

Abstract
Objective
Mutations in the colony‐stimulating factor 1 receptor gene (CSF1R) were identified as a cause of adult‐onset inherited leukoencephalopathy. The present study aims at investigating the frequency, clinical characteristics, and functional effects of CSF1R mutations in Taiwanese patients with adult‐onset leukoencephalopathy.
Methods
Mutational analysis of CSF1R was performed in 149 unrelated individuals with leukoencephalopathy by a targeted resequencing panel covering the entire coding regions of CSF1R. In vitro analysis of the CSF1‐induced autophosphorylation activities of mutant CSF1R proteins was conducted to assess the pathogenicity of the CSF1R mutations.
Results
Among the eight CSF1R variants identified in this study, five mutations led to a loss of CSF1‐induced autophosphorylation of CSF1R proteins. Four mutations (p.K586*, p.G589R, p.R777Q, and p.R782C) located within the tyrosine kinase domain of CSF1R, whereas the p.T79M mutation resided in the immunoglobulin‐like domain. The five patients carrying the CSF1R mutations developed cognitive decline at age 41, 43, 50, 79, and 86 years, respectively. Psychiatric symptoms and behavior changes were observed in four of the five patients. The executive function and processing speed were severely impaired at an early stage, and their cognitive function deteriorated rapidly within 3–4 years. Diffusion‐restricted lesions at the subcortical regions and bilateral corticospinal tracts were found in three patients.
Interpretation
CSF1R mutations account for 3.5% (5/149) of the adult‐onset leukoencephalopathy in Taiwan. CSF1R mutations outside the tyrosine kinase domain may also disturb the CSF1R function and lead to the clinical phenotype. Molecular functional validation is important to determine the pathogenicity of novel CSF1R variants.
Introduction
Adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is an inherited white matter disorder encompassing two clinic‐pathologically similar entities, namely hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). 1 , 2 Patients with ALSP usually develop early‐onset dementia, behavior changes, depression, gait ataxia and parkinsonism in the fourth or fifth decade of life, and rapidly deteriorate to bed‐ridden status within 3–5 years. 3 , 4 With the discovery of causative genes, 5 , 6 these two clinic‐pathologically similar conditions (i.e., POLD and HDLS) now can be clearly distinguished. 7 For patients diagnosed with POLD and some literature cases pathologically mislabeled as HDLS, the new nomenclature is “colony‐stimulating factor 1 receptor gene (CSF1R)‐related leukoencephalopathy.” On the other hand, the patients diagnosed with HDLS including the original Swedish family reported in 1984 are belonged to “alanyl‐transfer tRNA synthetase 2 gene (AARS2)‐related leukoencephalopathy.” 7 , 8 CSF1R‐related leukoencephalopathy has become increasingly recognized, and it is estimated to account for 10% of adult‐onset leukodystrophy in the Caucasian populations. 9 , 10
CSF1R encodes for a tyrosine kinase transmembrane receptor that regulates the survival, proliferation, and differentiation of mononuclear phagocytic cells, including microglia in the brain. 11 Upon stimulation by its ligand colony‐stimulating factor 1 (CSF1) or interleukin‐34, CSF1R protein would form a homodimer and autophosphorylate the tyrosine residues within its intracellular kinase domain and subsequently activate the downstream signaling pathways. 12 , 13 The original study identifying CSF1R mutations as the cause of ALSP also demonstrated that the pathogenic mutations abolished the CSF1‐induced autophosphorylation of CSF1R proteins. 5 The findings that haploinsufficiency of CSF1R genetic function is sufficient to cause leukoencephalopathy was again confirmed in the Csf1r +/− mouse model. 14 To date, more than 70 different CSF1R mutations have been reported worldwide, 3 , 4 , 15 and the majority of them are located within exons 12–22 of CSF1R, corresponding to the tyrosine kinase domain (TKD) of CSF1R protein. Among these mutations, only a small number of them had been proven to affect the CSF1R function by in vitro studies. 5 , 16 , 17 , 18 , 19 The arbitrariness to claim the pathogenicity without functional verification may raise the uncertainty of some of the CSF1R mutations in clinical reports.
To expand the knowledge of molecular spectrum and clinical phenotypes of CSF1R‐related leukoencephalopathy, the present study investigated the frequency, clinical features, and image characteristics of patients carrying a CSF1R mutation in a Taiwanese cohort with adult‐onset leukoencephalopathy of unknown causes. In vitro functional analysis of the CSF1R mutations was conducted to assess the pathogenicity.
Patients and Methods
Subjects
A consecutive series of 149 unrelated cases with adult‐onset leukoencephalopathy were enrolled from the Neurology Department of Taipei Veterans General Hospital between 2001 and 2020. Taipei Veterans General Hospital is a 2974‐bed tertiary medical center that serves both veterans and regular citizens in Taiwan. It accepts both self‐referred patients and referrals of difficult cases from other hospitals. The inclusion criteria were subjects with (1) marked leukoencephalopathy defined as Fazekas grade 2 or grade 3 on brain magnetic resonance imaging (MRI), 20 and (2) any of the following symptoms: cognitive dysfunction, psychiatric disorder, gait disturbance, or parkinsonism. The exclusion criteria were subjects (1) who ever developed ischemic stroke or intracerebral hemorrhage or (2) carrying pathogenic mutations in NOTCH3 or HTRA1.
All participants were of Han‐Chinese descent. Peripheral blood samples were collected after written informed consent was obtained. This study was approved by the Institutional Review Board of Taipei Veterans General Hospital.
Cognitive tests and neuroimaging studies
The demographic information, medical histories, and personal histories of the patients carrying the CSF1R mutations were obtained from the patient interview and medical record reviews. Global cognitive performance was assessed using the Mini‐Mental Status Examination (MMSE). 21 Tests specific to each cognitive domain were performed, including (1) memory (12‐item word recall test), 22 (2) language and executive function (category verbal fluency test), 23 (3) processing speed (triai making test A), 24 , 25 and (4) attention (forward and backward digit span test from the Wechsler Memory Scale IV). 26 The raw data and percentile score of each cognitive test were shown. The percentile score falling below 1.5 standard deviations (i.e., <16%) of the normative data was defined as significantly abnormal.
Brain MRI, including T 1‐weighted images (T 1WI), T 2‐weighted images (T 2WI), fluid‐attenuated inversion recovery (FLAIR) images, and diffusion‐weighted imaging (DWI), as well as brain computed tomography (CT) images were reviewed.
Mutation analysis
Sequence analysis of CSF1R was performed by utilizing a high‐throughput targeted resequencing panel covering the entire coding regions of CSF1R and other 183 genes associated with hereditary leukodystrophy and small vessel diseases (Table S1) on an Illumina HiSeq2500 platform. Alignment of sequenced reads and identification of variants were performed with the reference Human Genome version 38 (hg38/GRCh38) and the reference CSF1R coding sequence (NM_005211.3). Sanger sequencing was performed to confirm the identified CSF1R variants.
The putative pathogenic CSF1R mutations were discriminated by their absence or presence with an extremely rare allele frequency in the genome Aggregation Database (gnomAD version r2.1.1). 27 In silico analysis of pathogenicity of the CSF1R mutations was conducted using Combined Annotation Dependent Depletion (CADD), 28 PolyPhen‐2, 29 and MutationTaster. 30 Phylogenetic conservation of the mutated amino acid residues were analyzed by aligning the amino acid sequences of multiple CSF1R orthologs utilizing the UniProt website. 31
Expression plasmids and cell cultures
A human CSF1R cDNA clone was purchased from TransOMIC (BC047521; Huntsville, AL). The full‐length coding region of human CSF1R was cloned into pFLAG‐CMV‐5a (Sigma‐Aldrich, St. Louis, MO) to generate the wild‐type CSF1R expression plasmids. Nine CSF1R variants were introduced into the wild‐type expression plasmids, separately, using the QuikChange Site‐Directed Mutagenesis method (Stratagene; Agilent, Santa Clara, CA). These variants were p.T79M (c.236C > T), p.E478K (c.1432G > A), p.T507_H508insP (c.1520_1522dupCGC), p.K586* (c.1754dupT), p.G589R (c.1765G > A), p.R777Q (c.2330G > A), p.R782C (c.2344C > T), p.M875T (c.2624T > C), and p.A914T (c.2740G > A). CSF1R p.M875T was a well‐known pathogenic mutation 5 and served as a positive control in this study; the remaining eight variants were identified in the present study (Table 1). HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum in a humidified incubator at 37°C under 5% CO2.
Table 1.
The rare CSF1R variants identified in this study.
| CSF1R variant | Location | Bioinformatics prediction | Population controls | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | Exon | Domain | MutationTester | PolyPhen2 | CADD Phred score | ClinVar | gnomAD | TaiwanView |
| c.236C > T | p.T79M | 3 | IG | Disease causing | Damaging | 22.2 | Not reported | None | None |
| c.1432G > A | p.E478K | 10 | IG | Tolerated | Benign | 11.8 | Not reported | None | None |
| c.1520_1522dupCGC | p.T507_H508InsP | 11 | TM | Tolerated | NA. | NA | Not reported | None | None |
| c.1754dupT | p. K586* | 13 | TKD | Disease causing | NA | NA | Not reported | None | None |
| c.1765G > A | p.G589R | 13 | TKD | Disease causing | Damaging | 29.8 | Pathogenic | None | None |
| c.2330G > A | p.R777Q | 18 | TKD | Disease causing | Damaging | 32 | Pathogenic | None | None |
| c.2344C > T | p.R782C | 18 | TKD | Disease causing | Damaging | 24.1 | Not reported | None | None |
| c.2740G > A | p.A914T | 21 | C‐terminal | Tolerated | Benign | 3.4 | Not reported | EA: 1/19952 | 2/1513 |
ClinVar, a public archive of interpretations of clinically relevant variants (https://www.ncbi.nlm.nih.gov/clinvar/); CADD, Combined Annotation Dependent Depletion; IG, immunoglobulin‐like domain; TM, transmembrane domain; TKD, tyrosine kinase domain; C‐terminal, carboxyl‐terminal; EA, East Asian; gnomAD, genome Aggregation Database (http://gnomad.broadinstitute.org); TaiwanView, 1517 healthy Taiwanese control exomes were publically available in the Taiwan Biobank database (https://taiwanview.twbiobank.org.tw/index); NA, not applicable.
In vitro analysis of CSF1‐induced autophosphorylation of CSF1R
The plasmids expressing wild‐type or one of the eight mutant CSF1R proteins were transfected into HeLa cells, respectively, using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA). Forty‐eight hours posttransfection, the medium was changed to serum‐free medium for 16 h, and the transfected cells were treated with 100 ng/mL human recombinant CSF1 (R&D Systems, Minneapolis, MN) for 10 min. Treated cells were harvested immediately with RIPA buffer and analyzed by Western blotting. Transfected cells without CSF1 treatment were used as controls. Total CSF1R expression levels were analyzed using the CSF1R antibody (sc‐46662; Santa Cruz, Dallas, TX). CSF1R autophosphorylation ability was evaluated using CSF1R phospho‐tyrosine primary antibodies against p‐Tyr546 (#3083), p‐Tyr699 (#12251), p‐Tyr723 (#3155), and p‐Tyr923 (#3406), respectively (Cell Signaling Technology, Danvers, MA). Actin was used as a loading control to ensure an equal amount of protein loading (MAB1501; Merck Millipore, Burlington, MA).
Results
Identification of CSF1R mutations
Among the 149 unrelated patients with leukoencephalopathy, we found eight different CSF1R variants (Table 1). Except for CSF1R p.A914T presenting with an allele frequency of 0.00005 in the East Asian population in gnomAD, the other seven CSF1R variants were absent in the 123,136 exomes and 15,496 genome data of various populations from gnomAD, as well as the 1517 healthy Taiwanese exomes from Taiwan Biobank. Among them, p.G589R and p.R777Q mutations had been reported in patients with ALSP. 4 , 10 , 32 , 33 The p.R782C variant was novel, but CSF1R mutations altering the same amino acid residue (e.g., p.R782G and p.R782H) had been identified in patients with leukoencephalopathy. 34 , 35 The remaining five variants had never been reported before.
Six of the eight CSF1R variants were missense mutations, including three located within the TKD (p.G589R, p.R777Q, and p.R782C), two in the immunoglobulin‐like domain (p.T79M and p.E478K), and p.A914T at the carboxyl‐terminal domain of CSF1R (Table 1, Fig. 1A). The nonsense mutation (p.K586*) caused premature termination of TKD, whereas the p.T507_H508InsP mutation inserted an additional amino acid residue in the transmembrane domain without altering the reading frame. The three mutations located within TKD (p.G589R, p.R777Q, and p.R782C) and p.T79M were predicted to be pathogenic by PolyPhen‐2 and MutationTaster; the nonsense mutation (p.K586*) was also predicted as disease‐causing by MutationTaster (Table 1). The CADD Phred scores of these five mutations ranged from 22.2 to 32, ranking them in the top 6.0/1000 to 6.3/10,000 most deleterious variants in the genome. 28 The amino acids influenced by these four missense mutations (p.T79M, p.G589R, p.R777Q, and p.R782C) were evolutionally conserved across CSF1R orthologues from human to zebrafish (Fig. 1B). The remaining three mutations outside the TKD (p.E478K, p.T507_H508InsP, p.A914T) were predicted to be benign variants by the bioinformatics tools.
Figure 1.
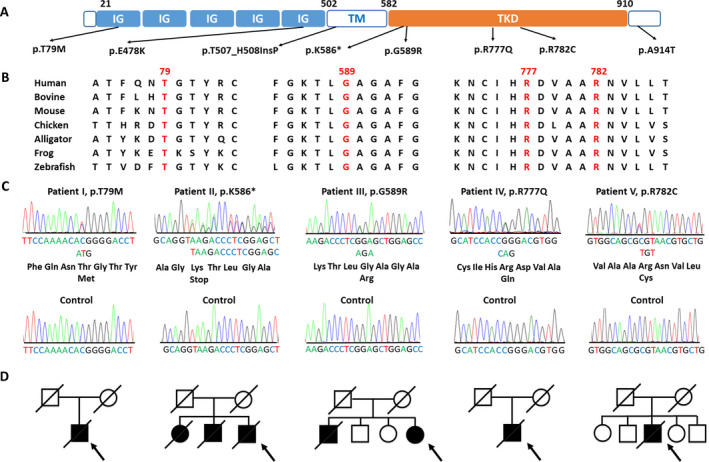
Genetic analysis of the CSF1R mutations identified in the Taiwanese patients with leukoencephalopathy. (A) Schematic illustration of the structure of CSF1R protein and position of the rare CSF1R variants identified in this study. IG = immunoglobulin‐like domain; TM = transmembrane domain; TKD = tyrosine kinase domain. (B) Alignment of multiple CSF1R protein orthologs showing evolutionary conservation of the amino acid residues altered by the putative pathogenic mutations. (C) Sanger sequencing trace demonstrating the five heterozygous CSF1R pathogenic mutations and corresponding wide‐type control sequences. (D) Pedigrees of the five index patients harboring a CSF1R pathogenic mutation. Open symbol: unaffected; filled symbol: affected; symbol with a diagonal line: deceased; arrow: proband; square: male; circle: female.
In vitro analysis of CSF1‐induced autophosphorylation of CSF1R proteins
To assess the functional consequence of CSF1R mutations identified in this study, we transfected HeLa cells with wild‐type or either one of the mutant CSF1R expression plasmids. The expression level and autophosphorylation ability of mutant and wide‐type proteins were compared using Western blotting. The expression of wild‐type and eight mutant CSF1R proteins were comparable (Fig. 2). However, the five CSF1R mutants, including T79 M, K586*, G589R, R777Q, and R782C CSF1R, as well as the positive control M875T CSF1R had a total or profound loss of their ability to phosphorylate the Tyr546, Tyr699, Tyr723, and Tyr923 residues upon CSF1 stimulation (Fig. 2). The remaining three CSF1R mutants, including E478K, T507_H508InsP, and A914T CSF1R, had preserved CSF1‐induced CSF1R tyrosine kinase activity to phosphorylate the tyrosine residues.
Figure 2.

In vitro analysis of CSF1‐induced autophosphorylation of CSF1R mutant proteins. Representative western blots of lysates from HeLa cells that were transfected with wide‐type (WT) or mutant CSF1R expressing constructs with or without colony‐stimulating factor 1 (CSF1) treatment. Immunoblotting analysis using anti‐CSF1R antibody revealed comparable expression between WT and mutant CSF1R proteins. To evaluate the CSF1‐induced autophosphorylation activity of each CSF1R mutant, protein lysates were compared between CSF1‐treated and untreated cells expressing either one of the CSF1R mutant proteins. The five CSF1R mutants (T79M, K586*, G589R, R777Q, and R782C CSF1R) and the positive control M875T CSF1R had lost their ability to phosphorylate the Tyr546, Tyr699, Tyr723, and Tyr923 residues within CSF1R upon CSF1 stimulation, whereas the other three CSF1R mutants (E478K, T507_H508InsP, and A914T CSF1R) had preserved function of the CSF1‐induced autophosphorylation. Experiments were repeated three times with similar results. The equivalent amount of protein loading is shown using Actin as a loading control.
Clinical features of patients harboring a CSF1R pathogenic mutation
The pathogenicity of the five CSF1R mutations (p.T79M, p.K586*, p.G589R, p.R777Q, and p.R782C) are supported by the absence in population databases, predictions of multiple bioinformatics programs, and well‐established in vitro functional analyses (Table 1 and Fig. 2). According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guideline, 36 the three novel CSF1R mutations (p.T79M, p.K586*, and p.R782C) match the PS3, PM2, PP3, and PP4 criteria and are classified as likely pathogenic variants. The other two known CSF1R mutations are categorized into pathogenic variants (PS1, PS3, PM2, PP3, and PP4).
Each of the five mutations was identified in one single index patient (Fig. 1C), suggesting that 3.4% (5 of 149) of the patients with adult‐onset leukoencephalopathy in Taiwan was attributed to CSF1R mutations. Among the five patients, two had a family history of dementia and three were sporadic cases (Fig. 1D).
All the five patients carrying a CSF1R mutation suffered from progressive cognitive decline (Table 2). The age at symptom onset ranged from 41 to 86 years, including three patients having symptoms at the fourth to fifth decade of life and two patients developing symptoms in their 80s. Patient IV carrying the CSF1R p.R777Q mutation had severe cognitive dysfunction with an MMSE score of 7, and could not perform other cognitive tests. For the remaining four patients, they had impaired cognition (MMSE scores = 17–22) and severe deficits in attention, executive function, and processing speed (Table 2). They performed poorly at the backward digit span test and category verbal fluency with percentile scores below 16% of the normative data. Their processing speed at Trial making test A fell below 2% of the normative data, suggesting severely impaired executive function. More importantly, the cognitive function deteriorated rapidly within 3–4 years. Patient II with CSF1R p.K586* mutation had an MMSE score of 22 at age 82 but a score of 5 4 years later; patient III with p.G589R had an MMSE score of 20 at age 46 but a score of 5 two years later.
Table 2.
Clinical features and cognitive tests of the patients carrying a CSF1R pathogenic mutation.
| Patient no | Patient I | Patient II | Patient III | Patient IV | Patient V |
|---|---|---|---|---|---|
| Mutation | p.T79M | p.K586* | p.G589R | p.R777Q | p.R782C |
| Age at onset, sex | 86 y, male | 79 y, male | 43 y, female | 41 y, female | 50 y, male |
| Family history of dementia | None | Brother, sister | Brother | None | None |
| Personal history | HTN, DM, Lipid, CKD, CHF, smoking | None | None | Sjogren's syndrome, Hashimoto thyroiditis | None |
| Cognitive tests, score (percentile score) | |||||
| Age at exam | 87 y | 82 y | 46 y | 44 y | 54 y |
| Education | 16 y | 12 y | 9 y | 12 y | 12 y |
| Mini‐mental status examination | 17 (≦1%) | 22 (≦1%) | 20 (≦1%) | 7 (≦1%) | 21 (≦1%) |
| Delayed recalls at 12‐item test | 1 (<2%) | 4 (5%–9%) | 5 (10%–30%) | ND | 5 (10%–30%) |
| Category verbal fluency | 5 (<3%) | 4 (<3%) | 6 (<3%) | ND | 8 (<11%) |
| Trial making test A | 360 sec (<1%) | 190 sec (<2%) | 150 sec (<2%) | ND | ND |
| Forward digit span test | 6 (<5%) | 5 (<1%) | 9 (>19%) | ND | 7 (5%–19%) |
| Backward digit span test | 2 (<2%) | 3 (4%–16%) | 2 (<2%) | ND | 2 (<2%) |
| Behavior changes | None | None | None | Repetition, stereotyping | Compulsive behavior |
| Psychiatric problems | None | Liable mood, anxiety | Depression | Apathy, social withdraw | Apathy, agitation |
| Parkinsonism | Postural tremor | Small steps, ataxic gait | Bradykinesia, rigidity, freezing gait | Action and postural tremor | None |
| Seizure | None | None | None | GTCS | None |
HTN, hypertension; DM, Diabetes mellitus; Lipid, hyperlipidemia; CKD, chronic kidney disease; CHF, congestive heart failure; ND, not done; GTCS, generalized tonic clonic seizure; y, years; sec, seconds. The raw score and percentile score of each cognitive test were shown. The percentile score falling below 1.5 standard deviations (i.e. <16%) of the normative data was defined as significantly impaired.
The majority of the patients (4 out of 5) also suffered from psychiatric symptoms and behavior changes, including apathy, liable mood, depression, agitation, and compulsive and repetitive behaviors (Table 2). The postural tremor was reported in two patients, while gait disturbance was noted in another two. For patient III who carried the p.G589R mutation, she was initially diagnosed with corticobasal degeneration for having bradykinesia, rigidity, and hyperreflexia on the right side of limbs.
Radiologic features of the patients with CSF1R‐related leukoencephalopathy
Confluent leukoencephalopathy was noted in all the five patients harboring a CSF1R pathogenic mutation (Fig. 3, Table 3). White matter hyperintensity on FLAIR images was most prominent at the frontal regions and gradually extended to the parietal lobe. Thinning of the corpus callosum was noted in all five patients, and hyperintensity at the genu or splenium of the corpus callosum was found in patients III and IV. Cavum septum pellucidum was present in three of the five patients (60%), whereas calcification was only detected in one of the three patients (30%) who ever received brain CT.
Figure 3.
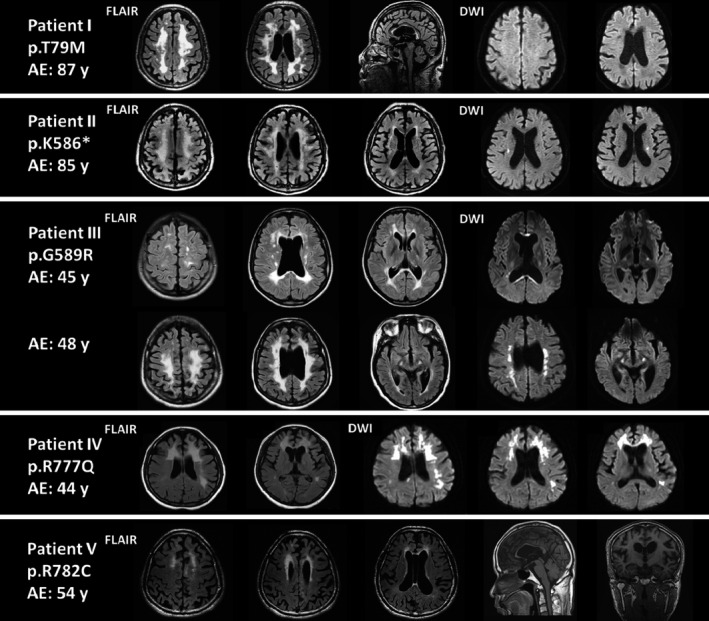
Representative magnetic resonance imaging of the patients with CSF1R‐related leukoencephalopathy. AE, age at the examination; FLAIR, fluid‐attenuated inversion recovery; DWI, diffusion‐weighted imaging.
Table 3.
Neuroimaging characteristics of the patients with CSF1R‐related leukoencephalopathy.
| Patient no/mutation | Patient I/p.T79M | Patient II/p.K586* | Patient III/p.G589R | Patient IV/p.R777Q | Patient V/p.R782C |
|---|---|---|---|---|---|
| T 2‐weighted or FLAIR images of MRI | |||||
| White matter hyperintensity | Frontoparietal region, anterior temporal pole | Frontoparietal region | Frontoparietal region | Frontal involvement predominant | Frontal involvement predominant |
| Thinning or hyperintensity of corpus callosum | Present | Present | Present | Present | Present |
| Cavum septum pellucidum | None | None | Present | Present | Present |
| DWI (diffusion‐weighted imaging) of MRI | |||||
| Diffusion‐restricted lesions | None | Bilateral corticospinal tracts | Bilateral corticospinal tracts, corpus callosum, periventricular white matter | Frontoparietal periventricular white matter | ND |
| CT (computed tomography) | |||||
| Calcification | None | None | Periventricular, parietal subcortical regions | ND | ND |
FLAIR, fluid‐attenuated inversion recovery; ND, not done.
Diffusion‐restricted lesions on DWI were found in three of the four patients (75%). Periventricular lesions were noted in patients III and IV, and a corpus callosum lesion was seen in patient III (Fig. 3). Of note, corticospinal tract involvement on DWI was found in two patients and might be a characteristic feature of CSF1R‐related leukoencephalopathy. Patient II had bilateral corticospinal tract lesions extending from corona radiate to internal capsule, whereas patient III initially presented with left pyramidal tract lesion at age 45 and then progressed to bilateral pyramidal tract involvement extending from corona radiate, internal capsule to cerebral peduncles at age 48.
Discussion
The present study identified five CSF1R pathogenic mutations, including two being reported in literatures and three novel ones, from 149 unrelated Taiwanese patients with molecularly unassigned leukoencephalopathy. All the five mutations compromised the CSF1‐induced autophosphorylation function of CSF1R proteins, including the one residing outside the TKD. There are four major findings that have important implications in this study. First, CSF1R‐related leukoencephalopathy accounts for 3.5% of adult‐onset leukoencephalopathy in the Han‐Chinese population in Taiwan. The onset age varies from 41 to 86 years in our cases, suggesting that mutation analysis of CSF1R is warranted not only in middle‐aged patients but also in the elderly with unexplained white matter lesions. Second, our study demonstrated variants outside the TKD of CSF1R still could disturb the kinase activity and cause phenotypes, which highlights the importance of functional validation in newly identified CSF1R variants. Third, in our patients, executive function and processing speed were severely impaired, psychiatric symptoms and behavior changes were frequently encountered, and the cognitive function deteriorated rapidly within 3–4 years. These neuropsychiatric features may be characteristic of CSF1R‐related leukoencephalopathy. Lastly, white matter abnormality with frontoparietal predominance was found in all patients, but such lesions were nonspecific and could be seen in other leukoencephalopathies. Restricted diffusion lesions in the periventricular regions, corpus callosum, and bilateral pyramidal tracts were detected in three cases, and these findings would be more specific to CSF1R‐related leukoencephalopathy.
More than 30 genes have been implicated in leukodystrophy and genetic leukoencephalopathy, 37 , 38 but each entity is extremely rare with no single disease having a prevalence >1/20,000. 39 CSF1R is recognized as one of the common genetic causes of adult‐onset leukoencephalopathy because it accounts for 4%–10.5% of leukoencephalopathy in the United Kingdom and European countries. 9 , 10 , 40 In our cohort, 3.4% (5/149) of the Taiwanese patients with leukoencephalopathy carried a CSF1R pathogenic mutation. Three of our patients suffered from cognitive decline since the early 40s, which is consistent with the average age of onset at 42 years in the literature. 4 , 41 Intriguingly, patient I carrying CSF1R p.T79M mutation became demented at age 86 years, and patient II with p.K586* mutation developed symptoms at age 79 years. A wide range of onset ages varying from 15 to 78 years had been reported in historical cases, 4 , 41 but our study further suggested that CSF1R mutation carriers might present the first symptom as late as 80s. In other words, screening for CSF1R mutations should be considered in patients of all ages with unexplained white matter lesions.
The majority of the CSF1R mutations reported in the literature locate within the TKD of CSF1R. 3 , 4 , 15 Only a small group of these variants had been investigated in vitro to confirm their deleterious effects on CSF1R function. 2 , 5 , 16 , 17 , 18 , 19 In the present study and previous ones, 2 , 5 , 16 , 17 missense mutations affecting amino acid residues in the TKD, as well as frameshift or nonsense mutations leading to TKD truncation, would abolish CSF1R kinase activity. Nevertheless, residing within the TKD does not assure the pathogenicity of individual mutation. CSF1R p.G747R mutation, located in the TKD, was associated with intact kinase activity of CSF1R. 18 On the contrary, the p.E573L mutation at the transmembrane domain caused impaired CSF1R autophosphorylation. 18 In the present study, the T79M mutant protein had lost its function to phosphorylate the tyrosine residues, supporting that variants outside the TKD still may play a pathogenic role. The patient with p.E573L mutation and our patient harboring p.T79M mutation both had a late‐onset disease with symptom onset at age 70 and 86, respectively. MRI of these two patients both showed diffuse leukoencephalopathy and atrophic corpus callosum, but they had neither calcification on CT nor periventricular lesions on DWI. 18 Our findings strengthen the importance of functional validation of CSF1R variants to determine their pathogenicity.
The present study is the first one to demonstrate the cognitive domains primarily affected in the early stage of CSF1R‐related leukoencephalopathy. Our patients showed severe impairments in processing speed, attention, and executive function. Such features (i.e., subcortical dementia with predominant frontal lobe dysfunction) may reflect the underlying pathogenesis of CSF1R‐related leukoencephalopathy, in which microglial dysfunction primarily results in axonopathy, followed by demyelination and neuronal degeneration. 3 , 42 In addition, white matter changes on MRI have been observed 6 years prior to symptom onset, 43 and cortical atrophy over the frontoparietal lobes occurs as disease progression. In addition, patients with CSF1R mutations usually have a rapid decline of cognition within 3–4 years and a short survival duration of 6.8 years. 4
Neuroimaging features can also help diagnose CSF1R‐related leukoencephalopathy. According to the recently proposed diagnostic criteria, 44 bilateral white matter lesions and thinning of the corpus callosum are the core features of CSF1R‐related leukoencephalopathy, and spotty calcification also supports the diagnosis. In our five patients, all had the two core features, and one of the three patients who ever received brain CT had calcification. To be noticed, thin‐section (1 mm) CT scans and reconstructed sagittal CT images are particularly helpful to detect small calcifications and the characteristic features of “stepping stone appearance in the pericallosal region.” 45 Since the brain CT scans were performed at 5‐mm section thickness in our three patients carrying the CSF1R mutations, small calcification might be overlooked. Previous studies showed that calcifications were detected in 21–67% of the patients with CSF1R‐related leukoencephalopathy. 46 , 47 Among these imaging features, thinning of the corpus callosum and diffusion‐restricted lesions are better predicting factors of CSF1R‐related leukoencephalopathy with odds ratios of 42.6 and 10.2, respectively. 46 In addition to the well‐known periventricular lesions on DWI, two of our patients had diffusion‐restricted lesions at the bilateral pyramidal tracts. The corticospinal tract lesion could be detected by T2WI, FLAIR, or DWI and was found in 63% of the 16 patients in an MRI study of ALSP. 47 The histopathologic analysis of a patient with T 2WI hyperintensity lesions involving bilateral corticospinal tracts from the motor cortex to the midbrain, pons, and medulla revealed vacuolization and reactive gliosis of the corresponding regions. 48
One limitation of the present study is a lack of pathologic data. However, with the advantage of genetic diagnosis, pathologic examination of brain biopsy tissue is no longer the golden standard to make a definite diagnosis of ALSP. All the five patients harboring CSF1R mutations in this study underwent a relentless deterioration in cognitive function and ambulation ability, and three of them had passed away. None of them ever received immunotherapy or other disease‐modifying therapy. Although no treatments have been approved for CSF1R‐related leukoencephalopathy, the latest study confirmed the therapeutic benefits of hematopoietic stem cell transplantation in six of seven patients, in whom the motor and cognitive functions, as well as MRI abnormalities, became stabilized after transplantation. 49
To conclude, the present study showed that 3.5% of the Taiwanese patients with adult‐onset leukoencephalopathy is attributed to CSF1R‐related leukoencephalopathy. Mutation analysis of CSF1R should be considered in patients of all age groups who have cognitive decline and white matter lesions on MRI. Slow processing speed, executive dysfunction, concomitant psychiatric and behavior symptoms, rapid progression of cognitive decline, and corticospinal tract lesions on MRI may be clues to differentiate CSF1R‐related leukoencephalopathy from other dementia etiologies.
Conflict of Interest
There is no conflict of interest.
Supporting information
Table S1. Genes in the targeted resequencing panel.
Acknowledgments
This study was supported by the grants from the Ministry of Science and Technology, Taiwan (107‐2314‐B075‐014‐MY3, 109‐2628‐B075‐025), Taipei Veterans General Hospital (V106D21‐004‐MY2, V108C‐076), and Brain Research Center, National Yang‐Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan. We thank the GenoInfo Core Facility (C1) funded by the National Core Facility Program of MOST Taiwan (MOST109‐2740‐B‐010‐002) for providing bioinformatics supports. We also thank the Clinical and Industrial Genomic Application Development Service Center funded by National Core Facility Program for Biotechnology, Taiwan (MOST 107‐2319‐B‐010‐002) for sequencing.
Funding Information
This study was supported by the grants from the Ministry of Science and Technology, Taiwan (107‐2314‐B075‐014‐MY3, 109‐2628‐B075‐025), Taipei Veterans General Hospital (V106D21‐004‐MY2, V108C‐076), and Brain Research Center, National Yang‐Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan. We thank the GenoInfo Core Facility (C1) funded by the National Core Facility Program of MOST Taiwan (MOST109‐2740‐B‐010‐002) for providing bioinformatics supports.
Funding Statement
This work was funded by Ministry of Science and Technology, Taiwan grants 107‐2314‐B075‐014‐MY3 and 109‐2628‐B075‐025; Taipei Veterans General Hospital grants V106D21‐004‐MY2 and V108C‐076; Brain Research Center; National Yang‐Ming University; Ministry of Education in Taiwan grant MOST109‐2740‐B‐010‐002.
Contributor Information
Yi‐Chung Lee, Email: yichunglee@gmail.com.
Yi‐Chu Liao, Email: yichu.liao@gmail.com.
References
- 1. Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. 2004;107(6):481‐488. [DOI] [PubMed] [Google Scholar]
- 2. Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80(11):1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R‐related leukoencephalopathy: a major player in primary microgliopathies. Neurology. 2018;91(24):1092‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Konno T, Yoshida K, Mizuno T, et al. Clinical and genetic characterization of adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. 2017;24(1):37‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lynch DS, Zhang WJ, Lakshmanan R, et al. Analysis of mutations in AARS2 in a series of CSF1R‐negative patients with adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. 2016;73(12):1433‐1439. [DOI] [PubMed] [Google Scholar]
- 7. Wszolek ZK. First Polish case of CSF1R‐related leukoencephalopathy. Neurol Neurochir Pol. 2021;55(3):239‐240. [DOI] [PubMed] [Google Scholar]
- 8. Sundal C, Carmona S, Yhr M, et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol Commun. 2019;7(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynch DS, Jaunmuktane Z, Sheerin U‐M, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to Parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. 2016;87(5):512‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerreiro R, Kara E, Le Ber I, et al. Genetic analysis of inherited leukodystrophies: genotype‐phenotype correlations in the CSF1R gene. JAMA Neurol. 2013;70(7):875‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stanley ER, Berg KL, Einstein DB, et al. Biology and action of colony–stimulating factor‐1. Mol Reprod Dev. 1997;46(1):4‐10. [DOI] [PubMed] [Google Scholar]
- 12. Pixley FJ, Stanley ER. CSF‐1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14(11):628‐638. [DOI] [PubMed] [Google Scholar]
- 13. Rohde CM, Schrum J, Lee AW. A juxtamembrane tyrosine in the colony stimulating factor‐1 receptor regulates ligand‐induced Src association, receptor kinase function, and down‐regulation. J Biol Chem. 2004;279(42):43448‐43461. [DOI] [PubMed] [Google Scholar]
- 14. Chitu V, Gokhan S, Gulinello M, et al. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult‐onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol Dis. 2015;74:219‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams SJ, Kirk A, Auer RN. Adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J Clin Neurosci. 2018;48:42‐49. [DOI] [PubMed] [Google Scholar]
- 16. Konno T, Tada M, Tada M, et al. Haploinsufficiency of CSF‐1R and clinicopathologic characterization in patients with HDLS. Neurology. 2014;82(2):139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miura T, Mezaki N, Konno T, et al. Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol. 2018;265(10):2415‐2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Konno T, Miura T, Harriott AM, et al. Partial loss of function of colony‐stimulating factor 1 receptor in a patient with white matter abnormalities. Eur J Neurol. 2018;25(6):875‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang X, Huang P, Tan Y, Xiao Q. A novel splicing mutation in the CSF1R gene in a family with hereditary diffuse leukoencephalopathy with axonal spheroids. Front Genet. 2019;10:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fazekas F, Barkhof F, Wahlund LO, et al. CT and MRI rating of white matter lesions. Cerebrovasc Dis. 2002;13(Suppl 2):31‐36. [DOI] [PubMed] [Google Scholar]
- 21. Folstein MF, Folstein SE, McHugh PR. "Mini‐mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189‐198. [DOI] [PubMed] [Google Scholar]
- 22. Vanderploeg RD, Schinka JA, Jones T, Small BJ, Graves AB, Mortimer JA. Elderly norms for the Hopkins verbal learning test‐revised. Clin Neuropsychol. 2000;14(3):318‐324. [DOI] [PubMed] [Google Scholar]
- 23. Harrison JE, Buxton P, Husain M, Wise R. Short test of semantic and phonological fluency: normal performance, validity and test‐retest reliability. Br J Clin Psychol. 2000;39(2):181‐191. [DOI] [PubMed] [Google Scholar]
- 24. Lu L, Bigler ED. Normative data on trail making test for neurologically normal, Chinese‐speaking adults. Appl Neuropsychol. 2002;9(4):219‐225. [DOI] [PubMed] [Google Scholar]
- 25. Gordon NG. The trail making test in neuropsychological diagnosis. J Clin Psychol. 1972;28(2):167‐169. [DOI] [PubMed] [Google Scholar]
- 26. Waters GS, Caplan D. The reliability and stability of verbal working memory measures. Behav Res Methods Instrum Comput. 2003;35(4):550‐564. [DOI] [PubMed] [Google Scholar]
- 27. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11(4):361‐362. [DOI] [PubMed] [Google Scholar]
- 31. UniProt C. Activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014;42(Database issue):D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inui T, Kawarai T, Fujita K, et al. A new CSF1R mutation presenting with an extensive white matter lesion mimicking primary progressive multiple sclerosis. J Neurol Sci. 2013;334(1‐2):192‐195. [DOI] [PubMed] [Google Scholar]
- 33. Daida K, Nishioka K, Li Y, Nakajima S, Tanaka R, Hattori N. CSF1R mutation p. G589R and the distribution pattern of brain calcification. Intern Med. 2017;56(18):2507‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foulds N, Pengelly RJ, Hammans SR, et al. Adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia caused by a novel R782G mutation in CSF1R. Sci Rep. 2015;15(5):10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci. 2012;318(1‐2):115‐118. [DOI] [PubMed] [Google Scholar]
- 36. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. 2015;114(4):501‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lynch DS, Wade C, Paiva ARBD, et al. Practical approach to the diagnosis of adult‐onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry. 2019;90(5):543‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahmed RM, Murphy E, Davagnanam I, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry. 2014;85(7):770‐781. [DOI] [PubMed] [Google Scholar]
- 40. Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain. 2017;140(5):1204‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wong JC, Chow TW, Hazrati LN. Adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia can present as frontotemporal dementia syndrome. Dement Geriatr Cogn Disord. 2011;32(2):150‐158. [DOI] [PubMed] [Google Scholar]
- 42. Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF‐1 receptor and its ligands in the nervous system. Trends Neurosci. 2016;39(6):378‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ayrignac X, Carra‐Dalliere C, Menjot de Champfleur N, et al. Adult‐onset genetic leukoencephalopathies: a MRI pattern‐based approach in a comprehensive study of 154 patients. Brain. 2015;138(Pt 2):284‐292. [DOI] [PubMed] [Google Scholar]
- 44. Konno T, Yoshida K, Mizuta I, et al. Diagnostic criteria for adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. 2018;25(1):142‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Konno T, Broderick DF, Mezaki N, et al. Diagnostic value of brain calcifications in adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. AJNR Am J Neuroradiol. 2017;38(1):77‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kondo Y, Matsushima A, Nagasaki S, Nakamura K, Sekijima Y, Yoshida K. Factors predictive of the presence of a CSF1R mutation in patients with leukoencephalopathy. Eur J Neurol. 2020;27(2):369‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Codjia P, Ayrignac X, Mochel F, et al. Adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia: an MRI study of 16 French cases. AJNR Am J Neuroradiol. 2018;39(9):1657‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S, Pawate S. Adult‐onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol. 2013;260(2):558‐571. [DOI] [PubMed] [Google Scholar]
- 49. Tipton PW, Kenney‐Jung D, Rush BK, et al. Treatment of CSF1R‐related leukoencephalopathy: breaking new ground. Mov Disord. 2021. 10.1002/mds.28734 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes in the targeted resequencing panel.