

Abstract
Oxysterols (OHCs) are hydroxylated cholesterol metabolites that play ubiquitous roles in health and disease. Due to the non-covalent nature of their interactions and their unique partitioning in membranes, the analysis of live-cell, proteome-wide interactions of OHCs remains an unmet challenge. In this manuscript, we present a structurally precise chemoproteomics probe for the biologically active molecule 20(S)-hydroxycholesterol (20(S)-OHC) and provide a map of its proteome-wide targets in the membranes of living cells. Our target catalogue consolidates diverse OHC ontologies and demonstrates that OHC-interacting proteins cluster with specific processes in immune response and cancer. Competition experiments reveal that 20(S)-OHC is a chemo-, regio-, and stereoselective ligand for the protein Tmem97 (the σ2 receptor), enabling us to reconstruct the 20(S)-OHC:Tmem97 binding site. Our results demonstrate that multiplexed, quantitative analysis of cellular target engagement can expose new dimensions of metabolite activity and identify actionable targets for molecular therapy.
Introduction
Cholesterol is the most complex metabolite produced de novo by humans.1 It serves as a substrate for the biosynthesis of an edifice of signaling molecules, including steroid hormones, glucocorticoids, bile acids, and vitamin D.2 Over one hundred human enzymes are dedicated to modifying cholesterol structure, whereas no catabolic enzymes are available to derive energy from its breakdown. Instead, the human body expresses a multitude of cholesterol metabolite receptors that control steroidogenesis, respiration, gut health, vision, and a repertoire of other essential processes. The gateway from cholesterol to all of these metabolites is a class of intermediates known as oxysterols (OHCs): specific mono-, di-, and polyhydroxylated cholesterol molecules that are barcoded by the configuration of their oxygen atoms.3,4 Although they serve as precursors to hundreds of metabolite structures, oxysterols are maintained at fleetingly low concentrations in the body and are rapidly converted to more metabolically stable products.5 A growing body of evidence has revealed that these ephemeral intermediates also function as sensitive, highly potent signaling molecules in their own right, coordinating critical processes such as neurotransmission, developmental signaling, and the innate immune response, where temporal control and specificity are paramount. Despite the fact that the chemical structures of OHCs directly reflect their receptor preferences, only a fraction of the proteins that interact with these molecules is known. To tap into their signaling functions and trace the circuits that regulate their activity, chemoproteomic technologies to fingerprint the cellular targets of specific OHC structures are required.
An accurate portrait of proteome-wide OHC interactions in live cells requires that chemoproteomics probes preserve the oxygenation pattern of the natural congener as well as physiochemical properties for native membrane partitioning.6 In this work, we describe our design, synthesis, and application of a structurally precise photoaffinity probe for 20(S)-hydroxycholesterol (20(S)-OHC), an agonist of the developmental signaling protein Smoothened.7,8 Using this probe, we perform an unbiased analysis of live-cell 20(S)-OHC targets in the membrane proteome of mammalian cells. Our data reveals over 100 enriched 20(S)-OHC interactors that cluster into discrete molecular functions. We elucidate a set of protein interactors that competably engage 20(S)-OHC and show that the orphan receptor Tmem97 is a regio-and stereoselective 20(S)-OHC target that links OHC binding to cholesterol homeostasis. Our work demonstrates the potential for chemoproteomic fingerprints to translate between the chemical structure of signaling metabolites and their biological functions.
Results
Design and synthesis of probe 2
Chemoproteomics strategies for identifying cellular OHC interactions face a unique combination of challenges. In live cells, subtle features of chemical structure dictate the membrane environments that OHCs traverse and the targets to which they are exposed.1,6 To capture the full complement of OHC interactions and the “fingerprint” of their cellular targets, chemoproteomics probes for cellular OHCs must preserve native membrane properties, retain oxygenation patterns recognized by specific receptors, and incorporate functionality to capture and identify live-cell targets. To address these requirements, we sought to modify natural OHC structures with a diazirine for live-cell photocrosslinking and an alkyne for downstream analysis. The diazirene functional group is benign to live cells, inert under ambient light, and can be photoactivated at 368 nm (long UV) to generate a reactive carbene that covalently crosslinks protein residues within a single-digit angstrom radius.9 Diazirines in general are favored for their small size, bio-orthogonality, and low background labeling; for OHC probes in particular, this non-polar functional group can be introduced at a variety of positions without significantly influencing OHC membrane behavior. Alkynes likewise provide sterically minimal, biorthogonal modifications for “click”-based appendage of crosslinked proteins to virtually any affinity tag (e.g. biotin), fluorescent reporter, or desired functionality. The complementary reactivities of diazirenes and alkynes underlie their emerging popularity as chemoproteomics handles and are especially suited to OHC probes.
We first chose to fashion a bifunctional probe for the OHC metabolite 20(S)-OHC (1). This molecule has been detected in the brain and placenta,10,11 and functions as a potent osteogenitor12 and teratogen.13 In addition to binding other proteins, 20(S)-OHC is a stereospecific ligand for the 7-transmembrane receptor Smoothened (Smo), a gatekeeper in developmental Hedgehog signaling.7,8 Notably, the 20(R) epimer of this molecule (20(R)-hydroxycholesterol, 20(R)-OHC) is inactive at Smo. Stimulation of endogenous Smo activity by 20(S)-OHC and congeners can be assayed in murine embryonic fibroblasts, providing a platform for us to evaluate the biological activity of a 20(S)-OHC probe relative to the parent molecule.
To enable an unbiased analysis of 20(S)-OHC targets in cells, we positioned the alkyne and diazirine handles to preserve the C20(S)-OHC pharmacophore, the α- and β-face topologies of the tetracyclic ring system, the accessible C3-hydroxyl group, and the length and hydrophobicity of the C17 side chain (Fig. 1a). Synthetically, we envisioned the use of an intramolecular C-H activation for conversion of the C19 methyl group to an alkyne14 and a stereoselective Grignard addition to introduce a diazirine side chain to a C17 methyl ketone.15 In the forward sense, stereoselective hydrobromination of pregnenolone acetate (3) and photoinduced C-H activation of the C19 methyl group in 414 provided ether 5 in 76% yield over two steps (Fig. 1b). Elaboration of the C19 primary alcohol to an alkyne via reductive elimination by elemental zinc in acetic acid, pyridinium chlorochromate oxidation to the C19 aldehyde 6, and homologation with the Seyferth-Glibert reagent provided the key intermediate 7. Gratifyingly, Grignard reaction to add the side chain at C20 afforded exclusively the 20(S) alcohol, whereupon liberation of the ketone in 8 and one-pot introduction of the C25 diazirine produced bifunctional photoaffinity probe 2 as an air- and ambient light-stable solid. In addition to furnishing 2, alkyne intermediate 7 represents a precursor to a library of bifunctional probes for side-chain oxysterols.
Fig. 1: Design, synthesis, and evaluation of 20(S)-OHC chemoproteomics probe 2.
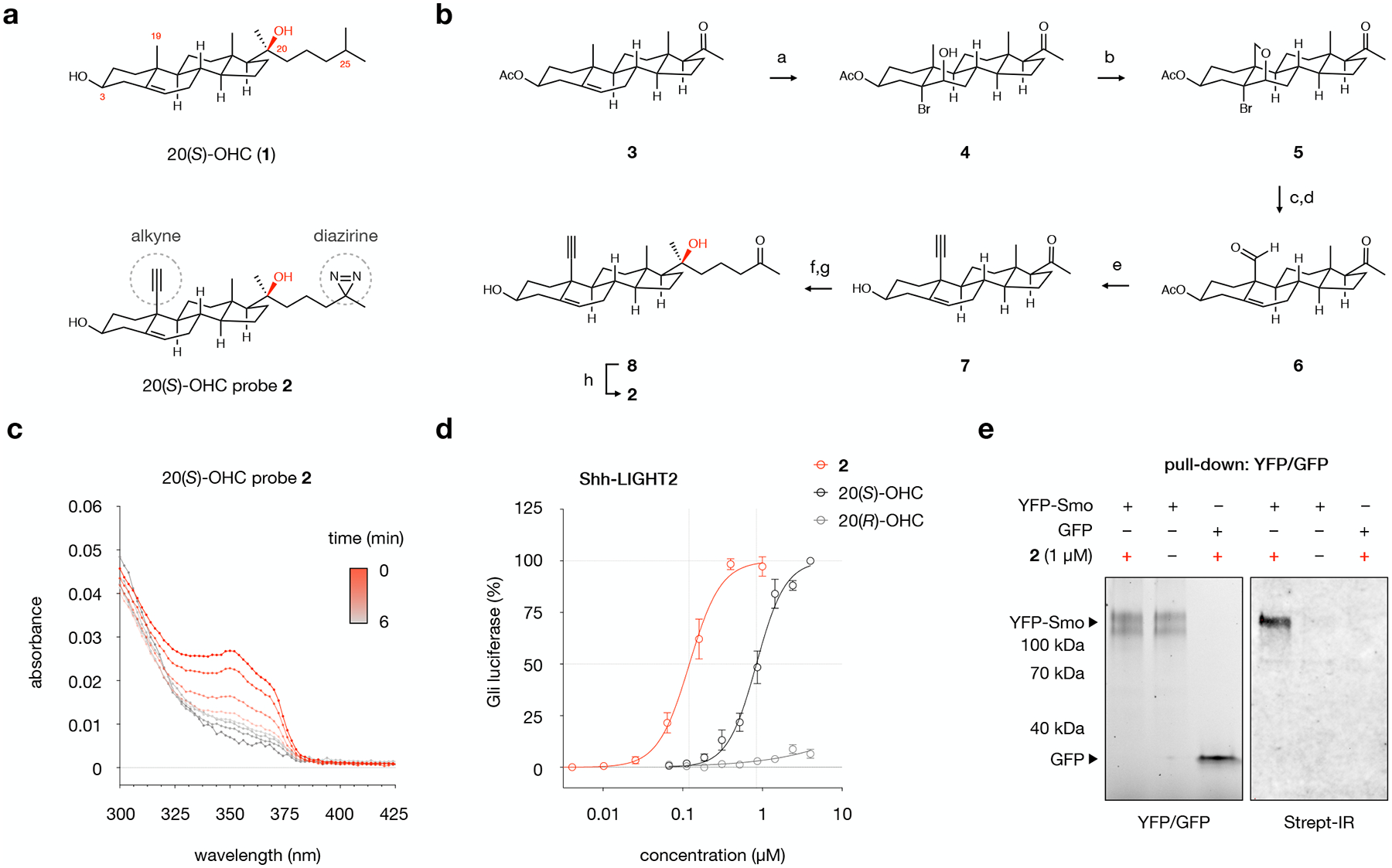
a, Structures of 20(S)-hydroxycholesterol (20(S)-OHC, 1) and probe 2, which contains a diazirine at C25 for photocrosslinking and an alkyne at C19 for click chemistry.
b, Chemical synthesis of probe 2. Conditions: a. NBA, aq. HClO4, 1,4-dioxane, 0 → 23 °C, 77%; b. Pb(OAc)4, CaCO3, I2, hν, cyclohexane, 80 °C, > 99%; c. Zn, AcOH-H2O, 45 °C, 88%; d. PCC, Celite, CH2Cl2, 23 °C, 87%; e. (i) Seyferth-Gilbert reagent, t-BuOK, THF, −78 °C, 80%; (ii) Cs2CO3, MeOH, 23 °C, >99%; f. 2-(3-bromopropyl)-2-methyl-1,3-dioxolane, Mg, THF, 0 → 23 °C, 61%; g. HCl, THF, 23 °C, 94%; h. (i) NH3, MeOH, 0 °C; (ii) NH2HSO3, 0 → 23 °C; (iii) I2, Et3N, THF, 23 °C, 46%.
c, Irradiation of probe 2 (10 mM, DMF) with 368 nm light results in loss of diazirine absorption at 353 nm with a half-life of 1.4 minutes. Values are the average of triplicate measurements.
d, Treatment of Shh-LIGHT2 cells with 20(S)-OHC, 20(R)-OHC, or probe 2 demonstrates that 20(S)-OHC and 2, but not the unnatural epimer 20(R)-OHC, activate the Smoothened (Smo)-regulated Gli transcription factors. Values are the average of 3 biological replicates ± s.d.
e, Live HEK293T cells overexpressing YFP-Smo or GFP were treated with 2 or DMSO and irradiated, then YFP-Smo or GFP was isolated from membrane fractions using a GFP nanobody that recognizes both fluorescent proteins. Isolated proteins were subjected to a click reaction for biotinylation. Left: In-gel fluorescence analysis of membrane fractions from cells overexpressing YFP-Smo or GFP. Right: Detection of biotinylated proteins using Streptavidin IRDye 800CW (Strept-IR), demonstrating that 2-treated YFP-Smo-expressing cells are labeled with biotin. YFP-Smo-expressing cells treated with DMSO and GFP-expressing cells treated with 2 show no biotin labeling. The experiment was repeated three times independently with similar results.
The structure of 2 was unambiguously established by 1D and 2D NMR spectroscopy. The presence of the diazirine was confirmed by a characteristic infrared N=N stretching frequency at 1644 cm−1 and ultraviolet absorption at 353 nm. Irradiation of 2 in organic solvent at 368 nm resulted in progressive extrusion of N2 with a half-life of 1.34 min (Fig. 1c). Thus, probe 2 differs from the parent structure by a count of only 4 atoms, ~1.5 Å in any dimension, and a logP of 0.48, and can be photoactivated to generate a reactive carbene for protein crosslinking.
To ensure that 2 preserves biological activity of 20(S)-OHC, we assessed its ability to activate Smo in Shh-LIGHT2 cells, a murine embryonic fibroblast line that bears a luminescent reporter of Smo-regulated Gli transcription (Fig. 1d). Commensurate with the known (S) stereoselectivity of 20-OHC:Smo binding, 20(S)-OHC induced reporter activity with an EC50 of 8.4 μM, whereas 20(R)-OHC was inactive. Probe 2 induced Smo activity with an EC50 of 1.2 μM, demonstrating that 2 retains activity at Smo. Here, the difference in response to 2 and 20(S)-OHC most likely reflects modest changes in solubility and/or membrane properties introduced by the probe functionalities, underscoring the sensitivity of certain OHC activities to subtle changes in structure. To assess biochemical engagement between 2 and Smo, we photocrosslinked 2 to YFP-Smo expressed in HEK293T cells and isolated Smo protein using a GFP/YFP nanobody (Fig. 1e). Click ligation of a biotin handle and visualization of crosslinked protein revealed that Smo, but not overexpressed GFP, was efficiently labeled by 2. These studies established that 2 retains activity at a stereospecific OHC receptor and can selectively crosslink target proteins in live cells, supporting its utility as a probe to map live-cell 20(S)-OHC interactions.
Establishment of OHC target labeling in live cells
To quantitatively catalogue the protein fingerprint interaction of 20(S)-OHC in live cells, we established conditions for live cell engagement, photochemical crosslinking, and biorthogonal tagging of target proteins (Fig. 2a). We performed these experiments in NIH-3T3 cells, which show an endogenous response to Smo activation and serve as a model to study osteogenesis and embryogenesis. For live cell engagement, we used an incubation time of 30 min to minimize effects of OHC-induced protein expression or degradation. To photocrosslink target proteins, we removed unbound probe and irradiated cells on ice at 368 nm for 5 min. To label crosslinked proteins, we lysed cells and performed a click reaction to ligate a TAMRA fluorophore to the alkyne handle in 2.
Fig. 2: Probe 2 labels membrane proteins in live NIH-3T3 cells.
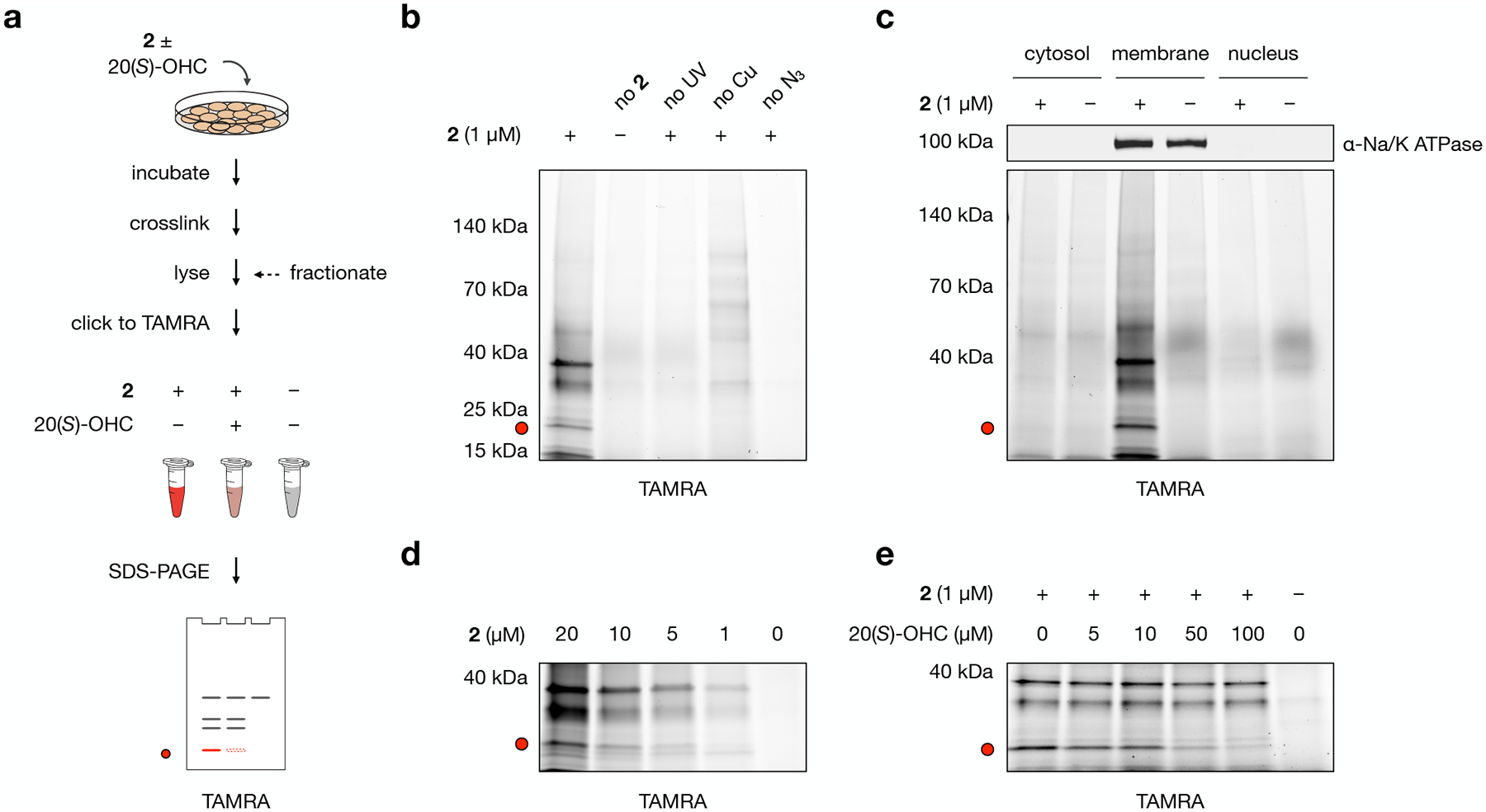
a, Workflow for gel-based profiling of probe 2 target proteins. Cells are incubated for 30 min with 1 μM 2, DMSO, or 1 μM 2 + 50 μM 20(S)-OHC and irradiated for 5 min with 368 nm light. Isolated membrane fractions are subjected to a click reaction with TAMRA azide, separated by SDS-PAGE, and visualized by in-gel fluorescence.
b, Control experiments demonstrate that fluorescent labeling requires treatment with 2, exposure to UV light, and click ligation to TAMRA. In the “no 2” condition, cells were incubated with DMSO only; in the “no UV” condition, probe-treated cells were not exposed to 368 nm light; in the “no Cu” condition, CuSO4 was omitted from the click catalyst mixture; in the “no N3” condition, TAMRA azide was omitted from the click reaction.
c, Cell fractionation shows that TAMRA-labeled proteins appear primarily in the membrane fraction as opposed to cytosolic or nuclear fractions. The sodium/potassium-transporting ATPase subunit alpha-1 (Na/K ATPase) was used as a membrane marker.
d, Proteins in the membrane fraction are labeled by 2 in a dose-dependent manner.
e, Competition by 20(S)-OHC during incubation of cells with 2 reduces TAMRA labeling of a 21 kDa band in a dose-dependent manner.
The red dot at 21 kDa in panels a–e marks the location of a competable TAMRA-labeled band. Experiments in b, c, d, and e were repeated two times independently with similar results.
SDS-PAGE and in-gel fluorescence analysis of labeled proteins revealed a discrete set of bands that appeared in a probe-, UV-, and copper-dependent manner, indicating that they represented 2-crosslinked target proteins (Fig. 2b). Cell fractionation revealed that the majority of labeled proteins, including prominent bands at approximately 13 kDa, 21 kDa, and 37 kDa, appeared exclusively in the membrane fraction (Fig. 2c). To focus on these enriched targets and identify novel 20(S)-OHC interactors in the membrane proteome, we employed this fractionation method for remainder of our experiments. Analysis of the membrane proteome demonstrated these bands were labeled in a manner that showed good signal-to-background at 1 μM (Fig. 2d). Competition with unlabeled 20(S)-OHC revealed that the 21 kDa band was competable in a dose-dependent manner, showing 85% reduction in signal at a competitor concentration of 50 μM (Fig. 2e). The ability to label membrane proteins in a probe, UV, and click catalyst-dependent fashion in a manner that could be dose-dependently competed by 20(S)-OHC established the utility of 2 as a live cell chemoproteomics probe.
Analysis of enriched OHC targets
To profile the entire complement of 2 labeling in the membrane proteome, we replaced TAMRA azide in the click reaction with biotin azide to label crosslinked proteins. To isolate crosslinked proteins using streptavidin enrichment, we removed excess biotin through acetone precipitation, resolubilized the membrane proteome in SDS/Igepal buffer, and isolated biotinylated proteins on streptavidin agarose beads (Fig. 3a and Extended Data Fig. 1). For global target profiling, we used this protocol in the presence and absence of 1 μM 2 at a normalized concentration of 0.01% DMSO. On-bead digestion of streptavidin-enriched proteins and tandem isotope labeling of replicate samples enabled multiplexed real-time search-enabled MS3-based mass spectrometry analysis (RTS-SPS-MS3) of 2 interactors in the membrane proteome.
Fig. 3: Protein targets of probe 2 converge in distinct gene ontologies and disease signatures.
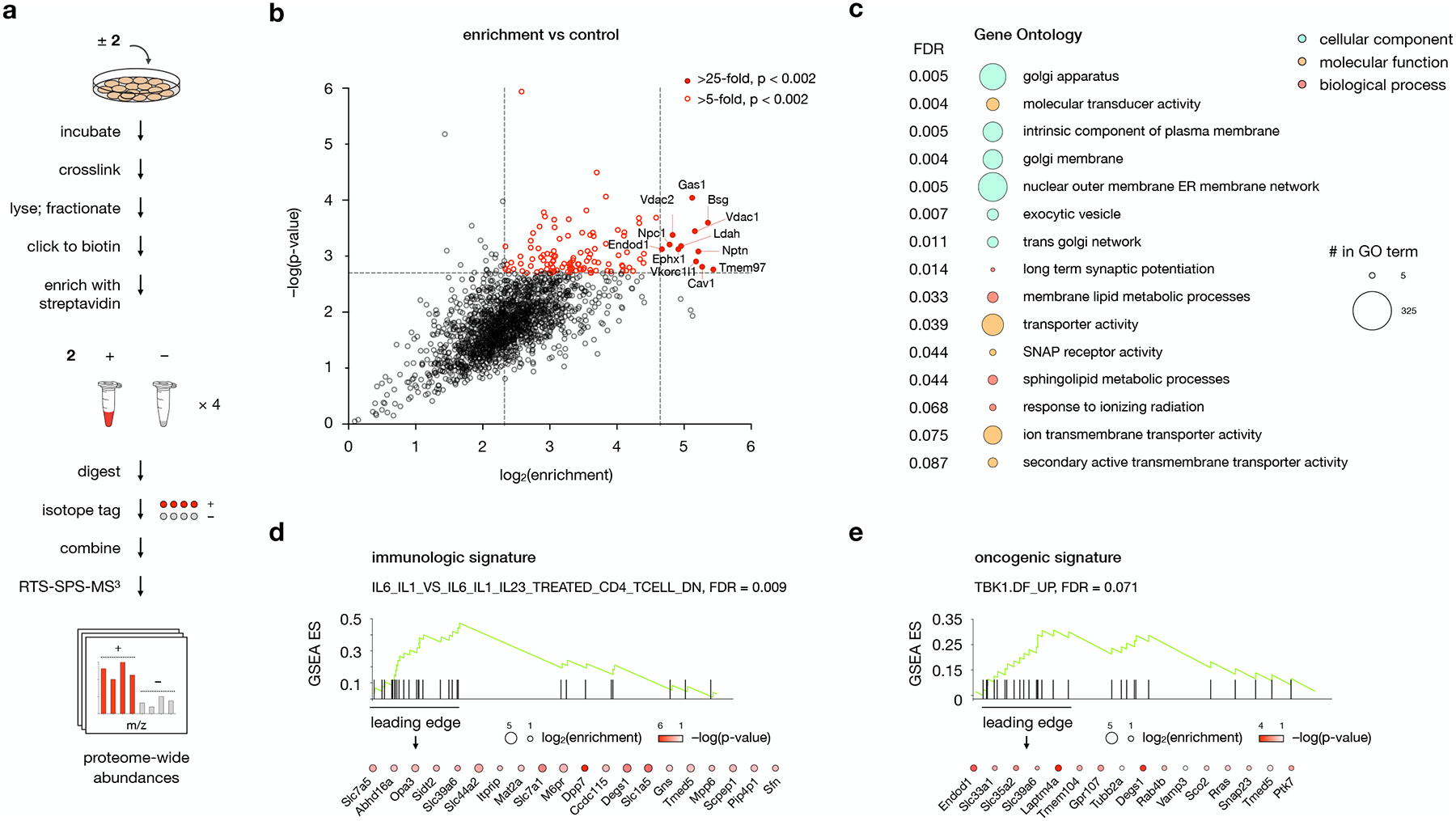
a, Workflow for MS-based profiling of probe 2 target proteins. Cells are incubated for 30 min with 1 μM 2 or DMSO and irradiated for 5 min with 368 nm light. Isolated membrane fractions are subjected to a click reaction with biotin picolyl azide, enriched with streptavidin agarose, and digested with trypsin. Digested peptides from each sample are labeled with unique isobaric mass tags, then samples are pooled and analyzed by LC/RTS-SPS-MS3.
b, Volcano plot showing statistical significance versus average log2(fold change) (“log2(enrichment)”) of peptides isolated from cells treated with 2 or DMSO alone. Cutoffs discussed in the text at a p-value of 0.002 and fold changes of 25 and 10 are indicated. Data represent 4 biological replicates consisting of matched experiments with 2 and DMSO. Statistical significance for each protein was calculated using a two-tailed paired t-test.
c, Gene Set Enrichment Analysis (GSEA) of 2-enriched proteins using the Gene Ontology (GO) gene set. The top 16 GO pathways ranked by NES (Normalized Enriched Score) are listed. Bubbles are colored according to GO term class; bubble size is proportional to the number of 2-enriched proteins found in each gene set.
d,e, GSEA using d, the immunologic and e, the oncogenic signature gene sets for 2-enriched proteins. Enriched proteins the leading edge of the gene set are listed; color and size of the bubbles represent log2(enrichment) and −log(p-value), respectively. For additional gene sets associated with 2-enriched proteins, see Supplementary Table 1.
For c, d, and e, statistical significance was assessed using the GSEA software.
Our results revealed 12 proteins that were enriched by 25-fold relative to DMSO alone, and 106 proteins that were enriched by 5- to 25-fold at a p-value of < 0.002 (Fig. 3b). Comparison with transcript levels from RNA-seq analysis16 revealed no significant correlation between target protein enrichment and expression, indicating that enhancement was not solely a reflection of abundance (Extended Data Fig. 2c). The most enriched targets consisted of proteins previously established to bind OHCs (NPC117 and Cav118), cholesterol-binding proteins (Vdac119), and proteins directly involved in cholesterol homeostasis (Tmem9720). The set also included metabolic proteins that act on sterol substrates (Ldah,21 Ephx122) or participate in redox complexes associated with cholesterol biosynthesis (Vkorc1l123). Three of the proteins (Nptn and Bsg,24 Endod125) function as adaptors that control trafficking and activity of specialized membrane protein complexes. Performing our affinity enrichment protocol using label-free instead of TMT analysis revealed a nearly identical target profile with respect to identity and enrichment, demonstrating that the method of quantification did not significantly influence the target set (Extended Data Fig. 2b).
A key aspect of OHC function involves the unique effect of these molecules on membrane proteins sensitive to cholesterol content.3,4,5,6 Accordingly, network analysis of OHC targets can reveal membrane topologies and cellular circuits that are responsive to low affinity and/or combinatorial OHC interactions. To identify ontologies associated with the 2 target fingerprint, we performed Gene Set Enrichment Analysis (GSEA) using 2-enriched proteins.26 GSEA compares genetic and/or proteomic data to a database of curated gene sets, termed molecular signatures, that are associated with specific ontologies and diseases. The analysis then provides a rank-ordered list of molecular signatures that are overrepresented in the input data. Using the Gene Ontology (GO) gene set collection for GSEA with 2-enriched targets revealed a top list of 15 GO terms for cellular components, molecular functions, and biological processes with an FDR of < 0.1 (Fig. 3c). Among the most highly enriched membrane components were the Golgi apparatus and plasma membrane-ER networks, consistent with the observed localization of 20(S)-OHC to the Golgi and perinuclear regions,27 and in contrast to the predominant localization of cholesterol to the plasma membrane.1 Correspondingly, protein interactors of 2 were associated with GO molecular functions that involved molecular signal transduction and transport across the membrane. The most highly represented GO biological processes included membrane lipid metabolism and sphingolipid metabolism in particular, in line with the observation that OHCs posttranslationally enhance the activity of enzymes involved in sphingomyelin biosynthesis.28
To identify associations between 2-binding proteins and processes in human health and disease, we performed GSEA using the immunologic and oncogenic signature gene set collections (Fig. 3d,e and Supplementary Table 1). The immunologic gene set with the highest representation of 2-enriched targets was GSE23505_IL6_IL1_VS_IL6_IL1_IL23_TREATED_CD4_TCELL_DN (NES = 2.92, FDR = 0.009). This gene set is downregulated in RORɣ-dependent, TGFβ-independent initiation of CD4+T cell differentiation into Th17 helper cells. The oncogenic gene set containing the most 2-enriched targets was TBK1.DF_UP (NES = 2.14, FDR = 0.071), a set of genes upregulated by TANK binding kinase 1 in KRAS-mutant lung adenocarcinoma cells. Since targeting of TBK1 in KRAS mutant cancer cells is synthetically lethal, inhibition of OHC-coordinated processes in these cells may represent an avenue for the treatment of refractory KRAS mutant cancers. The identification of OHC target proteins in immune system function and cancer signaling highlights new avenues of investigation for drug design and therapeutic targeting.
Identification of enriched and competable OHC targets
We next sought to determine which live-cell OHC interacting proteins could be out-competed by unlabeled 20(S)-OHC. To optimize the competability of 20(S)-OHC interactions, we focused on conditions that reduced non-specific labeling and enhanced sensitivity to competition. To do so, we pre-complexed competitor molecules with methyl-β-cyclodextrin (MβCD), a water-soluble cavitand that reversibly binds sterols and delivers them to cell membranes.29 Co-application of 0.5 mM MβCD with 1 μM 2 in the presence or absence of 50 μM 20(S)-OHC competitor reproducibly solubilized all sterols and provided robust competition of the 21 kDa band identified by in-gel analysis (Fig. 2e). Because MβCD can perturb cellular cholesterol content, we identified an MβCD concentration that preserved equal intensities of non-competable bands in the presence and absence of competitor (Extended Data Fig. 3).
Our competition experiments revealed 12 proteins that were enriched by 2 by more than 7-fold at a p-value of 0.05 (Fig. 4a,b). Comparison of our enriched targets in the presence and absence of MβCD demonstrated that, while MβCD reduced the magnitude of fold changes versus DMSO, neither the identity nor the relative ranking of probe targets were significantly altered (Fig. 4b and Extended Data Fig. 3c). We again observed the proteins Tmem97, Ephx1, Cav1, Vkorc1l1, Ldah, Vdac2, and Bsg in the top 5% of target enrichment, comprising ~60% of the most enriched proteins in our previous experiments (Fig. 3b). Another 89 targets were enriched at between 2.75 and 7-fold at a p-value of 0.05, representing a selection of transmembrane proteins, signal transduction proteins, lipid biosynthetic enzymes, and membrane trafficking/adaptor proteins identified in our previous GO analysis. Enriched proteins in these experiments also involved in Golgi membrane homeostasis (Tmem19930), transmembrane proteins of unannotated function (Tmem238, Smim4), and the caveolin isoform Cav2.
Fig. 4: 20(S)-OHC identifies enriched and competable probe 2 target proteins.
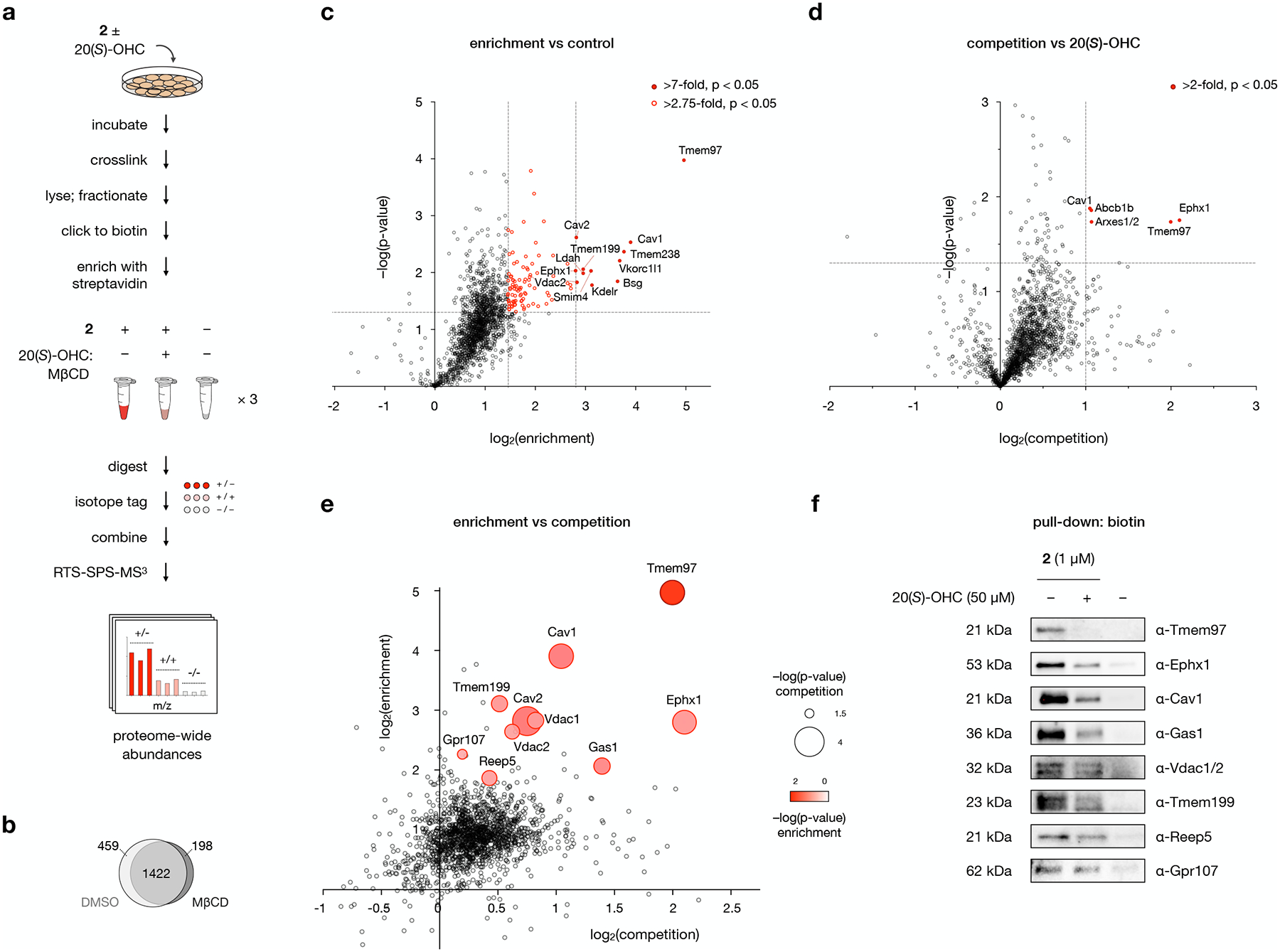
a, Workflow for MS-based profiling of probe 2 target proteins. Cells are pre-incubated with 50 μM 20(S)-OHC complexed with MβCD or with MβCD alone for 1 h before addition of 1 μM 2 or DMSO and further incubation for 30 min. Cells are irradiated for 5 min with 368 nm light, then isolated membrane fractions are subjected to a click reaction with biotin picolyl azide, enriched with streptavidin agarose, and digested with trypsin. Digested peptides from each sample are labeled with unique isobaric mass tags, then samples are pooled and analyzed by LC/RTS-SPS-MS3.
b, Venn diagram of proteins enriched by probe 2 in the presence and absence of MβCD. See also Extended Data Fig. 3.
c, Volcano plot showing statistical significance versus average log2(fold change) (“log2(enrichment)”) of peptides isolated from cells treated with 1 μM 2 or DMSO alone. Cutoffs discussed in the text at a p-value of 0.05 and fold changes of 7 and 2.75 are indicated.
d, Volcano plot showing statistical significance versus average log2(fold change) (“log2(competition)”) of peptides isolated from cells treated with 2 or 2 + 20(S)-OHC. Cutoffs discussed in the text at a p-value of 0.05 and a fold change of 2 are indicated.
e, Scatter plot of enrichment versus competition for 2 target proteins. Proteins selected for Western blot analysis are shown as bubbles, where size and color represent p-values for competition and enrichment, respectively.
For c, d, and e, data represent 3 biological replicates of matched experiments with 2, 2 + 20(S)-OHC, and DMSO. For c and d, statistical significance for each protein was calculated using a two-tailed paired t-test.
f, Western blot analysis of proteins labeled by 2 in the presence of 20(S)-OHC or DMSO, clicked to biotin, enriched on streptavidin, and resolved by SDS-PAGE. For quantitative analysis, see Extended Data Fig. 4.
As anticipated, only a subset of 2 interactions were competable by 20(S)-OHC (Fig. 4d,e). In this dataset, a sharp divide separated the top 0.3% of proteins from less competable interactors. The most competable proteins encompassed the highly enriched targets Tmem97, Ephx1, and Cav1. In addition, the list included Abcb1 (P-glycoprotein 1), an ATP-dependent multidrug/steroid transporter,31 and Arxes1/2, a retrotransposed protease gene on the X-chromosome that plays a role in adipogenesis.32 Both Abcb1b and Arxes1/2 also appear with in the top 2% of proteins enriched by 2.
The full fingerprint of direct OHC binding proteins provided us with an opportunity to probe the engagement of OHCs and proteins with relevant biological functions. Accordingly, we selected a set of functionally relevant 20(S)-OHC targets and validated their engagement with 2 by live-cell crosslinking, biotin-streptavidin enrichment, and Western blot detection using specific antibodies (Fig. 4f). A highly competable protein identified in our data, Gas1, is an upstream Smo regulator that coordinates the Hedgehog signaling pathway in embryonic development. Competable binding of Gas1 by 20(S)-OHC complements recent findings that Gas1 localizes to cholesterol microdomains as an obligate part of its Hh signaling activity,33 and suggests that endogenous OHCs may directly or indirectly contribute to this mechanism of regulation. The moderately competable protein isoforms Vdac 1 and 2 are known to control steroidogenesis by directing the transport of cholesterol from the outer to the inner mitochondrial membrane for conversion to pregnenolone by Cyp11a1,34 suggesting a possible regulatory role for OHCs. Potentially functional but less competable OHC binding was also observed with the scaffolding protein Reep5, which plays a structural role in the architecture of sarcolemmal membranes in muscle cells,35 the protein Gpr107, which chaperones receptors to organelle membranes,36 and the adaptor protein Tmem199, an assembly factor in vacuolar ATPase complexes that control energy-dependent acidification of cellular organelles.30 In each of these cases, 2 captured endogenous protein to an extent that correlated with MS-based abundance, verifying that our proteome-wide profiling can provide a quantitative catalogue of functional 2-interacting proteins in living cells (Extended Data Fig. 4).
Discovery of Tmem97 as a selective OHC target
The most enriched and competable protein identified in our studies was Tmem97, a transmembrane protein with a molecular weight of 21 kDa– approximately the same molecular weight as the competable band identified in our in gel fluorescence analysis (Fig. 2e). Tmem97, also known as the σ2 receptor, is a protein biomarker that is overexpressed in proliferative tumors and a target for PET imaging in the clinic.37 Originally characterized by its pharmacological profile, Tmem97 has also been investigated as a non-opioid target for psychiatric conditions and neuropathic pain.20 Identification of the Tmem97 gene in 2017 and genetic perturbation studies subsequently established an integral role for Tmem97 in cholesterol homeostasis, where it regulates levels of the cholesterol transporter protein NPC1.38,39,40 For its utility as a cancer radioimaging agent and its implication in lysosomal cholesterol storage diseases, Tmem97 is a valuable therapeutic target. However, no endogenous ligand for Tmem97 has been established.
To determine whether Tmem97 contributed to the 21 kDa band identified in our gel-based assays, we took advantage of ligands selective for Tmem97 or the structurally unrelated σ1 receptor. Using the σ1 ligand PRE-084 or the Tmem97 ligand BIMU-8 as competitors, live-cell crosslinking to 2, TAMRA labeling, and SDS-PAGE analysis revealed that the Tmem97 ligand, but not the σ1 receptor ligand, effectively out-competed labeling by 2 (Fig. 5a). Biotin labeling, streptavidin pulldown, and immunodetection using a Tmem97 antibody likewise confirmed that Tmem97 was recovered only in the presence of the σ1 receptor ligand, supporting the identification of Tmem97 as a 2-labeled target.
Fig. 5: Tmem97 is a selective protein target of probe 2.
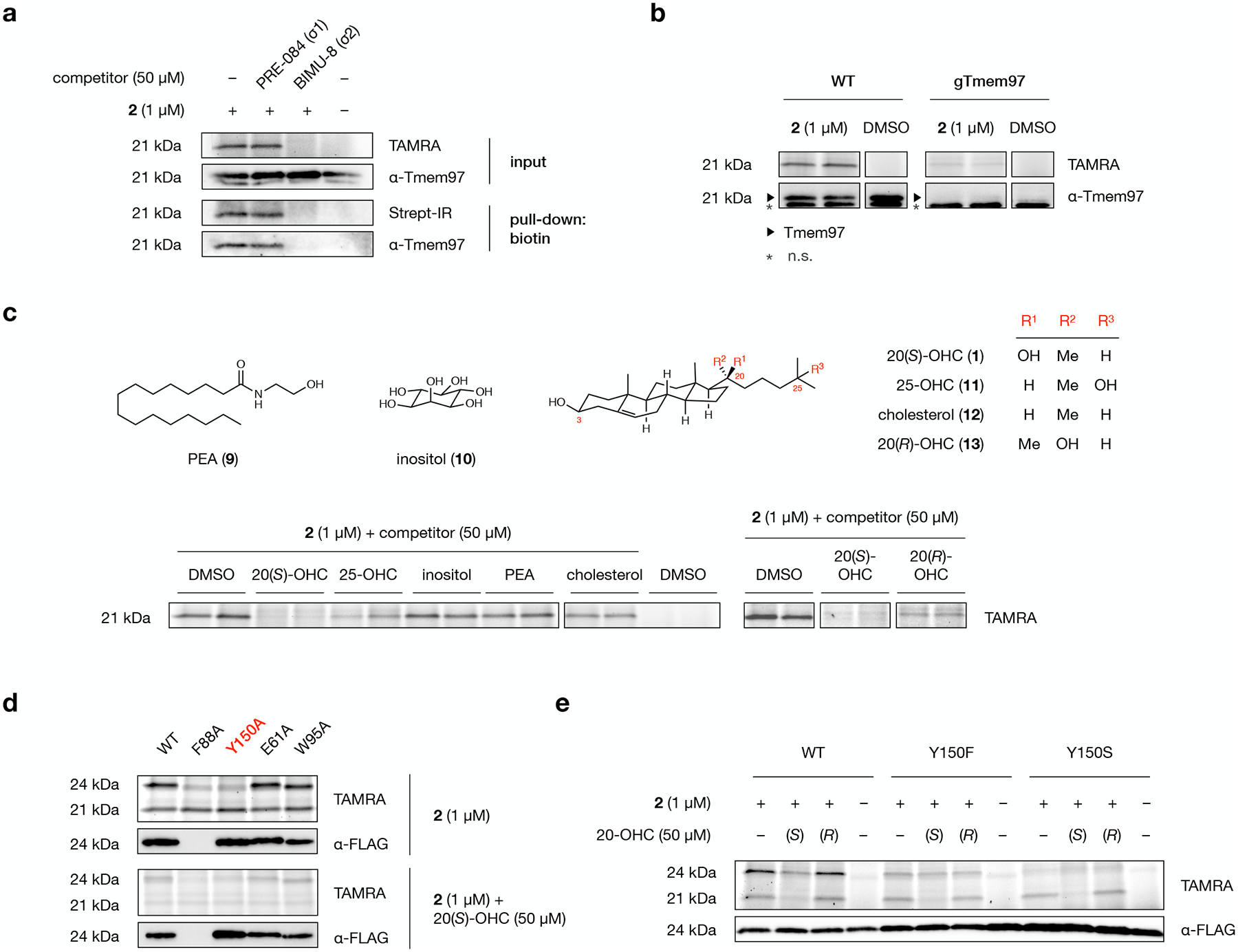
a, A TAMRA-labeled band at 21 kDa in the membrane proteome of 2-labeled NIH-3T3 cells is eliminated by competition with BIMU-8, a selective Tmem97 (σ2 receptor) ligand, but not PRE-084, a selective σ1 receptor ligand. Likewise, competition with BIMU-8, but not PRE-084, reduces labeling, biotin-streptavidin enrichment, and detection of the band at 21 kDa by a Tmem97 antibody.
b, TAMRA fluorescence and Tmem97 antibody signal from wild-type and Tmem97-KO NIH-3T3 cells treated with 2 (duplicate samples) or DMSO. An asterisk indicates non-specific (n.s.) protein detection by the Tmem97 antibody.
c, Representative in-gel analysis of probe 2 labeling at 21 kDa in the presence of 20(S)-OHC (1), 25-OHC (11), inositol (10), palmitoylethanolamine (PEA, 9), cholesterol (12), and 20(R)-OHC (13) as competitors in NIH-3T3 cells. For quantification, see Extended Data Fig. 5c.
d, Representative in-gel analysis of probe 2 labeling of Tmem97 mutants expressed as Tmem97-Myc-FLAG fusion constructs in HEK293T cells. Alanine substitution of the Y150 residue in the Tmem97:20(S)-OHC binding site eliminates labeling of overexpressed Tmem97. E61A and W95A mutants remain susceptible to labeling by 2, while an F88A mutant fails to express in cells.
e, Representative in-gel analysis of probe 2 labeling of wild-type Tmem97, Tmem97(Y150F), or Tmem97(Y150S) in expressed as Tmem97-Myc-FLAG fusion constructs in HEK293T cells. A Y150F mutation decreases labeling efficiency, reduces competition by 20(S)-OHC, and eliminates stereoselectivity for 20(S)- over 20(R)-OHC. A Y150S mutation eliminates probe labeling. For quantification, see Extended Data Fig. 6b.
Experiments in a, b, and d were repeated two times independently with similar results. In d and e, endogenous Tmem97 appears at 21 kDa; overexpressed Tmem97-Myc-FLAG appears at 24 kDa due to the epitope tags.
To confirm that Tmem97 was the major component of the 2-labeled 21 kDa band, we used CRISPR/Cas9 to ablate expression of Tmem97 in a population of NIH-3T3 cells (Fig. 5b). Photoaffinity crosslinking of 2 in wild-type and Tmem97-knockout cells demonstrated that loss of Tmem97 eliminated the prominent TAMRA-labeled band at 21 kDa. To evaluate specific competition of Tmem97, we crosslinked 2 to cells in the presence of varying amounts of 20(S)-OHC, enriched crosslinked proteins, and examined the intensity of Tmem97 immunodetection by Western blot (Extended Data Fig. 5a). Increasing concentrations of 20(S)-OHC dose-dependently reduced the strength of the Tmem97 band in a manner proportional to loss of the probe intensity at 21 kDa. To assess whether Tmeme97 labeling was selective for 2, we crosslinked cellular proteins using the commercially available cholesterol probe PhotoClick Cholesterol (14) (Extended Data Fig. 5b). Under identical conditions, 2, but not PhotoClick Cholesterol,41 competably labeled overexpressed Tmem97 in HEK293T cells. Together, these experiments indicate that Tmem97 is a dose-dependently competable, selective target of 2.
Next we sought to determine ability of structurally distinct small molecules and cholesterol metabolites to out-compete labeling by 2. Neither the long-chain lipid palmitoylethanolamide (PEA, 9) nor the polyhydroxylated carbocycle inositol (10) could effectively reduce labeling by 2, indicating that neither bound to Tmem97 at the same site as 2 and 20(S)-OHC (Fig. 5c and Extended Data Fig. 5c). To determine whether Tmem97 could distinguish hydroxylation patterns within the sterol structure, we evaluated competition by unmodified cholesterol (11) or the regioisomeric oxysterol 25-OHC (12). While cholesterol itself was a poor competitor of 2, 25-OHC could partially outcompete labeling by 2, indicating that Tmem97 is sensitive to the extent and pattern of sterol hydroxylation. Finally, we tested whether the configuration of the C20 alcohol could differentiate competition by 20(S)- and 20(R)-OHC (13). Notably, the 20(S) stereochemistry is produced endogenously by Cyp11a1 oxidation of cholesterol en route to pregnenolone.42 Competition with either the C20(S) or the C20(R) epimer revealed that 20(R)-OHC competed only 50% of probe labeling at a concentration of 50 μM, whereas competition by 50 μM 20(S)-OHC completely abolished labeling, indicating a stereochemical preference for the C20(S)-OH interaction.
Tmem97 is phylogenetically related to the enzyme 3-β-hydroxysteroid-Δ8,Δ7-isomerase (Emopamil-binding protein, EBP), which catalyzes migration of the C8-C9 double bond in zymosterol/zymostenol to its C7-C8 position in lathosterol/dehydrolathosterol.43 Despite their evolutionary relationship, Tmem97 lacks an acidic residue homologous to E122 in EBP (human numbering) and has no discernable enzymatic activity.20 To determine a binding site for 20(S)-OHC within Tmem97, we used the crystal structure of human EBP (PDB 6OHT)44 as a template to predict the structure of mouse Tmem97 (82% pairwise identity with human Tmem97) using the Robetta server.45 We embedded the resulting model within a POPC membrane and performed all-atom molecular dynamics simulations to equilibrate the membrane-bound Tmem97 structure. Docking of 20(S)-OHC into this model produced a stable pose in which 20(S)-OHC binds to several residues homologous to those in the active site of EBP (Extended Data Fig. 6a). Convincingly, while this manuscript was in revision, the first crystal structure of Tmem97 in complex with synthetic σ2 ligands was reported, evidencing a ligand binding site with striking resemblance to our homology-based model.46 As in EBP and other sterol-binding proteins, an array of hydrophobic and aromatic residues line a central Tmem97 ligand-binding cavity with a helical “cap” at the top. In our docked 20(S)-OHC:Tmem97 model, the C3 hydroxyl group of 20(S)-OHC forms a hydrogen bond to the imidazole side chain of H106, while the iso-octyl tail is accommodated by a hydrophobic pocket that includes F66, P113, V146, Y147, and Y150. Hydrophobic interactions between the iso-octyl side chain and these residues would disfavor introduction of a polar group at C25, providing a possible rationale for reduced competition of 2 by 25-OHC versus 20(S)-OHC.
To ascertain whether residues in the Tmem97 binding pocket influenced the extent of 2 labeling or degree of competition by 20(S)-OHC, we transiently overexpressed epitope-tagged Tmem97 and constructs containing mutations at residues near 20(S)-OHC in HEK293T cells (Fig. 5d). While the F88A mutant protein was not expressed, the Y150A, E61A, and W95A mutants were robustly produced, appearing ~3 kDa higher in apparent MW than endogenous Tmem97 due to the presence of a FLAG-Myc epitope tag. Interestingly, while the E61A and W95A mutants were labeled by 2 and competable by 20(S)-OHC to the same extent as the wild-type protein, the Y150A mutant was not labeled. Because Y150 is the closest polar residue to the C20-OHC alcohol in our docked model, we hypothesized that it might engage in polar and/or hydrogen bonding interactions via its phenolic hydroxyl group. To determine whether the Y150 side chain affected either the efficiency or structure-selectivity of Tmem97 labeling, we generated two additional Tmem97 mutants: Y150F, which retains an aromatic group but eliminates hydrogen bonding, and Y150S, which lacks aromaticity but retains the ability to form hydrogen bonds. Crosslinking and in-gel fluorescence analysis demonstrated that, while the Y150F mutant was labeled to approximately 60% of the wild-type protein, the Y150S showed no significant labeling (Fig. 5e and Extended Data Fig. 6b). Intriguingly, the Y150F mutation not only reduced competition by 20(S)-OHC, but eliminated stereoselective displacement by 20(S)-OHC over 20(R)-OHC. Together, these results suggest that the aromatic moiety of Y150 contributes to OHC binding affinity, while the phenolic hydroxyl group may confer a preference for engagement with the 20(S)-OHC epimer. These experiments identify discrete structural features of the Tmem97 binding site that dictate OHC:Tmem97 binding in cells, offering unique insight into the molecular details of their engagement.
20(S)-OHC enhances association of Tmem97 and NPC1
In 2009, Tmem97 was identified as a novel regulator of cholesterol homeostasis in a screen for proteins that influence cellular cholesterol content.39 This and a subsequent study established that genetic knockdown of Tmem97 inhibits internalization of low-density lipoprotein particles and reduces cholesterol concentrations in lysosomes.40 These effects coincide with an increase in the levels of NPC1 protein- a central coordinator of cholesterol distribution that exports free cholesterol from lysosomes. Tmem97 and NPC1 co-immunoprecipitate and function in a manner that relies the Tmem97 ER-targeting motif, consistent with a role for Tmem97 as an ER-localized NPC1 chaperone protein.
Localized modulation of protein-protein interactions is a hallmark of oxysterol metabolite signaling.3,4,5,6 In the most well-established of these mechanisms, selective ligation of INSIG by 25-OHC induces its association with the ER-localized sensor SCAP, activating expression of cholesterol biosynthetic genes.47 OHC-mediated protein-protein interactions also regulate the degradation of HMG-CoA reductase,48 the affinity of adrenodoxin-CYP11 complexes in pregnenolone synthesis,49 tumor-associated dimerization of surface receptors,50 and other processes. OHC regulation of protein activity extends to soluble and membrane-associated proteins, including members of the OSBP/OSBPL, StAR/StAr-related lipid transfer domain-containing, and Aster protein families, and nuclear hormone receptors such as LXRs, RORɣ, and ERα/β.4,5
Based on the pervasive examples of oxysterol-regulated protein-protein interactions, we hypothesized that ligation of Tmem97 by 20(S)-OHC may influence its association with NPC1. To test whether binding of 20(S)-OHC to Tmem97 affects its physical interaction with NPC1, we examined co-immunoprecipitation of Tmem97 and NPC1 in cells treated with varying concentrations of 20(S)-OHC. To avoid complications associated with OHC-induced upregulation of Tmem97 via SREBP,39 we overexpressed epitope-tagged Tmem97 and performed our experiments in HEK293T cells, which express robust levels of endogenous NPC1. Incubation of Tmem97-expressing cells with 20(S)-OHC followed by capture of NPC1 using Tmem97 as bait evidenced a robust increase in Tmem97:NPC1 association at 10 μM (Fig. 6a). Likewise, incubation of Tmem97-expressing cells with 10 μM 20(S)-OHC, and capture of Tmem97 using endogenous NPC1 as bait showed a significant enhancement of the Tmem97:NPC1 interaction (Fig. 6b). Importantly, whole-cell Tmem97:NPC1 ratios were unaffected by 20(S)-OHC, demonstrating that 20(S)-OHC selectively impacts the Tmem97:NPC1 interaction (Fig. 6a,b). As observed previously, overexpression of Tmem97 had no effect on overall cholesterol distributions, as determined by filipin staining,40 in either the absence or presence of 20(S)-OHC. The observation that 20(S)-OHC selectively influences Tmem97:NPC1 association without changing bulk cellular properties is consistent with a localized interaction between Tmem97 and NPC1.
Fig. 6: 20(S)-OHC enhances the interaction between Tmem97 and NPC1.
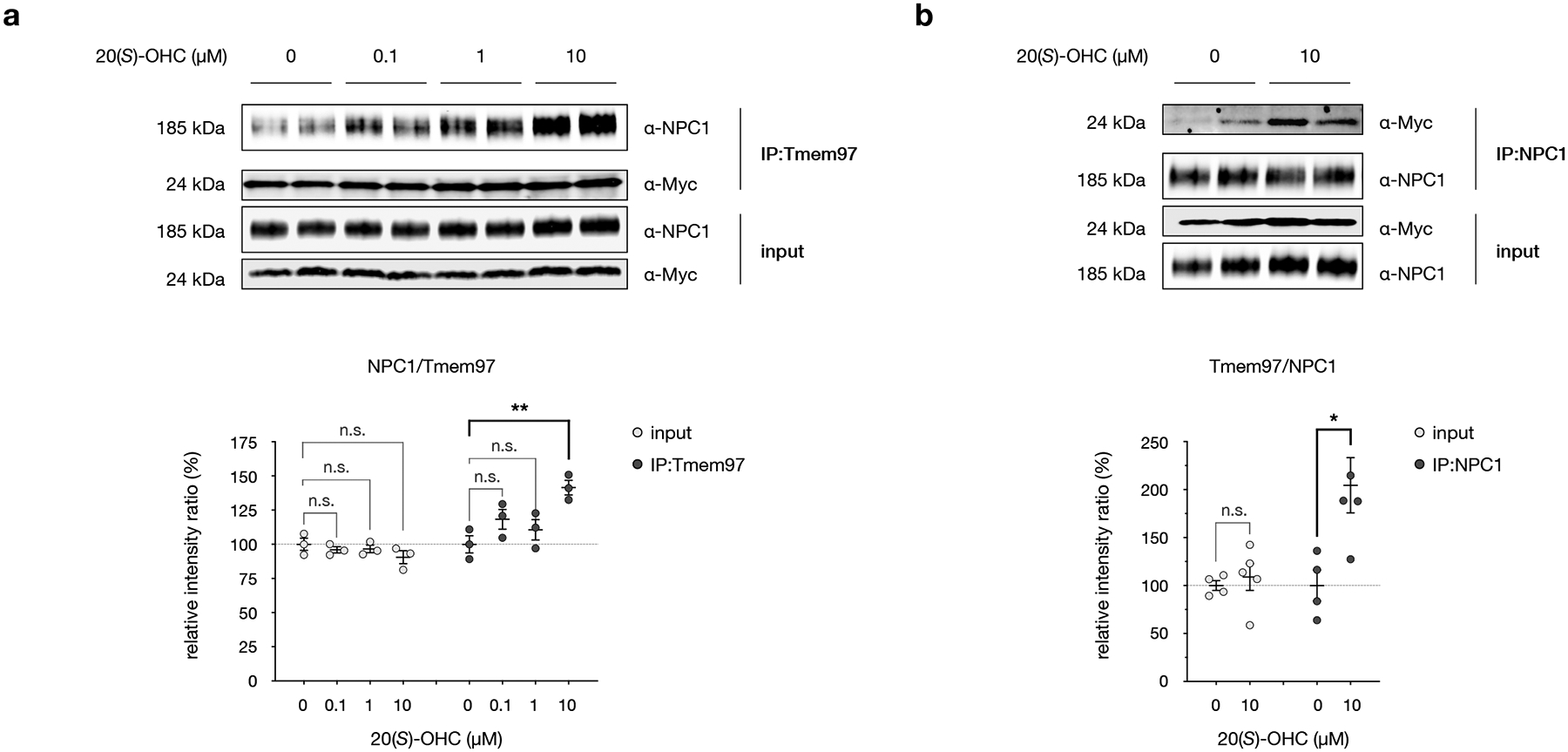
a, Top: Western blot analysis of the Tmem97:NPC1 interaction in HEK293T cells overexpressing Tmem97-Myc-FLAG. Cells were treated with indicated concentrations of 20(S)-OHC for 24 h, then endogenous NPC1 was captured from cell lysates using overexpressed Tmem97 as bait. Bottom: Quantitative Western blot analysis of NPC1 and Tmem97 in the sample input and after co-immunoprecipitation. Data are the average of 3 biological replicates ± s.e.m. Statistical significance, evaluated by two-way ANOVA with Dunnett’s multiple comparisons test, is shown, **Padj = 0.0056; n.s. = not significant.
b, Top: Western blot analysis of the Tmem97:NPC1 interaction in HEK293T cells overexpressing Tmem97-Myc-FLAG. Cells were treated with DMSO or 10 μM 20(S)-OHC for 24 h, then overexpressed Tmem97 was captured from cell lysates using endogenous NPC1 as bait. Bottom: Quantitative Western blot analysis of overexpressed Tmem97 and NPC1 in the sample input and after co-immunoprecipitation. Data are the average of 4 biological replicates ± s.e.m. Statistical significance, evaluated by a two-tailed unpaired t-test between indicated conditions, is shown, *p = 0.0217; n.s. = not significant.
Compellingly, other reports have demonstrated that OHCs act as chaperones for NPC1, although the exact mechanism of the OHC:NPC1 interaction remains undetermined.17 Our results raise the intriguing possibility this chaperoning activity may in part be mediated by a ternary interaction between OHCs, Tmem97, and NPC1 in a mechanism reminiscent of the 25-OHC:INSIG:SCAP interaction. We note that our studies are not without limitations: overexpressed Tmem97 may not fully recapitulate the behavior of endogenous Tmem97, and the relatively high 20(S)-OHC concentrations required to effect Tmem97:NPC1 perturbations might arise from the difference between applied and subcellular OHC concentrations. To more precisely examine local phenomena, we are currently extending our use of probe 2 as an imaging agent to visualize cellular interactions between 20(S)-OHC, Tmem97, and NPC1. Further studies to assess localized cholesterol concentrations, 20(S)-OHC-regulated trafficking and/or modification of NPC1, and structural features of the Tmem97:NPC1 interaction in cholesterol regulation are underway.
Discussion
While cholesterol is the most abundant lipid present in mammalian cell membranes, OHCs are transient species that exist at low concentrations in cells. Additional hydroxylation of OHCs relative to cholesterol confers these molecules with privileged access to cholesterol binding sites and membrane environments, where their presence can profoundly disrupt locales and resident proteins. The activity of OHCs in cells is a function of their affinity for specific receptors and their constellation of cellular targets. Well-known for their signaling roles, the complete set of OHC binding remain virtually unknown, and therapeutic opportunities associated with their direct targets and functions are largely untapped.
In this work, we describe our design, synthesis, and application of a chemoproteomics OHC probe to fingerprint OHC interactions in live mammalian cells. By incorporating alkyne and diazirine functional groups within the signaling molecule 20(S)-OHC, we establish a probe that precisely mimics the natural molecule and preserves its signaling activity in cells. We create a reproducible, quantitative protocol for competing targets of 2 in the native environments and perform multiplexed, proteome-wide analysis. We reveal OHC targets on a spectrum of competability that have functional roles in signal transduction, protein trafficking, and membrane lipid metabolism, and we identify specific processes in immune response and cancer that are functionally integrated with OHC target proteins. We demonstrate that the orphan receptor Tmem97 (also known as the σ2 receptor) is a stereo- and regioselective target of OHCs in live cells, evidencing its functional roles in NPC1-mediated cholesterol metabolism and providing a new template to design molecules that intervene in its functions.
We note that the membrane proteome examined in this study excludes nuclear hormone receptors that bind OHCs; we are currently profiling the nuclear fraction to quantify OHC engagement with known targets and yet-unidentified OHC receptors. In the future, chemoproteomics analysis of sterol and other hydrophobic metabolites will benefit from new reagents for small molecule delivery to cells, such as synthetic LDL particles and next-generation cavitands. Improved methods for membrane protein solubilization, combined with powerful advances in liquid chromatography/mass spectrometry of hydrophobic peptides, can likewise enhance the resolution and detection of target proteins. Critically, enhanced coverage hinges on fundamental improvements in diazirine photocrosslinking chemistry and continued optimization of click labeling protocols. In synergy with appropriate data analysis, this multiplexed chemoproteomic profiling provides a powerful platform to inform target prediction and aid the design of metabolite-inspired drugs. More broadly, the identification of metabolite targets in cells integrates chemical structure and biological activity, offering new dimensions to our understanding of human health.
Methods
Chemical synthesis
Synthetic procedures and spectral data for probe 2 and 20(R)-OHC are provided in Supplementary Note 1.
In vitro photoactivation
A solution of 10 mM probe 2 in DMF in a 0.2 mL PCR tube cut to a height of ~0.5 cm was irradiated using a UV crosslinker (Thermo Fisher, 13-245-221) equipped with a bank of five light tubes (F8T5/BLB, wavelength range 330–410 nm, peak wavelength 368 nm). At each time point, a 1 μL aliquot of the irradiated probe solution was transferred to a NanoQuant plate (Tecan). Absorbance was measured using a Spark M10 multimode plate reader (Tecan) with scanning from 280 nm to 550 nm.
Cell culture
NIH-3T3 cells (ATCC, CRL-1658) and Shh-LIGHT2 cells51 were cultured in high-glucose DMEM (Gibco, 11965118) containing 10% bovine calf serum (CS, ATCC, CRL-1658), 100 U/mL of penicillin-streptomycin (Gibco, 15140163), and 1 mM sodium pyruvate (Thermo Fisher, 11360070). HEK-293T cells (ATCC, CRL-3216) were cultured in high-glucose DMEM (Gibco, 11965118) containing 10% fetal bovine serum (FBS, Gibco, 26140079), 100 U/mL of penicillin-streptomycin, 1 mM sodium pyruvate, and 2 mM L-glutamine (Oakwood, M02960). Cells were seeded at an initial confluence of ~20%, passaged every 3–4 days upon reaching 75–85% confluence, and maintained in an atmosphere of 5% CO2 and 95% humidity at 37 °C.
Gel electrophoresis
Samples were loaded on a 4–15% PROTEAN TGX Stain-Free Protein Gel (Bio-Rad, 4568083 (Mini) or 5678084 (Midi)) with Tris/Glycine/SDS running buffer (Bio-Rad, 1610772), and proteins were resolved at a current of 125 V (Mini) or 150 V (Midi) for 1 h at room temperature. In-gel fluorescence and stain-free total protein signal were detected using a Bio-Rad Chemidoc MP Imaging System. Band intensities were quantified using Bio-Rad Image Lab Software v6.0.
Western blot analysis
Proteins resolved by SDS-PAGE were transferred to a PVDF membrane (0.22 μm, Bio-Rad, 1620177) using a Bio-Rad Trans-Blot Turbo Transfer System (Bio-Rad, 1704273). For detecting biotinylated proteins, membranes were blocked in 0.2 % I-Block (Thermo Fisher, T2015) in PBST overnight at 4 °C before incubating with IRDye 800CW Streptavidin (Licor, cat # 926–32230, 1:10,000) in 0.2% I-Block in PBST at room temperature for 1 h or at 4 °C overnight. For detection using primary antibodies, membranes were blocked in 2% non-fat milk (Nestle, Carnation Instant Nonfat Dry Milk) in PBST for 1 h at room temperature, incubated with primary antibody in 2% non-fat milk in PBST at 4 °C overnight, rinsed 3× with PBST, incubated with secondary antibody in 2% non-fat milk in PBST for 1 h at room temperature, and rinsed 3× with PBST. HRP-conjugated secondary antibodies were developed using Clarity Western ECL Substrate (Bio-Rad, 170506) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher, 34096). Fluorescence and chemiluminescence signals were collected using a Bio-Rad Chemidoc MP Imaging System. Band intensities were quantified using Bio-Rad Image Lab Software v6.0.
Antibodies
Rabbit monoclonal anti-Cav1 (clone D46G3, Cell Signaling 3267, 1:1000), Mouse monoclonal anti-Ephx1 (clone 17, Santa Cruz sc-135984, 1:200), Rabbit polyclonal anti-Gpr107 (Proteintech 25076–1-AP, 1:600), Goat polyclonal anti-Gas1 (Novus AF2644, 1:1000), Rabbit polyclonal anti-Reep5 (Proteintech 14643–1-AP, 1:5000), Rabbit polyclonal anti-Tmem199 (Sigma-Aldrich HPA027051, 1:1000), Rabbit polyclonal anti-Tmem97 (Proteintech 26444–1-AP, 1:500), Rabbit polyclonal anti-VDAC (Cell Signaling 4866, 1:1000),Mouse monoclonal anti-Na+/K+-ATPase α1 (clone 464.6, Novus NB300–146, 1:5000), Mouse monoclonal anti-β-Actin (clone C4, Santa Cruz sc-47778, 1:750), IRDye 800CW Streptavidin (Licor 926–32230, 1:10000), Rabbit polyclonal anti-NPC1 (Novus NB400–148, 1:1500), Mouse monoclonal anti-c-Myc (clone 9E10, Sigma-Aldrich M4439, 1:2500), Mouse monoclonal anti-Flag M2 (clone M2, Sigma-Aldrich F1804, 1:5000), Mouse monoclonal anti-FLAG (clone H-5, Santa Cruz sc-166355, 1:1000), HRP-conjugated donkey anti-mouse (Jackson ImmunoResearch 715-035-151, 1:10000), HRP-conjugated donkey anti-rabbit (Jackson ImmunoResearch 715-035-152, 1:10000), HRP-conjugated donkey anti-goat (Jackson ImmunoResearch 715-035-003, 1:10000)
Shh-LIGHT2 assays
Shh-LIGHT2 cells were seeded in 96-well plates at a density of 3.5 × 104 cells/well. After 24 h, growth media was replaced with serial dilutions of 2, 20(S)-OHC, 20(R)-OHC, or SAG (CAS no. 912545-86-9, Carbosynth, FS76762) in low-serum media (phenol red-free high-glucose DMEM, Gibco, 21063045) containing 0.5% bovine calf serum (CS, ATCC, CRL-1658), 100 U/mL of penicillin-streptomycin (Gibco, 15140163), and 1 mM sodium pyruvate) at a normalized DMSO concentration of 0.2%. After 30 h, cells were washed with PBS and treated with Passive Lysis Buffer (20 μL/well, Promega, E1941) at room temperature for 15 min with rocking. 10 μL lysate from each well was transferred to a white-bottomed assay plate (Corning, 3912) for Firefly and Renilla luciferase measurements using a Dual Luciferase Reporter kit (Promega, E1960) on a Tecan Spark M10 multimode plate reader. Gli activity was calculated as the ratio of Firefly/Renilla luciferase signal and percent Gli activation was assessed relative to DMSO-only control values. Dose-response curves were generated using GraphPad Prism software.
Preparation of MβCD complexes
20(S)-OHC, 20(R)-OHC, 25-OHC, cholesterol, and inositol were dissolved in MeOH-CHCl3 (2:1) in a glass vial. Solvent was removed under a stream of nitrogen to yield a thin film of solid on the bottom of the vial. A solution of 37.56 mM methyl-β-cyclodextrin (MβCD) in phenol red-free DMEM was added to a final concentration of 3.76 mM (10:1 molar ratio of MβCD:sterol), and the mixture was bath sonicated to obtain a clear solution (30~60 min). The solution was sterile-filtered through a 0.22 μm PES filter (CELLTREAT, 229746), aliquoted into glass vials, and stored at −20 °C until use.
Probe incubation and crosslinking
For in-gel fluorescence analysis, NIH-3T3 cells were seeded at a density of 3 × 105 cells/well in 6-well plates (Corning, 353046) and cultured until reaching 80–90% confluence (~24 h). For Western Blot analysis, cells were seeded at a density of 3 × 105 cells/well in a 10 cm dish (Corning, 353003) and cultured until reaching 80–90% confluence (~72 h). For mass spectrometry analysis, NIH-3T3 cells were seeded at a density of 8 × 105 cells cells/well in 15 cm dish (Corning, 353025) and cultured until reaching 80–90% confluence (~72 h).
For target profiling of 2 in the absence of MβCD, stock solutions of compounds in DMSO or DMSO alone were diluted in serum-free media to the concentrations indicated, with DMSO concentrations normalized to 0.01%. Cells were incubated with compound-containing media at 37 °C for 30 min, washed once with ice-cold PBS, and maintained in PBS on ice. Lids were removed from plates and cells were irradiated for 5 min on ice with 368 nm UV light using a UV-crosslinker (Thermo Fisher, 13-245-221). For no-UV control experiments, probe-treated cells were incubated on ice for 5 min in ambient light.
For target profiling of 2 or PhotoClick Cholesterol (Avanti Polar Lipids, 700147P) in the presence of MβCD, stock solutions of competitor compounds pre-complexed with MβCD were diluted in serum-free media to the concentrations indicated and a normalized concentration of 0.5 mM MβCD. Cells were incubated at 37 °C for 1 h before adding a 10× solution of probe 2 or DMSO to a normalized concentration of 0.01% DMSO. Cells were incubated at 37 °C for another 30 min, then rinsed and irradiated as above.
Cell harvest and subcellular fractionation
After crosslinking, cells were detached by scraping in cold PBS and collected by centrifugation at 750 × g for 5 min at 4 °C. Cell pellets were washed 2× by resuspension in ice-cold PBS and centrifugation, then snap-frozen in liquid N2 and stored at −80 °C before further processing. Subcellular fractionation was conducted according to Baghirova et al.52 Namely, cell pellets were resuspended in ice cold lysis buffer A (50 mM HEPES pH 7.4, 150 mM NaCl, 1 M hexylene glycol, 25 μg/mL digitonin, and 1× cOmplete EDTA-free Protease Inhibitor Cocktail (Roche, 11836170001)) and incubated using an end-over-end rotator for 10 min at 4 °C. Lysate was centrifuged at 2000 × g for 10 min at 4 °C and the supernatant was collected as the cytosolic fraction. Pellets were suspended in ice cold lysis buffer B (50 mM HEPES pH 7.4, 150 mM NaCl, 1 M hexylene glycol, 1% IGEPAL-CA630, and 1× cOmplete EDTA-free Protease Inhibitor Cocktail) and resuspended by vortexing. The suspension was incubated on an end-over-end rotator for 30 min at 4 °C and centrifuged at 7000 × g for 10 min at 4 °C. The supernatant was collected as the membrane fraction. The remaining pellet was suspended in ice cold lysis buffer C (50 mM HEPES pH 7.4, 150 mM NaCl, 1 M hexylene glycol, 0.5% sodium deoxycholate, 0.1% SDS, 25U/mL benzonase, and 1× cOmplete EDTA-free Protease Inhibitor Cocktail), resuspended by vortexing, and incubated using an end-over-end rotator for 20 min at 4 °C. The suspension was pelleted at 7800 × g for 10 min at 4 °C, and the resulting supernatant was collected as the nuclear fraction. Protein concentrations of each fraction were determined using the BCA protein assay (Thermo Fisher, 23227).
Click reaction
Membrane fractions from all samples in each experiment were normalized to the same protein concentration (~40–60 μg for gel-based analysis, ~600–900 μg for mass spectrometry analysis, ~100 μg for Western blot analysis) by dilution in fractionation buffer B. A solution of azide in DMSO was added to a concentration of 25 μM for TAMRA azide (Click Chemistry Tools, AZ109) or 100 μM for picolyl biotin azide (Click Chemistry Tools, 1167), followed by a freshly prepared catalyst mixture containing TBTA (102 μM, from a 1.7 mM stock in 4:1 DMSO:t-BuOH), CuSO4 (1 mM, from a 50 mM stock in H2O), and TCEP (1 mM, from a 50 mM stock in PBS, adjusted to pH 7~8 immediately before use). After incubating on an end-over-end rotator for 1 h at room temperature, each reaction was diluted with 4 volumes of cold acetone (–20 °C), vortexed briefly, and maintained at −20 °C for 30~60 min. Precipitate was pelleted at 20,000 × g for 10 min at 4 °C and the supernatant was discarded. The pellet was resuspended in acetone by brief bath-sonication, and maintained at −20 °C for 10~30 min, and centrifuged an additional 2×. After removing the last supernatant, the pellet was air dried at room temperature for 5~10 min.
In-gel fluorescence analysis
For fluorophore-conjugated samples, dried pellets from acetone precipitation were resuspended in equal volumes of 1% SDS in PBS and 1% IGEPEL CA-630 in PBS containing 1× cOmplete EDTA-free Protease Inhibitor Cocktail, and 1 × SDS sample buffer (from 6 × SDS sample buffer, which contains 300 mM Tris-HCl pH 6.8, 50% (v/v) glycerol, 12% (w/v) SDS, 600 mM DTT, and 0.6 g/L bromophenol blue). Samples were bath sonicated for 3~5 min, incubated on a Thermomixer (50 °C, 30 min, 950 rpm, Eppendorf, 5350) and subjected to SDS-PAGE.
Streptavidin enrichment
For biotin-conjugated samples, dried pellets from acetone precipitation were resuspended in equal volumes of 1% SDS in PBS and 1% IGEPEL CA-630 in PBS (containing 1× cOmplete EDTA-free Protease Inhibitor Cocktail) by bath sonication (3~5 min) before diluting to a final concentration of 0.1 % SDS and 1% IGEPEL CA-630 in PBS (containing 1× cOmplete EDTA-free Protease Inhibitor Cocktail). The diluted solution was bath-sonicated for 3~5 min and centrifuged at 7,000 × g for 3 min at room temperature to pellet any insoluble proteins. The supernatant was transferred to a 1.7 mL tube containing streptavidin agarose (Thermo Fisher, 20349 pre-washed twice with PBS). The mixture was incubated on an end-over-end rotator overnight (12~16 h) at room temperature. The suspension was transferred to a Pierce centrifuge column (Thermo Fisher, 0.8 mL, 89868) and centrifuged at 1,000 × g for 1 min at RT. The flow-through was discarded, and the resin was washed 3× with wash buffer (1% IGEPEL CA630 and 0.1% SDS in PBS) and transferred to a new 1.7 mL tube in wash buffer. The suspension was centrifuged at 2,500 × g for 1 min at room temperature and the supernatant was discarded.
For SDS-PAGE/Western blot analysis, proteins were eluted by incubating the resin in 1× SDS sample buffer containing 2 mM biotin on a Thermomixer (50 °C, 30 min, 650 rpm). For mass spectrometry analysis, proteins bound to the resin were directly subjected to on-bead digestion.
Mass spectrometry sample preparation
On-bead digestion.
After enrichment, streptavidin agarose was resuspended in 6 M urea in PBS, reduced with 10 mM DTT for 1 h at room temperature, and alkylated with 25 mM iodoacetamide in the dark using an end-over-end rotator for 30 min at room temperature. Beads were pelleted at 2,500 × g for 1 min, the supernatant was removed, and the beads were washed 2× with PBS and resuspended in 2 M urea in PBS. Trypsin (1.5 μg, Promega, V5111) and 1 mM CaCl2 were added to the samples and digestion was performed on a Thermomixer (37 °C, 12–16 h, 650 rpm). The supernatant was collected and the beads were washed with water and 50% acetonitrile. The supernatant and washes were combined and concentrated to dryness using a CentriVac concentrator (Labconco, 7310022).
Sample desalting.
Dried peptides from the trypsin digest were resuspended in buffer A (98% H2O, 2% acetonitrile, 0.2% formic acid) and desalted using a StageTip (homemade from Empore C18 Extraction disks, 3M, 2215, 200 μL Pipette tip, ~20 μg capacity) pre-equilibrated with acetonitrile and then with buffer A. The sample-loaded StageTip was washed with buffer A and subsequently eluted with elution buffer 1 (50% acetonitrile, 50% H2O, 0.2% formic acid). For TMT labeling, peptides were further eluted with elution buffer 2A (80% acetonitrile, 20% H2O, 0.2% formic acid). For label-free quantification, peptides were further eluted with elution buffer 2B (75% acetonitrile, 25% H2O, 0.2% formic acid). Desalted samples were concentrated to dryness using a CentriVac concentrator and stored at −80 °C.
TMT labeling.
Dried, desalted peptides from the trypsin digest were resuspended in 50 μL of 100 mM triethylammonium bicarbonate (TEAB, pH 8.5) and 20 μL of a 20 μg/μL solution of TMT 6-plex (Thermo Fisher, 90061) or 10-plex isobaric label reagent (Thermo Fisher, 90110) dissolved in anhydrous acetonitrile. Labeling reactions were incubated at room temperature on a Thermomixer (room temperature, 2 h, 650 rpm). Label efficiency was checked by withdrawing a 0.4 μL aliquot from each sample within a single plex to ensure at least 98% labeling of all N-termini and lysine residues. Reactions were quenched with 4 μL of 5% hydroxylamine at room temperature for 15 min. TMT-labeled peptides from all samples were combined and concentrated to dryness using a CentriVac concentrator.
Sample desalting and TMT reagent removal.
The dried, combined, TMT-labeled peptides were resuspended in 200 μL buffer A (98% H2O, 2% acetonitrile, 0.2% formic acid) and desalted using an Agilent 1100 HPLC with an Optimized Technologies C4 peptide Macrotrap (3 × 8 mm; 200 μg maximum capacity). The mobile phase consisted of solvent A (98% H2O, 2% acetonitrile, 0.2% formic acid) and solvent B (95% acetonitrile/0.1% formic acid) delivered at a flow rate of 250 μL/min with the following gradient: 0–10 min, 0% B; 10–12 min, 0–85% B; 12–17 min, 85% B; 17–18 min, 85–90% B; 18–23 min 90% B; 23–24 min, 90–100% B; 24–30 min, 100% B (post time: 5 min, 0% B and 200 μL/min). The fraction between 12–15 min was collected and concentrated to dryness using a CentriVac concentrator and stored at −80 °C before LC-MS/MS analysis at the Beckmann Institute Proteome Exploration Laboratory at Caltech. Full details of LC-MS/MS and LC-MS/MS/MS are provided in Supplementary Note 2.
TMT labeling quantification.
Proteomics data analysis was performed in Proteome Discoverer 2.4 (Thermo Scientific). Spectra were filtered for HCD spectra with signal-to-noise > 1.5. Spectra were searched against Reviewed proteins in the Uniprot mouse proteome (UP000000589, downloaded July 2020) and common contaminant proteins (Maxquant contaminants database, downloaded July 2020) using the SEQUEST HT search algorithm. Full search parameters are provided in Supplementary Note 2.
Data analysis.
Non-normalized abundance values of proteins from Proteome Discoverer were log2-transformed before further analysis. Missing values were imputed within treatment groups using the MinProb (Probabilistic Minimum Imputation) method embedded in the imputeLCMD R package.53 Log2 fold changes were calculated from the differences in the means of replicates between groups. The statistical significance of differences between groups was assessed using a two-tailed unpaired t-test.
Analysis of probe-labeled YFP-mSmo in HEK-293T cells
Protein from the membrane fraction of each sample was normalized to ~60 μg and transferred to a 1.7 mL tube containing pre-washed GFP-Trap® magnetic agarose beads (Chromotek, gtma, pre-washed twice with PBS). The suspension was incubated on an end-over-end rotator for 3 h at 4 °C. The beads were separated with a magnetic rack (Thermo Fisher, DynaMag-2 Magnet, 12321D) and washed 3× with ice-cold 0.1% IGEPAL CA-630 in PBS. The beads were resuspended in 1% SDS in PBS and subjected click reaction using the conditions described above. After the reaction, beads were separated with a magnet and washed three times with PBS. Proteins were eluted by resuspending the beads in 1% SDS in PBS and 2× SDS sample buffer, bath-sonication (3~5 min), and incubation using a Thermomixer (50 °C, 20 min, 450 rpm). The eluate was was transferred to a new tube and the elution was repeated once. The eluate fractions were combined, YFP-Smo and GFP expression was assessed by SDS-PAGE and in-gel fluorescence, and proteins were transferred to a PVDF membrane for detection of biotinylated proteins.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed using GSEA desktop version 4.1.0.54 Datasets with log2-transformed fold changes were analyzed using the C5 (gene ontology), C6 (oncogenic signatures) or C7 (immunologic signatures) gene set collections in the Molecular Signatures Database (MSigDB v. 7.2). The number of permutations was set to 1000 for p-value calculation and permutation type was set to gene_set. All basic and advanced fields were set to default.
Generation of Tmem97 knockout cell lines
Guide plasmid cloning.
A guide RNA target sequence in exon 1 of mouse Tmem97 was identified using Benchling tools.55 Oligonucleotides with the sgRNA sequence and adaptor overhangs were ordered from IDT and annealed in annealing buffer (100 mM potassium acetate, 30 mM HEPES-KOH pH 7.4, 2 mM magnesium acetate) for 5 min at 95 °C with cooling to room temperature over 45 min. Annealed oligos were phosphorylated using T4 polynucleotide kinase (New England Biolabs, M0201) and ligated into the BbsI restriction site of the linearized pU6-(BbsI)_CBh-Cas9-T2A-mCherry plasmid (Addgene, 64324) using T4 DNA ligase (New England Biolabs, M0202). Oligonucleotide sequences are provided in Supplementary Table 2.
NIH-3T3 cell guide transfection and FACS.
NIH-3T3 cells were seeded at a density of 1.5 × 106 cells/10 cm plate 24 h before transfection. After reaching approximately 80~85% confluence, cells were transfected with 14 μg plasmid DNA, which was precomplexed with Lipofectamine 2000 (Thermo Fisher, 11668019) at a ratio of 1:2.5 DNA:Lipo2000 (w/w) in Opti-MEM reduced serum media (Thermo Fisher, 51985034) for 10 min before adding dropwise to the cells. Cells were cultured for an additional 42–48 h, trypsinized, and sorted by mCherry fluorescence on a Sony SY3200 Cell Sorter at the Caltech Flow Cytometry Core. Cells in the top 1% mCherry fluorescence were re-seeded into 96-well plates to achieve ~1 cell/well and expanded.
TIDE analysis of knockout efficiency.
Genomic DNA was extracted from cells using the DNeasy blood and tissue kit (QIAGEN, 69504) according to manufacturer instructions. The primers for TIDE analysis were designed according to protocol recommendations56 (see Supplementary Table 2 for primer sequences). PCR reactions were performed on 50 ng isolated genomic DNA extract using Phusion High-Fidelity DNA Polymerase in Phusion HF buffer (New England Biolabs, M0531) in a final volume of 25 μL. PCR products were purified using QIAquick PCR Purification Kit (QIAGEN, 28104) according to manufacturer instructions and analyzed by Sanger sequencing. The publicly available TIDE program was used to determine knockout efficiency for each cell line.57
Site-directed mutagenesis
Myc-DDK-tagged mouse transmembrane protein 97 (Tmem97, NP_133706) in the pCMV6 vector (Tmem97-Myc-FLAG) was obtained from Origene (MR201535) and used for site-directed mutagenesis to create the E61A, F88A, W95A, Y150A, Y150F, and Y150S mutants. PCR reactions were performed using Phusion High-Fidelity 2X Master Mix (New England Biolabs, M0531) and parent constructs were digested with DpnI (New England Biolabs, R0176). All primer sequences are available in Supplementary Table 2.
Overexpression of Tmem97 and mutants in HEK-293T cells
HEK-293T cells were seeded at a density of 2.5 × 105 cells/well in a 6-well plate 24 h before transfection. After reaching approximately 60~70% confluence, cells were transfected with 2 μg DNA precomplexed with polyethylenimine (PEI) at a ratio of 1:3 DNA:PEI (w/w) in Opti-MEM reduced serum media for 30 min. For mutant expression and probe 2 or PhotoClick Cholesterol labeling analysis, transfected cells were cultured for 42–48 h before compound treatment. For Tmem97-NPC1 co-immunoprecipitation, transfected cells were cultured for 24 h before compound treatment.
Co-immunoprecipitation of NPC1 and Tmem97-MycDDK
24 h after transfection, transfection media was replaced with fresh culture media containing the indicated concentrations of 20(S)-OHC diluted from DMSO stock at a normalized DMSO concentration of 0.04%. After an additional 24 h, cells were detached by scraping in cold PBS and collected by centrifugation at 750 × g for 5 min at 4 °C. Cell pellets were washed 2× by resuspension in ice-cold PBS and centrifugation. Washed cell pellets were immediately resuspended in 100 μL of ice cold lysis buffer (1% Triton X-100 and 2% IGEPAL-CA630 in PBS, supplemented with 1× cOmplete EDTA-free Protease Inhibitor Cocktail (Roche, 11836170001) and 1× PhosSTOP phosphatase inhibitor (Roche, 4906845001)), gently pipetted up and down, and incubated using an end-over-end rotator for 30 min at 4 °C. Lysate was centrifuged at 13,000 × g for 10 min at 4 °C, supernatants were collected, and protein concentration was determined using the BCA protein assay (Thermo Fisher, 23227). Total protein in each sample was normalized to 300~400 μg and diluted 1:1 with PBS supplemented with cOmplete EDTA-free Protease Inhibitor Cocktail. For input analysis, 15 μg of the lysate was removed, resuspended in 1× SB, incubated at 37 °C for 15 min, and stored at −80 °C until SDS-PAGE analysis.
For immunoprecipitation of Tmem97-Myc-FLAG and co-immunoprecipitation of endogenous NPC1, the diluted lysate was transferred to a 1.7 mL tube containing 25 μL Pierce Anti-DYKDDDDK magnetic agarose beads (ThermoFisher, A36798) pre-washed twice with 0.1% IGEPAL-CA630 in PBS. The suspension was incubated using an end-over-end rotator overnight at 4 °C. Beads were separated using a magnetic rack (Thermo Fisher, DynaMag-2 Magnet, 12321D) and washed 5× with cold 0.1% IGEPAL CA-630 in PBS. Washed beads were resuspended in 30 μL of 50 mM glycine (pH 2.8) and 2× SDS sample buffer and incubated using a Thermomixer (50 °C, 20 min, 950 rpm), then eluted proteins were resolved by SDS-PAGE.
For immunoprecipitation of endogenous NPC1 and co-immunoprecipitation of Tmem97-Myc-FLAG, 1 μg NPC1 antibody (Novus, NB400–148) was added to the diluted lysate and the mixture was incubated using an end-over-end rotator for 2 h at 4 °C. The mixture was transferred to a 1.7 mL tube containing 25 μL of Dynabeads Protein G magnetic beads (ThermoFisher, 10003D) pre-washed once with 0.1% IGEPAL-CA630 in PBS. The suspension was incubated using an end-over-end rotator overnight at 4 °C. Beads were separated using a magnetic rack (Thermo Fisher, DynaMag-2 Magnet, 12321D) and washed 5× with cold 0.1% IGEPAL CA-630 in PBS. Washed beads were resuspended in 30 μL of 50 mM glycine (pH 2.8) and 2× SDS sample buffer and incubated using a Thermomixer (50 °C, 20 min, 950 rpm), then eluted proteins were resolved by SDS-PAGE.
Tmem97 homology modeling, 20(S)-OHC docking, and molecular dynamics analysis
Homology modeling and ligand docking.
The Robetta prediction server was used to generate a homology model for TMEM97 based on the cryo-EM structure of human 3β-hydroxysteroid-Δ(8),Δ(7)-isomerase (NP_006570.1) complexed with the inhibitor U18666A (PDB 6OHT). Ligand-docking studies were performed using the Glide software in Maestro (Schrödinger, Version 12.5.139). Protein energy minimization and assignment of hydrogen bonds were performed using the OPSL3e force field. The 20(S)-OHC binding site was refined using the Receptor Grid Generation script in Glide.
Ten conformers of 20(S)-OHC were generated using the Python library ETKDG,58 included in the cheminformatics Python library rdkit.59 Docking poses for 20(S)-OHC conformers in the Tmem97 model were predicted using Glide via the Ligand Docking script. Rigid ligand sampling was specified to retain the ligand conformations obtained from ETKDG and standard precision was used. The ligand conformers were permitted to dock anywhere within the cavity or on the exterior surface of the protein, and up to five docking poses per ligand were generated. The docking pose with the best Glide score, which docked the ligand in the internal cavity of the protein, was selected for molecular dynamics analysis.
Molecular dynamics simulations.
The Tmem97 protein, POPC lipids, water molecules, and ions were parameterized as described previously.60 MD simulations were performed in a canonical ensemble, where the system was heated from 0 K to 310 K for 155 ps. The system was equilibrated by performing an MD simulation at a temperature of 310 K and pressure of 1 bar for 10 ns.
Extended Data
Extended Data Fig. 1: Endogenous Smoothened protein is inefficiently resolubilized after acetone precipitation.
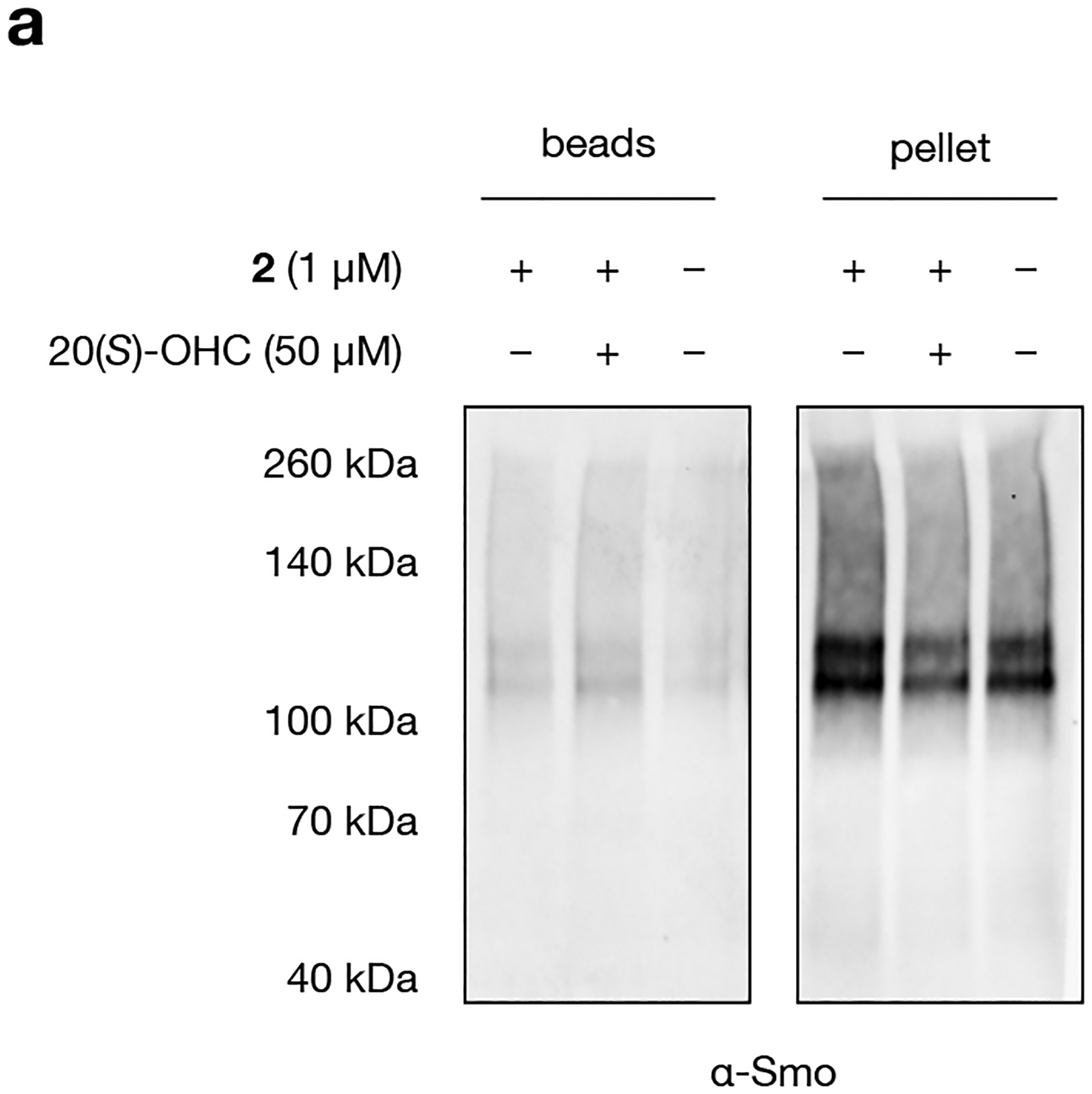
a, Western blot of Smoothened protein eluted from streptavidin beads after live-cell photocrosslinking to 2, biotinylation, acetone precipitation (to remove excess biotin), and streptavidin enrichment. Analysis of precipitate removed before incubation with streptavidin resin shows that Smoothened is largely insoluble in incubation buffer (1% SDS and 1% IGEPEL CA-630 in PBS) after acetone precipitation. The experiment was repeated five times independently with similar results.
Extended Data Fig. 2: The enrichment profile of 2 is independent of quantification method and mRNA transcript abundance.
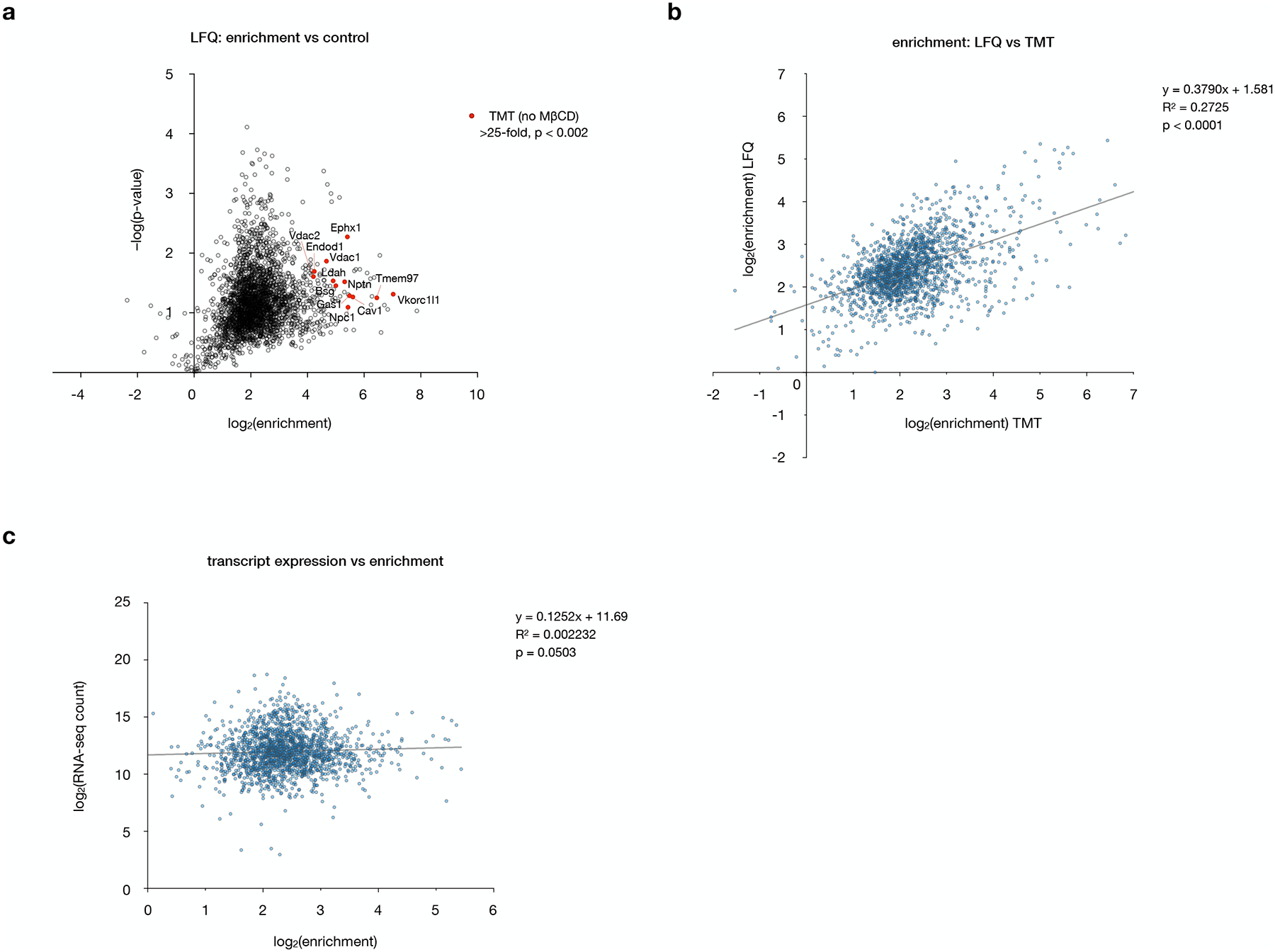
a, Volcano plot of proteins enriched by 2 versus DMSO alone, performed as in Fig. 3a but quantified by label-free quantification (LF Q) instead of TMT analysis. The top 12 enriched proteins identified by TMT analysis (labeled in Fig. 3b) are highlighted in red. Data represent two biological replicates consisting of matched experiments with 2 and DMSO. Statistical significance for each protein was calculated using a two-tailed paired t-test.
b, Plot of enrichment values from label-free versus TMT quantification, showing a positive correlation. Statistical significance, assessed by linear regression in GraphPad Prism, is shown (R2 = 0.2725, p < 0.0001).
c, Plot of RNA transcript abundances in NIH-3T3 cells (ref 16) versus enrichment of the corresponding proteins by TMT quantification, showing no significant correlation. Statistical significance, assessed by linear regression in GraphPad Prism, is shown (R2 = 0.002232, p = 0.0503).
Extended Data Fig. 3: Co-treatment with MβCD enhances 20(S)-OHC competition without altering 2-enrichment profiles.
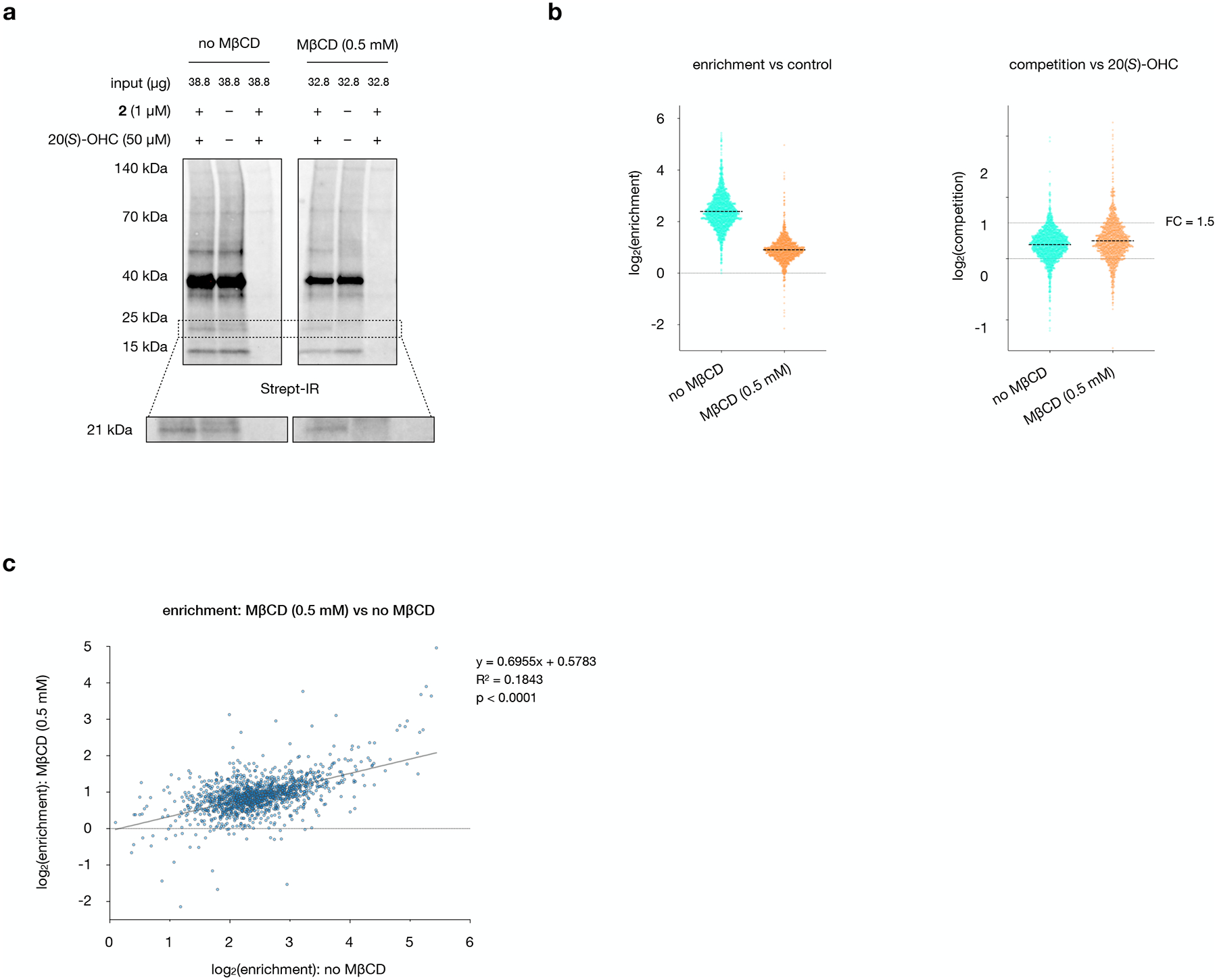
a, Streptavidin IRDye 800CW signal for biotinylated proteins in aliquots from mass spectrometry samples. MβCD reduces overall labeling, enhances competition of the probe-labeled 21 kDa band, and preserves equal intensity of non-competable bands in the presence and absence of 20(S)-OHC competitor. The experiments were repeated four times independently with similar results.
b, Left: Enrichment profiles in the presence and absence of MβCD show a uniform ~2.8-fold decrease. Right: Competition by 20(S)-OHC in the absence and presence of MβCD shows that a greater fraction of proteins are competable in the presence of MβCD. A horizonal line at 1.5-fold competition is provided for reference.
c, Plot of 2 enrichment values in the presence and absence of MβCD, demonstrating that enrichment profiles are not dramatically influenced by MβCD. Statistical significance, assessed by linear regression in GraphPad Prism, is shown (R2 = 0.1843, p < 0.0001).
Extended Data Fig. 4: Comparison of Western blot and mass spectrometry quantification.
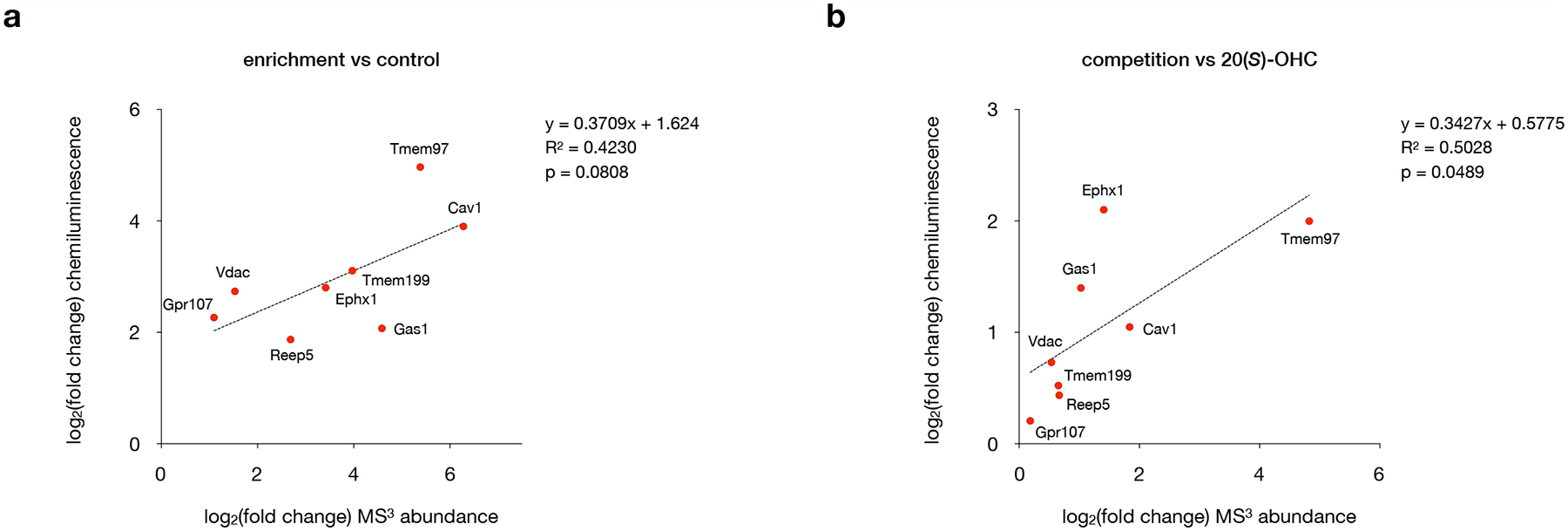
a, Log2(fold change) values for target protein enrichment obtained from mass spectrometry analysis versus quantitative Western blot. Statistical significance, assessed by linear regression in GraphPad Prism, is shown (R2 = 0.4230, p = 0.0808).
b, Log2(fold change) values for 20(S)-OHC competition obtained from mass spectrometry analysis versus quantitative Western blot. Statistical significance, assessed by linear regression in GraphPad Prism, is shown (R2 = 0.5028, p = 0.0489).
For a and b, data points represent quantification of one set of Western blots, where two independent experiments were performed with similar results, plotted against average mass spectrometry values from four biological replicates.
Extended Data Fig. 5: Tmem97 is competably labeled by probe 2 but not alternative sterol probes.
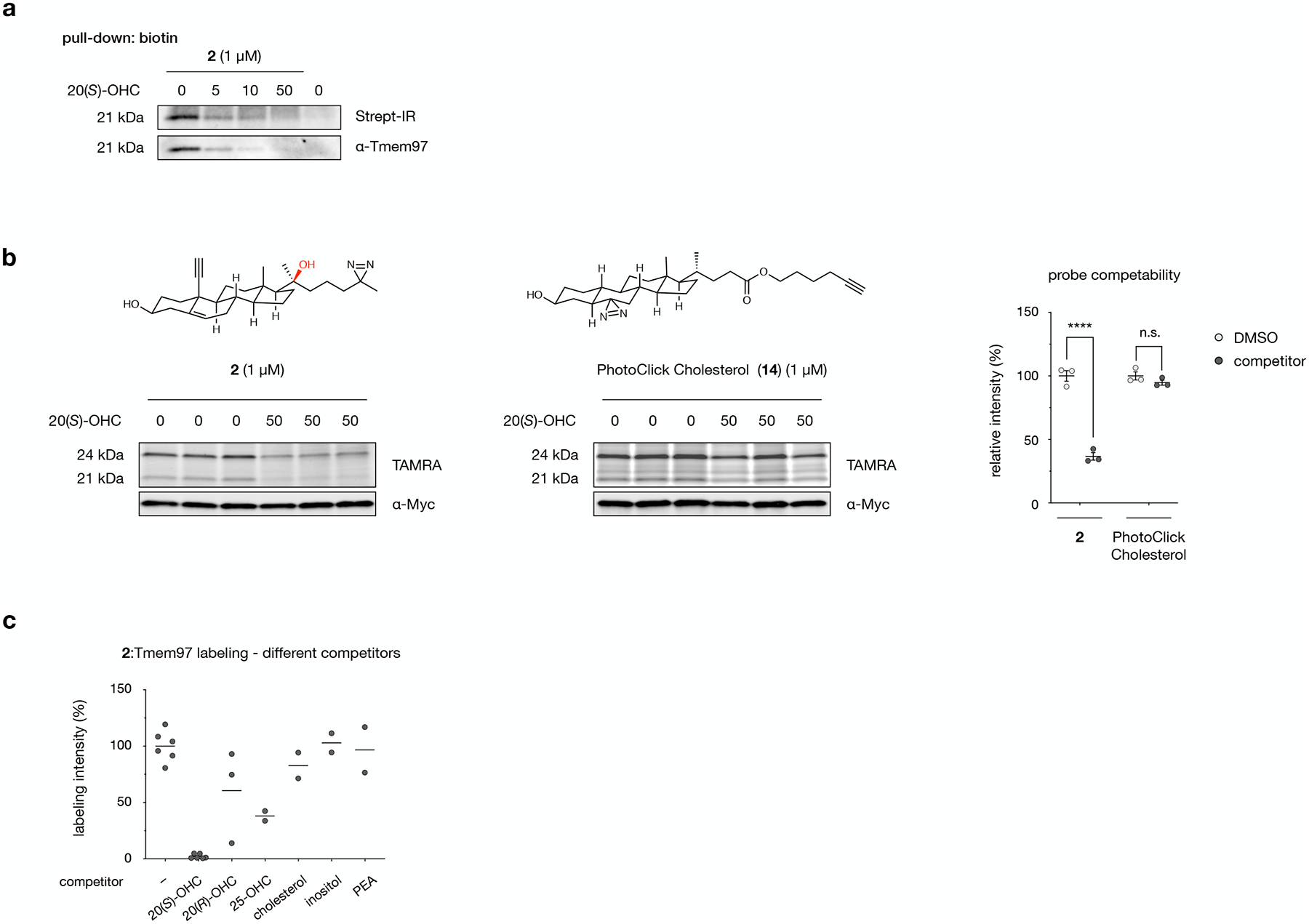
a, Crosslinking of probe 2 in NIH-3T3 cells, biotinylation of crosslinked proteins, streptavidin enrichment, and Western blot detection of endogenous Tmem97. Addition of 20(S)-OHC as a competitor reduces the intensity of whole-cell biotin labeling at 21 kDa and streptavidin-enriched Tmem97 in a dose-dependent manner. The experiment was repeated two times independently with similar results.
b, Crosslinking of probe 2 or PhotoClick Cholesterol in HEK293T cells overexpressing Tmem97-Myc-FLAG. Labeling of overexpressed Tmem97 is competable for probe 2 but not PhotoClick Cholesterol (14). Data are the average of 3 biological replicates ± s.e.m. Statistical significance, evaluated by two-tailed unpaired t-tests between indicated conditions, is shown, ***p = 0.0007; n.s. = not significant.
c, Quantitative fluorescence analysis of probe 2 labeling at 21 kDa by competitors shown in Fig. 5c, normalized to DMSO. Values are the average of 2 biological replicates.
Extended Data Fig. 6: Y150 mediates 20(S)-OHC:Tmem97 binding.
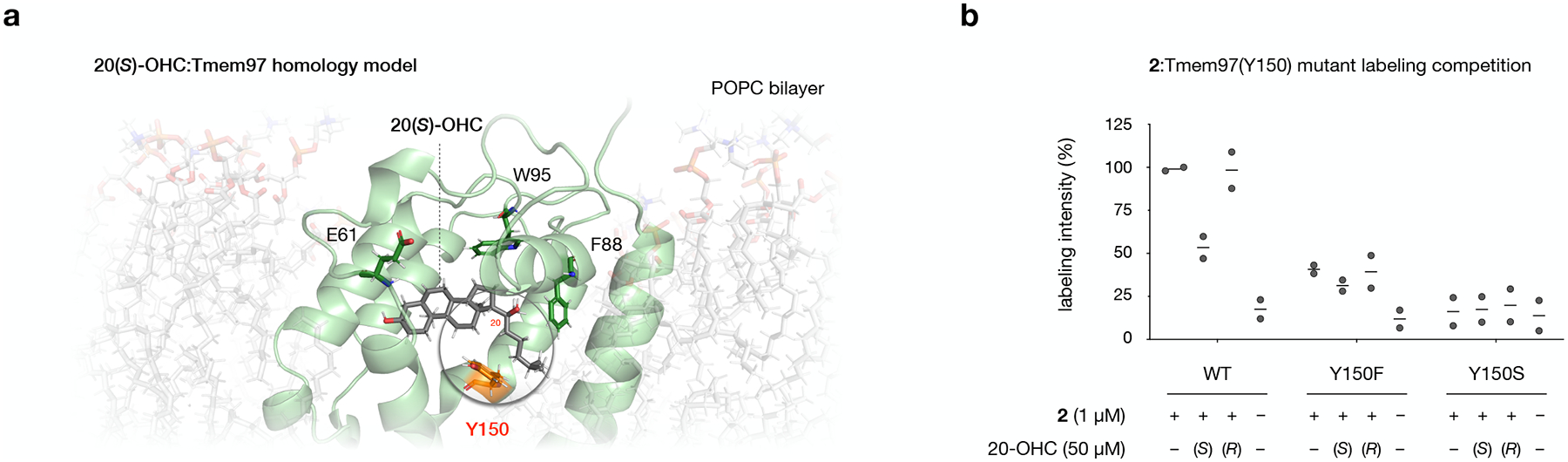
a, Homology model of mouse Tmem97 based on the cryo-EM structure of human EBP (PDB 6OHT) embedded in a phosphatidylcholine (POPC) bilayer. 20(S)-OHC docks in a central binding pocket.
b, Quantitative fluorescence analysis of probe 2 labeling with wild-type and Tmem97 and Y150 mutants shown in Fig. 5e, normalized to labeling of the wild-type Tmem97 construct in the presence of DMSO alone. Values are the average of 2 biological replicates.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank A. Li for synthesizing 20(R)-OHC intermediates, the Beckman Institute/Caltech Flow Cytometry Cell Sorting Facility for cell sorting, and A.K. Menon and F.R. Maxfield for helpful discussions. This work was funded by the Margaret Early Foundation Research Trust, (A.E.O.) a John Stauffer Charitable Trust SURF Fellowship (T.Z.), a William N. Lacy SURF fellowship (S.P.), and NIH training grant NIH GM07616 (G.C.Z.). The Proteome Exploration Laboratory was supported by NIH OD010788, NIH OD020013, the Betty and Gordon Moore Foundation through grant GBMF775 and the Beckman Institute at Caltech.
Footnotes
Competing interests
The authors declare no competing interests.
Code availability
Code used for mass spectrometry data analysis is available on GitHub: https://github.com/alex-ondrus/mass_spec_data_imputation.
Data availability
Raw mass spectrometry data for this study has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD027787. Raw gel and Western blot images are provided as Source Data files.
References
- 1.Maxfield FR & van Meer G Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol 22, 422–429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller WL & Auchus RJ The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine Rev 32, 81–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroepfer GJ Oxysterols: Modulators of cholesterol metabolism and other processes. Physiol Rev 80, 361–554 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Olkkonen Vesa M., M. V, Béaslas O & Nissilä E Oxysterols and their cellular effectors. Biomolecules 2, 76–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Yutuc E & Griffiths WJ Cholesterol metabolism pathways – are the intermediates more important than the products? FEBS J 288, 3727–3745 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bielska AA, Schlesinger P, Covey DF & Ory DS Oxysterols as non-genomic regulators of cholesterol homeostasis. Trends Endocrinol Metab 23, 99–106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nachtergaele S et al. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol 8, 211–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nedelcu D, Liu J, Xu Y, Jao C & Salic A Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol 9, 557–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T, Ondrus AE Structure, bonding, and photoaffinity labeling applications of dialkyldiazirines. Synlett 32, A–G (2021). [Google Scholar]
- 10.Li T et al. Ion mobility-based sterolomics reveals spatially and temporally distinctive sterol lipids in the mouse brain. Nature Commun 12, 4343 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin YY, Welch M & Lieberman S The detection of 20(S)-hydroxycholesterol in extracts of rat brains and human placenta by a gas chromatograph/mass spectrometry technique. J Steroid Biochem Mol Biol 85, 57–61 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Johnson JS et al. Novel oxysterols have pro‐osteogenic and anti‐adipogenic effects in vitro and induce spinal fusion in vivo. J Cell Biochem 112, 1673–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pratt HP, Keith J & Chakraborty J Membrane sterols and the development of the preimplantation mouse embryo. J Embryol Exp Morph 60, 303–19 (1980). [PubMed] [Google Scholar]
- 14.Jao CY et al. Bioorthogonal probes for imaging sterols in cells. Chembiochem 16, 611–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makino T, Shibata K, Rohrer DC & Osawa Y Steroid conformations in solid and solution: Stereoselectivity of Grignard addition to 20-oxo steroids. J Org Chem 43, 276–280 (1978). [Google Scholar]
- 16.Tran U, Zhang GC, Eom R, Billingsley KL & Ondrus AE Small molecule intervention in a Protein Kinase C–Gli transcription factor axis. ACS Chem Biol 15, 1321–1327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohgane K, Karaki F, Dodo K & Hashimoto Y Discovery of oxysterol-derived pharmacological chaperones for NPC1: Implication for the existence of second sterol-binding site. Chem Biol 20, 391–402 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Okamoto Y, Ninomiya H, Miwa S & Masaki T Cholesterol oxidation switches the internalization pathway of Endothelin Receptor Type A from caveolae to clathrin-coated pits in Chinese Hamster Ovary cells. J Biol Chem 275, 6439–6446 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Budelier MM et al. Photoaffinity labeling with cholesterol analogues precisely maps a cholesterol-binding site in voltage-dependent anion channel-1. J Biol Chem 292, 9294–9304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt HR & Kruse AC The molecular function of σ receptors: Past, present, and future. Trends Pharmacol Sci 40, 636–654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goo Y-H, Son S-H, Kreienberg PB & Paul A Novel lipid droplet–associated serine hydrolase regulates macrophage cholesterol mobilization. Arterio Thromb Vasc Biol 34, 386–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel‐Bindel U, Bentley P & Oesch F Endogenous role of microsomal epoxide hydrolase. Eur J Biochem 126, 425–431 (1982). [PubMed] [Google Scholar]
- 23.Schaafhausen A, Rost S, Oldenburg J & Müller C Identification of VKORC1 interaction partners by split-ubiquitin system and coimmunoprecipitation. Thromb Haemostasis 105, 285–294 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Suzuki J, Imanishi E & Nagata S Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc Natl Acad Sci USA 113, 9509–9514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fenech EJ et al. Interaction mapping of endoplasmic reticulum ubiquitin ligases identifies modulators of innate immune signalling. Elife 9, e57306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. P Natl Acad Sci USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peyrot SM et al. Tracking the subcellular fate of 20(S)-hydroxycholesterol with click chemistry reveals a transport pathway to the Golgi. J Biol Chem 289, 11095–11110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridgway ND 25-Hydroxycholesterol stimulates sphingomyelin synthesis in Chinese Hamster Ovary cells. J Lipid Res 36, 1345–1358 (1995). [PubMed] [Google Scholar]
- 29.Zidovetzki R & Levitan I Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochimica Et Biophysica Acta BBA - Biomembr 1768, 1311–1324 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen JC et al. TMEM199 Deficiency is a disorder of Golgi homeostasis characterized by elevated aminotransferases, alkaline phosphatase, and cholesterol and abnormal glycosylation. Am J Hum Genetics 98, 322–330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Kalken C et al. Cortisol is transported by the multidrug resistance gene product P-glycoprotein. Brit J Cancer 67, 284–289 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prokesch A et al. Arxes: Retrotransposed genes required for adipogenesis. Nucleic Acids Res 39, 3224–3239 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wierbowski BM et al. Hedgehog pathway activation requires coreceptor-catalyzed, lipid-dependent relay of the Sonic Hedgehog ligand. Dev Cell 55, 450–467.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rone MB et al. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol 26, 1868–1882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao L et al. REEP5 (Receptor Accessory Protein 5) acts as a sarcoplasmic reticulum membrane sculptor to modulate cardiac function. J Am Heart Assoc 7, e007205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou GL, Na S-Y, Niedra R & Seed B Deficits in receptor-mediated endocytosis and recycling in cells from mice with Gpr107 locus disruption. J Cell Sci 127, 3916–3927 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Zeng C, Riad A & Mach RH The biological function of sigma-2 receptor/TMEM97 and its utility in PET imaging studies in cancer. Cancers 12, 1877 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alon A et al. Identification of the gene that codes for the σ2 receptor. Proc Natl Acad Sci USA 114, 7160–7165 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartz F et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab 10, 63–75 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Ebrahimi-Fakhari D et al. Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells. Hum Mol Genet 25, 3588–3599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE & Cravatt BF Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods 10, 259–264 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morisaki M, Shikita M & Ikekawa N Side-chain cleavage of cholesterol by gas chromatography-mass spectrometry in a selected ion monitoring mode. Methods Enzymol 111, 352–364 (1985). [DOI] [PubMed] [Google Scholar]
- 43.Moebius FF et al. Histidine 77, glutamic acid 81, glutamic acid 123, threonine 126, asparagine 194, and tryptophan 197 of the human Emopamil Binding Protein are required for in vivo sterol Δ8−Δ7 isomerization. Biochemistry 38, 1119–1127 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Moebius FF et al. Histidine 77, glutamic acid 81, glutamic acid 123, threonine 126, asparagine 194, and tryptophan 197 of the human Emopamil Binding Protein are required for in vivo sterol Δ8−Δ7 isomerization. Biochemistry 38, 1119–1127 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Raman S et al. Structure prediction for CASP8 with all‐atom refinement using Rosetta. Proteins Struct Funct Bioinform 77, 89–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alon A et al. Crystal structures of the σ2 receptor template large-library docking for selective chemotypes active in vivo. bioRxiv (2021) doi: 10.1101/2021.04.29.441652. [DOI] [Google Scholar]
- 47.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS & Goldstein JL Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA 104, 6511–6518 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang TY, Limanek JS & Chang CC Evidence indicating that inactivation of 3-hydroxy-3-methylglutaryl coenzyme A reductase by low density lipoprotein or by 25-hydroxycholesterol requires mediator protein(s) with rapid turnover rate. J Biol Chem 256, 6174–6180 (1981). [PubMed] [Google Scholar]
- 49.Yablokov EO et al. Substrate-induced modulation of protein-protein interactions within human mitochondrial cytochrome P450-dependent system. J Steroid Biochem Mol Biol 208, 105793–105799 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Alpy F et al. Functional characterization of the MENTAL domain. J Biol Chem 280, 17945–17952 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Taipale J et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406, 1005–1009 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Baghirova S, Hughes BG, Hendzel MJ & Schulz R Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2, 440–445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.https://cran.rstudio.com/web/packages/imputeLCMD/index.html, accessed November 2020.
- 54.http://www.broad.mit.edu/gsea/, accessed November 2020.
- 55.http://benchling.com, accessed August 2020.
- 56.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.https://tide.nki.nl, accessed December 2020.
- 58.Riniker S & Landrum GA Better informed distance geometry: Using what we know to improve conformation generation. J Chem Inf Model 55, 2562–2574 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Landrum G RDKit: Open-Source Cheminformatics http://www.rdkit.org, accessed November 2020.
- 60.Mafi A et al. Hedgehog proteins create a dynamic cholesterol interface. PLOS ONE 16, e0246814 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data for this study has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD027787. Raw gel and Western blot images are provided as Source Data files.