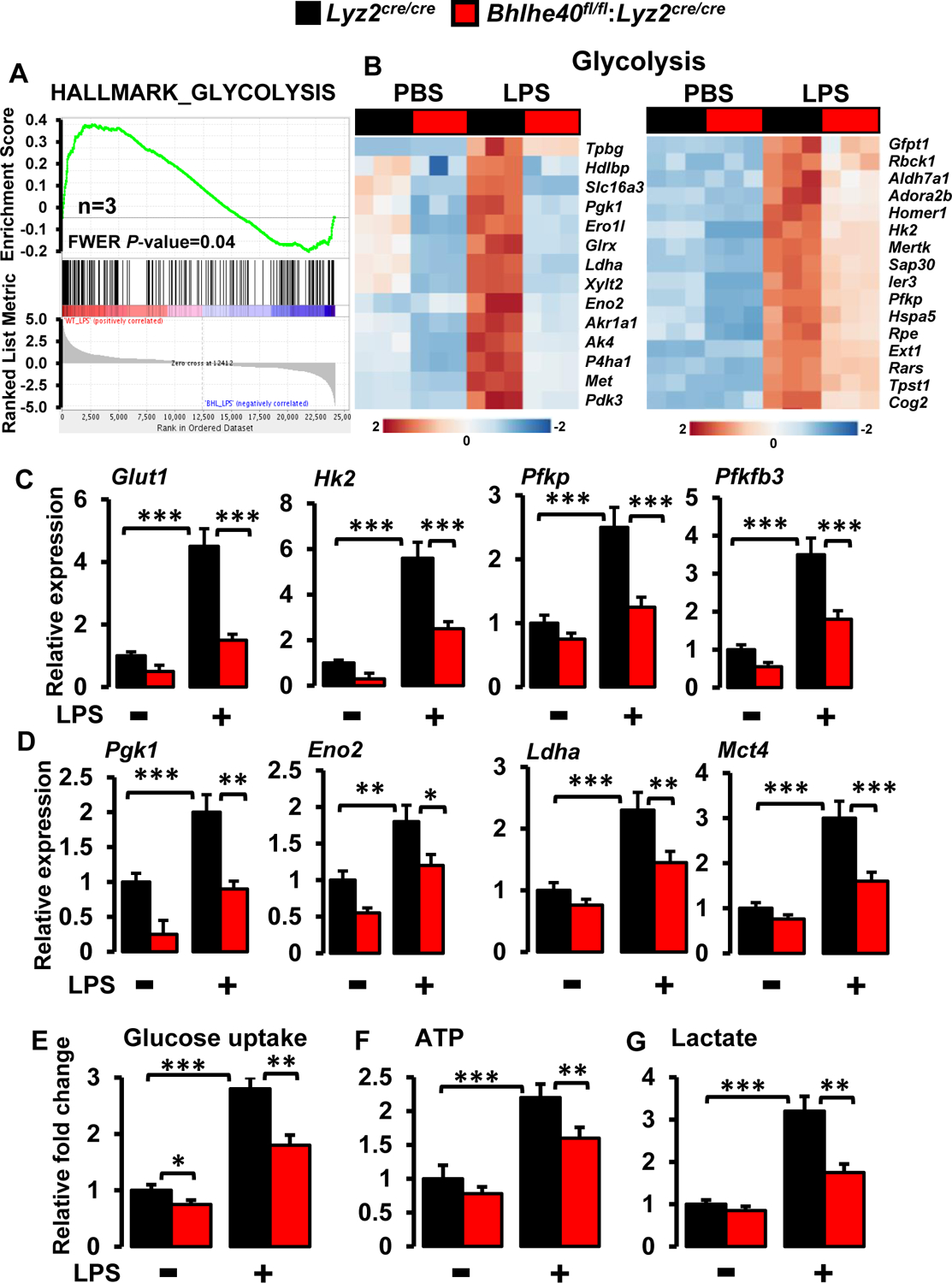
(A and B) Enrichment plot of glycolysis gene set obtained by GSEA (A) and heatmap showing clustering of glycolysis genes (B) generated by comparing RNAseq data from Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs following LPS treatment (n=3). (C and D) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100 ng/ml LPS for 4 hours. Total RNA from these experiments were evaluated for the expression of Glut1, Hk2, Pfkp, Pfkfb3 (C), and Pgk1, Eno2, Ldha, Mct4 (D) expression by RT-qPCR (n=4). 36B4 was used as a housekeeping gene for RT-qPCR analysis. (E-G) Lyz2cre/cre and Bhlhe40fl/fl:Lyz2cre/cre mice PMs were stimulated with 100ng/ml LPS. These cells were evaluated glucose uptake (E), ATP generation (F), and extracellular lactate production (G) by commercially available assay kits (n=3). Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. *p < 0.05, ** p< 0.01 and ***p < 0.001.
BHLHE40 utilizes HIF1α to promotes LPS-induced inflammatory and glycolytic gene expression
Previous reports have illustrated that HIF1α is a central regulator of inflammatory and glycolytic gene expression in macrophages (10, 34). Our analyses thus far show that BHLHE40 promotes inflammatory, hypoxia response, and glycolytic gene expression in macrophages (Figures 3–8). Thus, we hypothesize that BHLHE40 may utilize HIF1α signaling to elevate LPS-induced inflammatory and glycolytic gene expression. To test this hypothesis, we used genetic approaches to overexpress the oxygen stable form of HIF1α (ΔHif1α -P402A/P564A) in macrophages (36). Accordingly, RAW264.7 macrophages were transfected with a combination of siBhlhe40 siRNA and/or ΔHif1α plasmid. These cells were stimulated with LPS, and total RNA samples were evaluated for inflammatory and glycolytic gene expression by RT-qPCR.
Expression of ΔHIF1α in RAW264.7 cells were confirmed by western blot analysis (Figure 9A).
Figure 9. BHLHE40 promotes LPS-induced inflammatory and glycolytic gene expression through HIF1α.
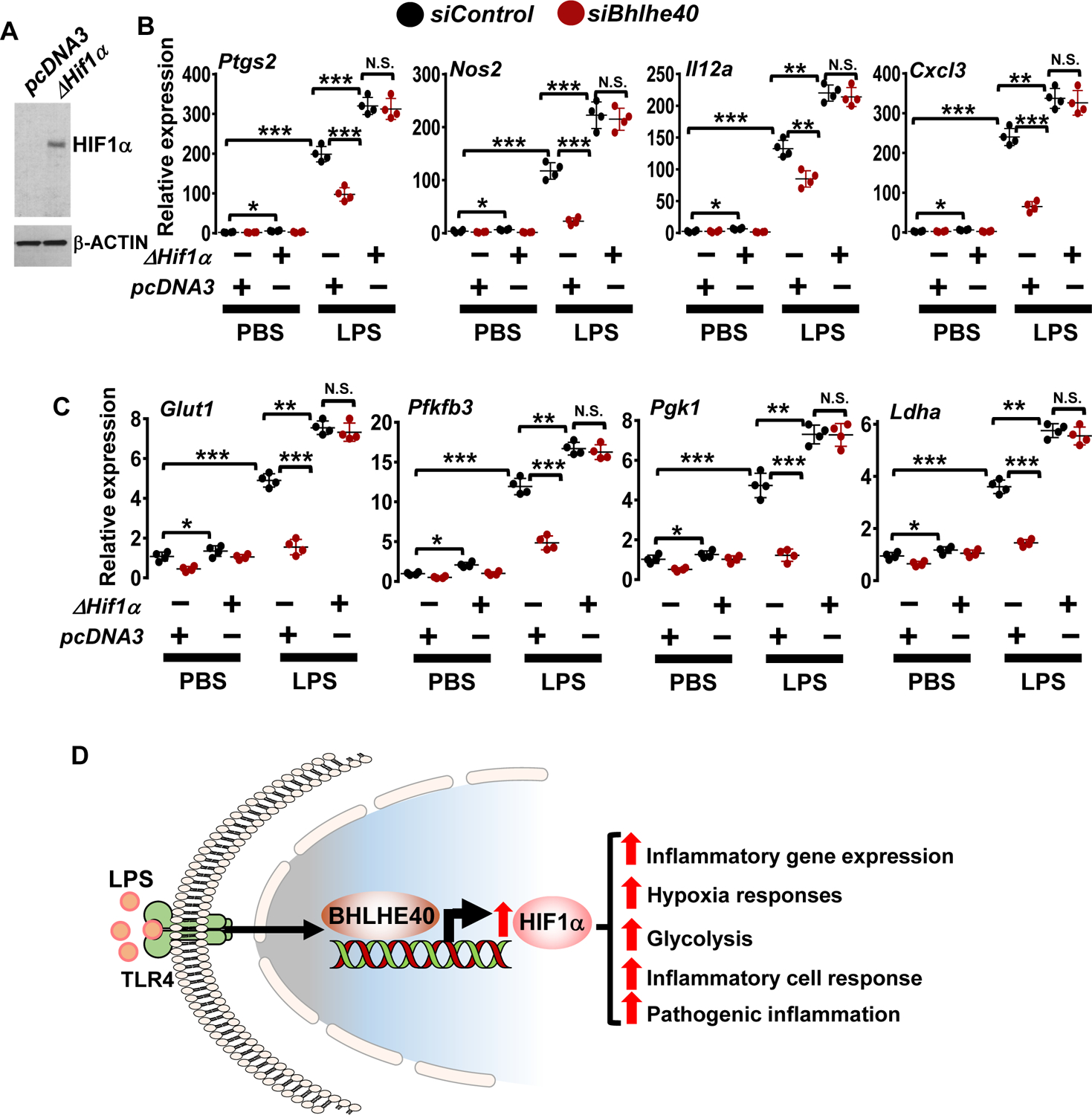
(A) RAW264.7 cells were transfected with pcDNA3 or pcDNA3-ΔHif1α plasmid. Total protein extracts from these cells were evaluated for HIF1α expression by western blots.
(B and C) RAW264.7 cells were transfected with Bhlhe40-specific siRNA or/and ΔHif1α plasmid. These cells were stimulated with 100 ng/ml LPS for 4 hours. Total RNA from these experiments were evaluated for expression of Ptgs2, Nos2, Il12a, Cxcl3 (B) and Glut1, Pfkfb3, Pgk1, Ldha (C) by RT-qPCR (n=4). Actin and 36B4 were used as housekeeping genes for western blot and RT-qPCR analyses, respectively. (D) BHLHE40 promotes macrophage inflammatory gene programs, hypoxia response, glycolytic gene expression, and pathogenic inflammatory responses by elevating HIF1α expression. All values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. N.S., not significant; ** p < 0.01; ***F p < 0.001.
As shown in Figures 9B and C, LPS challenge significantly elevated inflammatory (Ptgs2, Nos2, Il12a, and Cxcl3) and glycolytic (Glut1, Pfkfb3, Pgk1, and Ldha) gene expression in macrophages. As anticipated, BHLHE40 deficiency significantly curtailed LPS-induced inflammatory (Figure 9B) and glycolytic (Figure 9C) gene targets in macrophages. Surprisingly, overexpression of oxygen stable form of HIF1α fully reversed abbreviated inflammatory (Figure 9B) and glycolytic (Figure 9C) gene targets in BHLHE40 deficient macrophages. Overexpression of ΔHIF1α also modestly elevated basal level of inflammatory and glycolytic gene expression in unstimulated RAW264.7 cells (Figures 9B and C). Collectively, our analyses demonstrate that BHLHE40 promotes inflammatory and glycolytic gene expression by elevating HIF1α signaling in macrophages (Figure 9D).
Discussion
Our studies identify BHLHE40 as a novel molecular toggle that promotes broad pro-inflammatory and glycolytic gene expression by elevating HIF1α expression in macrophages. The key findings of this study are as follows: (1) BHLHE40 is abundantly and broadly expressed across macrophage cell lines, and primary macrophages; (2) Myeloid-BHLHE40 deficiency is protective against zymosan-induced lung inflammation; (3) BHLHE40 deficiency curbs broad pro-inflammatory gene programs in macrophages; (4) BHLHE40 deficiency attenuates inducible pro-inflammatory gene expression in macrophages; (5) BHLHE40 promotes LPS-induced HIF1α expression in macrophages; (6) BHLHE40 deficiency attenuates inducible glycolytic gene expression in macrophages; (7) BHLHE40 utilizes HIF1α to promotes LPS-induced inflammatory and glycolytic gene expression. Collectively, these findings demonstrate that BHLHE40 promotes inducible inflammatory and glycolytic gene programs by elevating HIF1α expression in macrophages (Figure 9D)
Monocyte-derived macrophages are the professional phagocytes that play a central role in maintaining human health and the development of inflammatory diseases. The protective and pathogenic nature of the macrophages are primarily determined by the magnitude of inflammatory gene expression at the sites of inflammation. The inflammatory cytokines, chemokines, and foreign agents predominately induce the expression of these inflammatory genes at the transcriptional levels. The transcriptional regulation of inducible inflammatory gene expression involves the coordinated regulation of multiple transcription factors and cofactors. In this context, our analyses show that BHLHE40 is abundantly expressed in macrophages. In addition, exposure to inflammatory agents such as LPS abruptly induced BHLHE40 mRNA and protein expression in macrophages. Previous studies have implicated that BHLHE40 may be expressed in several immune cell populations, including alveolar macrophages, B-cells, T-cells, and dendritic cells (37). BHLHE40 systemic knockout mice were viable, fertile, and did not exhibit any discernible phenotypic differences from their wild-type littermates (38). In addition, detailed evaluation of thymus, spleen, lymph node, and bone marrow cells demonstrated that no major differences in the populations of lymphoid and myeloid lineages between wild type and BHLHE40 knockout mice (38). Similarly, our analyses show that myeloid-specific deletion of Bhlhe40 did not significantly alter the hematopoietic cells compartment. Prior studies have shown that BHLHE40 deficient CD4+ T-cells exhibited significantly attenuated IFNγ production and displayed defects in their effector functions (38). In addition, Bhlhe40 knockout mice are protected from immunization-induced autoimmune neuroinflammation (17). Bhlhe40 knockout mice challenged with myelin oligodendrocyte glycoprotein (MOG) peptide had reduced myeloid, CD4+ T, and γδ T cells compared to wild-type mice (17). These BHLHE40 deficient CD4+ T cells also exhibited defects in GM-CSF, IFNγ, and IL17A production (17). Draining lymph nodes (DLNs) of immunized Bhlhe40 knockout mice had reduced cellularity relative to wild-type mice (17). DLNs from Bhlhe40 knockout mice showed a significantly reduced frequency of MOG-specific GM-CSF-producing T cells relative to wild-type mice (17). Similarly, CD4+ T cells from Bhlhe40 knockout mice expressed lower levels of Csf2, Il3, Il1a, Ifitm3, and Ptgs2 transcripts (17). Pertussis toxin (PTX) is a classical co-adjuvant for actively induced autoimmune encephalomyelitis. PTX exposure strongly promotes myeloid cells to produce IL1β in DLNs and serves as a potent stimulus for BHLHE40 expression in Th cells (18). Interestingly, BHLHE40 deficiency significantly attenuated PTX-induced encephalomyelitis (18). In this direction, our analyses show that myeloid-BHLHE40 deficiency significantly attenuates macrophage and neutrophil abundance following the zymosan challenge. Further, our analyses here show BHLHE40 deficiency markedly diminished pro-inflammatory gene expression in responding immune cells. Likewise, BHLHE40+-GM-CSF+-CD4+ T-cells are required for establishing pathogenic phenotypes during alloantigen presentation in the gastrointestinal tract during acute graft-versus-host disease (22). Recent studies have also demonstrated that BHLHE40 is required for repression of IL10 expression during Mycobacterium tuberculosis infection and inhibition of IL10 signaling in BHLHE40 null mice reversed disease phenotypes (21). In addition, BHLHE40 has been shown to regulate Th2 cell transcriptional program during helminth infection to support the normal expression of genes required for host protection functions (24). It is important to note that we have utilized homozygous Lyz2cre mice as a control group in our studies. The Lyz2cre mice line were created by inserting Cre recombinase to the endogenous locus of Lyz2 gene. This results in the complete abolition of Lyz2 gene expression in these mice lines and could modulate disease phenotypes observed in these mice. Taken together, these discoveries provide the basis for future investigations focused on the molecular role of myeloid-BHLHE40 in the regulation of chronic and acute inflammatory disease pathogenesis.
Macrophage-mediated inflammation is characterized by elevated levels of inflammatory cytokine and chemokine production. An earlier report has shown that BHLHE40 is required for experimentally induced periodontal inflammation in vivo (39). In this regard, our analyses show that myeloid-BHLHE40 deficiency significantly attenuates zymosan-induced lung inflammation. Similarly, our transcriptomics and GSEA studies show that BHLHE40 deficiency significantly attenuated LPS-induced broad pro-inflammatory gene programs in macrophages. Specifically, BHLHE40 deficiency curtailed interferon-gamma/alpha signaling, inflammatory response signaling, complement, reactive oxygen species pathway, allograft rejection, and MTORC1 signaling in macrophages. Prior studies have indicated that BHLHE40 could interact with STAT3 to modulate STAT3-dependent targets (40). In this context, our analyses show that BHLHE40 deficiency markedly attenuates LPS-induced IL6-JAK-STAT3 signaling and STAT3-regulated pro-inflammatory gene expression in macrophages. Past studies have shown that BHLHE40 as a classical target of HIF1α and BHLHE40 expression is diminished in HIF1α-null cells (41). This increase in BHLHE40 expression by HIF1α could be essential for modulating a number of genes that are involved in hypoxia-induced cellular processes. However, whether BHLHE40 regulates HIF1α or the hypoxia response gene has never been investigated. In this respect, our GSEA studies show that BHLHE40 deficiency greatly diminished hypoxia response signaling in macrophages. Interestingly, BHLHE40 elevated LPS-induced HIF1α mRNA and protein expression in macrophages. Likewise, BHLHE40 deficiency substantially attenuated HIF1α transcriptional activity and classical hypoxia response gene expression in macrophages. Previous studies have demonstrated that HIF1α deficiency markedly attenuates pro-inflammatory and glycolytic gene expression in macrophages (34). In this case, our analyses show that BHLHE40 deficiency broadly attenuated pro-inflammatory gene expression in macrophages. Further, BHLHE40 deficiency also attenuated LPS-induced glycolytic gene expression in macrophages. In addition, forced overexpression of oxygen stable form of HIF1α reversed attenuated pro-inflammatory and glycolytic gene expression in BHLHE40 deficient macrophages. It is important to note that our GSEA studies show that BHLHE40 deficiency significantly attenuated allograft rejection and complement responses. However, these pathways are not examined in this study and our future studies will explore these pathways.
Collectively, our studies uncovered that BHLHE40 promotes inflammatory and glycolytic gene expression by elevating HIF1α expression in macrophages. In summary, our in vivo, ex vivo, and molecular studies presented here highlight the importance of myeloid-BHLHE40 in initiating broad inflammatory gene expression and initiation of inflammatory disease pathogenesis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant HL126626, HL141423, and Crohn’s and Colitis Foundation of America Senior Research Award 421904 (to G. H. M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- BHLHE40
Basic helix-loop-helix family member e40
- BMDMs
Bone marrow-derived macrophages
- PMs
Thioglycollate-elicited peritoneal macrophages
- LPS
Lipopolysaccharides
- GSEA
Gene set enrichment analysis
- HIF1α
Hypoxia-inducible factor 1 alpha
- IFNγ
Interferon-gamma
- FWER
Family-wise error rate
- RT-qPCR
Quantitative real-time PCR
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Accession number
The accession number for the sequencing data reported in this manuscript is GEO: GSE176174.
References
- 1.Locati M, Curtale G, and Mantovani A (2020) Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annual review of pathology 15, 123–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, and Mosser DM (2017) Macrophages and the Recovery from Acute and Chronic Inflammation. Annual review of physiology 79, 567–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monticelli S, and Natoli G (2017) Transcriptional determination and functional specificity of myeloid cells: making sense of diversity. Nature reviews. Immunology 17, 595–607 [DOI] [PubMed] [Google Scholar]
- 4.Nizet V, and Johnson RS (2009) Interdependence of hypoxic and innate immune responses. Nature reviews. Immunology 9, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadiku P, and Walmsley SR (2019) Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO reports 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL, Roth PH, Fang HM, and Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. The Journal of biological chemistry 269, 23757–23763 [PubMed] [Google Scholar]
- 7.Palazon A, Goldrath AW, Nizet V, and Johnson RS (2014) HIF transcription factors, inflammation, and immunity. Immunity 41, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhry H, and Harris AL (2018) Advances in Hypoxia-Inducible Factor Biology. Cell metabolism 27, 281–298 [DOI] [PubMed] [Google Scholar]
- 9.Lin N, and Simon MC (2016) Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. The Journal of clinical investigation 126, 3661–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, and Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bäcker V, Cheung FY, Siveke JT, Fandrey J, and Winning S (2017) Knockdown of myeloid cell hypoxia-inducible factor-1α ameliorates the acute pathology in DSS-induced colitis. PloS one 12, e0190074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim GD, Ng HP, Chan ER, and Mahabeleshwar GH (2021) Macrophage-Hypoxia-Inducible Factor-1α Signaling in Carotid Artery Stenosis. The American journal of pathology 191, 1118–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim GD, Ng HP, Chan ER, and Mahabeleshwar GH (2020) Kruppel-like factor 6 promotes macrophage inflammatory and hypoxia response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 34, 3209–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, and Karin M (2008) NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453, 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ow JR, Tan YH, Jin Y, Bahirvani AG, and Taneja R (2014) Stra13 and Sharp-1, the non-grouchy regulators of development and disease. Current topics in developmental biology 110, 317–338 [DOI] [PubMed] [Google Scholar]
- 16.Kiss Z, Mudryj M, and Ghosh PM (2020) Non-circadian aspects of BHLHE40 cellular function in cancer. Genes & cancer 11, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin CC, Bradstreet TR, Schwarzkopf EA, Sim J, Carrero JA, Chou C, Cook LE, Egawa T, Taneja R, Murphy TL, Russell JH, and Edelson BT (2014) Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nature communications 5, 3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin CC, Bradstreet TR, Schwarzkopf EA, Jarjour NN, Chou C, Archambault AS, Sim J, Zinselmeyer BH, Carrero JA, Wu GF, Taneja R, Artyomov MN, Russell JH, and Edelson BT (2016) IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. The Journal of experimental medicine 213, 251–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, Konno H, Guo X, Ye Y, Gao S, Wang S, Hu X, Ren X, Shen Z, Ouyang W, and Zhang Z (2018) Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 [DOI] [PubMed] [Google Scholar]
- 20.Yu F, Sharma S, Jankovic D, Gurram RK, Su P, Hu G, Li R, Rieder S, Zhao K, Sun B, and Zhu J (2018) The transcription factor Bhlhe40 is a switch of inflammatory versus antiinflammatory Th1 cell fate determination. The Journal of experimental medicine 215, 1813–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huynh JP, Lin CC, Kimmey JM, Jarjour NN, Schwarzkopf EA, Bradstreet TR, Shchukina I, Shpynov O, Weaver CT, Taneja R, Artyomov MN, Edelson BT, and Stallings CL (2018) Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. The Journal of experimental medicine 215, 1823–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piper C, Zhou V, Komorowski R, Szabo A, Vincent B, Serody J, Alegre ML, Edelson BT, Taneja R, and Drobyski WR (2020) Pathogenic Bhlhe40+ GM-CSF+ CD4+ T cells promote indirect alloantigen presentation in the GI tract during GVHD. Blood 135, 568–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cook ME, Jarjour NN, Lin CC, and Edelson BT (2020) Transcription Factor Bhlhe40 in Immunity and Autoimmunity. Trends in immunology 41, 1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarjour NN, Bradstreet TR, Schwarzkopf EA, Cook ME, Lai CW, Huang SC, Taneja R, Stappenbeck TS, Van Dyken SJ, Urban JF Jr., and Edelson BT (2020) BHLHE40 Promotes T(H)2 Cell-Mediated Antihelminth Immunity and Reveals Cooperative CSF2RB Family Cytokines. Journal of immunology (Baltimore, Md. : 1950) 204, 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarjour NN, Schwarzkopf EA, Bradstreet TR, Shchukina I, Lin CC, Huang SC, Lai CW, Cook ME, Taneja R, Stappenbeck TS, Randolph GJ, Artyomov MN, Urban JF Jr., and Edelson BT (2019) Bhlhe40 mediates tissue-specific control of macrophage proliferation in homeostasis and type 2 immunity. Nature immunology 20, 687–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rauschmeier R, Gustafsson C, Reinhardt A, N AG, Tortola L, Cansever D, Subramanian S, Taneja R, Rossner MJ, Sieweke MH, Greter M, Månsson R, Busslinger M, and Kreslavsky T (2019) Bhlhe40 and Bhlhe41 transcription factors regulate alveolar macrophage self-renewal and identity. The EMBO journal 38, e101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanda M, Yamanaka H, Kojo S, Usui Y, Honda H, Sotomaru Y, Harada M, Taniguchi M, Suzuki N, Atsumi T, Wada H, Baghdadi M, and Seino K (2016) Transcriptional regulator Bhlhe40 works as a cofactor of T-bet in the regulation of IFN-γ production in iNKT cells. Proceedings of the National Academy of Sciences of the United States of America 113, E3394–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Date D, Das R, Narla G, Simon DI, Jain MK, and Mahabeleshwar GH (2014) Kruppel-like transcription factor 6 regulates inflammatory macrophage polarization. The Journal of biological chemistry 289, 10318–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim GD, Das R, Rao X, Zhong J, Deiuliis JA, Ramirez-Bergeron DL, Rajagopalan S, and Mahabeleshwar GH (2018) CITED2 Restrains Proinflammatory Macrophage Activation and Response. Molecular and cellular biology 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metsalu T, and Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic acids research 43, W566–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newson J, Stables M, Karra E, Arce-Vargas F, Quezada S, Motwani M, Mack M, Yona S, Audzevich T, and Gilroy DW (2014) Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood 124, 1748–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, and Blackwell TS (2010) NADPH oxidase limits innate immune responses in the lungs in mice. PloS one 5, e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim GD, Ng HP, Chan ER, and Mahabeleshwar GH (2021) Macrophage-Hypoxia-Inducible Factor-1α Signaling in Carotid Artery Stenosis. The American journal of pathology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van den Bossche J, O’Neill LA, and Menon D (2017) Macrophage Immunometabolism: Where Are We (Going)? Trends in immunology 38, 395–406 [DOI] [PubMed] [Google Scholar]
- 36.Yan Q, Bartz S, Mao M, Li L, and Kaelin WG Jr. (2007) The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Molecular and cellular biology 27, 2092–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, and Glass CK (2014) Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun H, Lu B, Li RQ, Flavell RA, and Taneja R (2001) Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nature immunology 2, 1040–1047 [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Suzuki M, Kim IS, Kobayashi R, Hamada N, Sato F, and Bhawal UK (2018) Transcription factor DEC1 is required for maximal experimentally induced periodontal inflammation. Journal of periodontal research 53, 883–893 [DOI] [PubMed] [Google Scholar]
- 40.Ivanova AV, Ivanov SV, Zhang X, Ivanov VN, Timofeeva OA, and Lerman MI (2004) STRA13 interacts with STAT3 and modulates transcription of STAT3-dependent targets. Journal of molecular biology 340, 641–653 [DOI] [PubMed] [Google Scholar]
- 41.Yun Z, Maecker HL, Johnson RS, and Giaccia AJ (2002) Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Developmental cell 2, 331–341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.