

Abstract
Objective
To define the transcriptomic changes responsible for the histologic alterations in skeletal muscle and their progression in collagen VI‐related muscular dystrophy (COL6‐RD).
Methods
COL6‐RD patient muscle biopsies were stratified into three groups based on the overall level of pathologic severity considering degrees of fibrosis, muscle fiber atrophy, and fatty replacement of muscle tissue. Using microarray and RNA‐Seq, we then performed global gene expression profiling on the same muscle biopsies and compared their transcriptome with age‐ and sex‐matched controls.
Results
COL6‐RD muscle biopsy transcriptomes as a group revealed prominent upregulation of muscle extracellular matrix component genes and the downregulation of skeletal muscle and mitochondrion‐specific genes. Upregulation of the TGFβ pathway was the most conspicuous change across all biopsies and was fully evident even in the mildest/earliest histological group. There was no difference in the overall transcriptional signature between the different histologic groups but polyserial analysis identified relative changes along with COL6‐RD histological severity.
Interpretation
Overall, our study establishes the prominent dysregulation of extracellular matrix genes, TGFβ signaling, and its downstream cellular pathways at the transcriptomic level in COL6‐RD muscle.
Introduction
Collagen VI‐related dystrophies (COL6‐RDs) are a form of congenital muscular dystrophy with a phenotypic continuum of variable clinical severity characterized by muscle weakness, large joint contractures, and concurrent distal joint hyperlaxity. 1 , 2 , 3 Similar to the notable variability in severity of COL6‐RD clinical phenotypes, the severity of histologic findings in skeletal muscle in patients with COL6‐RD can also vary widely. Histologic changes limited to myofiber atrophy and fiber type predominance typify the mildly affected samples, while more severely affected muscle biopsies show dystrophic changes, that is, endomysial fibrosis, fatty infiltration, fiber size variability, and myofiber degeneration and regeneration. In addition, histologic severity does not always correlate with overall clinical severity. Available serial biopsies in a few patients support the notion that histologic findings evolve from predominant myofiber atrophy without overt fibrosis 4 to dystrophic and fibrotic changes over time (unpublished observations). However, it remains unclear how the skeletal muscle transcriptome, proteome, and structure evolve over time in COL6‐RD.
Collagen type VI is a microfibrillar component of the muscle extracellular matrix (ECM) that is primarily secreted by non‐myogenic, interstitial fibro‐adipogenic progenitors (FAPs) 5 , 6 in the muscle tissue. The three collagen VI alpha chains encoded by COL6A1, COL6A2, and COL6A3, undergo a complex assembly into tetramers of heterotrimeric monomers before secretion into the extracellular space. In the extracellular space, collagen VI tetramers assemble head‐to‐head to form a beaded, microfibrillar polymer with an apparent 100 nm periodicity. 7 Pathogenic variants in one of the three major collagen 6 genes result in COL6‐RD with both dominant and recessive inheritance patterns. 7 , 8 , 9 , 10 , 11 , 12 , 13 Disease‐causing variants result either in the absence or mislocalization of collagen VI in the muscle ECM. How collagen VI loss or mislocalization in the muscle ECM results in muscle weakness, atrophy, and dystrophic change is not fully understood.
Both RNA‐Seq and microarray‐based gene expression profiling have been used to shed light on the molecular changes in a limited number of COL6‐RD patient muscle biopsies and patient‐derived fibroblasts with somewhat incongruous results. 14 , 15 , 16 , 17 To complement and improve on prior studies, we used a muscle transcriptome‐wide approach in a relatively large cohort of COL6‐RD patients (n = 22) compared to age‐matched controls (n = 14). To circumvent the limitations imposed by the variable disease and histologic severity in COL6‐RD, we first stratified the COL6‐RD muscle biopsies into three groups based on the subjective severity of histologic features, for example, myofiber size, fibrosis, and overall appearance. We then comprehensively characterized and compared the gene expression profiles by both microarray and RNA‐Seq.
Materials and Methods
Patients and muscle biopsy samples
Ethical approval for human subject research included in this study was obtained from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NINDS) Institutional Review Board for this study, protocol 12‐N‐0095. Subjects (and/or parents, for minors under the age of 18 years) who agreed to participate provided informed consent (and/or assent if applicable) to provide access to their medical records.
Muscle biopsies used in this study were obtained as part of standard clinical care and excess material was used for the research studies. For COL6‐RD patients, the variants in collagen 6 genes and immunostaining for collagen VI in muscle and fibroblasts were used for confirmation of the clinical diagnosis (Table S1). Two individuals (UC106 and UC158) had equivocal confirmatory testing and were also outliers in the microarray dataset (see microarray in Materials and Methods) and thus were not included in data analysis. The control muscle biopsies were obtained from individuals who underwent muscle biopsies as part of their clinical care and were chosen from individuals who had no clinical or pathological suspicion for muscle disease after their diagnostic workup was completed. The control muscle biopsies were all obtained from the quadriceps muscle (Table S2).
Availability of data and materials
The datasets generated and/or analyzed during this study are available in the NCBI repository, https://www.ncbi.nlm.nih.gov/geo. The RNA‐Seq data generated as part of this analysis has been deposited in the NCBI Short Read Archive (SRP117086) and NCBI Gene Expression Omnibus (GSE103975). The microarray data generated as part of this analysis have been deposited in the NCBI Gene Expression Omnibus (GSE103972).
Microarray analysis
Coding RNA (cRNA) libraries were prepared according to Illumina protocols (Illumina, Inc, San Diego, CA). For RNA labeling, 500 ng of total RNA was hybridized to the HumanHT‐12 v4.0 Expression BeadChip. After washing and staining, BeadChips were scanned using the Illumina BeadArray Reader and the Illumina GenomeStudio Gene Expression Module was used to generate summarized expression measurements. The expression data were then imported into R (http://cran.r‐project.org/), Log2 transformed, quantile normalized, quality inspected via Tukey box plot, covariance‐based principal component analysis (PCA) scatter plot, and correlation‐based heatmap. Post quality inspection, two samples were removed as outliers (UC106 and UC158) which belonged to patients with variants of uncertain significance in collagen VI genes and an equivocal molecular diagnosis of COL6‐RD. After outlier removal, quantile normalization was repeated, mean expression and coefficient of variation (CV) were calculated for each gene per group and modeled using locally weighted scatter plot smoothing (CV ˜mean). The resulting model fits were used to define system noise as the mean expression value across groups at which the linear relationship between CV (i.e., noise) and mean expression (i.e., signal) is grossly lost. Genes that did not have at least one normalized value (Quantile(Log2(RPKM+2))) greater than system noise were discarded as noise‐biased. The normalized values for remaining genes that were less than system noise were floored to equal system noise. To identify differences in gene expression between COL6‐RD samples and control samples, the Welch‐modified t‐test was applied under Benjamin–Hochberg (BH) false‐discovery rate (FDR) multiple comparison correction (MCC) condition. Genes with a corrected p < 0.05 and an absolute linear fold change ≥1.5× between groups were considered to be differentially expressed. Annotations for the genes were obtained from IPA (http://www.ingenuity.com) which was also used for further analysis. Comparison and intersection of results with those obtained by RNA‐Seq analysis were accomplished using the IPA‐assigned gene symbol.
RNA‐Seq analysis
cDNA libraries were prepared according to Illumina protocols (Illumina, Inc, San Diego, CA) and sequenced by Beckman Coulter Genomics (Danvers, Massachusetts). Briefly, mRNA libraries were constructed from 150 ng of total RNA using the Illumina TruSeq RNA Sample Prep Kits, version 2. The cDNA was fragmented using a Covaris E210 and amplified using 12–15 cycles. Unique barcode adapters were applied to each library. Equal volumes of individual libraries were pooled and run on a MiSeq. The libraries were then repooled based on the MiSeq demultiplexing results and sequenced on a HiSeq 2000 with version 3 flow cells and sequencing reagents. On average, 69 million 100‐base read pairs were generated for each individual library. Quality inspection of the reads showed an expected per‐sample GC content peak around 60%, ambiguity base content at nearly 0%, and average PHRED score near 40 across all the samples, indicative of high quality of the sequencing. Data were processed using RTA 1.13.48 and deplexed via CASAVA 1.8.2. After deplexing, resulting fastq files were clipped to remove 3’ occurring adaptor sequences using the FASTQ/A Clipper tool (http://hannonlab.cshl.edu). Adaptor‐trimmed fastq files were then imported into the CLCbio Genomics Workbench (www.clcbio.com) for quality inspection, quality trimming, filtering, and mapping. Per quality trimming, 15 bp from the 5’ end of each read for each sample was globally removed along with 1bp from the 3’ end. Bases with a call accuracy <95% were also removed. Per filtering, read pairs having more than two ambiguities in at least one read were discarded along with pairs <15 bp in length. Filtering and trimming of the reads were uniform throughout the samples and, most importantly, preserved 70%–80% of the total reads sequenced. Per mapping, the RNA‐Seq tool was used under default parameters to align reads in pairs to the human genome (hg19). Ninety nine percent of the post‐trimmed and filtered reads were successfully mapped to the human genome.
Post mapping, the number of aligned reads falling in each annotated gene were enumerated and converted into number of Reads Per Kilobase of transcript Million mapped reads (RPKM). RPKM values were then imported into R (http://www.r‐project.org/), pedestalled by 2 (RPKM + 2), log (base 2) transformed (Log2(RPKM + 2)), and filtered to remove genes not having a transformed value >1 for at least one sample. Post filtering, transformed values for remaining genes were quantile normalized (Quantile(Log2(RPKM + 2))) and quality inspected via Tukey box plot, covariance‐based principal component analysis (PCA) scatter plot, and correlation‐based heatmap. Noise modeling and statistical analysis were performed similar to the microarray dataset analysis and genes with a corrected p < 0.05 and an absolute fold change ≥1.5× were annotated (using IPA) and analyzed further as described. Approximately 30% of the total read depth was used to sequence just 16 genes (mainly myosins and mitochondrial genes) that were highly expressed but not differentially regulated in COL6‐RD. This, in effect, reduced the reading depth available to sequence less abundant genes and in part explains why we identified a higher total number of differentially expressed genes by microarray compared to RNA‐Seq.
See Supplementary text (Data S1) for additional description of the methods used in this study.
Results
Muscle biopsy samples and histological severity stratification
Skeletal muscle biopsies of patients with a diagnosis of COL6‐RD were included in this study (Table S1). The majority of the patients were children at the time of the muscle biopsy (mean age ± SD = 7.6 ± 12.4; male/female = 16/11). The biopsy slides were evaluated by a physician trained in neuromuscular pathology and subjectively stratified into three histologic groups of increasing severity: COL6‐RD‐1 (mild), COL6‐RD‐2 (moderate), and COL6‐RD‐3 (severe) (Fig. 1A). To validate this classification approach, we performed an unbiased, automated quantification of myofiber atrophy, fiber size variability, and fibrosis on a subset of the samples in each stratified group, which confirmed a progressive increase in fiber size variability (assessed by coefficient of variability, normal <250 18 ) and fibrosis, in correlation with increasing histologic severity (Fig. 1B–D) (see Data S1). Even though the ideal control group would include muscle biopsies of age‐ and sex‐matched patients with other muscular dystrophies, due to the rarity of these samples in children, we were unable to use the ideal disease control samples for comparison analysis. Thus, we focused our analysis on comparing control biopsy samples of age‐ and sex‐matched controls (mean age ± SD = 8.4 ± 4.7; male/female = 8/6) who had undergone diagnostic muscle biopsies that were deemed to be normal by the interpreting pathologist (Table S2).
Figure 1.
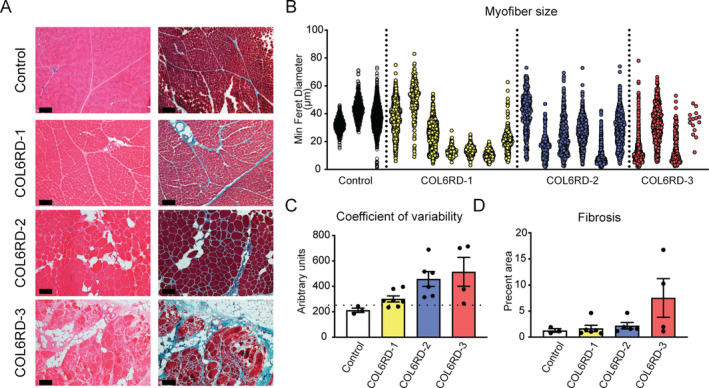
Muscle histologic group classification and morphometry. (A) Representative hematoxylin & eosin and Gömöri trichrome staining of frozen muscle sections used to qualitatively stratify the biopsies into three histologic severity groups: COL6‐RD‐1, COL6‐RD‐2, and COL6‐RD‐3. (B–D) Morphometric quantification of fiber size (B), fiber size variability (C), and fibrosis (D) of a subset of biopsies confirm the expected trends of histologic severity and qualitative classification. Note the severe myofiber atrophy in COL6‐RD muscle biopsies (B). The dotted line in panel C indicates the cutoff for normal coefficient of variability (250). The error bars represent standard error of the mean. Scale bar = 50 μm.
Comparison of RNA‐Seq and microarray expression profiling
We performed both microarray‐based gene expression profiling and RNA‐Seq to compare the transcriptome of the COL6‐RD muscle biopsies to age‐matched controls (Microarray: control n = 14, COL6‐RD n = 20; RNA‐Seq: control n = 14, COL6‐RD n = 22). Microarray analysis identified 648 differentially regulated genes (n = 394 upregulated and n = 254 downregulated). In the RNA‐Seq dataset, a total of 248 genes were differentially expressed (n = 169 upregulated and n = 79 downregulated) (Fig. 2A). There was a strong concordance between microarray and RNA‐Seq in direction (R 2= 1) and magnitude of fold change (R 2 = 0.71) in differentially expressed transcripts identified by both platforms (n = 138) (Fig. 2B). In addition, we independently performed bioinformatic analysis using ingenuity pathway analysis (IPA) on the results obtained from either platform. Microarray identified 325 and RNA‐Seq identified 268 canonical pathways (overlap rate 59.2%; 220 of 371 unique pathways). None of the pathways exclusively identified by one platform reached statistical significance (p < 0.05). Thus, to facilitate our discovery‐based approach, we used a combined list of differentially expressed genes by at least one platform for the remainder of data analysis. For genes detected by both methods, the higher absolute value of the fold change was used. Hierarchical heatmaps and PCA of either method clearly differentiated controls and COL6‐RD muscle biopsies (Fig. 2C and D), but no distinct differences between the different COL6‐RD histologic groups were identified. Consequently, we focused our analysis on all COL6‐RD patients as a single group compared to controls.
Figure 2.
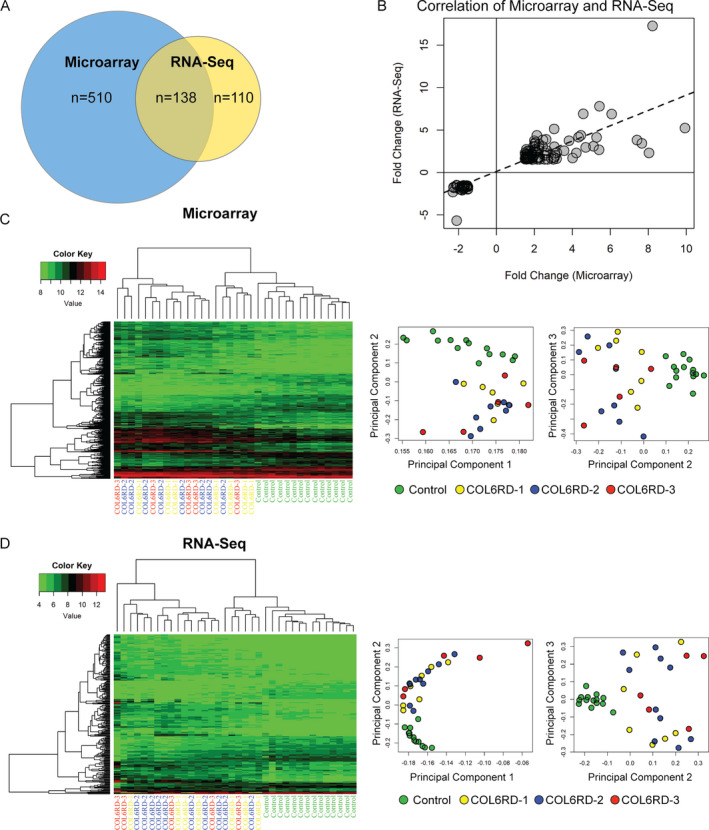
Summary of microarray and RNA‐Seq data. (A) Comparison of the number of differentially expressed genes (p‐value <0.05; fold change magnitude ≥ 1.5) detected by microarray and RNA‐Seq. (B) Correlation of fold change in expression between differentially regulated genes detected by microarray and RNA‐Seq. There was high concordance of direction (R 2 = 1) and magnitude (R 2 = 0.71) of fold change. Hierarchical heatmap and principle component analysis of microarray (C) and RNA‐Seq (D) gene expression profiling. Control samples cluster together and segregate from the COL6‐RD samples, while there is no clear clustering of the COL6‐RD samples by histology severity group.
Gene ontology analysis, functional annotation, and individual genes
We used the Database for Annotation, Visualization, and Integrated Discovery (DAVID) 19 , 20 for gene ontology analysis of the differentially regulated genes (n = 758) in our dataset. Ranked by p‐value, extracellular matrix‐related terms were the most prominent finding (Fig. 3A; Table S3), driven in large part by upregulation of genes that encode fibril‐forming collagens, such as COL1A1 (fold change = 7.79; p = 8.41E‐07), COL1A2 (fold change = 6.87; p = 6.55E‐08), and COL3A1 (fold change = 6.90; p = 4.66E‐07) and the ECM proteoglycans BGN (fold change = 4.33; p = 3.36E‐07), DCN (fold change = 3.35; p‐value = 4.10E‐08), and LUM (fold change = 4.44; p = 2.56E‐06). In contrast, gene ontology analysis of the downregulated genes identified several terms related to myofibrils (driven in part by MYH1 [fold change = −5.69; p = 7.23E‐4], MYL1 [fold change = −2.57; p = 5.89E‐05], TTN [fold change = −2.07; p = 0.002], and MYLK2 [fold change = −2.08; p = 0.007]) as well as to mitochondria (driven in part by ATP5D [fold change = −1.53; p = 4.02E‐4], ATP5L [fold change = −1.51; p = 1.93E‐4], and PTGES2 [fold change = 1.66; p = 1.69E‐4]) (Fig. 3B; Table S3).
Figure 3.
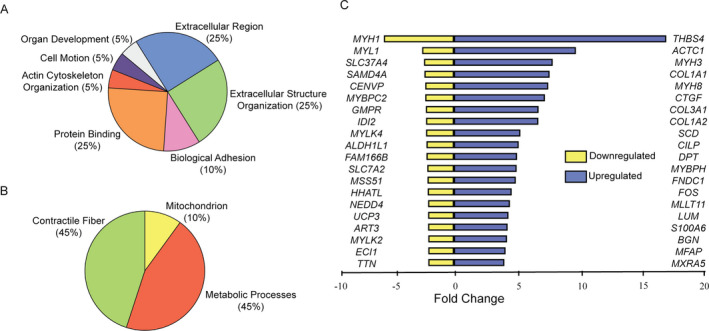
Gene Ontology functional categories most represented among the upregulated genes (A) and the downregulated genes (B) in COL6‐RD skeletal muscle biopsies. (C) A list of top 20 individual upregulated and downregulated genes. The scale represents the linear fold change.
Cellular function analysis of the differentially regulated genes using IPA was enriched in annotations relating to cell movement in COL6‐RD muscle biopsies (Table S4). This was again largely driven by the predominance of upregulated ECM genes implicated in these functions. The downregulated cellular function annotations were far fewer and related to cell metabolism (Table S4).
At the individual gene level, THBS4 (encoding thrombospondin‐4, a matrix‐associated protein) had the greatest difference between COL6‐RD and control muscle biopsies (fold change = 17.28; corrected p‐value = 2.59E‐09). Out of the 20 most highly upregulated genes in the combined microarray/RNA‐Seq list, two were involved in cartilage and bone formation, (CILP and LUM), and four were associated with connective tissue and fibrosis, including CTGF, COL3A1, COL1A1, and COL1A2 (Fig. 3C). The proteoglycans decorin (fold change = 3.35; p‐value = 4.10E‐08) and biglycan (fold change = 4.33; p‐value = 3.36E‐07) were also significantly upregulated in the COL6‐RD samples.
Upstream regulator analysis
To assess the dataset at a more global level and to predict cellular pathways that orchestrate the differentially expressed gene signatures in COL6‐RD skeletal muscle, we used the upstream regulator analysis tool in the IPA platform (Table 1). When sorted by z‐score, TGFβ signaling pathway was by far the most activated upstream regulator (p‐value = 6.48E‐18; z‐score = 5.627) which was driven by a robust upregulation of its downstream effector genes. Upstream regulator analysis also found downstream targets of TGFβ that are in turn regulators of distinct downstream pathways, such as CTGF (p‐value = 1.77E‐05; z‐score = 2.402), SMAD1 (p‐value = 5.14E‐05; z‐score = 1.982), SMAD4 (p‐value = 3.25E‐03; z‐score = 1.866), and SMAD3 (p‐value = 9.13E‐12; z‐score = 1.652). This consistent and multilevel complexity suggest that a TGFβ‐driven cascade of gene regulatory events may be involved in the pathogenesis of COL6‐RD. Consistent with this finding, TGFβ‐related genes were consistently found to be dysregulated in COL6‐RD samples, regardless of histologic severity (Fig. 4).
Table 1.
Upstream regulator analysis results of the differentially regulated genes obtained from the combination of microarray and RNA‐Seq data.
| Upstream regulator | p‐value | Activation z‐score | Upstream regulator | p‐value | Activation z‐score |
|---|---|---|---|---|---|
| TGFB1 | 6.48E‐18 | 5.627 | HDAC6 | 7.63E‐07 | 2.157 |
| ERK | 6.21E‐06 | 3.553 | PGR | 1.30E‐08 | 2.116 |
| ERBB2 | 1.78E‐07 | 3.313 | EGFR | 1.71E‐04 | 2.109 |
| SYVN1 | 2.29E‐07 | 3.298 | IL5 | 9.10E‐02 | 2 |
| P38 MAPK | 3.25E‐07 | 3.146 | RLIM | 1.75E‐04 | 2 |
| PDGF BB | 1.41E‐12 | 3.112 | SMARCD3 | 1.59E‐03 | 2 |
| JNK | 5.30E‐04 | 2.765 | MAP3K14 | 1.87E‐02 | 2 |
| GLI1 | 3.24E‐08 | 2.746 | TAZ | 5.14E‐05 | 2 |
| PKC | 2.87E‐04 | 2.574 | NEDD9 | 6.69E‐03 | 2 |
| HIF1A | 4.18E‐10 | 2.552 | RBPJ | 2.00E‐03 | −2 |
| CCL5 | 3.11E‐03 | 2.449 | MAP3K7 | 2.88E‐02 | −2 |
| RELA | 1.11E‐02 | 2.425 | miR‐29b‐3p | 2.38E‐03 | −2.219 |
| PRKCD | 1.35E‐03 | 2.414 | HDAC | 3.57E‐05 | −2.273 |
| CTGF | 1.77E‐05 | 2.402 | COL18A1 | 1.08E‐04 | −2.538 |
| TWIST1 | 3.13E‐04 | 2.236 | WISP2 | 2.91E‐08 | −2.714 |
| EIF2AK2 | 4.87E‐02 | 2.236 | FBN1 | 2.75E‐09 | −2.797 |
| MYB | 1.20E‐04 | 2.215 | FOXA1 | 1.08E‐04 | −2.802 |
| AKT | 3.52E‐03 | 2.213 | IgG | 2.25E‐06 | −3.5 |
| TWIST2 | 1.26E‐04 | 2.207 | MGEA5 | 1.30E‐13 | −3.55 |
| CSF2 | 1.62E‐01 | 2.2 | SPDEF | 6.24E‐14 | −3.935 |
| LDL | 8.56E‐03 | 2.186 | ER | 1.38E‐15 | −4.062 |
The top 30 predicted upstream regulators, sorted by p‐value, are reported in this table.
Figure 4.
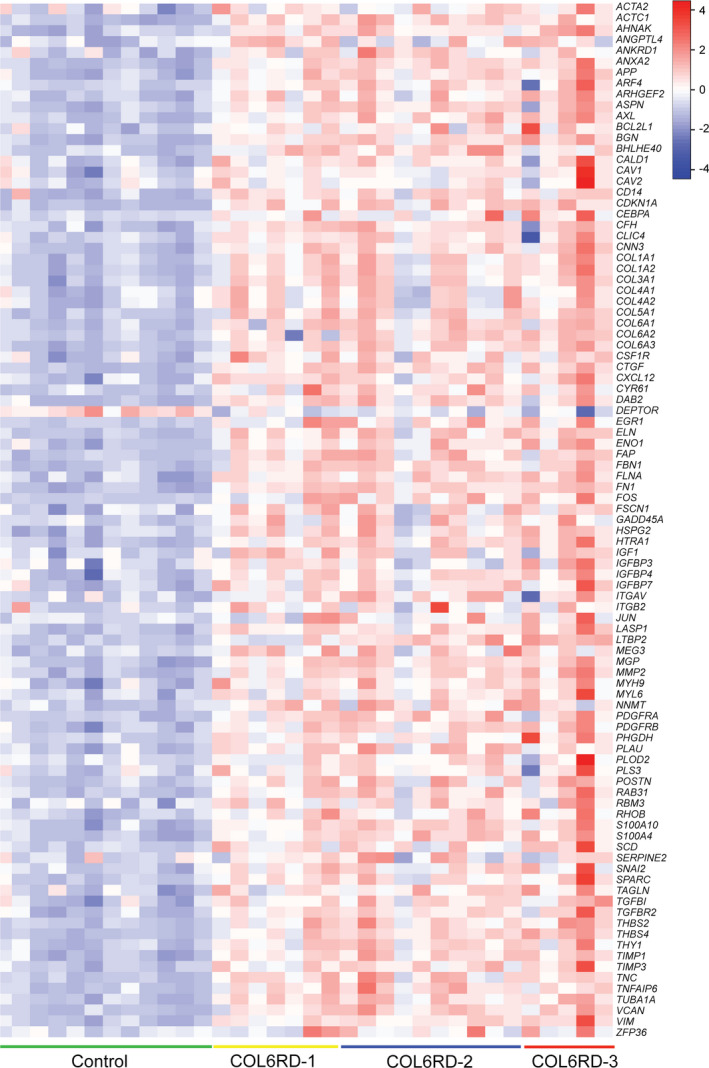
Expression levels of 95 TGFβ‐related genes in the RNAseq dataset are depicted in a heatmap format using row Z‐scores derived from Log2(RPKM + 2) for each sample. Nearly all of these selected genes, with rare exceptions (e.g., DEPTOR), are upregulated in the COL6‐RD samples.
A few pathways were predicted to be inhibited, including estrogen receptor signaling (ESR, p‐value = 1.38E‐15; z‐score = −4.062), the transcription regulator SPDEF (p‐value = 6.24E‐14; z‐score = −3.935), and the growth factor WISP2 (p‐value = 2.91E‐08; z‐score = −2.714).
Polyserial correlation analysis
Polyserial correlation analysis helps to investigate genes that show a consistent behavior (increasing or decreasing expression level) in correlation with another variable (worsening histologic severity). Using this data analysis method, we identified 1154 genes (410 positively correlated and 744 negatively correlated) in correlation with the increasing severity of the histologic stratification (correlation coefficient r ≥ 0.4) (Table S5). However, consistent with our prior results, the majority of the magnitude of differences were noted when comparing controls with any of the COL6‐RD histologic groups (Fig. 5A). Gene ontology and upstream regulator analysis of the list of genes identified by polyserial correlation analysis did not uncover any previously overlooked terms or pathways (Table S5).
Figure 5.
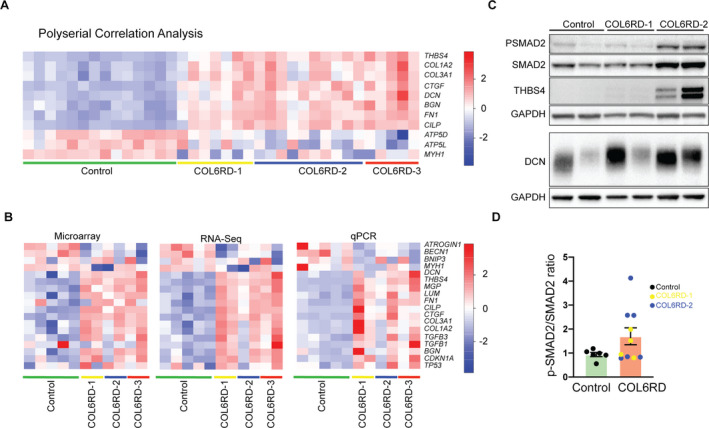
(A) Expression level of selected genes identified by polyserial correlation analysis. Expression levels are depicted in a heatmap format using row Z‐scores derived from Log2(RPKM + 2) for each gene. RPKM = Reads per kilobase of transcript per million (B) Technical validation of microarray and RNA‐Seq data by qPCR on 17 genes performed on a randomly selected subset of controls (n = 5) COL6‐RD (n = 6, two from each histologic severity group) muscle biopsies. The relative expression levels are plotted in a heatmap using row Z‐scores: microarray and RNA‐Seq: Log2(RPKM + 2), qPCR: linear fold change normalized to a control sample). (C) Representative western blot of human muscle biopsy lysates for detection of P‐SMAD2, SMAD2, thrombospondin‐4 (THBS4), and decorin (DCN). (D) Densitometry of p‐SMAD2/SMAD2 ratio in control (n = 6) and COL6‐RD (n = 10) skeletal muscle samples normalized to control samples in each group of analysis. P‐SMAD2/SMAD2 ratio trends higher in COL6‐RD samples with a high degree of variability.
Validation of differentially expressed genes by qRT‐PCR and western blotting
To validate the RNA‐Seq and microarray methodology and findings, we selected 18 target genes (BECN1, CILP, COL1A2, COL3A1, CTGF, DCN, FN1, LUM, MGP, MYH1, CDKN1A, TGFB1,TGFB3, THBS4, BGN, TP53, ATROGIN1, and BNIP3), performed quantitative reverse transcription PCR (qRT‐PCR), and evaluated the gene expression levels across the different methods, that is, microarray, RNA‐Seq, and qRT‐PCR (Fig. 5B). The different methods showed strong concordance; however, qRT‐PCR fold change was almost always more pronounced for the genes assessed.
Validation at the protein level was limited due to scarcity of samples but we confirmed that thrombospondin‐4, with the highest fold change, and decorin, an ECM proteoglycan in the top 20 upregulated genes, were increased in COL6‐RD samples at the protein level, consistent with the transcriptomic data (Fig. 5C). To further evaluate the TGFβ pathway at the protein level, we compared phosphorylated Smad2 (P‐Smad2) to total Smad2 protein levels in muscle biopsies to assess this canonical effector of this pathway. P‐Smad2/Smad2 ratio was variable but trended higher in COL6‐RD biopsies relative to controls, especially in a subset of biopsies with a higher degree of histologic severity (Fig. 5C and D).
Discussion
In this study, we set out to investigate relevant molecular pathways that contribute to the pathogenesis of COL6‐RD. Cellular pathways such as apoptosis and ultrastructural abnormalities of mitochondria have been reported in collagen VI‐deficient mice (Col6a1 −/−) and myoblasts derived from muscle biopsies of COL6‐RD patients. 21 , 22 Dysregulation of the autophagy pathway is suggested to be the underlying cause for both the mitochondrial alterations and increased apoptosis seen in collagen VI deficiency. 23 In a more recent study, dysregulation of the circadian rhythm genes was found in correlation with defective skeletal muscle regeneration. 16 Impaired satellite cell self‐renewal has also been reported, in particular relation to changes in stiffness of the satellite cell niche. 24 However, a direct mechanistic link between collagen VI loss from the muscle matrix and downstream cellular pathways is not conclusively identified to date.
In muscular dystrophies, including COL6‐RD, the relative severity of disease manifestations in different individual muscles within the same patient can be heterogeneous 8 , 25 , 26 , 27 and change over time. Hence, when it comes to staging the disease, muscle biopsy specimens are prone to significant variability due to sampling variations. It is reasonable to surmise that the histologic changes observed in a muscle biopsy specimen represent the severity of the underlying pathogenic molecular processes directly in the sampled tissue. Using this rationale, we set out to circumvent the inherent sampling variability in COL6‐RD muscle biopsies by stratifying them into three groups of increasing histologic severity before transcriptomic analysis. We hoped to see correlations between the transcriptomic data and the severity of muscle pathological changes.
We were surprised to find an essentially similar pattern of altered gene expression among the different histologic stages of COL6‐RD. The prominent ECM transcriptomic signature, typically associated with fibrosis, was already apparent in the least severe histologic group (COL6‐RD‐1), which lacked overt fibrosis, thus representing very early stages of muscle remodeling. Extracellular matrix genes were the most prominent group of upregulated genes based on gene ontology analysis (155 of 462 total). Among these genes, fibril‐forming collagens, such as COL1A1, COL1A2, and COL3A1 and the ECM proteoglycans BGN, DCN, and LUM were consistently upregulated. Fewer genes were downregulated than upregulated (296 vs. 462). These genes were predominantly related to myofibrils (e.g., MYH1, MYL1, TTN, and MYLK2) as well as to mitochondria (e.g., ATP5D, ATP5L, and PTGES2).
Remarkably, none of the classical atrophy pathways, including the ubiquitin−proteasome system, autophagy−lysosome system, nor apoptosis stood out as prominently dysregulated in our analysis. In particular, FOXO3 and its mediators, ATROGIN1 and BECN1, responsible for ubiquitination and lysosomal degradation, respectively, were unchanged between COL6‐RD and controls at the transcript level. The apparent discrepancy between differential regulation of BECN1 in our dataset and other reports 23 may reflect the rapid changes in selected transcript levels depending on fasting status at the time of biopsy. Dysregulation of circadian rhythm genes in Col6a1 −/− mouse muscle as well as a small number of COL6‐RD patient muscle biopsies has also been recently reported. 16 However, in our dataset, circadian rhythm or the AKT1 pathway was not identified in the upstream regulator analysis. In another transcriptomic study of muscle biopsies in COL6‐RD (n = 6), activation of an immune response attributed to M2 macrophages was reported. 14 We did not identify a similar activation of immune response in our dataset. There are a number of potential reasons for seeming discrepancies in results obtained by us and other studies. Our data were obtained from the analysis of a comparatively larger set of COL6‐RD patient biopsies and age‐matched controls, providing greater statistical power and making it less prone to sampling bias and statistical drift. 28 However, our cohort was also intentionally more broad in disease severity. In addition, methodologic differences in both transcriptomic approaches and sequencing platforms may account for a portion of the differences noted between the studies. In view of these differences, additional comparative studies are needed to identify common disease mechanism/networks across these different datasets. For example, M2 macrophages identified in another report 14 may be activated by TGFβ, 29 , 30 especially in tissue repair in response to chronic injury, 31 which is entirely consistent with our finding of TGFβ pathway upregulation in COL6‐RD muscle biopsies.
TGFβ and some of its downstream effectors (e.g., ERK, P38 MAPK, and CTGF) were the most prominently dysregulated pathways based on upstream regulator analysis of our dataset (TGFβ; z‐score = 5.627; p‐value = 6.48E‐18) (Table 1, Fig. 4). Increased p‐SMAD2/SMAD2 ratio, the canonical effector in the TGFβ pathway in COL6‐RD muscle biopsies, was also consistent with increased TGFβ activity (Fig. 5C and D). Increased TGFβ activity has been reported in other muscular dystrophies, possibly reflecting the progressive development of fibrosis, a prominent characteristic of late stages of chronic muscle disease in general. 32 , 33 , 34 For example, in Duchenne muscular dystrophy (DMD), ECM‐related genes (COL1A1, COL1A2, COL3A2, and FN1), and TGFB1 are prominently upregulated. However, in DMD, this upregulation appears in later stages of the disease, while inflammatory pathways (e.g., TNFa or NF‐kB) dominate the abnormalities early in the disease course. 33 , 35 , 36 Thus, the prominent upregulation of TGFβ‐regulated genes in COL6‐RD diverges from other muscular dystrophies such as DMD in its early prominence. The significance of this finding remains unclear, and additional studies are necessary to further investigate its mechanistic implications.
A conspicuous finding emerging from our study was the early and prominent upregulation of two small leucine‐rich proteoglycans, decorin (fold change = 3.35; p‐value = 4.10E‐08) and biglycan (fold change = 4.33; p‐value = 3.36E‐07), which was consistent with a prior study of a few COL6‐RD muscle biopsies. 14 Dysregulation of decorin and biglycan has also been reported in other muscular dystrophies such as DMD, LGMD 2A/LGMD R1 (calpainopathy), and Pompe disease, likely reflecting the development of fibrosis and muscle ECM remodeling in these disorders. 35 , 37 , 38 , 39 , 40 Decorin and biglycan are known to directly interact with collagen VI, 41 , 42 and while the functional implications of these interactions are complex, they may involve the regulation of growth factors including TGFβ. Decorin in particular has been shown to inhibit TGFβ activity in skeletal muscle, counteracting the stimulating effect of TGFβ and other members of the TGFβ superfamily (e.g., TGFβ2 and myostatin) on fibroblast proliferation, thus enhancing muscle regeneration and decreasing the amount of fibrosis, both in vitro and in vivo. 43 , 44 Of note, the actual composition of the surrounding ECM plays a crucial role in determining the cellular response to decorin 45 ; therefore, the development of fibrosis over time might in turn modulate decorin–TGFβ interactions and their effects on cells. Biglycan also binds TGFβ with similar affinity to decorin in vitro. 46 However, unlike decorin, biglycan overexpression in vivo does not prevent the pro‐fibrotic effects of TGFβ1. 46 Additional mechanistic studies are necessary to investigate the role of biglycan and decorin in COL6‐RD and their contribution to fibrotic changes in muscle.
Another novel finding in our study was the upregulation of two cartilage‐specific genes, CILP (fold change = 5.27; p‐value = 1.31E‐09) and MGP (fold change = 3.83; p‐value = 1.12E‐07), suggestive of transdifferentiation of muscle‐resident cells to chondrocytes or adipocytes. Under normal conditions, cartilage intermediate layer protein (CILP) is exclusively expressed in articular cartilage and is synthesized and deposited into the ECM by chondrocytes. 47 , 48 CILP protein has been implicated in calcium crystal deposition in articular cartilage in patients with osteoarthritis and rheumatoid arthritis during aging and in response to increased concentrations of TGFβ. 49 , 50 , 51 Matrix Gla Protein (MGP) is expressed in a broad range of tissues and cells, 52 , 53 but accumulates to significant levels only in cartilage, bone, and calcified cartilage. 52 MGP‐deficient mice show extensive calcification of the aorta and articular cartilage, 54 , 55 suggesting that MGP inhibits calcification of soft connective tissues. MGP and CILP thus seem to have opposite effects when secreted in the cartilage ECM, possibly suggesting that they counterbalance each other. CILP transcriptional regulation is mediated by the canonical TGFβ/Smad2/3 pathway via SK1 and S1P3, 56 , 57 , 58 as well as non‐canonical TGFβ signaling pathways involving PI3K, MEK/ERK, and p38. 59 , 60 Taken together, with apparent upregulation of TGFβ in COL6‐RD muscle, the TGFβ‐induced transdifferentiation of muscle‐resident cells may thus contribute to the pathogenesis of COL6‐RD or secondary histologic changes such as fatty replacement and ECM remodeling. Since we did not perform single‐cell or single‐nuclei transcriptome analysis, it remains unclear which cell type in muscle (e.g., myonuclei, FAPs, endothelial cells, tenocytes, inflammatory cells, etc), primarily drives the upregulation in cartilage and adipose transdifferentiation genes. Future studies that use single‐cell/nuclei isolation followed by transcriptome analysis will help to answer these important questions.
THBS4 was the most highly upregulated gene in our dataset (THBS4; fold change = 17.28; p‐value = 2.59E‐09), which is also upregulated in other muscular dystrophies such as in DMD and in dy3k /dy3k mice, a mouse model of LAMA2‐related muscular dystrophy. 35 , 61 Expression of THBS4 is normally restricted to skeletal and cardiac muscle, and the nervous system. In cardiac muscle, thrombospondin‐4 is mainly localized to the perimysium and endomysium and may play a role in mechanosignaling, protecting against cardiac fibrosis. 62 , 63 Thrombospondin‐4 is also part of the endoplasmic reticulum (ER) stress response mediated through ATF6. 64 In skeletal muscle, thrombospondin‐4 is particularly abundant in endomysium and perimysium of oxidative muscles (e.g., soleus), where it is thought to stabilize the muscle membranes, 65 and in tendon, where it is thought to regulate the structural organization of collagen fibrils. 66 The significant upregulation of THBS4 mRNA and protein in our cohort of COL6‐RD patients did not correspond to upregulation of ER stress response genes or proteins (e.g., ATF6 and BiP) perhaps suggesting a more classical role in regulating mechanosignaling in skeletal muscle. Mechanistic studies are necessary to further elucidate the role of thrombospondin‐4 in the pathogenesis of COL6‐RD.
Microarray‐based and RNA‐Seq transcriptomic analyses each have technical advantages and disadvantages. 67 Given our discovery‐based approach, by combining both datasets, we set out to circumvent the limitations of each method. The two methods performed concordantly in our dataset (Fig. 2A), and both were validated by the gold‐standard of qPCR (Fig. 5A). Analysis of the differentially expressed transcripts using each platform independently gave very similar results.
In summary, in this comparatively large transcriptomic study of histologically staged COL6‐RD muscle biopsies, we demonstrate that ECM‐related and TGFβ‐responsive genes are highly upregulated in this disease. Furthermore, this upregulation is established early in the disease process, preceding histologically evident fibrosis. Given the direct role of ECM in regulation of TGFβ activity, we propose that dysregulation of TGFβ may be an early and primary driver of disease in COL6‐RD as a direct result of collagen VI absence or mislocalization in the ECM. The direct mechanistic connection between collagen VI and TGFβ activity regulation is under active investigation, and, if confirmed, would posit a divergent role for the TGFβ pathway compared to what occurs in other muscular dystrophies with secondary engagement of the pathway. Thus, we define the TGFβ signaling pathway as a rational choice for the future exploration of biomarkers and therapeutic targets in COL6‐RD.
Conflict of Interest
The authors declare that they have no conflict of interest.
Authors Contribution
EG, PM, and CGB conceived the project and designed the experiments. JD (clinical and histological classification), AD and MRC (microarray), and EG (all of the remainder) performed the experiments. MS characterized and provided control biopsy samples for the study. EG, PM, KRJ, PU, YL, YH, and CGB analyzed the data. EG and PM drafted the manuscript and are co‐first authors. All authors contributed to writing of the final version of the manuscript and read and approved its publication.
Supporting information
Table S1. The list of COL6‐RD biopsy samples used for analysis, corresponding genetic variants, age at the time of biopsy, and sex.
Table S2. The list of control biopsy samples used for analysis, corresponding age at the time of biopsy, sex, and pathologist's interpretation.
Table S3. Gene ontology analysis using DAVID database.
Table S4. Cellular function annotations using ingenuity pathway analysis and presented classified by p‐value and activation z‐score.
Table S5. Results from the polyserial correlation analysis, the corresponding gene ontology analysis for positively and negatively regulated genes, and ingenuity pathway upstream regulator analysis.
Data S1. Supplemental methods of transcriptomic data analysis, histology, automated image analysis, RNA isolation, RT‐qPCR, immunoblotting, and the list of primers used in this study.
Acknowledgments
This study was supported in part by the Intramural Research Program of National Institutes of Health, National Institute of Aging, and by funds of the National Institutes of Neurological Disorders and Stroke intramural research program to CGB. The image analysis was performed by CytoInformatics.com, a National Institutes of Health funded small business focused on data/image analysis. CytoInformatics is funded in part by the grant #9R42AG055375‐03 from the National Institutes of Health.
Funding information
This study was supported in part by the Intramural Research Program of National Institutes of Health, National Institute of Aging, and by funds of the National Institutes of Neurological Disorders and Stroke intramural research program. The image analysis was performed by CytoInformatics.com, a National Institutes of Health funded small business focused on data/image analysis. CytoInformatics is funded in part by the grant #9R42AG055375‐03 from the National Institutes of Health.
Funding Statement
This work was funded by Intramural Research Program of National Institutes of Health, National Institute of Aging; National Institutes of Neurological Disorders and Stroke intramural research program; National Institutes of Health grant 9R42AG055375‐03.
References
- 1. Camacho Vanegas O, Bertini E, Zhang R‐Z, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA. 2001;98(13):7516‐7521. doi: 10.1073/pnas.121027598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonnemann CG. The collagen VI‐related myopathies: muscle meets its matrix. Nat Rev Neurol. 2011;7(7):379‐390. doi: 10.1038/nrneurol.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foley AR, Mohassel P, Donkervoort S, Bolduc V, Bonnemann CG. Collagen VI‐related dystrophies. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle, WA: University of Washington;1993–2021. [Google Scholar]
- 4. Schessl J, Goemans NM, Magold AI, et al. Predominant fiber atrophy and fiber type disproportion in early ullrich disease. Muscle Nerve. 2008;38(3):1184‐1191. doi: 10.1002/mus.21088 [DOI] [PubMed] [Google Scholar]
- 5. Zou Y, Zhang RZ, Sabatelli P, Chu ML, Bonnemann CG. Muscle interstitial fibroblasts are the main source of collagen VI synthesis in skeletal muscle: implications for congenital muscular dystrophy types Ullrich and Bethlem. J Neuropathol Exp Neurol. 2008;67(2):144‐154. doi: 10.1097/nen.0b013e3181634ef7 [DOI] [PubMed] [Google Scholar]
- 6. Rubenstein AB, Smith GR, Raue U, et al. Single‐cell transcriptional profiles in human skeletal muscle. Sci Rep. 2020;10(1):229. doi: 10.1038/s41598-019-57110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lamande SR, Bateman JF. Collagen VI disorders: insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018;72:348‐367. doi: 10.1016/j.matbio.2017.12.008 [DOI] [PubMed] [Google Scholar]
- 8. Demir E, Sabatelli P, Allamand V, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet. 2002;70(6):1446‐1458. doi: 10.1086/340608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in‐frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73(2):355‐369. doi: 10.1086/377107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14(2):279‐293. doi: 10.1093/hmg/ddi025 [DOI] [PubMed] [Google Scholar]
- 11. Lampe AK, Zou Y, Sudano D, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat. 2008;29(6):809‐822. doi: 10.1002/humu.20704 [DOI] [PubMed] [Google Scholar]
- 12. Butterfield RJ, Foley AR, Dastgir J, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 2013;34(11):1558‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohassel P, Foley AR, Bonnemann CG. Extracellular matrix‐driven congenital muscular dystrophies. Matrix Biol. 2018;72:188‐204. doi: 10.1016/j.matbio.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 14. Paco S, Kalko SG, Jou C, et al. Gene expression profiling identifies molecular pathways associated with collagen VI deficiency and provides novel therapeutic targets. PLoS One. 2013;8(10):e77430. doi: 10.1371/journal.pone.0077430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paco S, Casserras T, Rodríguez MA, et al. Transcriptome analysis of ullrich congenital muscular dystrophy fibroblasts reveals a disease extracellular matrix signature and key molecular regulators. PLoS One. 2015;10(12):e0145107. doi: 10.1371/journal.pone.0145107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scotton C, Bovolenta M, Schwartz E, et al. Deep RNA profiling identified CLOCK and molecular clock genes as pathophysiological signatures in collagen VI myopathy. J Cell Sci. 2016;129(8):1671‐1684. doi: 10.1242/jcs.175927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Butterfield RJ, Dunn DM, Hu Y, Johnson K, Bonnemann CG, Weiss RB. Transcriptome profiling identifies regulators of pathogenesis in collagen VI related muscular dystrophy. PLoS One. 2017;12(12):e0189664. doi: 10.1371/journal.pone.0189664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oertel G. Morphometric analysis of normal skeletal muscles in infancy, childhood and adolescence. An autopsy study. J Neurol Sci. 1988;88(1‐3):303‐313. doi: 10.1016/0022-510X(88)90227-4 [DOI] [PubMed] [Google Scholar]
- 19. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44‐57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 20. da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1‐13. doi: 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irwin WA, Bergamin N, Sabatelli P, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35(4):367‐371. doi: 10.1038/ng1270 [DOI] [PubMed] [Google Scholar]
- 22. Merlini L, Angelin A, Tiepolo T, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci USA. 2008;105(13):5225‐5229. doi: 10.1073/pnas.0800962105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16(11):1313‐1320. doi: 10.1038/nm.2247 [DOI] [PubMed] [Google Scholar]
- 24. Urciuolo A, Quarta M, Morbidoni V, et al. Collagen VI regulates satellite cell self‐renewal and muscle regeneration. Nat Commun. 2013;4:1964. doi: 10.1038/ncomms2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mercuri E, Cini C, Counsell S, et al. Muscle MRI findings in a three‐generation family affected by Bethlem myopathy. Eur J Paediatr Neurol. 2002;6(6):309‐314. doi: 10.1053/ejpn.2002.0618 [DOI] [PubMed] [Google Scholar]
- 26. Mercuri E, Cini C, Pichiecchio A, et al. Muscle magnetic resonance imaging in patients with congenital muscular dystrophy and Ullrich phenotype. Neuromuscul Disord. 2003;13(7‐8):554‐558. doi: 10.1016/S0960-8966(03)00091-9 [DOI] [PubMed] [Google Scholar]
- 27. Mercuri E, Lampe A, Allsop J, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303‐310. doi: 10.1016/j.nmd.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 28. Alberti A, Belser C, Engelen S, et al. Comparison of library preparation methods reveals their impact on interpretation of metatranscriptomic data. BMC Genom. 2014;20(15):912. doi: 10.1186/1471-2164-15-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451‐483. doi: 10.1146/annurev.immunol.021908.132532 [DOI] [PubMed] [Google Scholar]
- 30. Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. 2013;120:163‐184. [DOI] [PubMed] [Google Scholar]
- 31. Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Adv Wound Care. 2012;1(1):10‐16. doi: 10.1089/wound.2011.0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. 1996;107(3):404‐411. doi: 10.1111/1523-1747.ep12363389 [DOI] [PubMed] [Google Scholar]
- 33. Chen YW, Nagaraju K, Bakay M, et al. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65(6):826‐834. [DOI] [PubMed] [Google Scholar]
- 34. Kharraz Y, Guerra J, Pessina P, Serrano AL, Munoz‐Canoves P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int. 2014;2014:965631. doi: 10.1155/2014/965631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haslett JN, Sanoudou D, Kho AT, et al. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci USA. 2002;99(23):15000‐15005. doi: 10.1073/pnas.192571199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pescatori M, Broccolini A, Minetti C, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21(4):1210‐1226. doi: 10.1096/fj.06-7285com [DOI] [PubMed] [Google Scholar]
- 37. Bakay M, Zhao P, Chen J, Hoffman EP. A web‐accessible complete transcriptome of normal human and DMD muscle. Neuromuscul Disord. 2002;12(Suppl 1):S125‐S141. doi: 10.1016/S0960-8966(02)00093-7 [DOI] [PubMed] [Google Scholar]
- 38. Zanotti S, Negri T, Cappelletti C, et al. Decorin and biglycan expression is differentially altered in several muscular dystrophies. Brain. 2005;128(Pt 11):2546‐2555. doi: 10.1093/brain/awh635 [DOI] [PubMed] [Google Scholar]
- 39. Sáenz A, Azpitarte M, Armañanzas R, et al. Gene expression profiling in limb‐girdle muscular dystrophy 2A. PLoS One. 2008;3(11):e3750. doi: 10.1371/journal.pone.0003750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Palermo AT, Palmer RE, So KS, et al. Transcriptional response to GAA deficiency (Pompe disease) in infantile‐onset patients. Mol Genet Metab. 2012;106(3):287‐300. doi: 10.1016/j.ymgme.2012.05.004 [DOI] [PubMed] [Google Scholar]
- 41. Wiberg C, Hedbom E, Khairullina A, et al. Biglycan and decorin bind close to the n‐terminal region of the collagen VI triple helix. J Biol Chem. 2001;276(22):18947‐18952. doi: 10.1074/jbc.M100625200 [DOI] [PubMed] [Google Scholar]
- 42. Rafii MS, Hagiwara H, Mercado ML, et al. Biglycan binds to alpha‐ and gamma‐sarcoglycan and regulates their expression during development. J Cell Physiol. 2006;209(2):439‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukushima K, Badlani N, Usas A, Riano F, Fu F, Huard J. The use of an antifibrosis agent to improve muscle recovery after laceration. Am J Sports Med. 2001;29(4):394‐402. doi: 10.1177/03635465010290040201 [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Li J, Zhu J, et al. Decorin gene transfer promotes muscle cell differentiation and muscle regeneration. Mol Ther. 2007;15(9):1616‐1622. doi: 10.1038/sj.mt.6300250 [DOI] [PubMed] [Google Scholar]
- 45. Goetsch KP, Niesler CU. The extracellular matrix regulates the effect of decorin and transforming growth factor beta‐2 (TGF‐beta2) on myoblast migration. Biochem Biophys Res Commun. 2016;479(2):351‐357. [DOI] [PubMed] [Google Scholar]
- 46. Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF‐beta‐mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1327‐L1334. [DOI] [PubMed] [Google Scholar]
- 47. Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU. In vitro chondrogenesis of bone marrow‐derived mesenchymal progenitor cells. Exp Cell Res. 1998;238(1):265‐272. doi: 10.1006/excr.1997.3858 [DOI] [PubMed] [Google Scholar]
- 48. Palmer GD, Steinert A, Pascher A, et al. Gene‐induced chondrogenesis of primary mesenchymal stem cells in vitro. Mol Ther. 2005;12(2):219‐228. doi: 10.1016/j.ymthe.2005.03.024 [DOI] [PubMed] [Google Scholar]
- 49. Lorenzo P, Bayliss MT, Heinegard D. A novel cartilage protein (CILP) present in the mid‐zone of human articular cartilage increases with age. J Biol Chem. 1998;273(36):23463‐23468. doi: 10.1074/jbc.273.36.23463 [DOI] [PubMed] [Google Scholar]
- 50. Hirose J, Masuda I, Ryan LM. Expression of cartilage intermediate layer protein/nucleotide pyrophosphohydrolase parallels the production of extracellular inorganic pyrophosphate in response to growth factors and with aging. Arthritis Rheum. 2000;43(12):2703‐2711. doi: [DOI] [PubMed] [Google Scholar]
- 51. Mori M, Nakajima M, Mikami Y, et al. Transcriptional regulation of the cartilage intermediate layer protein (CILP) gene. Biochem Biophys Res Commun. 2006;341(1):121‐127. doi: 10.1016/j.bbrc.2005.12.159 [DOI] [PubMed] [Google Scholar]
- 52. Cancela L, Hsieh CL, Francke U, Price PA. Molecular structure, chromosome assignment, and promoter organization of the human matrix Gla protein gene. J Biol Chem. 1990;265(25):15040‐15048. doi: 10.1016/S0021-9258(18)77221-9 [DOI] [PubMed] [Google Scholar]
- 53. Fraser JD, Price PA. Lung, heart, and kidney express high levels of mRNA for the vitamin K‐dependent matrix Gla protein. Implications for the possible functions of matrix Gla protein and for the tissue distribution of the gamma‐carboxylase. J Biol Chem. 1988;263(23):11033‐11036. doi: 10.1016/S0021-9258(18)37912-2 [DOI] [PubMed] [Google Scholar]
- 54. Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386(6620):78‐81. doi: 10.1038/386078a0 [DOI] [PubMed] [Google Scholar]
- 55. Gheduzzi D, Boraldi F, Annovi G, et al. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab Investig. 2007;87(10):998‐1008. doi: 10.1038/labinvest.3700667 [DOI] [PubMed] [Google Scholar]
- 56. Wu Y, Zhang X, Salmon M, Lin X, Zehner ZE. TGFbeta1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP‐1 and Sp1 family members. Biochem Biophys Acta. 2007;1773(3):427‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cencetti F, Bernacchioni C, Nincheri P, Donati C, Bruni P. Transforming growth factor‐beta1 induces transdifferentiation of myoblasts into myofibroblasts via up‐regulation of sphingosine kinase‐1/S1P3 axis. Mol Biol Cell. 2010;21(6):1111‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bruno G, Cencetti F, Pertici I, et al. CTGF/CCN2 exerts profibrotic action in myoblasts via the up‐regulation of sphingosine kinase‐1/S1P3 signaling axis: implications in the action mechanism of TGFbeta. Biochem Biophys Acta. 2015;1851(2):194‐202. [DOI] [PubMed] [Google Scholar]
- 59. Agley CC, Rowlerson AM, Velloso CP, Lazarus NR, Harridge SD. Human skeletal muscle fibroblasts, but not myogenic cells, readily undergo adipogenic differentiation. J Cell Sci. 2013;126(Pt 24):5610‐5625. doi: 10.1242/jcs.132563 [DOI] [PubMed] [Google Scholar]
- 60. Alexeev V, Arita M, Donahue A, Bonaldo P, Chu ML, Igoucheva O. Human adipose‐derived stem cell transplantation as a potential therapy for collagen VI‐related congenital muscular dystrophy. Stem Cell Res Ther. 2014;5(1):21. doi: 10.1186/scrt411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hager M, Bigotti MG, Meszaros R, et al. Cib2 binds integrin alpha7Bbeta1D and is reduced in laminin alpha2 chain‐deficient muscular dystrophy. J Biol Chem. 2008;283(36):24760‐24769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frolova EG, Sopko N, Blech L, et al. Thrombospondin‐4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 2012;26(6):2363‐2373. doi: 10.1096/fj.11-190728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frolova EG, Drazba J, Krukovets I, et al. Control of organization and function of muscle and tendon by thrombospondin‐4. Matrix Biol. 2014;37:35‐48. doi: 10.1016/j.matbio.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lynch JM, Maillet M, Vanhoutte D, et al. A thrombospondin‐dependent pathway for a protective ER stress response. Cell. 2012;149(6):1257‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vanhoutte D, Schips TG, Kwong JQ, et al. Thrombospondin expression in myofibers stabilizes muscle membranes. Elife. 2016;26:5. doi: 10.7554/eLife.17589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arber S, Caroni P. Thrombospondin‐4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995;131(4):1083‐1094. doi: 10.1083/jcb.131.4.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao S, Fung‐Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA‐Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9(1):e78644. doi: 10.1371/journal.pone.0078644 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The list of COL6‐RD biopsy samples used for analysis, corresponding genetic variants, age at the time of biopsy, and sex.
Table S2. The list of control biopsy samples used for analysis, corresponding age at the time of biopsy, sex, and pathologist's interpretation.
Table S3. Gene ontology analysis using DAVID database.
Table S4. Cellular function annotations using ingenuity pathway analysis and presented classified by p‐value and activation z‐score.
Table S5. Results from the polyserial correlation analysis, the corresponding gene ontology analysis for positively and negatively regulated genes, and ingenuity pathway upstream regulator analysis.
Data S1. Supplemental methods of transcriptomic data analysis, histology, automated image analysis, RNA isolation, RT‐qPCR, immunoblotting, and the list of primers used in this study.
Data Availability Statement
The datasets generated and/or analyzed during this study are available in the NCBI repository, https://www.ncbi.nlm.nih.gov/geo. The RNA‐Seq data generated as part of this analysis has been deposited in the NCBI Short Read Archive (SRP117086) and NCBI Gene Expression Omnibus (GSE103975). The microarray data generated as part of this analysis have been deposited in the NCBI Gene Expression Omnibus (GSE103972).