

Abstract
Introduction:
Missense variants and multiplications of the alpha-synuclein gene (SNCA) are established as rare causes of autosomal dominant forms of Parkinson’s Disease (PD).
Methods:
Two families of Turkish origins with PD were studied; the SNCA coding region was analyzed by Sanger sequencing, and by whole exome sequencing (WES) in the index patient of the first and the second family, respectively. Co-segregation studies and haplotype analysis across the SNCA locus were carried out. Functional studies included in vitro thioflavin-T aggregation assay and in silico structural modelling of the alpha-synuclein (α-syn) protein.
Results:
We identified a novel heterozygous SNCA variant, c.215C > T (p.Thr72Met), segregating with PD in a total of four members in the two families. A shared haplotype across the SNCA locus was found among variant carriers, suggestive of a common ancestor. We next showed that the Thr72Met α-syn displays enhanced aggregation in-vitro, compared to the wild-type species. In silico analysis of a tetrameric α-syn structural model revealed that Threonine 72 lies in the tetrameric interface, and substitution with the much larger methionine residue could potentially destabilize the tetramer.
Conclusion:
We present clinical, genetic, and functional data supporting a causative role of the SNCA c.215C > T (p.Thr72Met) variant in familial PD. Testing for this variant in patients with PD, especially of Turkish origin, might detect additional carriers. Further functional analyses might offer new insights into the shared biochemical properties of the PD-causing SNCA missense variants, and how they lead to neurodegeneration.
Keywords: SNCA, α-syn, Variant, Thr72Met, Phenotype, Late-onset, Parkinsonism
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by loss of neurons in the substantia nigra and other brain areas, and accumulation of intracellular inclusions containing misfolded alpha-synuclein (α-syn) protein, termed Lewy bodies and Lewy neurites [1]. The etiological landscape of PD is complex and incompletely known; however, the identification of rare disease-causing genes has contributed substantially to illuminate the underlying disease mechanisms and pathways [2].
Rare, highly penetrant variants in the gene encoding α-syn (SNCA) were the first identified cause of dominantly inherited PD. Since the first reported and most intensively studied missense substitution (p. Ala53Thr) in the “Contursi kindred” and additional Greek families [3], additional missense SNCA variants have been identified in patients with PD or related neurodegenerative disorders from several populations (Table 1). However, a disease-causing role has not been established for several of these variants, particularly those identified in single patients and without evidence of intra-familial co-segregation with disease. Genomic multiplications (duplications and more rarely triplications) encompassing the whole SNCA locus are a more frequent cause of PD than are the missense variants [4]. Last, common non-coding SNCA variants are one of the most relevant risk factors for the sporadic forms of PD [5].
Table 1.
Summary of clinical features in previously reported cases and families with parkinsonism carrying SNCA missense variants and multiplications. AAO: onset age of parkinsonism in reported symptomatic cases; Cases: number of symptomatic SNCA variant carriers (genotyped) described in the corresponding study; hom: homozygous; −: absent; +: present; NA: not available. Additional references for the studies included in this table are provided in the Supplementary Appendix
| Variant | Study | Descent | AAO range (mean ± SD) | Families/Cases | Family history | Cognitive decline | Hallucinations/psychosis | Autonomic | Additional clinical features | Response to levodopa |
|---|---|---|---|---|---|---|---|---|---|---|
| Ala53Thr | Golbe 1996 [31]; Polymeropoulos1997 [3] | Italian, Greek | 20–85 (45.6 ± 13.8) | 4 families | + | +/− | +/− | +/− | depression | good |
| Papadimitriou 1999 [21] | Greek | 39–49 (43± 4.32) | 2 families (4 cases) | + | +/− | − | +/− | − | good | |
| Athanassiadou1999 [32] | Greek | 40–58 (48 ± 6.32) | 4 families (6 cases) | + | NA | NA | NA | NA | NA | |
| Markopoulou 1995 [33]; Scott 1999 [34]; Markopoulou 1999 [35]; | Greek | 31–71 | 1 family (12 cases) | + | +/− | − | +/− | sleep disorder, myoclonus | +/NA | |
| Markopoulou 2008 [24] Spira 2001 [36] | Greek- Australian | 42–46 (44.33 ± 2.08) | 1 family (3 cases) | + | + | +/− | + | myoclonus, sleep disorder, apathy | moderate/good | |
| Papapetropoulos 2001 [37] | Greek | 25–64 (40.2 ± 15.67) | 3 families (5 cases) | + | +/− | − | +/− | depression | good | |
| Bostantjopoulou 2001 [38] | Greek | 32–50 (39.7 ± 7.6) | 6 families (8 cases) | + | +/− | − | − | olfactory impairment, depression | +/NA | |
| Kobayashi 2003 [39] | Greek | 39–42 (40.5 ± 2.12) | 2 families (9 cases) | + | +/− | NA | NA | NA | +/transient | |
| Michell 2005 [40] | Polish | 74 | 1 sporadic case | − | NA | NA | NA | NA | + | |
| Berg 2005 [41] | Greek | NA | 1 familial case | + | NA | NA | NA | NA | + | |
| Morfis 2006 [42] | Greek | 71 | 1 familial case | + | + | + | + | myoclonic jerks, dysphagia, sleep disorder | − | |
| Ki 2007 [43]; Choi 2008 [44] | Korean | 35–63 (49 ± 19.8) | 1 family (2 cases) | + | NA | NA | NA | − | good | |
| Bostantjopoulou 2008 [45] | Greek | NA | 9 familial cases | + | NA | NA | NA | NA | NA | |
| Puschmann 2009 [46] | Swedish | <31-<40 | 1 family (2 cases) | + | + | − | + | speech difficulties, myoclonus | + | |
| Bozi 2014 [47] | Greek | 31–61 (43.6 ± 11.65) | 5 familial cases | + | NA | NA | NA | NA | NA | |
| Xiong 2016 [48] | Chinese | 22 | 1 sporadic case | − | − | − | − | olfactory impairment | good | |
| Tambasco 2016 [49] | Italian | 58 | 1 familial case | + | − | − | + | sleep disorder, olfactory impairment | + | |
| Bougea 2017 [8] | Greek | 30–55 (45.25 ± 10.72) | 2 families (3 cases) | + | + | − | + | eye lid opening apraxia, speech deficits, hyperreflexia, frontal release signs, pseudoeuphoria, apathy, anxiety, myoclonus | moderate/NA | |
| Breza 2018 [7] | Greek | 30–44 (39 ± 7.81) | 3 familial cases | + | +/− | +/− | − | apathy | NA | |
| Blauwendraat 2018 [50] | European, Korean | 19–49 (34 ± 21.21) | 2 cases | NA | NA | NA | NA | NA | NA | |
| Wilson 2019 [51] | Greek, Italian | NA | 7 cases | NA | NA | NA | NA | NA | NA | |
| Lesage 2020 [52] | French | +/− | +/− | NA | +/− | mild to good | ||||
| 26–40 (34.67 ± 7.57) | 1 familial case & 2 sporadic cases | dysarthria, depression/psychiatric disorders | ||||||||
| Simitsi 2021 [53] | Greek | NA | 10 cases | NA | +/− | NA | NA | sleep disorder, olfactory impairment | NA | |
| Ala30Pro | Krüger 1998 [54]; Krüger 2001 [55] | German | 54–76 (59.75 ± 10.84) | 1 family (5 cases) | + | +/− | +/− | − | − | good/NA |
| Glu46Lys | Zarranz 2004 [56]; Zarranz 2005 [57] | Spanish | 44–81 (59.43 ± 13.07) | 1 family (5 cases) | + | +/− | +/− | +/− | sleep disorder, behavioral changes, depression | +/−/NA/transient |
| Pimentel 2015 [20] | Bolivian | 50–75 (57.2 ± 10.47) | 1 family (3 cases) | + | − | − | + | sleep disorder, olfactory impairment, anxiety, depression | NA | |
| Gly51Asp | Kiely 2013 [9]; Kiely 2015 [58] | British | 19–40 (32.67 ± 11.85) | 1 family (2 cases) | + | +/− | + | + | dysarthria, dystonia, myoclonus seizures, pyramidal signs, anxiety | good |
| Lesage 2013 [59] | French | 31–60 (40.25 ± 13.3) | 1 family (3 cases) | + | − | +/− | +/− | pyramidal signs, anxiety, depression | mild/moderate/NA | |
| Tokutake 2014 [60] | Japanese | 28 | 1 familial case | + | + | + | + | pyramidal signs, myoclonus, tonic seizures | good | |
| Kiely 2015 [58] | British | 46–69 (57.5 ± 16.26) | 1 family (2 cases) | + | + | + | + | pyramidal signs, vertical supranuclear gaze palsy, apraxia of eyelid opening, blepharospasm, dysphagia, anxiety, depression, apathy | transient | |
| Blauwendraat 2018 [50] | European | 41 | 1 case | NA | NA | NA | NA | NA | NA | |
| His50Gln | Proukakis 2013 [61]; Kiely 2015 [58] | British | 71 | 1 sporadic case | − | + | − | − | blepharospasm | good |
| Appel-Cresswell 2013 [62] | British | 56–60 (58 ± 2.83) | 1 familial case | + | + | − | − | dystonia, anxiety, apathy | + | |
| Blauwendraat 2018 [50]; Lesage 2020 [52] | French | 32 | 1 sporadic case (hom) | − | − | − | + | dystonia, dysarthria | + | |
| Ala18Thr | Hoffman-Zacharska 2013 [63] | Polish | 50 | 1 sporadic case | − | + | − | + | − | good (diminishing) |
| Ala29Ser | Hoffman-Zacharska 2013 [63] | Polish | 60 | 1 sporadic case | − | − | − | − | anxiety, depression, restless legs syndrome, dysphagia | good |
| Ala53Glu | Pasanen 2014 [64] | Finnish | 32–62 (43.33 ± 16.29) | 1 family (3 cases) | + | − | − | + | pyramidal signs, myoclonus, sleep disorder, anxiety, panic disorder | + |
| Martikainen 2015 [65] | Finnish | 25–52 (39.67 ± 13.65) | 1 family (2 cases) | + | − | − | -/unclear | pyramidal signs, dysarthria, depression, panic disorder | good | |
| Pasanen 2017 [66] | Finnish | 41 | 1 familial case | + | NA | NA | NA | dysarthria, dysphagia | NA | |
| Picillo 2018 [67] | Canadian (Dutch-Scottish-Irish) | 25–58 (40± 16.7) | 1 family (3 cases) | + | + | + | − | myoclonus | + | |
| Ala53Val | Yoshino 2017 [68] | Japanese | 55–57 (56 ± 1.41) | 1 familial case (hom) | + | + | + | − | sleep disorder | good |
| Yang 2019 [69] | Chinese | NA | 1 sporadic case | − | NA | NA | NA | NA | NA | |
| Chen 2020 [70] | Chinese | +/− | − | NA | NA | sleep disorder, olfactory impairment, depression | good | |||
| 35–39 (37.12 ± 1.75) | 1 familial case & 2 sporadic cases | |||||||||
| Glu57Asp | Youn 2019 [71] | Korean | 48 | 1 sporadic case | − | + | − | + | dystonia, olfactory impairment | NA |
| Val15Ala | Cali 2019 [72] | NA | 59 | 1 familial case | + | + | + | NA | sleep disorder, olfactory impairment, apathy, abulia, emotional lability, agitation, anxiety | partial |
| Leu8Ile | Chen 2020 [70] | Chinese | 37 | 1 sporadic case | − | + | NA | NA | − | good |
| Val15Asp | Zheng 2020 [73] | Chinese | NA | 1 sporadic case | − | NA | NA | NA | NA | NA |
| Met127Ile | Zheng 2020 [73] | Chinese | NA | 1 sporadic case | − | NA | NA | NA | NA | NA |
| Pro117Ser | Zhao 2020 [74] | Chinese | 50 | 1 familial case | + | NA | NA | NA | NA | NA |
| Met5Thr | Zhao 2020 [74] | Chinese | 45 | 1 familial case | + | NA | NA | NA | NA | NA |
| Gly93Ala | Zhao 2020 [74] | Chinese | 45 | 1 familial case | + | NA | NA | NA | NA | NA |
| Glu83Gln | Kapasi 2020 [75] | NA | 59 | 1 familial case | + | + | intermittent clonus, seizures, possible sleep disorder, depression, anxiety, behavioral changes, dysphagia | NA | ||
| Ala30Gly | Liu 2021 [76] | Greek | 36–80 (58.44 ± 12.8) | 3 families (5 cases) | + | +/NA | +/− | +/NA | depression, anxiety, apathy, disinhibition, sleep disorder, impulse control disorder | +/NA |
| Triplication | Muenter 1998 [77]; Singleton 2003 [78]; Gwinn 2011 [79] | Iowan | 24–48 (33.15 ± 8.43) | 1 family (4 cases) | + | +/− | +/− | +/− | sleep disorder, depression, myoclonus, dysarthria | moderate/good/NA |
| Farrer 2004 [80]; Fuchs 2007 [81] | Swedish-American | 31-early 60s | 1 familial case | + | + | + | + | olfactory impairment, depression, anxiety | + | |
| Ibáñez 2009 [4] | French | 36–61 (48.3 ± 12.5) | 1 familial case | + | + | − | + | − | limited | |
| Sekine 2010 [82] | Japanese | 28–49 (33.67 ± 10.97) | 1 familial case | + | +/− | − | + | depression | mild | |
| Keyser 2010 [83] | French-Italian | 46 | 1 familial case | + | + | + | + | − | + | |
| Byers 2011 [84] | NA | 38 | 1 familial case | + | + | − | +/unclear | sleep disorder, diplopia, olfactory impairment, anxiety, depression | + | |
| Olgiati 2015 [85] | Italian | 28–42 (33.3 ± 7.57) | 1 family (2 cases) | + | +/− | +/− | − | sleep disorder, behavioral changes, depression | +/NA | |
| Ferese 2015 [86] | Italian | 28–42 (35 ± 9.9) | 1 family (2 cases) | + | + | +/NA | +/NA | dysarthria, ataxia, sleep disorder, depression, aggressive behavior, dysphagia, motor apraxia | +/NA | |
| Youn 2019 [71] | Korean | 44–45 (44.5 ± 0.71) | 2 sporadic cases | + | + | dystonia | NA | |||
| Duplication | Chartier-Harlin 2004 [87] | French | 39–65 (48.4 ± 10.45) | 1 family (4 cases) | + | − | − | − | depression | mild/NA |
| Ibááez 2004 [88] | Italian, French | 46–50 (48 ± 2.83) | 2 familial cases | + | − | − | − | epilepsy, depression | good | |
| Nishioka 2006 [89] | Japanese | 38–48 (44.3 ± 5.51) | 2 families (3 cases) | + | +/− | +/− | − | olfactory impairment, sleep disorder, depression | mild to good | |
| Fuchs 2007 [81] | Swedish | 40–71 (58.8 ± 11.43) | 1 familial case | + | + | + | + | myoclonus, depression, anxiety | poor | |
| Ikeuchi 2008 [90] | Japanese | + | + | + | +/− | − | initially + | |||
| 28–71 (49.75 ± 19.72) | 1 family (4 cases) (1 case hom) | |||||||||
| Ahn 2008 [19]; Seo 2020 [91] | Korean | 40–65 (51.67 ± 12.58) | 1 familial case & 2 sporadic cases | +/− | +/− | +/NA | + | depression, pyramidal signs | good | |
| Brueggemann 2008 [92] | German | 36 | 1 sporadic case (de novo) | − | − | − | − | frontal release signs, olfactory impairment, horizontal nystagmus | good | |
| Troiano 2008 [93] | European/North African | 35 | 1 sporadic case | − | − | − | + | − | NA | |
| Uchiyama 2008 [94] | Japanese | 47–73 (60 ± 18.38) | 1 family (2 cases) | + | + | + | − | anxiety, depression | + | |
| Ibáñez 2009 [4] | French, Italian | 38–65 (46 ± 8.7) | 4 families (7 cases) | + | +/− | − | − | − | moderate | |
| Nuytemans 2009 [95] | Belgian | 68 | 1 case | NA | + | − | − | − | + | |
| Nishioka 2009 [18] | Japanese | 31–62 (47.5 ± 10.89) | 4 families (7 cases) & 1 sporadic case | +/− | +/− | +/− | − | olfactory impairment, sleep disorder, depression | poor to good | |
| Sironi 2010 [96]; Antonini 2012 [97] | Italian | 41–47 (44 ± 4.24) | 1 familial case | + | +/− | − | + | dystonia, depression, anxiety-panic attacks, compulsive behavior | good | |
| Shin 2010 [98]; Seo 2020 [91] | Korean | 48–55 (51.5 ± 4.95) | 2 sporadic cases | − | + | + | + | sleep disorder, depression, hypometric saccade | +/NA | |
| Pankratz 2011 [99] | NA | 44 | 1 familial case | + | NA | NA | NA | NA | NA | |
| Kojovic 2012 [100] | Pakistani | 31 | 1 sporadic case (hom) | − | + | − | − | postpartum psychosis, depression | good | |
| Garraux 2012 [101] | NA | 30 | 1 sporadic case | − | − | − | − | mental retardation, developmental delay, ataxic gait | good | |
| Meeus 2012 [102] | Belgian | 77 | 1 sporadic case | − | + | + | − | behavioral changes | NA | |
| Itokawa 2013 [17] | Asian | 20s-50s | 1 familial case | + | − | − | − | dystonia | + | |
| Elia 2013 [103] | Argentinian, Italian | 32–44 (39.67 ± 6.66) | 2 families (4 cases) | + | + | +/− | +/− | depression, sleep disorder, aggressiveness, dysphagia | good/modest | |
| Kara 2014 [23] | British | 38 | 1 familial case | + | + | + | + | anxiety, panic disorder, behavioral changes, blepharospasm, cervical dystonia, pyramidal signs, sleep disorder, dysarthria, seizures | transient | |
| Konno 2016 [104] | American | 46 | 1 familial case | + | + | + | + | sleep disorder, foot dystonia, square-wave jerks, frontal release signs, depression, abulia, palilalia, cerebellar dysfunction |
+ | |
| Benitez 2016 [105] | European-American | 67 | 1 familial case | + | − | − | − | − | NA | |
| Takamura 2016 [106] | Japanese | 53 | 1 familial case | + | − | + | − | − | NA | |
| Lahut 2017 [15] | Turkish | NA | 1 family (5 cases) | + | − | NA | NA | − | NA | |
| Kessler 2018 [14] | Turkish | 41–46 (43.5 ± 3.54) | 2 familial cases | + | +/− | NA | NA | NA | NA | |
| Book 2018 [107] | NA | NA | 25 families | + | NA | NA | NA | NA | NA | |
| Bentley 2018 [108] | Australian | 39–51 (45 ± 8.49) | 2 familial cases | + | − | +/− | +/− | speech defect, muscular skeletal dysfunction, sleep disorder, anxiety | NA | |
| Tan 2019 [109] | NA | <60 | 1 familial case | + | NA | NA | NA | NA | NA | |
| Urso 2019 [110] | NA | 57 | 1 familial case | + | + | + | + | mild | ||
| sleep disorder, olfactory impairment, loss of consciousness episodes, coat-hanger pain, anxiety, depression | ||||||||||
| Du 2019 [111] | Chinese | 34–69 (51.57 ± 12.29) | 2 families (4 cases) | + | + | +/− | +/− | olfactory impairment, sleep disorder, depression, dystonia | good | |
| Lesage 2020 [52] | Turkish, Moroccan, French | 36–56 (45.3 ± 6.3) | 6 familial cases & 3 sporadic cases | +/− | +/− | +/− | dystonia, neuropsychiatric signs | +/NA | ||
| Nan 2020 [112] | Japanese | 42–69 (52 ± 12.59) | 1 family (3 cases) | + | +/− | +/NA | + | depression | +/NA | |
| Seo 2020 [91] | Korean | 51 | 1 sporadic case | + | + | + | sleep disorder, ocular flutter, hypometric saccade, depression | NA | ||
| Zhao 2020 [74] | Chinese | 34–46 (39.67 ± 6.03) | 3 familial cases | + | +/− | NA | +/− | depression, fatigue | NA | |
| Robak 2020 [113] | Hispanic-Native American | NA | 1 familial case | + | +/− | hyperreflexia, clonus | NA |
The phenotypic spectrum associated with the rare, disease-causing SNCA variants is broad, encompassing PD (with or without atypical clinical signs), PD-dementia, dementia with Lewy bodies, frontotemporal dementia, and, more rarely, multiple system atrophy [6–9]. The mechanisms by which SNCA variants lead to neurodegeneration remain incompletely understood. Therefore, identification of additional PD-causing variants in this gene might provide new and important clues. Here, we report clinical, genetic, and protein expression data of a novel rare SNCA variant: c.215C > T (p.Thr72Met), identified in two families of Turkish origin with dominantly transmitted, late-onset PD and concomitant cognitive decline.
2. Materials and methods
Two unrelated families of Turkish origins with multiple members affected by PD and compatible with dominant inheritance were identified and clinically characterized, one (Family 1) at the Istanbul Faculty of Medicine, Turkey, and the other (Family 2) at the Erasmus MC, Rotterdam, The Netherlands.
Genomic DNA from 292 unrelated Turkish individuals free from PD was used to test for the frequency of the SNCA variant identified in the two PD families. Some of these samples (n = 56) were also used for haplotype analyses. These study procedures were approved by the relevant ethical authorities, and informed consent was obtained from all participating subjects.
2.1. Genetic studies
Genomic DNA was extracted from peripheral blood using standard protocols. In Family 1 (F1), Sanger sequencing of the coding and the flanking intronic regions of SNCA, LRRK2, and GBA was performed (PCR protocols and primers for SNCA Sanger sequencing are listed in Supplementary Table 1). The PCR products were then loaded on an ABI 3730XL Genetic Analyzer (Thermo Fisher Scientific). Sequences were analyzed using the software packages Seqscape v3⋅0 (Thermo Fisher Scientific) and Sequencing Analysis v6⋅0 (Thermo Fisher Scientific). SNCA variants were annotated according to the Gene Bank transcript accession number NM_000345 (NACP140). Variant annotation with Varcards was used for the in silico pathogenicity predictions [10]. The presence of copy number variations in known PD-causing genes was tested by Multiplex Ligation-dependent Probe Amplification (MLPA) technique, according to the MLPA General Protocol of MRC-Holland (https://www.mrcholland.com/). We used the MRC Holland kits P051 and P052B that contain probes targeting multiple PD-causing genes. In Family 2 (F2), subject III-1 underwent testing by Whole Exome Sequencing (WES), filtered for variants in known PD-causing or PD-related genes (the list of genes tested can be found on Supplementary Table 2). In addition, one of the offspring of subject II-1 underwent NGS-based gene panel testing for inherited neuropathies.
For haplotype analysis, six short tandem repeat (STR) markers distributed in a region of~11 Mb and containing the SNCA gene were selected. The markers were amplified by PCR as described elsewhere [11]. PCR products were mixed with the GeneScan 500-LIZ Size Standard (Applied Biosystems), separated on an ABI 3730xl capillary sequencer (Applied Biosystems), and analyzed with GeneMarker (v2.4.0) (Softgenetics LLC, State College, PA, USA). PCR primers are provided in Supplementary Table 3. Four markers, tagging a haplotype shared between F1 and F2, were also typed in 56 unrelated Turkish controls to estimate the frequency of the shared haplotype in the Turkish population.
2.2. Alpha-synuclein (α-syn) protein studies
The expression and purification of GST-fused α-syn carrying the Thr72Met variant (α-syn-Thr72Met) were carried out as previously described for the wild-type α-syn [12]. The p. Thr72Met substitution was created by site-directed mutagenesis using PCR and confirmed by DNA sequencing. Briefly, α-syn-Thr72Met was expressed in Rosetta 2 (DE3) E. coli (Novagen) by inducing with 1 mM iso-propyl-β-D-thiogalactopyranoside for 16 h at 20 °C. Cells were harvested by centrifugation and lysed mechanically with an emulsifier (Avestin). The GST-fusion protein was purified by GST affinity chromatography on a glutathione-Sepharose column (Pharmacia). The N-terminal GST tag was then removed by overnight digestion with Prescission protease (GE Biosciences) at 4 °C. Cleavage with Prescission protease left 10 residues (GPLGSPEFPG) of the protease recognition site on the N-terminal of α-syn-Thr72Met. The α-syn-Thr72Met protein was separated from the GST tag and from the uncleaved fusion protein on a glutathione-Sepharose column. The purified protein was then “polished” by passing through a size-exclusion column (Sephacryl 200 HR column; GE Healthcare) in buffer containing 100 mM Hepes (pH 7.4), 150 mM NaCl, 0.1% BOG, and 10% glycerol. The purified protein was then concentrated to ~5 mg/mL, flash-frozen in liquid nitrogen, and stored at −80 °C. For α-syn protein aggregation studies (thioflavin T [ThT] assay), 0.6 mg of α-syn was added to 200 μL of 100 mM Hepes (pH 7.4), 150 mM NaCl, 10% glycerol, 0.1% BOG, and 5 μM ThT and incubated at 37 °C with frequent agitation. The fluorescence of ThT was measured with a FlexStation (Molecular Devices) at an excitation wavelength of 440 nm, an emission wavelength of 490 nm, and a cutoff wavelength of 475 nm.
3. Results
3.1. Clinical reports
Fig. 1A depicts the pedigree of Family 1, originating from the region of Istanbul. The index case (II-1) presented with bradykinesia and cramps in the upper and lower extremities at the age of 57 years. At the age of 59, she manifested asymmetrical resting tremor of the hand and postural instability. L-dopa treatment resulted in significant improvement of her motor symptoms. Neuropsychological evaluation revealed cognitive impairment [Mini-Mental State Examination (MMSE) 22/30 and Addenbrooke’s Cognitive Examination-Revised (ACE-R) 67/100]; hallucinations or severe dysautonomia were not reported. Brain MRI showed cortical atrophy. Medical history revealed obesity (BMI: 31), diabetes, and cataract surgery. The mother of the index case (I-2) was reported to be nearly bedridden with a tremor-predominant form of PD and severe cognitive impairment by the age of 80 years old. Information regarding the age at onset of her symptoms was not available. Her medical history included diabetes and arterial hypertension. One sister of the index case (II-2) presented at age 56 with memory problems and mild cognitive impairment on testing (MMSE 27/30 and ACE-R 73/100), but no signs of parkinsonism. Brain MRI showed bilateral frontal subcortical white matter lesions, which typically occur in the context of cardiovascular disease. Another sister of the index case (II-3) died at the age of 52 years due to an infectious disease. She was examined, and DNA was sampled at the age of 51 years; at that time, her clinical examination was normal.
Fig. 1.
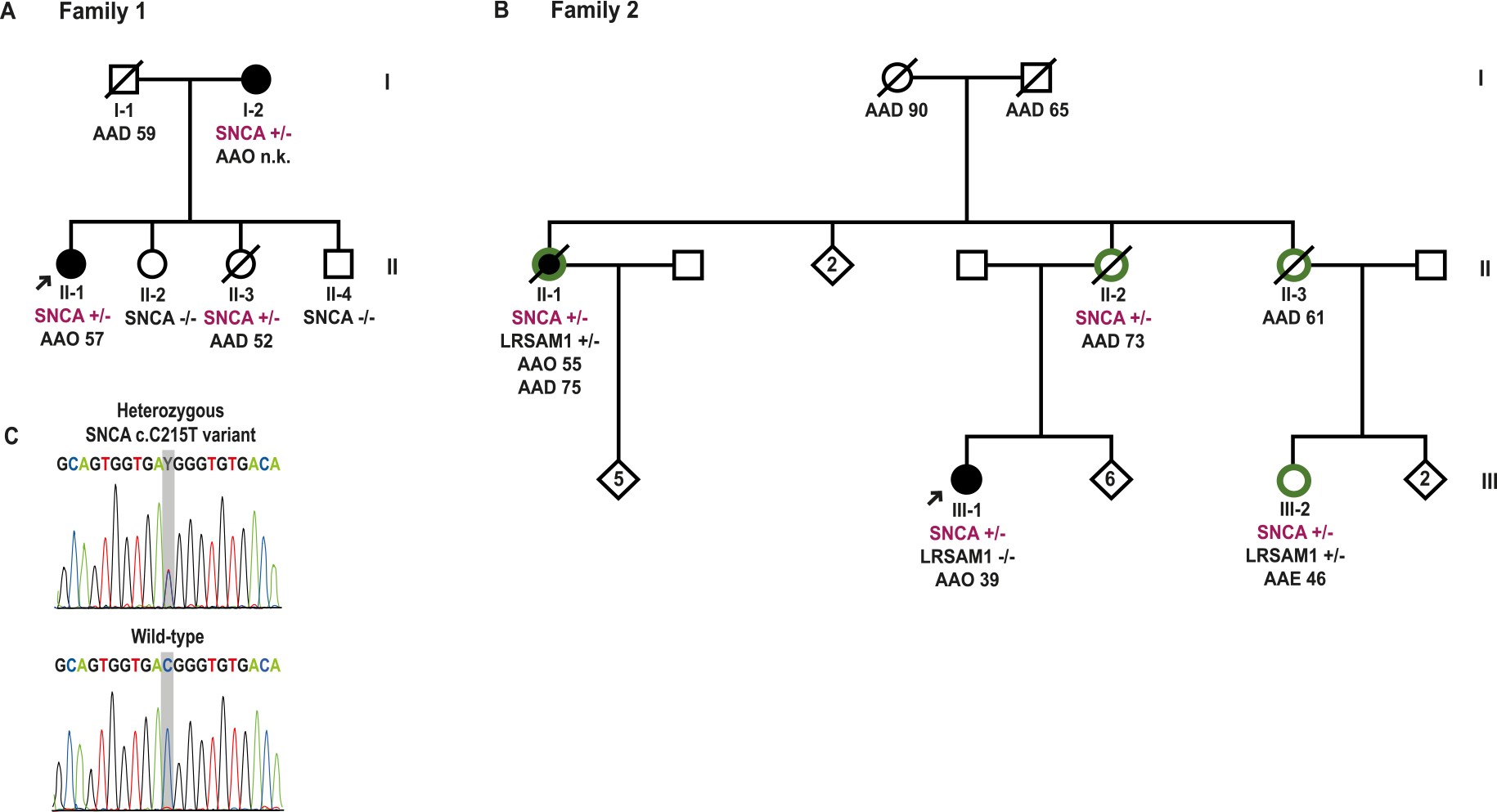
Pedigrees and segregation analysis. A- and B- Pedigrees of Family 1 (F1) and Family 2 (F2), respectively, harbouring the SNCA c.215C > T (p.Thr72Met) variant. Filled black symbols indicate PD patients, white symbols unaffected members, and green halos around symbols show subjects with polyneuropathy (HMSN2). Arrows indicate the index cases. All the available genotypes for the SNCA c.215C and LRSAM1 c.2005G positions are given for each of the tested family members. AAO: onset age of PD; AAD: age at death; AAE: age at last examination; n. k.: not known; SNCA: SNCA c.215C > T (p.Thr72Met); LRSAM1: LRSAM1 c.2005G > T (p.Glu669*); +/−: heterozygous carrier; −/−: non-carrier. C- Representative electropherogram of one PD case shows the heterozygous SNCA c.215C > T (p.Thr72Met) variant, as compared to reference (wild-type) sequence.
The pedigree of Family 2 (F2), originating from the region of Karaman in central-south Turkey, is shown in Fig. 1B. The index case (III-1) presented with a progressive hypokinetic-rigid parkinsonian syndrome, along with pyramidal signs, cognitive decline, and hallucinations at 39 years of age. Neurological examination revealed bradykinesia, rigidity, reduced arm swing, and altered postural reflexes, without tremor. The brain MRI, performed at the age of 41 years, showed no structural abnormalities. By the age of 46 years, the patient had become bedridden and dysphagic. In the past medical history, congenital deafness was reported. The maternal aunt of the index case (II-1) was also diagnosed with PD at the age of 55. At the age of 64, she showed dysphagia and hallucinations; 5 years later she became cognitively impaired. No other members of F2 were diagnosed with PD.
Of note, several members of F2 suffered from axonal sensorimotor neuropathy (Charcot–Marie–Tooth, CMT, type 2, HMSN2). The mother of the index case (II-2) suffered from HMSN2, which led to wheelchair dependence at the age of 70 years and later to the need for mechanical ventilation due to bilateral diaphragm paralysis. She also showed cognitive impairment at the age of 71 years. Whether signs and symptoms of PD were examined systematically in this individual remains unclear. Subject II-3 presented with HSMN2 at the age of 45 years, and with a pyramidal syndrome, increased muscle tone, and pseudobulbar symptoms a few years later. Brain imaging showed enlarged ventricles and cortical atrophy. Finally, subject III-2 was diagnosed with HMSN2 and cognitive impairment at the age of 43 but no signs or symptoms of PD were reported up to the age at last examination (46 years old). Brain atrophy predominantly in the cerebellum was seen in the brain MRI.
3.2. Genetic studies
In the index case of Family 1 (F1), Sanger sequencing revealed a heterozygous C/T transition in SNCA exon 4 (c.215C > T), predicted to lead to a threonine (Thr) to methionine (Met) amino acid change at codon 72 (p.Thr72Met). Screening for rare variants with coding or putative splicing effect in LRRK2 and GBA by Sanger sequencing as well as testing for copy number variants in SNCA, PARK2, PINK1, PARK7, ATP13A2, LRRK2 and GCH1 by MPLA were negative in this patient. Subsequent screening in F1 by Sanger sequencing showed the heterozygous SNCA c.215C > T (p.Thr72Met) variant in the other subject with overt parkinsonian phenotype (subject I-2), as well as in the subject II-3, who was free from PD symptoms and signs until she died at the age of 52 years, which was younger than the age of onset in the index case in this family. The SNCA variant was not present in the other examined and clinically unaffected members, II-2 and II-4.
In Family 2 (F2), inspection of the WES for the known PD-causing and PD-related genes in the index case (III-1) revealed the same SNCA c.215C > T (p.Thr72Met) variant, and no possible pathogenic variants in any of the other examined genes. Sanger sequencing confirmed the SNCA variant in the subject III-1 and yielded 3 additional heterozygous carriers: the second patient diagnosed with PD (II-1), and two more relatives, II-2 and III-2, who were not reported with parkinsonian signs. Additional family members could not be tested for the SNCA p. Thr72Met variant. As several members in F2 suffered from HMSN2, the known neuropathy-causing genes were also tested. A heterozygous c. G2005T/p.Glu669* disease-causing variant was identified in LRSAM1 (NM_138361) in the subjects II-1 and III-2. The index case with PD was also tested but did not carry the LRSAM1 variant.
Sanger sequencing of SNCA exon 4 in a series of 292 unrelated Turkish individuals detected no carriers of the c.215C > T variant. Haplotype analysis of the SNCA region in F1 and F2 revealed allele sharing among all the tested subjects carrying the p. Thr72Met variant for 4 markers (D4S2460, CGR784, D4S414, and CGR795) located closest to the SNCA gene (Fig. 2), suggesting a common ancestor. We also estimated the frequency of the haplotype on which the SNCA variant likely originated by typing the same DNA markers in 56 individuals of Turkish origin (Fig. 2). Of note, for the D4S414 marker, closely flanking the SNCA variant, the allele linked to the variant in the PD patients was present in only 3.6% of the 112 tested chromosomes, further supporting the contention of a single common origin of the SNCA variant.
Fig. 2.
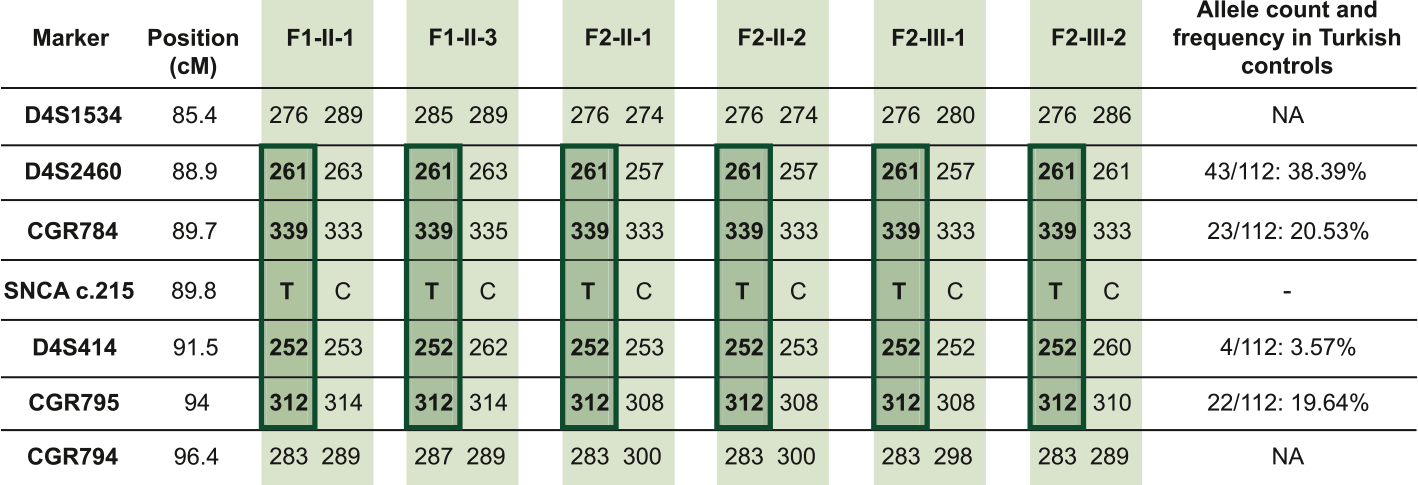
Haplotype analysis. Genotyping across the SNCA locus reveals a haplotype shared among the tested SNCA carriers, indicated in dark green. Allele counts and frequencies in the 56 Turkish individuals are also shown. Genomic positions are annotated according to the Genome Reference Consortium human genome build 38 (GRCh38). NA: not available; -: absent.
3.3. Protein studies
In protein aggregation studies, we observed that α-syn-Thr72Met began to aggregate robustly after 27 h of incubation, reaching maximum intensity at 45 h, while the aggregation of the wild-type protein remained relatively flat (Fig. 3A, and Supplementary Figure). Substituting in silico the threonine 72 (Thr72), which in the structure of the α-syn fibril (PDB ID: 6RT0) [13] is surrounded by three proximal valine residues, for methionine, we observed no steric clashes (Fig. 3B). Additionally, mapping the location of Thr72 on the tetrameric α-syn model of Wang et al. [12] reveals that Thr72 lies in the tetrameric interface (Fig. 3C).
Fig. 3.
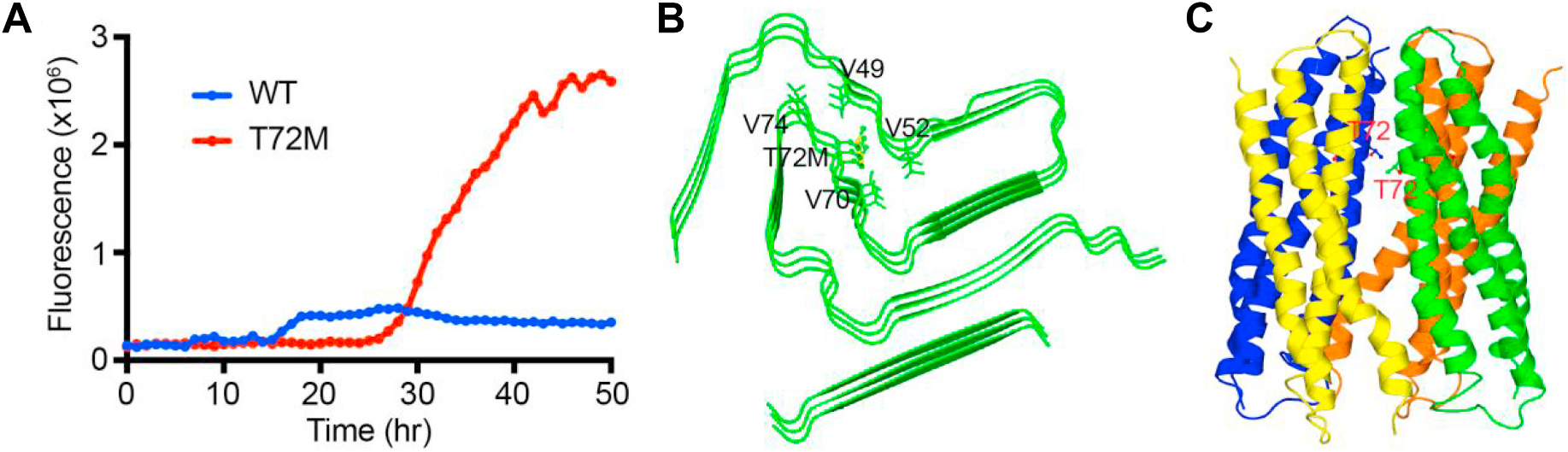
alpha-Synuclein (α-syn) aggregation assay and in silico structural modelling. A- Thioflavin-T aggregation assay of wild-type α-syn (blue) and α-syn-Thr72Met variant (red). B- and C- Structural models of the α-syn monomer and tetramer, showing the position of the Thr72 residue.
4. Discussion
Here, we describe clinical, genetic, and functional studies in two families of Turkish origin with PD associated with the SNCA c.215C > T (p.Thr72Met) variant, which, to our knowledge, has not been reported before in patients with neurodegenerative disorders.
The p. Thr72Met variant (rs767026129) is present in the gno-mADv2.1.1 database in only 2 out of 282798 alleles (allelic frequency <0.001%) and is absent in the 1000 Genomes database and in 292 Turkish non-PD controls screened in this study. This variant is considered deleterious by the majority of the available in silico prediction tools (CADD score 32), and the position c.215C is highly conserved (GERP score 4.21; PhyloP 7.864; PhastCons 1; SiPhy 17.886). Frequency in public databases and in-silico pathogenicity predictions of this novel variant as well as previously published SNCA missense variants can be found in Supplementary Table 4.
We also provide evidence of a shared SNCA haplotype in the affected members of these two Turkish families. Our data suggest that the background haplotype on which the SNCA variant occurs is present in only a small fraction (<4%) of chromosomes obtained from the Turkish population. These results strongly suggest that the SNCA p. Thr72Met variant has originated from a single founder. Data regarding the presence and frequency of SNCA variants in the Turkish population are scarce. Two studies identified SNCA duplications in Turkish families [14,15], while another study failed to identify any SNCA variants [16]. This scarcity of data is not surprising, given the rarity of the SNCA variants in PD and related neurodegenerative disorders (an overview is provided in Table 1). Regarding the other missense SNCA variants identified in PD, only a very few can be considered as definitely disease-causing, while many others, detected in isolated cases or without convincing evidence of co-segregation with disease, remain of unclear significance (Table 1).
The clinical phenotype in the patients reported here with the p. Thr72Met variant shows variability regarding the occurrence of non-motor features as well as the age at onset. Cognitive decline is present in both families, whereas hallucinations and pyramidal signs are only reported in the second family. Furthermore, the onset age of PD [39–57 years (50.33 ± 9.87)] spanned almost 2 decades in these patients, i.e. a rather broad range, similar to many of the previously identified SNCA missense variants. Carriers of SNCA p. Ala53Thr display a variable age at onset, in most cases around the fourth to fifth decade, i.e. earlier than that observed for the SNCA p. Thr72Met. Similarly, carriers of SNCA p. Ala53Glu and p. Gly51Asp usually manifest PD around the fourth to fifth decade. In carriers of p. Ala30Pro and p. Glu46Lys, however, PD onset most often occurs during the sixth decade. Finally, carriers of p. Ala30Gly show an average age at onset in the sixth decade. Marked variability in age at onset, clinical phenotype, and progression is observed in carriers of SNCA variants among different families and even within the same family (Table 1).
In both families reported here, there is evidence of age-related, probably incomplete penetrance of the SNCA p. Thr72Met variant. In family 2, one carrier of the variant (III-2) has not manifested signs of parkinsonism by the age of 46, but she suffered from cognitive impairment. Another mutation carrier (II-2) was reported with cognitive decline since the age of 71, without parkinsonian signs by the age of 73. However, the severe weakness due to the co-existing severe HMSN2 may have made it difficult to appreciate possible subtle signs of parkinsonism. Reduced penetrance has been extensively reported, especially for SNCA duplications but also in missense variants, while SNCA triplications appear to be more penetrant [7,17–22]. The reasons for this incomplete penetrance and variable expressivity remain largely unknown. Multiple genetic and/or non-genetic modifiers might be involved [23,24], and much more work remains ahead to fill these important knowledge gaps.
An LRSAM1 c. G2005T/p.Glu669* variant was also identified in members of family 2 affected by HMSN2. LRSAM1 disruptive variants are an established cause of autosomal dominant axonal sensorimotor neuropathy [25]. Of note, in a previously published pedigree with HMSN2, three out of five affected members developed PD in addition to neuropathy [26], suggesting a possible link between LRSAM1 defects and the development of PD. In family 2, however, the LRSAM1 variant did not segregate with PD, as one of the two clinically affected parkinsonian cases (the index case, III-1) did not carry this variant. We, therefore, argue that the SNCA substitution is the PD-causing variant in these two Turkish pedigrees.
The disease-causing SNCA missense variants reported so far are located in the amphipathic alpha-helical domain of α-syn, responsible for binding to lipid membranes. It has been considered that the aforementioned variants might exert their effects either by hampering the binding of α-syn to lipid membrane structures or by stimulating α-syn aggregation propensity, or both, but such mechanisms might not be shared by all of these variants [27]. In contrast, the p. Thr72Met variant is positioned in the central hydrophobic region of the α-syn protein, known as the non-amyloid-β component (NAC) domain. This domain is required for polymerization into amyloid filaments [28] and appears to be unique to α-syn among the synuclein family [29,30]. A viable hypothesis as to how the novel variant might cause PD would therefore be by enhancing α-syn aggregation potential. In our protein expression studies, we compared the aggregation kinetics of α-syn-Thr72Met with that of wild-type α-syn using a thioflavin-T aggregation assay. We observed that α-syn-Thr72Met began to aggregate robustly and much earlier than the wild-type (Fig. 3A). This result is not surprising as a substitution of a small hydrophilic sidechain (threonine) for one that is large and hydrophobic (methionine) is expected to result in significant structural alterations and thereby in changes in the aggregation propensity.
In order to understand potential reasons for the observed increase in aggregation propensity, we substituted in silico threonine 72 for methionine in fibrillar and tetrameric structural models of α-syn. We observed no steric clashes in the fibrillar model, with the hydrophobic sidechain of Met 72 potentially contributing to the stability of the hydrophobic pocket of the α-syn fibril (Fig. 3B). Additionally, mapping the location of Thr72 on the tetrameric α-syn model of Wang et al. [12] reveals that Thr72 lies in the tetrameric interface, and the substitution with the much larger methionine residue could potentially lead to destabilization of the complex (Fig. 3C).
In conclusion, we report a novel SNCA missense variant associated with PD in two Turkish families. Testing for this variant in patients with familial or sporadic PD of Turkish origins might detect additional carriers. Further functional analyses might offer new insights into the biochemical properties of p. Thr72Met and other PD-causing missense variants in α-syn, and to their mechanisms of action leading to neurodegeneration.
Supplementary Material
Acknowledgements
We are indebted to all participating subjects and their families. This work was supported by research grants from the Stichting Parkinson-Fonds (The Netherlands) to Dr. Bonifati. Dr. Hoang acknowledges funding from the NIH (R21NS079881, R01GM111639, R01GM115844) and the Michael J. Fox Foundation; Dr. Petsko has also received past support from the Michael J. Fox Foundation.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.parkreldis.2021.06.023.
Disclosures
Vincenzo Bonifati receives honoraria from Elsevier Ltd, for serving as co-Editor-in-Chief of Parkinsonism & Related Disorders. He also received speaking honoraria from the International Parkinson and Movement Disorder Society, and as Chair of the MDS International Congress Program Committee. Gregory A. Petsko is on the Scientific Advisory Boards of MeiraGTx, Proclara Biosciences, and Annovis Bio, and is a co-founder of Retromer Therapeutics; these companies work on treatments for a variety of neurodegenerative diseases. Murat Emre serves on the Advisory Boards of Abdi İbrahim, AC Immune, ARIS, Britannia, Lundbeck and PD Neurotechnology and receives honoraria.
References
- [1].Spillantini MG, Schmidt ML, Lee VM, et al. , Alpha-synuclein in Lewy bodies, Nature 388 (6645) (1997) 839–840, 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- [2].Kumar KR, Lohmann K, Klein C, Genetics of Parkinson disease and other movement disorders, Curr. Opin. Neurol. 25 (4) (2012) 466–474, 10.1097/WCO.0b013e3283547627. [DOI] [PubMed] [Google Scholar]
- [3].Polymeropoulos MH, Lavedan C, Leroy E, et al. , Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease, Science 276 (5321) (1997) 2045–2047, 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- [4].Ibáñez P, Lesage S, Janin S, et al. , Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms, Arch. Neurol. 66 (1) (2009) 102–108, 10.1001/archneurol.2008.555. [DOI] [PubMed] [Google Scholar]
- [5].Nalls MA, Blauwendraat C, Vallerga CL, et al. , Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies, Lancet Neurol. 18 (12) (2019) 1091–1102, 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nussbaum RL, Genetics of synucleinopathies, Cold Spring Harb Perspect Med 8 (6) (2018) a024109, 10.1101/cshperspect.a024109. Published 2018 Jun 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Breza M, Koutsis G, Karadima G, et al. , The different faces of the p. A53T alpha-synuclein mutation: a screening of Greek patients with parkinsonism and/or dementia, Neurosci. Lett. 672 (2018) 136–139, 10.1016/j.neulet.2017.12.015. [DOI] [PubMed] [Google Scholar]
- [8].Bougea A, Koros C, Stamelou M, et al. , Frontotemporal dementia as the presenting phenotype of p.A53T mutation carriers in the alpha-synuclein gene, Park. Relat. Disord. 35 (2017) 82–87, 10.1016/j.parkreldis.2016.12.002. [DOI] [PubMed] [Google Scholar]
- [9].Kiely AP, Asi YT, Kara E, et al. , α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 125 (5) (2013) 753–769, 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li J, Shi L, Zhang K, et al. , VarCards: an integrated genetic and clinical database for coding variants in the human genome, Nucleic Acids Res. 46 (D1) (2018) D1039–D1048, 10.1093/nar/gkx1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Olgiati S, Doğu O, Tufekcioglu Z, et al. , The p.Thr11Met mutation in c19orf12 is frequent among adult Turkish patients with MPAN, Park. Relat. Disord. 39 (2017) 64–70, 10.1016/j.parkreldis.2017.03.012. [DOI] [PubMed] [Google Scholar]
- [12].Wang W, Perovic I, Chittuluru J, et al. , A soluble α-synuclein construct forms a dynamic tetramer, Proc. Natl. Acad. Sci. U. S. A. 108 (43) (2011) 17797–17802, 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guerrero-Ferreira R, Taylor NM, Arteni AA, et al. , Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy, Elife 8 (2019), e48907, 10.7554/eLife.48907. Published 2019 Dec 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kessler C, Atasu B, Hanagasi H, et al. , Role of LRRK2 and SNCA in autosomal dominant Parkinson’s disease in Turkey, Park. Relat. Disord. 48 (2018) 34–39, 10.1016/j.parkreldis.2017.12.007. [DOI] [PubMed] [Google Scholar]
- [15].Lahut S, Gispert S, Ömür Ö, et al. , Blood RNA biomarkers in prodromal PARK4 and rapid eye movement sleep behavior disorder show role of complexin 1 loss for risk of Parkinson’s disease, Dis Model Mech 10 (5) (2017) 619–631, 10.1242/dmm.028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Erer S, Egeli U, Zarifoglu M, et al. , Mutation analysis of the PARKIN, PINK1, DJ1, and SNCA genes in Turkish early-onset Parkinson’s patients and genotype-phenotype correlations, Clin. Neurol. Neurosurg. 148 (2016) 147–153, 10.1016/j.clineuro.2016.07.005. [DOI] [PubMed] [Google Scholar]
- [17].Itokawa K, Sekine T, Funayama M, et al. , A case of α-synuclein gene duplication presenting with head-shaking movements, Mov. Disord. 28 (3) (2013) 384–387, 10.1002/mds.25243. [DOI] [PubMed] [Google Scholar]
- [18].Nishioka K, Ross OA, Ishii K, et al. , Expanding the clinical phenotype of SNCA duplication carriers, Mov. Disord. 24 (12) (2009) 1811–1819, 10.1002/mds.22682. [DOI] [PubMed] [Google Scholar]
- [19].Ahn TB, Kim SY, Kim JY, et al. , alpha-Synuclein gene duplication is present in sporadic Parkinson disease, Neurology 70 (1) (2008) 43–49, 10.1212/01.wnl.0000271080.53272.c7. [DOI] [PubMed] [Google Scholar]
- [20].Pimentel MM, Rodrigues FC, Leite MA, et al. , Parkinson disease: α-synuclein mutational screening and new clinical insight into the p.E46K mutation, Park. Relat. Disord. 21 (6) (2015) 586–589, 10.1016/j.parkreldis.2015.03.011. [DOI] [PubMed] [Google Scholar]
- [21].Papadimitriou A, Veletza V, Hadjigeorgiou GM, et al. , Mutated alpha-synuclein gene in two Greek kindreds with familial PD: incomplete penetrance? Neurology 52 (3) (1999) 651–654, 10.1212/wnl.52.3.651. [DOI] [PubMed] [Google Scholar]
- [22].Trinh J, Guella I, Farrer MJ, Disease penetrance of late-onset parkinsonism: a meta-analysis, JAMA Neurol 71 (12) (2014) 1535–1539, 10.1001/jamaneurol.2014.1909. [DOI] [PubMed] [Google Scholar]
- [23].Kara E, Kiely AP, Proukakis C, et al. , A 6.4 Mb duplication of the α-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations, JAMA Neurol 71 (9) (2014) 1162–1171, 10.1001/jamaneurol.2014.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Markopoulou K, Dickson DW, McComb RD, et al. , Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Variability in familial Parkinson’s disease, Acta Neuropathol. 116 (1) (2008) 25–35, 10.1007/s00401-008-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Weterman MA, Sorrentino V, Kasher PR, et al. , A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy, Hum. Mol. Genet. 21 (2) (2012) 358–370, 10.1093/hmg/ddr471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aerts MB, Weterman MA, Quadri M, et al. , A LRSAM1 mutation links Charcot-Marie-Tooth type 2 to Parkinson’s disease, Ann Clin Transl Neurol 3 (2) (2015) 146–149, 10.1002/acn3.281. Published 2015 Dec 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rosborough K, Patel N, Kalia LV, α-Synuclein and parkinsonism: updates and future perspectives, Curr. Neurol. Neurosci. Rep. 17 (4) (2017) 31, 10.1007/s11910-017-0737-y. [DOI] [PubMed] [Google Scholar]
- [28].Waxman EA, Mazzulli JR, Giasson BI, Characterization of hydrophobic residue requirements for alpha-synuclein fibrillization, Biochemistry 48 (40) (2009) 9427–9436, 10.1021/bi900539p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Uchihara T, Giasson BI, Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies, Acta Neuropathol. 131 (1) (2016) 49–73, 10.1007/s00401-015-1485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giasson BI, Murray IV, Trojanowski JQ, Lee VM, A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly, J. Biol. Chem. 276 (4) (2001) 2380–2386, 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.