

Abstract
Allosteric modulation of membrane receptors has been intensively studied in the past three decades and is now considered to be an important indirect mechanism for the control of receptor function. The allosteric site on the GABAA receptor is the target for the most widely prescribed sleep medicines, the benzodiazepines. Cinacalcet, an allosteric enhancer of the calcium-sensing receptor, is used to treat secondary hyperparathyroidism. Allosteric ligands might be especially valuable to control receptors for which the design of selective orthosteric agonists or antagonists has been elusive, such as muscarinic acetylcholine receptors.
Modulation of membrane receptor function is crucial for controlling cellular processes. An important mechanism of controlling receptor function is allosteric modulation, in which modulators bind to a regulatory site on a receptor distinct from the orthosteric site, which is the site of binding of the native ligand. Allosteric modulators induce conformational changes that profoundly influence the behavior of a membrane receptor in response to its native ligand [1–3]. There are positive, negative and neutral allosteric modulators of membrane receptors.
Christian Bohr [4] made the initial observation of the phenomenon of allostery. He carefully measured the oxygenation of hemoglobin and found that the binding curve was sigmoidal instead of hyperbolic, indicating cooperativity of binding of oxygen molecules to hemoglobin; he also found that carbon dioxide lowers oxygen affinity for hemoglobin. These properties make hemoglobin an efficient transporter of oxygen from the lungs to the tissues and of carbon dioxide from the tissues to the lungs.
Before the 1960s, hemoglobin was one of the few known examples of proteins that displayed allosteric properties. It was later demonstrated that many enzymes in bacteria and in higher organisms are subject to allosteric modulation. Researchers later discovered that in most allosteric proteins, indirect interactions between distinct specific binding sites account for the performance of their modulatory function [1]. An extension of the allosteric theory to membrane receptors was proposed in the late 1960s [3,5].
Although the history of allostery spanned over a century, it is still relevant to current research on protein function and signal transduction mechanisms [3]. Significant progress has been made in the investigation of allosteric modulation of protein action. The presence of allosteric sites on membrane receptors offers a novel pharmacological means of modulating receptor function. Allosteric sites have been found on ligand-gated ion channels (LGICs). An allosteric site on the γ-aminobutyric acid (GABA)A receptor provides the basis for the therapeutic effects of benzodiazepines [6]. Another germane example is the anticholinesterase inhibitor galantamine. In addition to its anticholinesterase activity, galantamine is also a positive allosteric modulator of α4β2 and α7 nicotinic receptors. Its successful use in the treatment of Alzheimer’s disease might be an impetus for further development of more-potent and selective allosteric modulators [7]. Allosteric sites have also been demonstrated in G-protein-coupled receptors (GPCRs) [2]. Cinacalcet,a positive allosteric modulator of the calcium-sensing receptor (CaR), has been successfully used for the treatment of secondary hyperparathyroidism [8,9].
Therapy using receptor agonists is often prone to side effects owing to the widespread distribution of the target receptor in the body. An advantage of a positive allosteric modulator of a membrane receptor over its native, orthosteric activator is that, in principle, greater selectivity can be achieved. The positive allosteric modulator would enhance the action of the native agonist but might have no effect of its own on the unoccupied receptor. Thus, the agonist effect, which might be insufficient in a particular disease state, might be magnified through allosteric modulation. The higher subtype selectivity commonly exerted by allosteric modulators, and the fact that the allosteric action is ideally coupled to the simultaneous presence of the endogenous ligand, both help to prevent over-dosage compared with the administration of a conventional nonselective orthosteric agonist.
GPCRs
GPCRs, which constitute the largest family of cell-surface receptors, are major targets of drugs currently in clinical use. The GPCRs display the characteristic motif of seven transmembrane helices (TMs) and thus are also referred to as 7TM receptors. The 7TM proteins account for ~4% of the human genome. Mammalian GPCRs can be divided into three major subfamilies: class A, rhodopsin-like receptors; class B, secretin-like receptors; and class C, metabotropic glutamate-like and pheromone receptors. The rhodopsin-like receptors represent the predominant class of GPCRs. Members of a particular subfamily feature a substantial degree of amino acid homology, whereas different subfamilies demonstrate very low amino acid homology. Among the class A GPCRs, only the crystal structure of bovine rhodopsin has been determined [10].
The orthosteric ligand-binding site of a GPCR would be located either among TM helices or within the extracellular domain [3,10,11]. The third intracellular loop and carboxy-terminal end could interact with a G protein. Originally, a single receptor unit was thought to mediate the allosteric interactions between a modulator and the receptor. However, recent experiments suggest that the active forms of GPCRs can occur as GPCR dimers [12]. A dimerized nature of GPCRs is possibly associated with a diversification in their interactions with different G proteins, as well as in their desensitization and sequestration properties. It can also be argued that the functional unit of a GPCR is monomeric rather than dimeric or oligomeric [13]. Indeed, dimerization does not seem to be required for functional coupling of some GPCRs to G proteins but rather for its intracellular trafficking [14,15]. Thus, it is possible that, at least in some cases, the monomer is sufficient as a functional unit for some of the class A GPCRs but this might not be the case for other classes of GPCRs. For example, a heterodimer is essential for the GABAB receptor to function properly [16]. Taste receptors also exhibit this property [16]. Evidence suggests that the T1R1 and T1R3 taste receptors, which respond to sweet taste, combine as one functional unit [17].
An increasing percentage of GPCRs has been found to be modulated allosterically by various compounds. These receptors include all five subtypes of muscarinic receptors [18], at least three of the four subtypes of adenosine receptors [19], dopamine receptors [20], several class B GPCRs [21] and class C GPCRs [11]. The flexible nature of the interactions between the receptors and various allosteric modulators, combined with the potential for subtype selectivity, makes allosteric sites attractive for therapeutic intervention [2,22]. An allosteric modulator of CaR, cinacalcet, has been successfully administered for the treatment of secondary hyperparathyroidism [8].
A detailed mathematical analysis of the action of allosteric modulators of GPCRs has been described by Christopoulos [3], and Birdsall and Lazareno [18]. The allosteric ternary complex model (ATCM) features a term for the degree of cooperativity (α). The cooperativity factor α>1 denotes a positive cooperativity; α<1 results in negative cooperativity; neutral cooperativity occurs in cases where α = 1. It should be noted that ATCM details the action of all allosteric modulators (not just enhancers) on the binding of orthosteric ligands to GPCRs but not on the signaling. The ATCM has been validated by direct binding measurements at an allosteric site on the muscarinic M2 receptor using an allosteric radioligand [23]. The model has recently been extended by Hall [24], Price et al. [25] and Ehlert [26] to accommodate the effects of allosteric modulators on efficacy. The effect of an allosteric modulator on agonist affinity and efficacy has been described in detail. A given allosteric modulator might have different effects on each of these parameters.
Class A GPCRs: rhodopsin-like receptors
Adenosine receptors
The A1 adenosine receptor (AR) was the first subtype in the AR family for which selective orthosteric and allosteric ligands were developed. The aminobenzoylthiophene derivative 2-amino-4,5-dimethyl-3-thienyl-[3-trifluoromethylphenyl]methanone (PD81723; Figure 1) was the first allosteric enhancer in the GPCR field, as originally observed by Bruns and Fergus [27]. They found that PD81723 enhances the binding of agonist radioligand [3H]cyclohexyladenosine to A1 ARs and decreases the rate of dissociation of [3H]cyclohexyladenosine from these receptors [27], suggesting that these compounds act at an allosteric site distinct from the orthosteric adenosine binding site to stabilize agonist-receptor-G-protein complexes. The aminobenzoylthiophenes were found to be highly selective for A1 ARs, with little or no effect on the binding of agonists to other ARs or other GPCRs [27].
FIGURE 1.
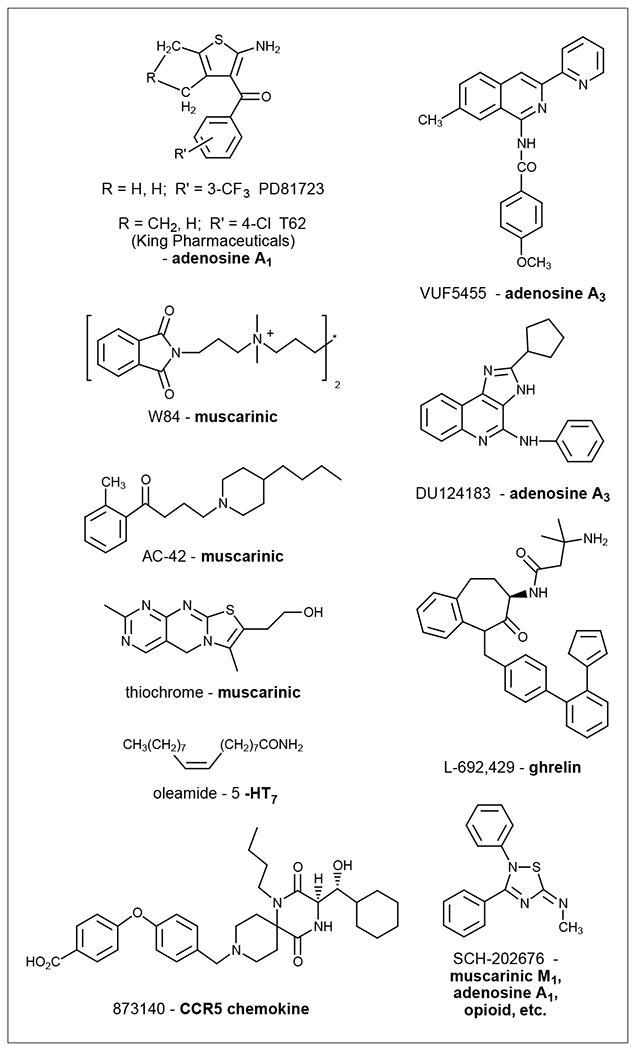
Structures of representative allosteric modulators for class A GPCRs that are in clinical trials or used as research tools.
The behavior of A1 AR enhancers has traditionally been complicated by both enhancement at low concentrations and inhibition at high concentrations [27]. Recently, several new series of 2-amino-3-benzoylthiophenes have been synthesized [28,29]. Several compounds proved to be superior to PD81723: they are more potent than PD81723 in enhancing the binding of A1 AR agonist N6-cyclopentyladenosine to the receptor and their antagonistic activity on the A1 ARs was diminished. A 4-chloro analog, in which the methyl groups of PD81723 have been cyclized, T62 (King Pharmaceuticals) (Figure 1), has been in preclinical development for the treatment of chronic pain [30,31] and is now in Phase I clinical trials. The benzoylthiophene allosteric enhancers also appear to act as coactivators of the A1 AR – that is, it is capable of activating the receptor through binding to an allosteric site, even in the absence of an orthosteric agonist [32]. The thiadiazole SCH-202676 (Figure 1) and its analogs are also modulators for A1 ARs and several other GPCRs [33,34]. It was recently demonstrated that the thiadiazoles are sulfhydryl-modifying agents rather than allosteric modulators because they appear reversibly to modify the sulfhydryl groups of cysteine residues in cell membrane preparations [35].
The allosteric modulation of A2A and A3 ARs has also been studied recently [36–39]. Although an allosteric enhancer for the A2A ARs has not yet been developed, amiloride and analogs were demonstrated to be allosteric inhibitors for the A2A AR. There are at least three classes of allosteric modualtors for the A3 AR, including DU124183 (Figure 1), VUF5455 (Figure 1) and amiloride analogs [19]. All of these modulators influence the dissociation kinetics of the radioligand from the A3 AR. They also influence the A3 AR function, as demonstrated by in vitro cyclic AMP functional assays in intact cells [19]. It should be noted that the functional enhancement of these allosteric modulators can only be observed in the presence of a high concentration of competitive antagonist, thus overwhelming their antagonistic activity. An exception is DU124183 and some of its analogs, which enhance agonist efficacy even in the absence of a competitive antagonist. DU124183 achieved a 30% potentiation of the maximal efficacy of a potent A3 receptor agonist. Thus, functional enhancement by DU124183 and its analogs could be further pursued for eventual therapeutic applications. A3 allosteric enhancers might be predicted to be useful against ischemic conditions.
A3AR mutagenesis studies have implicated F1825.43 and N2747.45 in the action of the enhancers DU124183 and VUF5455 [39]. The mutagenesis data were interpreted using a rhodopsin-based A3AR molecular model, suggesting multiple binding modes of the enhancers either at the orthosteric site or at a distinct putative allosteric site.
Muscarinic receptors
Central muscarinic cholinergic neurotransmission has been implicated in psychosis and cognitive disorders [40]. Both acetylcholine esterase inhibitors and weakly selective muscarinic agonists improved neuropsychiatric symptoms and cognitive function in Alzheimer’s disease patients. However, the lack of truly selective drugs for the five muscarinic receptors has prevented an unambiguous determination of the role of each receptor subtype in human behavior. Thus, the muscarinic receptors are prime candidates for the development of allosteric modulators to fill an unmet medical need [18].
An allosteric site on a GPCR was first discovered in muscarinic receptors through studies with isolated organs [41,42]. Subsequent receptor-binding studies confirmed the nature of the allosteric modulation of muscarinic receptors by gallamine [43]. Allosteric modulation of GPCRs has been most extensively studied in muscarinic receptors [18]. The existence of a secondary allosteric site on the muscarinic receptor suggests that it might be possible to develop novel allosteric enhancers to potentiate the effects of endogenous acetylcholine in the same way that benzodiazepines potentiate the action of GABA at the GABAA receptor complex [6].
None of the orthosteric muscarinic receptor agonists or antagonists is highly selective for a given subtype, whereas highly subtype-selective allosteric modulators have been identified. Thus, the allosteric site could be a promising target in drug development. Therapeutically, allosteric enhancement of the effect of endogenous acetylcholine on a given subtype could be an effective way to relieve several disorders. For example, an M1 receptor enhancer could be applied for treatment of Alzheimer’s disease by compensating for the local acetylcholine deficit.
A wide range of molecules have been reported to be allosteric modulators of muscarinic receptors, including strychnine, neuromuscular blockers, staurosporine and analogs, and other amine derivatives of alkaloids [18]. One of the allosteric modulators, dimethyl-W84 (Figure 1), has been radiolabeled to study the allosteric site on the muscarinic receptor [44]. A recently synthesized positive allosteric modulator for the M2 muscarinic receptor was shown to be ~100-fold more potent than W84 [45]. Potential allosteric agonists of this receptor include AC-42 (Figure 1) [46,47] and N-desmethylclozapine [48]. Some pirenzepine analogs were reported to interact with both binding domains for acetylcholine and the allosteric modulator brucine [49]. Brucine and its analogs act as allosteric modualtors of various subtypes of muscarinic receptors in a complex manner. For example, N-chloromethylbrucine allosterically enhances and inhibits the binding and function of acetylcholine at M3 and M2 muscarinic receptors, respectively [50]. Analogs of the steroid derivative WIN 62,577 define a second allosteric site on the M3 muscarinic receptor that is nonoverlapping and pharmacologically distinct from the strychnine site [51]. Thiochrome (Figure 1), a metabolite of thiamine, enhances acetylcholine affinity selectively at the M4 receptor subtype via positive cooperativity but shows neutral cooperativity at M1, M2 and M3 receptors [52]. Felder et al. [53] have identified a novel selective positive allosteric modulator of the M4 receptor, which displayed an α of >20 in binding experiments and also enhanced functional responses. Trankle et al. [54] and Wess [55] recently described evidence suggesting that two allosteric agents and one orthosteric ligand might be able to bind to the M2 muscarinic receptor simultaneously. It has been suggested that the complex behavior might be rationalized by allosteric interactions transmitted within a receptor dimer [54,55].
Dopamine receptors
Dopamine receptors are intimately involved in several diseases, including Parkinson’s disease and schizophrenia. Evidence suggests that dopamine receptors are subject to allosteric modulation [20]. Allosteric modulators of dopamine receptors include sodium ions, zinc ions, amiloride and its analogs and the tripeptide l-proline-l-leucine-l-glycine. All three types of allosteric modulation (positive, negative and neutral cooperativity), previously demonstrated in muscarinic receptors, have also been shown to occur in dopamine receptors. The ability of an allosteric modulator to fine tune the pharmacological response has attracted much interest in discovering new clinical applications. In the case of Parkinson’s disease, an allosteric modulator that enhances the effect of the residual amount of endogenous dopamine could be extremely helpful. One of the promising allosteric modulators, l-proline-l-leucine-l-glycine, has been extensively modified for the potential treatment of Parkinson’s disease [56].
Growth hormone secretagogue receptors
Growth hormone secretagogue receptors (GHSR) have been suggested as an important drug target for several diseases, including obesity and diabetes. Ghrelin is the endogenous agonist for the GHSR. Recently, Holst et al. [57] reported that a nonpeptide agonist, MK-0677, behaved essentially like ghrelin in activating the GHSR in all systems tested. However, it was found that another nonpeptide agonist, L-692,429 (Figure 1), and a peptide agonist, GH-releasing peptide 6, behave as positive and negative allosteric modulators of GHSR, respectively, in addition to their allosteric agonist activity. It is suggested that an optimal drug candidate could be an agonist which is also a positive modulator of ghrelin receptor signaling [57].
Chemokine receptors
Chemokines are a family of structurally related low molecular weight proteins acting on at least 18 GPCR members [58]. Their function is predominantly related to leukocyte trafficking [59]. Both allosteric agonists and allosteric antagonists have recently been found, and are of potential clinical application. Two peptide allosteric agonists have been pinpointed for the CXCR4 chemokine receptor, a co-receptor for T-tropic strains of HIV-1 [60]. Studies demonstrated that noncompetitive allosteric inhibitors of CXCR1 and CXCR2 chemokine receptors, including repertaxin, were protective against reperfusion-induced organ injury [61]. Targeting the allosteric site on the CXCR1 receptor has been suggested as a general strategy to modulate the activity of chemoattractant receptors. The allosteric noncompetitive HIV entry inhibitor 873140 (Figure 1) exerts its effect by interacting with an allosteric site on the CCR5 chemokine receptor [62].
Allosteric modulation of other members of class A GPCRs has also been described. These members include α1-adrenoceptor [63,64], serotonin 5-HT2C [65], CB1 cannabinoid [25], purinergic P2Y1 [66,67] and oxytocin receptors [68]. Interestingly, oleamide (Figure 1), which is an allosteric modulator of the 5-HT receptor, was recently found also to be an endogenous agonist for the CB1 cannabinoid receptor [69].
Class B GPCRs: secretin-like receptors
Class B GPCRs (also known as the secretin-like receptor family) are a family of peptide-binding receptors comprising 15 members which share little sequence homology with class A (rhodopsin-like) or class C (mGlu) GPCRs. Several class B GPCRs, including corticotropin-releasing factor (CRF), growth hormone-releasing hormone, glucagon, secretin, calcitonin and parathyroid hormone (PTH) receptors, are crucially involved in controlling numerous important physiological processes [21].
Unlike some of the class A GPCRs in which the binding of small-molecule ligands occurs within the TM region, peptides bind to the class B GPCRs in a putative two-domain model. The carboxy-terminus of the peptide ligand binds to the extracellular amino-terminal domain (~100–160 residues) of the receptor. Binding causes the interaction of the amino-terminus of ligands with the juxtamembrane domain (J-domain) of the receptor. Nonpeptide ligands can bind either the amino-terminus or the J-domain and allosterically modulate the peptide binding [21]. Thus, although it is unknown if all peptides bind orthosterically, allosteric modulation using small molecules seems to be a classical means of controlling the class B receptor function.
Hoare et al. [70] quantitatively evaluated the allosteric inhibitory mechanism of the nonpeptide modulators for the CRF1 receptor using an allosteric ternary complex model. Mutagenesis studies of the CRF1 receptor indicated that nonpeptide antagonists predominantly bind to the J-domain of the receptor. Two mutations (H199V and M276I) in the J-domain affected the nonpeptide but not peptide binding [71]. It should be noted that the experimental evidence from mutagenesis is not unequivocal. Hoare et al. [70] argued that there should be caution in the interpretation of the interaction as allosteric because of a lack of conclusive evidence. The nonpeptide glucagon receptor antagonist L-168,049 (Figure 2) also binds to the J-domain of the receptor and allosterically modulates glucagon binding [72]. Nonpeptide agonists have also been found for the calcitonin receptor [73] and the glucagon-like protein-1 receptor [74].
FIGURE 2.
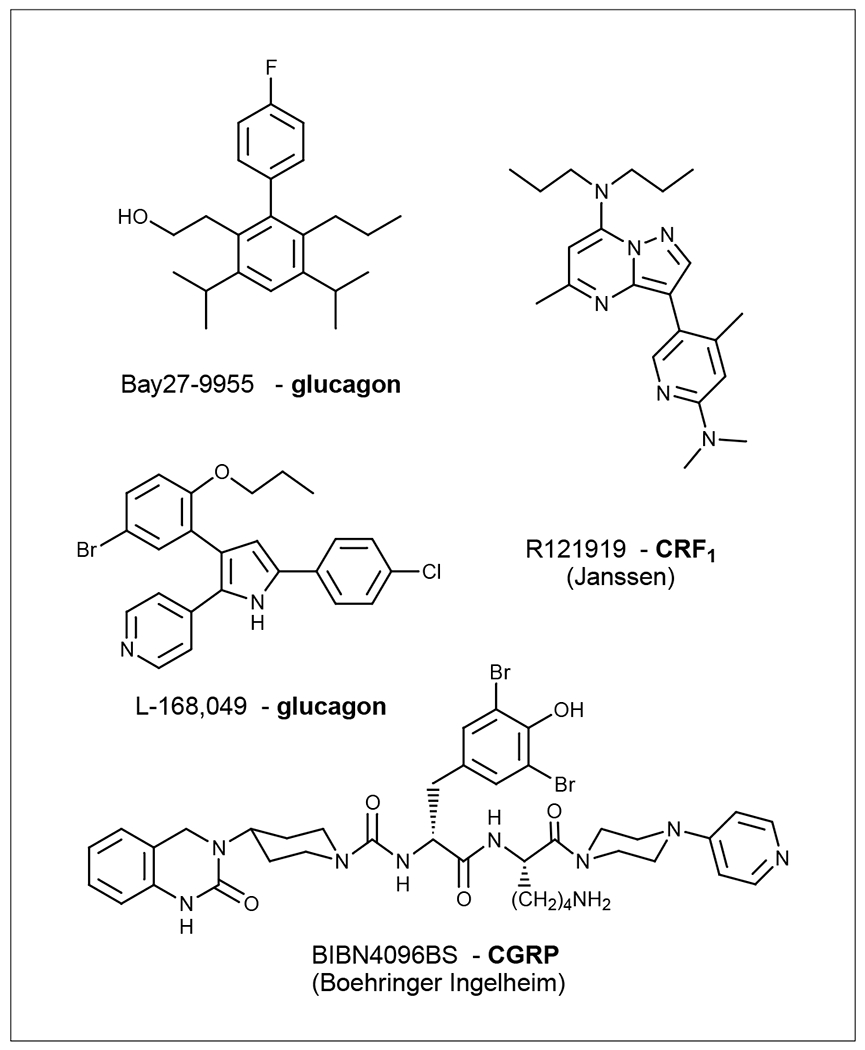
Structures of representative allosteric modulators for class B GPCRs that are in clinical trials or used as research tools.
The first-generation nonpeptide CRF1 antagonist R121919 (Figure 2) is being developed by Janssen Pharmaceuticals NV and Neurocrine Biosciences. It is in Phase II clinical trials for treating depression and anxiety [75]. Neurocrine Biosciences and GlaxoSmithKline (GSK) jointly initiated Phase I clinical trials of GSK 876008 (structure not disclosed) in late 2004. This compound will be entering Phase II clinical trials in the first half of 2006. The calcitonin gene-related peptide receptor antagonist BIBN4096BS (Boehringer Ingelheim Pharma, Germany and University of Copenhagen, Denmark) (Figure 2) is in Phase II clinical trials for migraine relief [76]. A glucagon antagonist (Bay27-9955; Figure 2) has been proposed for the treatment of type 2 diabetes [77].
Class C GPCRs: metabotropic glutamate-like receptors
Glutamate is the major excitatory transmitter in the brain and binds to both LGICs and a family of class C GPCRs known as metabotropic glutamate receptors (mGluRs). Unlike most class A and class B GPCRs, class C GPCRs contain three domains: the extracellular Venus flytrap domain (VFD), which contains the agonist-binding site, the cysteine-rich domain (CRD) and the heptahelical domain (HD) involved in G protein activation [11]. In addition, the carboxy-terminal tail can be unusually long – for example, 376 residues in the mGluR5b. The fact that in class C receptors the native agonists bind in an entirely separate domain of the receptor, with respect to class A GPCRs, provides an opportunity to modulate receptor action through interactions of small molecules within the HD region.
Class C GPCRs are constitutive homo- or heterodimers. mGluRs exist in homodimers but GABAB receptors form heterodimers (GABAB1 and GABAB2). Full activation of the homodimers requires two agonists, whereas one agonist is sufficient to activate the heterodimeric receptors. In the case of GABAB receptors, the GABAB1 VFD binds to an agonist but cannot activate the G protein, whereas GABAB2 is responsible for G protein activation. It was recently demonstrated that, although GABA binds to the VFD of GABAB1 through allosteric transition to activate the receptor, the GABAB2 HD can be activated directly by an allosteric modulator, CGP7930 [78] (Figure 3).
FIGURE 3.
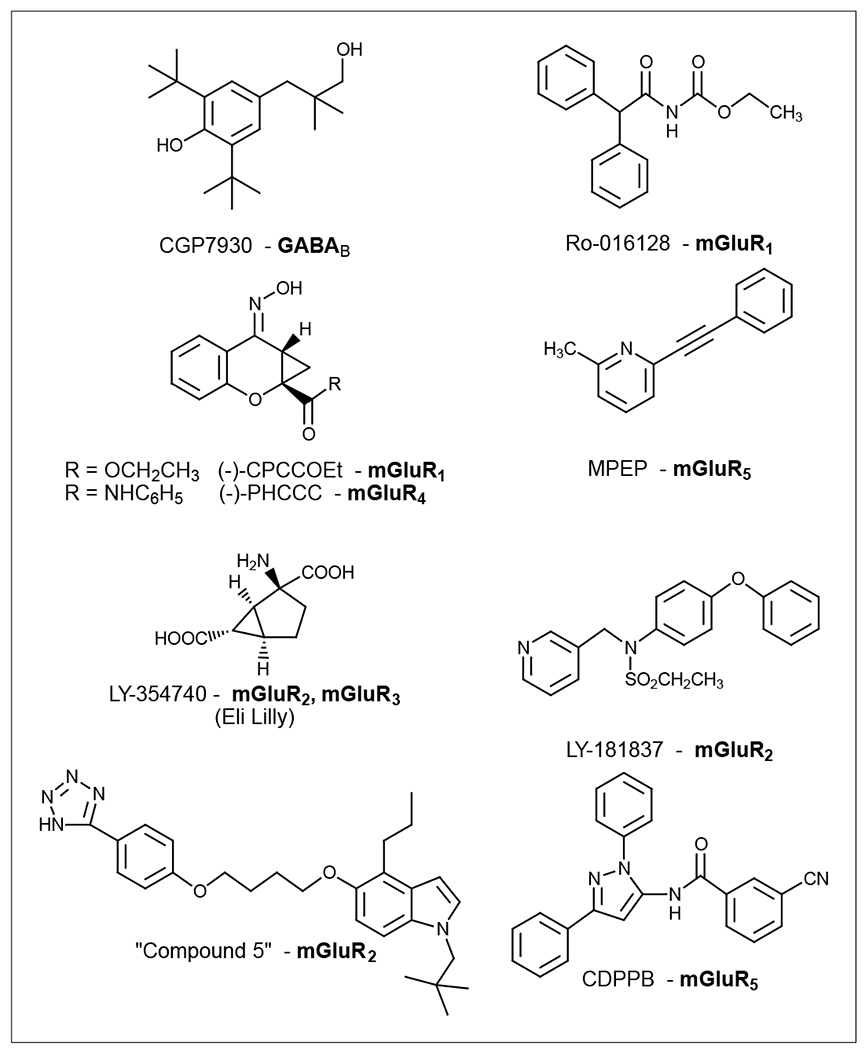
Structures of representative allosteric modulators for class C GPCRs (GABAB and mGlu) that are in clinical trials or used as research tools.
Experiments have indicated that some class C receptors exhibit agonist-independent constitutive activity. Spontaneous closure of the VFD was observed for the GABAB receptors but competitive antagonists behave as inverse agonists to block the spontaneous closure of the VFD. However, in the case of mGluR1 and mGluR5, the spontaneous closure of the VFD was not blocked by competitive antagonists. Noncompetitive antagonists were found to bind to the HD and showed inverse agonist properties. In addition, the positive allosteric modulators of the class C GPCRs can also bind to HD, potentiating efficacy and potency of agonists, although they themselves did not show any agonist activity [79]. Interestingly, when the extracellular domain was removed from the receptors, the positive allosteric modulators acted as full agonists [80], suggesting that the HD of class C GPCRs might behave independently, similarly to the class A receptors. Thus, Pin et al. [11] concluded that the HD of mGluRs can be either inhibited or activated by synthetic small molecules. Two noncompetitive antagonists are required per receptor dimer to block receptor activity, whereas a single positive allosteric modulator is sufficient for activation. This analysis supports the view that in GPCR dimers, a single monomeric HD is turned on at a given time.
Although the role of the CRD in receptor activation is less clear, it was recently found that the CRD of T1R3 receptors determines responses to intensely sweet proteins [81]. Also, it appears to be necessary for the activation of mGluRs and the CaR [82]. Thus, all three domains of class C GPCRs are amenable to orthosteric and allosteric modulation.
As a result of the emerging roles of class C GPCRs in numerous physiological and pathophysiological conditions, allosteric modulators have emerged as a classical means of control for several the class C GPCRs, and are the subject of intensive research owing to their therapeutic potential [83].
mGluRs
The mGluRs comprise eight members in three classes. Group I includes mGluR1 and mGluR5, which are predominantly coupled to Gq proteins, the activation of which results in an increase in intracellular calcium levels. Group I mGluRs are mainly located post-synaptically, generally enhancing postsynaptic signaling. Both group II (mGluR2 and mGluR3) and group III (mGluR4, -6, -7 and -8) are coupled to Gi/o proteins. Groups II and III are located presynaptically and have an inhibitory effect on neurotransmitter release.
Allosteric modulation has been well studied in the mGluRs. Glutamate and other known agonists and competitive antagonists bind to the amino-terminal region. All known positive and negative allosteric modulators bind to the 7TM domain. Development of subtype-selective allosteric modulators and their potential therapeutic applications have recently been reviewed [11,84–88].
The first allosteric modulators characterized for the mGluRs, 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester [(−)-CPCCOEt] and 2-methyl-6-(phenylethynyl)-pyridine (MPEP), are negative allosteric modulators for mGluR1 and mGluR5, respectively [89,90]. Selective positive allosteric modulators have recently been reported for mGluR1, mGluR2, mGluR4 and mGluR5 [91]. Ligands acting at orthosteric sites have not been demonstrated to be highly selective, suggesting that the allosteric sites might be able to be readily targeted by ligand optimization, a strategy that has been less successful for orthosteric glutamate binding sites. This finding supports the observation that putative allosteric sites generally have less sequence conservation between subtypes relative to orthosteric agonist-binding sites.
Positive allosteric modulators for mGluR1 include Ro-016128 (Figure 3), Ro-677476 and Ro-674853. These compounds are selective for mGluR1, with the exception of Ro-674853, which also showed some effect on mGluR5. Species differences between humans and rats are typical for these compounds, which are generally more potent at rat mGluR5 than at the human homolog. LY-181837 (Figure 3) was the first selective mGluR2 positive enhancer [92]. Subsequently, more-potent and selective enhancers, such as LY-487379, were developed. Other series of compounds acting as positive allosteric modulators for mGluR2 were also recently developed [85,93]. (−)-CPCCOEt (Figure 3) and MPEP (Figure 3) were initially found to be negative allosteric modulators for mGluR1 and mGluR5, respectively, and were later found to be positive for mGluR4. An analog of (−)-CPCCOEt, (−)-N-phenyl-7-(hydroxylimino)cyclopropa[b]chromen-1a-carboxamide [(−)-PHCCC] (Figure 3), was found to be potentially useful for the treatment of Parkinson’s disease [94]. 3,3’-Difluorobenzaldazine was the first positive allosteric modulator to be identified for mGluR5 [95]. Interestingly, two close analogs, 3,3’-dimethoxybenzaldazine and 3,3’-dichlorobenzaldazine, showed a negative allosteric effect and neutral cooperativity, respectively. Other novel classes of positive modulators for mGluR5, including N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide (CPPHA) and 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB; Figure 3), were later identified [96,97]. Benzazoles, such as compound 5 (Figure 3), display efficacy as allosteric potentiators of mGlu2 in animal model for schizophrenia [98]. CDPPB exhibited a significant antipsychotic effect. These modulators have the potential to treat a range of central nervous system diseases, including anxiety, Parkinson’s disease, schizophrenia and cognition disorders.
LY-354740 (Eli Lilly) (Figure 3), an allosteric enhancer of mGlu2 and mGlu3 receptors, is in Phase II clinical trials for the treatment of anxiety-related disorders [99,100].
GABAB receptors
GABAB receptors are emerging drug targets. Agonists can be used in the treatment of pain, drug-dependence and anxiety, whereas antagonists can prove beneficial in cognitive disorders and depression. To date, drug development has been impeded by the difficulty in achieving receptor subtype selectivity. Allosteric modulators of GABAA channels, including diazepam, have been administered successfully, whereas directly acting GABAA agonists have not been applied clinically, owing to the severe side effects [6]. Thus, the same approach might be applied to the GABAB receptor. Indeed, recently synthesized compounds acting via an allosteric mechanism have been identified and are providing hope that future drugs might be developed to target this receptor [16]. CGP7930 and CGS13501 are the first positive allosteric modulators of the GABAB receptor to be identified. These compounds do not activate the receptor on their own; rather, they potentiate the agonist efficacy and affinity, as observed with diazepam on GABAA receptors.
CaRs
Secondary hyperparathyroidism (HPTH) is a common, life-threatening complication of chronic kidney disease, and can lead to cardiovascular calcification. The CaR is a class C GPCR that regulates the secretion of PTH and is most highly expressed in parathyroid, thyroid parafollicular cells and renal tubular cells. It is downregulated in chronic renal failure. Medicinal chemical studies of allosteric modulators of the CaR have led to the novel treatment of HPTH with calcimimetic agents.
Certain phenylalkylamine compounds, typified by the first-generation compounds NPS467 and NPS568, are among the positive allosteric modulators identified for the CaR [101]. At nanomolar concentrations, they were found to increase the concentration of cytoplasmic calcium in bovine parathyroid cells and to inhibit PTH secretion. Acidic residues in the extracellular loops of the CaR have been shown to be crucial for the response to both Ca2+ and NPS568 [102]. Both positive and negative CaR allosteric modulators in this chemical class interact with the HD [103]. The second-generation calcimimetic cinacalcet (Figure 4) (NPS Pharmaceuticals) was the first allosteric modulator for a GPCR to be marketed for the treatment of HPTH [8,9]. Certain l-amino acids were also shown to be allosteric activators of the CaR [104]. The VFD of CaR has been shown to be the site of action of l-amino acid modulators [105].
FIGURE 4.
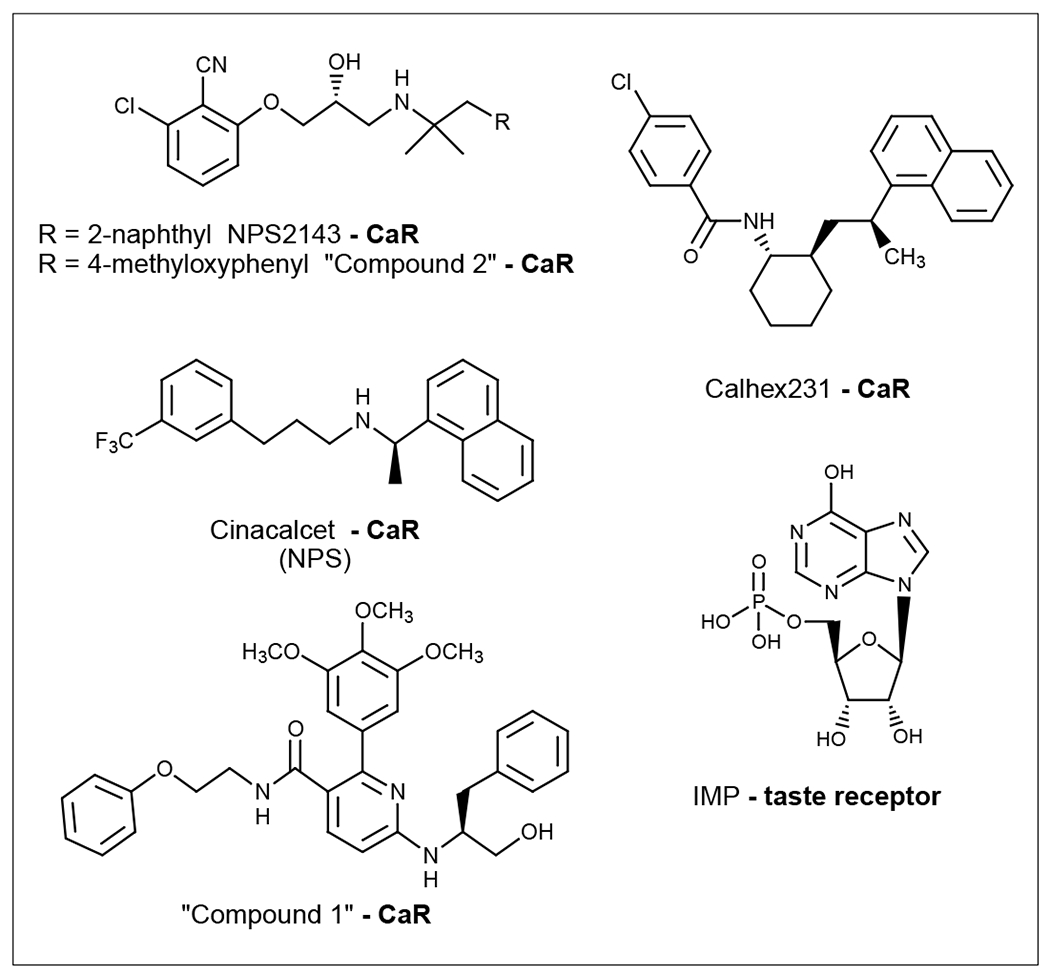
Structures of representative allosteric modulators for class C GPCRs (CaR) that are used in the clinic or as research tools.
Calcilytic agents, such as NPS2143 and CalHex231 (Figure 4), have also been designed [106,107]. Mutagenesis of the CaR has identified a mutation (P823A) at the TM6-TM7 junction that mimics the action of the negative allosteric modulator NPS2143, blocking the activation of the CaR occuring in autosomal dominant hypocalcemia. The finding that NPS2143 corrects a molecular defect associated with autosomal dominant hypocalcemia suggests that such negative allosteric modulators could offer an effective form of treatment for this disease [108].
Arey et al. [109] have designed a novel class of orally active calcilytic agents that increase serum PTH levels in vivo. Compound 1 (Figure 4) possesses potent antagonist activity at the human CaR, with an IC50 value of 64 nM in inhibiting intracellular Ca2+ flux. Such compounds might offer an improved treatment for osteoporosis.
Taste receptors
The perception of taste depends, to a large extent, upon an individual’s sensitivity to taste, including sweet, bitter, sour, salty and unami. It has been proposed that sodium chloride stimulates taste receptors in an allosteric manner [110]. Activation of the unami receptor by disodium 5’-guanylate (GMP), but not by monosodium glutamate (MSG), was blocked by amiloride in a competitive manner [111]. MSG has been shown to be an allosteric modulator of the GMP-stimulated taste response [112]. The family of taste receptors has recently been cloned and functionally characterized [113]. It was suggested that T1R1 and T1R3 receptors, which respond to sweet taste, combine into one functional unit [17]. Certain nucleotides, such as inosine 5’-monophosphate (IMP; Figure 4), do not activate the functional unit but potentiate the ability of l(+)-2-amino-4-phosphonobutanoic acid (L-AP4), MSG and other amino acids to activate the receptor [17].
LGICs
LGICs are membrane receptors responsible for rapid synaptic transmission and modulation of cellular activity. These channels are proteins spanning the cell membrane and forming both the binding site for the natural ligand and the ion-conducting pore, which can be opened or closed upon ligand binding. Upon activation, ion channels open to enable ion flux across the cell membrane. The flux can cause depolarization or hyperpolarization, depending on the charge and concentration of the ions. Allosteric modulators of LGICs are important therapeutic agents [114,115].
In mammals, LGICs comprise three subfamilies, based on the number of TMs. The first family is the P2X receptors, activated by ATP, which are cationic channels containing three subunits. Each subunit contains one large extracellular loop and two TMs with both amino- and carboxy-termini in the cytosol. The second family is glutamate-activated cationic channels, including N-methyl-d-aspartate (NMDA), alpha-amino-3-hydroxy-5-methylisoxazole-propionate (AMPA) and kainate receptors, all comprising four subunits. The amino-terminus is located extracellularly and the carboxy-terminus is located intracellularly. The third family is the largest family, and includes nicotinic, glycine, 5-HT3, GABAA, GABAC, zinc-activated ion channel receptors and several others. Members of the third family are made up of five subunits, each containing an amino-terminus and four TMs. Both the carboxy-and amino-termini are located extracellularly. The ligand-binding site lies on the interface between adjacent subunits. Almost all members of the LGIC family are subject to allosteric modulation.
P2X receptors
The allosteric sites on the P2X receptors have been well defined [116]. In the rat dorsal root ganglion neuron, calcium ions selectively modulate the P2X3 receptor, which is inhibited by magnesium [117]. Cibacron blue, which is an antagonist of P2X1 and P2X2 receptors, enhanced the P2X3-mediated nociceptive responses induced by 3’-O-(4-benzoylbenzoyl)-ATP and attenuated by the effects of the antagonist trinitrophenyl-ATP [118]. Tetramethylpyrazine used in Chinese medicine as an analgesic negatively modulates ATP-activated currents in rat dorsal root ganglion neurons [119]. Ethanol has also been found to act as a negative modulator of P2X receptors by right-shifting the activation curve [120]. Ivermectin is a positive allosteric modulator of P2X4 receptors [121]. Zinc has been shown to act as allosteric modulator of P2X2 receptors, potentiating channel opening [122]. Although these molecules have not translated to therapeutic application, they could represent the optimizable leads for eventual clinical use [123].
Glutamate receptors
Glutamate is the major excitatory neurotransmitter in the brain [124]. Glutamate receptors are the only LGICs for which multiple high-resolution crystal structures have been solved. The agonist-binding site is between two regions of the extracellular domain. NMDA receptors are cation-permeable channels and can be modulated by the positive allosteric modulator glycine and by the negative allosteric modulators zinc and ifenprodil (Figure 5) [125]. ATP was recently shown to act as both a competitive antagonist and a positive allosteric modulator at the NMDA receptor [126]. Positive and negative allosteric modulators have also been identified for the AMPA receptors [124].
FIGURE 5.
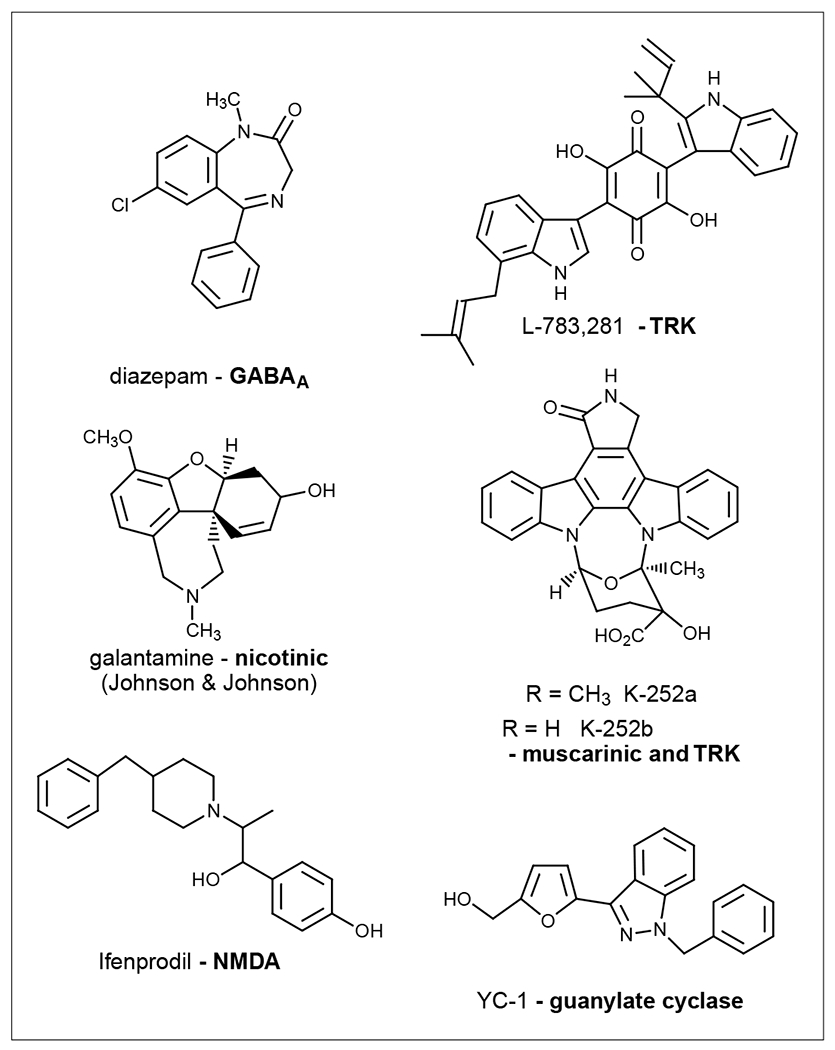
Structures of representative allosteric modulators for LGICs,Trk and guanylate cyclase receptors that are used in the clinic or as research tools.
GABAA receptors
GABA, acting on LGICs, GABAA and GABAC, and G-protein-coupled GABAB receptors, is the principal inhibitory neurotransmitter in the brain [6,127]. GABAA receptors are important drug targets for anxiety, epilepsy and sleep disorders. The GABAA receptors are the target for the most widely prescribed sleep medications, the benzodiazepines.
Activation of GABAA receptors leads to opening of channels that are permeable to HCO3− and Cl−. Multiple allosteric sites have been found in GABAA receptors [127,128]. Directly acting GABAA agonists have not found clinical applications, owing to various side effects. An alternative approach was to design ligands that could potentiate the effect of the endogenous agonist, and thereby gain selectivity. Benzodiazepines, including diazepam (Figure 5), were among the first generation of allosteric modulators for a membrane receptor to be used clinically. Benzodiazepines bind to a site on the receptor distinct from the site where GABA binds, potentiating the functional response upon receptor activation by GABA. Drugs acting at the allosteric sites have shown important advantages in comparison with conventional agonists or antagonists.
It was found recently that benzodiazepines show binding selectivity for a subset of GABAA receptors [128,129], which is encouraging from the perspective of drug discovery. Efforts have been made to develop more selective ligands for a given binding site on the receptor [129,130].
Nicotinic receptors
Nicotinic receptors were among the first membrane receptors in which allosterism was studied [3,114]. Various molecules were shown to be allosteric modulators of the nicotinic receptors. Positive modulators included divalent cations, steroid hormones, ivermectin, cocaine methiodide and 5-hydroxyindole [131]. Galantamine (Johnson and Johnson) (Figure 5) is a positive allosteric modulator of α4β2 and α7 nicotinic receptors [132,133], in addition to its anticholinesterase activity. Galantamine has been successfully used in the clinical treatment of Alzheimer’s disease [7].
Other membrane receptors
Tyrosine kinase receptors
Tyrosine kinases are classified as transmembrane tyrosine kinase receptors (or receptor tyrosine kinases) and intracellular nonreceptor tyrosine kinases. Tyrosine kinase receptors are transmembrane proteins with a ligand-binding extracellular domain and a catalytic intracellular kinase domain. Emerging targets for cancer therapy [134], tyrosine kinase receptors have a central role in cellular growth, differentiation and oncogenesis. The activation of tyrosine kinase receptors by growth factors leads to dimerization and activation of an intrinsic tyrosine-specific kinase activity by an allosteric mechanism [135]. The insulin receptor, which belongs to the tyrosine kinase receptor family, functions as an allosteric protein in which the binding of the hormone to the α-subunit leads to a series of conformational changes resulting in the activation of the β-subunit tyrosine kinase [136].
Neurotrophins activate two types of cell-surface receptor, the Trk receptor tyrosine kinase and the shared p75 neurotrophin receptor [137,138]. p75 binds along the homodimeric interface of nerve growth factor, which disables the symmetry-related second p75 binding site of nerve growth factor through an allosteric conformational change [137]. Moreover, p75 affects Trk subdomain utilization in ligand-dependent activation, possibly by conformational or allosteric control [139]. L-783,281 (Figure 5) is a prototype for a small-molecule neurotrophin mimetic that activates Trk by interacting at a site different from the neurotrophin-binding site and induces conformational change [140].
Nerve growth factors as Trk receptor activators have not met with clinical success owing to their poor pharmacokinetic behavior, whereas small-molecule mimetics have shown promise for future treatment of neurodegenerative diseases [141]. These compounds include allosteric modulators and other indirectly acting agents. Given the difficulty of designing directly acting agonists, an alternative approach is to identify allosteric modulators that might potentiate the endogenous agonists, neurotrophins, which are physiologically secreted at low levels. Ganglioside GM1 and related compounds were among the first to be identified to potentiate the action of neurotrophins following Trk receptor activation [142]. Two staurosporine-like compounds, K-252a and K-252b (Figure 5), which are also allosteric modulators for muscarinic receptors [143], were found either to inhibit or to enhance the action of the Trk receptor, depending on the concentrations used [141]. It was found that the staurosporine-like compound L-753,000 potentiates the neurotrophic effects of neurotrophin 3 by acting selectively at the TrkA receptor [144]. Additionally, Trk receptors in PC12 cells have been demonstrated to be activated indirectly by an adenosine A2A receptor agonist, CGS21680, and this effect was blocked by a selective A2A antagonist, ZM241385. The effect could also be blocked by the Trk inhibitor K252a. Thus, indirect activation by small molecules could open a new avenue for manipulating Trk receptors.
Guanylate cyclase receptors
Guanylate cyclases (GCs) are membrane receptors which, upon activation, catalyze the conversion of GTP to cGMP, and which can be classified into two subgroups: soluble and particulate GCs. The soluble GC is a ubiquitous receptor for nitric oxide (NO), which is involved in neurotransmission and vasodilation. The atrial natriuretic peptide receptor (particulate GC) is one of the best-studied membrane GCs and is a target for congestive heart failure [145].
3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) (Figure 5) is an allosteric activator of soluble GC that increases the GC catalytic rate and sensitizes GC towards its gaseous activators, NO and carbon monoxide [146]. Bay58–2667 activates soluble GC in a NO-independent manner and its binding site is distinct from the binding site of the analog BAY41–2272, which activates soluble GC [147,148]. Thus, drug development for membrane GCs could target a distinct allosteric site to increase the prospect of selectivity.
Concluding remarks
Increasing evidence suggests that GPCR dimerization is common to all three classes of GPCRs. Original models describing the activation of a GPCR, especially the class A GPCRs, is generally related to the conformational change within a monomer. In the case of a dimer, it can be speculated that an orthosteric agonist for one dimerized monomer could be a positive or negative allosteric modulator for another monomer. From this point of view, most, if not all, membrane receptors could be naturally allosterically modulated. Thus, allosteric modulation is a classical controlling mechanism for receptor behavior and cellular processes.
Allosteric sites are an increasingly attractive target for drug development; thus, future screening assay systems should be re-engineered to accommodate the requirement of allosteric sites. It is necessary to note the use of dissociation kinetics to detect the allosteric phenomenon. It has been demonstrated that some molecules, such as SCH202626 and its analogs, which do not act through an allosteric mechanism, might also increase or decrease the dissociation rate. Therefore, multiple binding and functional assays should be utilized for the study and validation of a potential allosteric modulator.
Allosteric modulators could be particularly useful to control receptors for which the design of selective orthosteric agonists or antagonists has been elusive, such as muscarinic receptors. Allosteric modulators for chemokine receptors are poised to join the next generation of anti-HIV drugs. Neurotrophins have not proven successful as therapeutic agents owing to their poor pharmacokinetics and lack of selectivity. Thus, identification of small-molecule allosteric modulators to produce a conformation change in the receptor tyrosine kinase structure to attenuate or potentiate its interaction with a large endogenous ligand is likely to be an intensive future effort. Owing to the fact that increasing effort has been invested in the study of the allosteric sites, and that increasing numbers of allosteric modulators are in clinical trials, it can be speculated that more drugs targeting allosteric sites will be used therapeutically in the near future.
Acknowledgements
We thank the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases for support and Srikar Rao for textual editing.
Biographies
ZHAN-GUO GAO
Zhan-Guo Gao is a Staff Scientist in the Molecular Recognition Section, Laboratory of Bioorganic Chemistry,NIDDK, NIH.After studying medicine, he became interested in the mechanisms of action of G-protein-coupled receptors.Both his current field of research and his PhD thesis work at the Leiden–Amsterdam Centre for Drug Research in The Netherlands concern adenosine receptors.He has published extensively on allosteric modulation of adenosine receptors.

KENNETH A.JACOBSON
Kenneth Jacobson is Chief of the Molecular Recognition Section of the Laboratory of Bioorganic Chemistry, NIDDK,NIH. He is a medicinal chemist with a research focus on receptors for nucleosides and nucleotides. Jacobson completed his PhD studies at the University of California,San Diego, USA,in 1981 and subsequent postdoctoral work at the Weizmann Institute of Science, Rehovot,Israel.He recently served as Chair of the Medicinal Chemistry Division of the American Chemical Society.He is a ‘highly cited researcher’in Pharmacology and Toxicology (Institute for Scientific Information) and received the first Giuliana Fassina Award from the Purine Club in 1996,as well as the Hillebrand Prize of the Chemical Society of Washington in 2003 for ‘outstanding research contributions in the medicinal chemistry of G-protein-coupled receptors’.

References
- 1.Monod J et al. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 2.Christopoulos A (2002) Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discov 1, 198–210 [DOI] [PubMed] [Google Scholar]
- 3.Changeux JP and Edelstein SJ (2005) Allosteric mechanisms of signal transduction. Science 308, 1424–1428 [DOI] [PubMed] [Google Scholar]
- 4.Bohr C et al. (1904) Ubereinen in biologischen Beziehung wichtigen Einfluss, den die Kohlen-sauerspannung des Blütes auf dessen Sauerstoffbindung ubt. Skand. Arch. Physiol 15, 401–412 [Google Scholar]
- 5.Changeux JP et al. (1967) On the cooperativity of biological membranes. Proc. Natl. Acad. Sci. U. S. A 57, 335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macdonald RL and Olsen RW (1994) GABAA receptor channels. Annu. Rev. Neurosci 17, 569–602 [DOI] [PubMed] [Google Scholar]
- 7.Bullock R (2004) Galantamine: use in Alzheimer’s disease and related disorders. Expert Rev. Neurother 4, 153–163 [DOI] [PubMed] [Google Scholar]
- 8.Poon G (2005) Cinacalcet hydrochloride (Sensipar). Proc. (Bayl. Univ. Med. Cent.) 18, 181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindberg JS et al. (2005) Cinacalcet HCl, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study. J. Am. Soc. Nephrol 16, 800–807 [DOI] [PubMed] [Google Scholar]
- 10.Palczewski K et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 11.Pin JP et al. (2005) Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J 272, 2947–2955 [DOI] [PubMed] [Google Scholar]
- 12.George SR et al. (2002) G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov 1, 808–820 [DOI] [PubMed] [Google Scholar]
- 13.Chabre M and le Maire M (2005) Monomeric G-protein-coupled receptor as a functional unit. Biochemistry 44, 9395–9403 [DOI] [PubMed] [Google Scholar]
- 14.Terrillon S and Bouvier M (2004) Receptor activity-independent recruitment of betaarrestin2 reveals specific signalling modes. EMBO J 23, 3950–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulenger S et al. (2005) Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci 26, 131–137 [DOI] [PubMed] [Google Scholar]
- 16.Pin JP et al. (2004) Activation mechanism of the heterodimeric GABAB receptor. Biochem. Pharmacol 68, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 17.Nelson G et al. (2002) An amino-acid taste receptor. Nature 416, 199–202 [DOI] [PubMed] [Google Scholar]
- 18.Birdsall NJ and Lazareno S (2005) Allosterism at muscarinic receptors: ligands and mechanisms. Mini Rev. Med. Chem 5, 523–543 [DOI] [PubMed] [Google Scholar]
- 19.Gao ZG et al. (2005) Allosteric modulation of the adenosine family of receptors. Mini Rev. Med. Chem 5, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schetz JA (2005) Allosteric modulation of dopamine receptors. Mini Rev. Med. Chem 5, 555–561 [DOI] [PubMed] [Google Scholar]
- 21.Hoare SR (2005) Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 22.Soudijn W et al. (2004) Allosteric modulation of G protein-coupled receptors: perspectives and recent developments. Drug Discov. Today 9, 752–758 [DOI] [PubMed] [Google Scholar]
- 23.Trankle C et al. (1999) Using a radioalloster to test predictions of the cooperativity model for gallamine binding to the allosteric site of muscarinic acetylcholine M(2) receptors. Mol. Pharmacol 56, 962–965 [DOI] [PubMed] [Google Scholar]
- 24.Hall DA (2000) Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol. Pharmacol 58, 1412–1423 [DOI] [PubMed] [Google Scholar]
- 25.Price MR et al. (2005) Allosteric modulation of the cannabinoid CB1 receptor. Mol. Pharmacol 68, 1484–1495 [DOI] [PubMed] [Google Scholar]
- 26.Ehlert FJ (2005) Analysis of allosterism in functional assays. J. Pharmacol. Exp. Ther 315, 740–754 [DOI] [PubMed] [Google Scholar]
- 27.Bruns RF and Fergus JH (1990) Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol 38, 939–949 [PubMed] [Google Scholar]
- 28.van der Klein PA et al. (1999) Allosteric modulation of the adenosine A1 receptor. Synthesis and biological evaluation of novel 2-amino-3-benzoylthiophenes as allosteric enhancers of agonist binding. J. Med. Chem 42, 3629–3635 [DOI] [PubMed] [Google Scholar]
- 29.Tranberg CE et al. (2002) 2-Amino-3-aroyl-4,5-alkylthiophenes: agonist allosteric enhancers at human A1 adenosine receptors. J. Med. Chem 45, 382–389 [DOI] [PubMed] [Google Scholar]
- 30.Li X et al. (2002) Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain 97, 117–125 [DOI] [PubMed] [Google Scholar]
- 31.Li X et al. (2003) Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J. Pharmacol. Exp. Ther 305, 950–955 [DOI] [PubMed] [Google Scholar]
- 32.Figler H et al. (2003) Allosteric enhancers of A1 adenosine receptors increase receptor-G protein coupling and counteract guanine nucleotide effects on agonist binding. Mol. Pharmacol 64, 1557–1564 [DOI] [PubMed] [Google Scholar]
- 33.Lanzafame A and Christopoulos A (2004) Investigation of the interaction of a putative allosteric modulator, N-(2,3-diphenyl-1,2,4-thiadiazole-5-(2H)-ylidene) methanamine hydrobromide (SCH-202676), with M1 muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther 308, 830–837 [DOI] [PubMed] [Google Scholar]
- 34.Fawzi AB et al. (2001) SCH-202676: an allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol. Pharmacol 59, 30–37 [DOI] [PubMed] [Google Scholar]
- 35.Göblyös A et al. (2005) Synthesis and biological evaluation of a new series of 2,3,5-substituted [1,2,4]-thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J. Med. Chem 48, 1145–1151 [DOI] [PubMed] [Google Scholar]
- 36.Gao ZG and IJzerman AP (2000) Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodium ions. Biochem. Pharmacol 60, 669–676 [DOI] [PubMed] [Google Scholar]
- 37.Gao ZG et al. (2001) Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol. Pharmacol 60, 1057–1063 [PMC free article] [PubMed] [Google Scholar]
- 38.Gao ZG et al. (2002) Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol. Pharmacol 62, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao ZG et al. (2003) Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol. Pharmacol 63, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sur C and Kinney GG (2005) Selective targeting of muscarinic receptors: novel therapeutic approaches for psychotic disorders. Curr. Neuropharmacol 3, 63–71 [Google Scholar]
- 41.Lullmann H et al. (1969) Inhibition of the actions of carbachol and DFP on guinea pig isolated atria by alkane-bis-ammonium compounds. Eur. J. Pharmacol 6, 241–247 [DOI] [PubMed] [Google Scholar]
- 42.Clark AJ and Mitchelson F (1976) The inhibitory effect of gallamine on muscarinic receptors. Br. J. Pharmacol 58, 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stockton JM et al. (1983) Modification of the binding properties of muscarinic receptors by gallamine. Mol. Pharmacol 23, 551–557 [PubMed] [Google Scholar]
- 44.Trankle C et al. (2003) Interactions of orthosteric and allosteric ligands with [3H]dimethyl-W84 at the common allosteric site of muscarinic M2 receptors. Mol. Pharmacol 64, 180–190 [DOI] [PubMed] [Google Scholar]
- 45.Muth M et al. (2005) Muscarinic allosteric enhancers of ligand binding: pivotal pharmacophoric elements in hexamethonio-type agents. J. Med. Chem 48, 2212–2217 [DOI] [PubMed] [Google Scholar]
- 46.Spalding TA et al. (2002) Discovery of an ectopic activation site on the M1 muscarinic receptor. Mol. Pharmacol 61, 1297–1302 [DOI] [PubMed] [Google Scholar]
- 47.Langmead CJ et al. (2006) Probing the molecular mechanism of interaction between AC-42 and the muscarinic M1 receptor: Direct pharmacological evidence that AC-42 is an allosteric agonist. Mol. Pharmacol 69, 236–246 [DOI] [PubMed] [Google Scholar]
- 48.Sur C et al. (2003) N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. U. S. A 100, 13674–13679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tahtaoui C et al. (2004) Fluorescent pirenzepine derivatives as potential bitopic ligands of the human M1 muscarinic receptor. J. Med. Chem 47, 4300–4315 [DOI] [PubMed] [Google Scholar]
- 50.Birdsall NJ et al. (1999) Subtype-selective positive cooperative interactions between brucine analogs and acetylcholine at muscarinic receptors: functional studies. Mol. Pharmacol 55, 778–786 [PubMed] [Google Scholar]
- 51.Lazareno S et al. (2002) Analogs of WIN 62,577 define a second allosteric site on muscarinic receptors. Mol. Pharmacol 62, 1492–1505 [DOI] [PubMed] [Google Scholar]
- 52.Lazareno S et al. (2004) Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Mol. Pharmacol 65, 257–266 [DOI] [PubMed] [Google Scholar]
- 53.Felder CC et al. (2004) Pharmacological and molecular characterisation of a positive allosteric modulator selective for the muscarinic M4 receptor being developed for the treatment of psychosis. Neuropsychopharmacol 29, S115–S115 [Google Scholar]
- 54.Trankle C et al. (2005) Atypical muscarinic allosteric modulation: cooperativity between modulators and their atypical binding topology in muscarinic M2 and M2/M5 chimeric receptors. Mol. Pharmacol 68, 1597–1610 [DOI] [PubMed] [Google Scholar]
- 55.Wess J (2005) Allosteric binding sites on muscarinic acetylcholine receptors. Mol. Pharmacol 68, 1506–1509 [DOI] [PubMed] [Google Scholar]
- 56.Fisher A et al. (2006) Design and synthesis of photoaffinity-labeling ligands of the L-prolyl-L-leucylglycinamide binding site involved in the allosteric modulation of the dopamine receptor. J. Med. Chem 49, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holst B et al. (2005) Non-peptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol. Endocrinol 19, 2400–2411 [DOI] [PubMed] [Google Scholar]
- 58.White FA et al. (2005) Chemokines: integrators of pain and inflammation. Nat. Rev. Drug Discov 4, 834–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bendall L (2005) Chemokines and their receptors in disease. Histol. Histopathol 20, 907–926 [DOI] [PubMed] [Google Scholar]
- 60.Sachpatzidis A et al. (2003) Identification of allosteric peptide agonists of CXCR4. J. Biol. Chem 278, 896–907 [DOI] [PubMed] [Google Scholar]
- 61.Bertini R et al. (2004) Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc. Natl. Acad. Sci. U. S. A 101, 11791–11796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watson C et al. (2005) The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol. Pharmacol 67, 1268–1282 [DOI] [PubMed] [Google Scholar]
- 63.Waugh DJ et al. (1999) Binding, partial agonism, and potentiation of alpha(1)-adrenergic receptor function by benzodiazepines: a potential site of allosteric modulation. J. Pharmacol. Exp. Ther 291, 1164–1171 [PubMed] [Google Scholar]
- 64.Sharpe IA et al. (2003) Allosteric alpha 1-adrenoreceptor antagonism by the conopeptide rho-TIA. J. Biol. Chem 278, 34451–34457 [DOI] [PubMed] [Google Scholar]
- 65.Im WB et al. (2003) Positive allosteric modulator of the human 5-HT2C receptor. Mol. Pharmacol 64, 78–84 [DOI] [PubMed] [Google Scholar]
- 66.Spedding M et al. (2000) Antagonists and the purinergic nerve hypothesis: 2, 2’-pyridylisatogen tosylate (PIT), an allosteric modulator of P2Y receptors. A retrospective on a quarter century of progress. J. Auton. Nerv. Syst 81, 225–227 [DOI] [PubMed] [Google Scholar]
- 67.Gao ZG et al. (2004) 2,2’-Pyridylisatogen tosylate antagonizes P2Y1 receptor signaling without affecting nucleotide binding. Biochem. Pharmacol 68, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gimpl G and Fahrenholz F (2001) The oxytocin receptor system: structure, function, and regulation. Physiol. Rev 81, 629–683 [DOI] [PubMed] [Google Scholar]
- 69.Leggett JD et al. (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br. J. Pharmacol 141, 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoare SR et al. (2003) Mechanism of corticotropin-releasing factor type I receptor regulation by nonpeptide antagonists. Mol. Pharmacol 63, 751–765. Erratum in: Mol Pharmacol. 2005 68, 260 [DOI] [PubMed] [Google Scholar]
- 71.Liaw CW et al. (1997) Localization of agonist- and antagonist-binding domains of human corticotropin-releasing factor receptors. Mol. Endocrinol 11, 2048–2053 [DOI] [PubMed] [Google Scholar]
- 72.Cascieri MA et al. (1999) Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J. Biol. Chem 274, 8694–8697 [DOI] [PubMed] [Google Scholar]
- 73.Katayama T et al. (2001) Discovery of a non-peptide small molecule that selectively mimics the biological actions of calcitonin. Biochim. Biophys. Acta 1526, 183–190 [DOI] [PubMed] [Google Scholar]
- 74.Teng M et al. (2000) Nonpeptide GLP-1 agonists. WO 00/42026
- 75.Zobel AW et al. (2000) Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J. Psychiatr. Res 34, 171–181 [DOI] [PubMed] [Google Scholar]
- 76.Olesen J et al. (2004) Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med 350, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 77.Petersen KF and Sullivan JT (2001) Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 44, 2018–2024 [DOI] [PubMed] [Google Scholar]
- 78.Binet V et al. (2004) Molecular mechanisms of GABAB receptor activation: new insights from the mechanism of action of CGP7930, a positive allosteric modulator. Biochem. Soc. Trans 32, 871–872 [DOI] [PubMed] [Google Scholar]
- 79.Carroll FY et al. (2001) BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol. Pharmacol 59, 965–973 [PMC free article] [PubMed] [Google Scholar]
- 80.Goudet C et al. (2004) Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc. Natl. Acad. Sci. U. S. A 101, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang P et al. (2004) The cysteine-rich region of T1R3 determines responses to intensely sweet proteins. J. Biol. Chem 279, 45068–45075 [DOI] [PubMed] [Google Scholar]
- 82.Hu J et al. (2000) Human Ca2+ receptor cysteine-rich domain. Analysis of function of mutant and chimeric receptors. J. Biol. Chem 275, 16382–16389 [DOI] [PubMed] [Google Scholar]
- 83.Shipe WD et al. (2005) Recent advances in positive allosteric modulators of metabotropic glutamate receptors. Curr. Opin. Drug Discov. Devel 8, 449–457 [PubMed] [Google Scholar]
- 84.Layton ME (2005) Subtype-selective noncompetitive modulators of metabotropic glutamate receptor subtype 1 (mGluR1). Curr. Top. Med. Chem 5, 859–867 [DOI] [PubMed] [Google Scholar]
- 85.Rudd MT and McCauley JA (2005) Positive allosteric modulators of the metabotropic glutamate receptor subtype 2 (mGluR2). Curr. Top. Med. Chem 5, 869–884 [DOI] [PubMed] [Google Scholar]
- 86.Marino MJ et al. (2005) Targeting the metabotropic glutamate receptor mGluR4 for the treatment of diseases of the central nervous system. Curr. Top. Med. Chem 5, 885–895 [DOI] [PubMed] [Google Scholar]
- 87.Williams DL Jr, and Lindsley CW (2005) Discovery of positive allosteric modulators of metabotropic glutamate receptor subtype 5 (mGluR5). Curr. Top. Med. Chem 5, 825–846 [DOI] [PubMed] [Google Scholar]
- 88.Niswender CM et al. (2005) New therapeutic frontiers for metabotropic glutamate receptors. Curr. Top. Med. Chem 5, 847–857 [DOI] [PubMed] [Google Scholar]
- 89.Litschig S et al. (1999) CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol. Pharmacol 55, 453–461 [PubMed] [Google Scholar]
- 90.Gasparini F et al. (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503 [DOI] [PubMed] [Google Scholar]
- 91.Kew JN (2004) Positive and negative allosteric modulation of metabotropic glutamate receptors: emerging therapeutic potential. Pharmacol. Ther 104, 233–244 [DOI] [PubMed] [Google Scholar]
- 92.Johnson MP et al. (2003) Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J. Med. Chem 46, 3189–3192 [DOI] [PubMed] [Google Scholar]
- 93.Pinkerton AB et al. (2004) Phenyl-tetrazolyl acetophenones: discovery of positive allosteric potentiatiors for the metabotropic glutamate 2 receptor. J. Med. Chem 47, 4595–4599 [DOI] [PubMed] [Google Scholar]
- 94.Marino MJ et al. (2003) Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc. Natl. Acad. Sci. U. S. A 100, 13668–13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Brien JA et al. (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol. Pharmacol 64, 731–740 [DOI] [PubMed] [Google Scholar]
- 96.O’Brien JA et al. (2004) A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J. Pharmacol. Exp. Ther 309, 568–577 [DOI] [PubMed] [Google Scholar]
- 97.Kinney GG et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J. Pharmacol. Exp. Ther 313, 199–206 [DOI] [PubMed] [Google Scholar]
- 98.Govek SP et al. (2005) Benzazoles as allosteric potentiators of metabotropic glutamate receptor 2 (mGluR2): Efficacy in an animal model for schizophrenia. Bioorg. Med. Chem. Lett 15, 4068–4072 [DOI] [PubMed] [Google Scholar]
- 99.Grillon C et al. (2003) Anxiolytic effects of a novel group II metabotropic glutamate receptor agonist (LY354740) in the fear-potentiated startle paradigm in humans. Psychopharmacology (Berl.) 168, 446–454 [DOI] [PubMed] [Google Scholar]
- 100.Kellner M et al. (2005) Effects of a metabotropic glutamate (2/3) receptor agonist (LY544344/LY354740) on panic anxiety induced by cholecystokinin tetrapeptide in healthy humans: preliminary results. Psychopharmacology (Berl.) 179, 310–335 [DOI] [PubMed] [Google Scholar]
- 101.Nemeth EF et al. (1998) Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc. Natl. Acad. Sci. U. S. A 95, 4040–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hu J et al. (2002) Identification of the acidic residues in the extracellular loops of the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ and a positive allosteric modulator. J. Biol. Chem 29, 46622–46631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Petrel C et al. (2004) Positive and negative allosteric modulators of the Ca2+-sensing receptor interact within overlapping but not identical binding sites in the transmembrane domain. J. Biol. Chem 279, 18990–18997 [DOI] [PubMed] [Google Scholar]
- 104.Conigrave AD et al. (2000) L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc. Natl. Acad. Sci. U. S. A 97, 4814–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mun HC et al. (2004) The Venus Fly Trap domain of the extracellular Ca2+-sensing receptor is required for L-amino acid sensing. J. Biol. Chem 279, 51739–51744 [DOI] [PubMed] [Google Scholar]
- 106.Nemeth EF et al. (2001) Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J. Pharmacol. Exp. Ther 299, 323–331 [PubMed] [Google Scholar]
- 107.Petrel C et al. (2003) Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca2+-sensing receptor. J. Biol. Chem 278, 49487–49494 [DOI] [PubMed] [Google Scholar]
- 108.Hu J et al. (2005) A cluster of naturally occurring, activating mutations identifies a region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+. J. Biol. Chem 280, 5113–5120 [DOI] [PubMed] [Google Scholar]
- 109.Arey BJ et al. (2005) A novel calcium-sensing receptor antagonist transiently stimulates parathyroid hormone secretion in vivo. Endocrinology 146, 2015–2022 [DOI] [PubMed] [Google Scholar]
- 110.Mooser G (1980) Sodium and potassium salt stimulation of taste receptor cells: an allosteric model. Proc. Natl. Acad. Sci. U. S. A 77, 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura M and Kurihara K (1991) Canine taste nerve responses to monosodium glutamate and disodium guanylate: differentiation between umami and salt components with amiloride. Brain Res 541, 21–28 [DOI] [PubMed] [Google Scholar]
- 112.Kurihara K and Kashiwayanagi M (1998) Introductory remarks on umami taste. Ann. N. Y. Acad. Sci 855, 393–397 [DOI] [PubMed] [Google Scholar]
- 113.Adler E et al. (2000) A novel family of mammalian taste receptors. Cell 100, 693–702 [DOI] [PubMed] [Google Scholar]
- 114.Changeux JP and Edelstein SJ (1998) Allosteric receptors after 30 years. Neuron 21, 959–980 [DOI] [PubMed] [Google Scholar]
- 115.Hogg RC et al. (2005) Allosteric modulation of ligand-gated ion channels. Biochem. Pharmacol 70, 1267–1276 [DOI] [PubMed] [Google Scholar]
- 116.Alexander K et al. (1999) Allosteric modulation and accelerated resensitization of human P2X3 receptors by cibacron blue. J. Pharmacol. Exp. Ther 291, 1135–1142 [PubMed] [Google Scholar]
- 117.Giniatullin R et al. (2003) Modulation of P2X3 receptors by Mg2+ on rat DRG neurons in culture. Neuropharmacology 44, 132–140 [DOI] [PubMed] [Google Scholar]
- 118.Jarvis MF et al. (2001) Modulation of BzATP and formalin induced nociception: attenuation by the P2X receptor antagonist, TNP-ATP and enhancement by the P2X(3) allosteric modulator, cibacron blue. Br. J. Pharmacol 132, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liang SD et al. (2005) Tetramethylpyrazine inhibits ATP-activated currents in rat dorsal root ganglion neurons. Brain Res 1040, 92–97 [DOI] [PubMed] [Google Scholar]
- 120.Weight FF et al. (1999) Alcohol action on membrane ion channels gated by extracellular ATP (P2X receptors). Neurochem. Int 35, 143–152 [DOI] [PubMed] [Google Scholar]
- 121.Khakh BS et al. (1999) Allosteric control of gating and kinetics at P2X4 receptor channels. J. Neurosci 19, 7289–7299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nagaya N et al. (2005) An intersubunit zinc binding site in rat P2X2 receptors. J. Biol. Chem 280, 25982–25993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kennedy C (2005) P2X receptors: targets for novel analgesics? Neuroscientist 11, 345–356 [DOI] [PubMed] [Google Scholar]
- 124.Mayer ML (2005) Glutamate receptor ion channels. Curr. Opin. Neurobiol 15, 282–288 [DOI] [PubMed] [Google Scholar]
- 125.Rachline J et al. (2005) The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J. Neurosci 25, 308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kloda A et al. (2004) Adenosine triphosphate acts as both a competitive antagonist and a positive allosteric modulator at recombinant N-methyl-D-aspartate receptors. Mol. Pharmacol 65, 1386–1396 [DOI] [PubMed] [Google Scholar]
- 127.Bateson AN (2004) The benzodiazepine site of the GABAA receptor: an old target with new potential? Sleep Med 5, S9–15 [DOI] [PubMed] [Google Scholar]
- 128.Rudolph U et al. (1999) Benzodiazepine actions mediated by specific gamma-aminobutyric acidA receptor subtypes. Nature 401, 796–800 [DOI] [PubMed] [Google Scholar]
- 129.Whiting PJ. (2003) GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov. Today 8, 445–450 [DOI] [PubMed] [Google Scholar]
- 130.Street LJ et al. (2004) Synthesis and biological evaluation of 3-heterocyclyl-7,8,9,10-tetrahydro-(7,10-ethano)-1,2,4-triazolo[3,4-a]phthalazines and analogues as subtype-selective inverse agonists for the GABA(A)alpha5 benzodiazepine binding site. J. Med. Chem 47, 3642–3657 [DOI] [PubMed] [Google Scholar]
- 131.Hogg RC and Bertrand D (2004) Nicotinic acetylcholine receptors as drug targets. Curr. Drug Targets CNS Neurol. Disord 3, 123–130 [DOI] [PubMed] [Google Scholar]
- 132.Samochocki M et al. (2000) Galantamine is an allosterically potentiating ligand of the human alpha4/beta2 nAChR. Acta Neurol. Scand. Suppl 176, 68–73 [DOI] [PubMed] [Google Scholar]
- 133.Texido L et al. (2005) Effect of galantamine on the human alpha7 neuronal nicotinic acetylcholine receptor, the Torpedo nicotinic acetylcholine receptor and spontaneous cholinergic synaptic activity. Br. J. Pharmacol 145, 672–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Krause DS and Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med 353, 172–187 [DOI] [PubMed] [Google Scholar]
- 135.Chantry A (1995) The kinase domain and membrane localization determine intracellular interactions between epidermal growth factor receptors. J. Biol. Chem 270, 3068–3073 [PubMed] [Google Scholar]
- 136.Van Obberghen E (1994) Signalling through the insulin receptor and the insulin-like growth factor-I receptor. Diabetologia 37, S125–S134 [DOI] [PubMed] [Google Scholar]
- 137.He XL and Garcia KC (2004) Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 304, 870–875 [DOI] [PubMed] [Google Scholar]
- 138.Lu B et al. (2005) The yin and yang of neurotrophin action. Nat. Rev. Neurosci 6, 603–614 [DOI] [PubMed] [Google Scholar]
- 139.Zaccaro MC et al. (2001) p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J. Biol. Chem 276, 31023–31029 [DOI] [PubMed] [Google Scholar]
- 140.Wilkie N et al. (2001) The non-peptidyl fungal metabolite L-783,281 activates TRK neurotrophin receptors. J. Neurochem 78, 1135–1145 [DOI] [PubMed] [Google Scholar]
- 141.Pollack SJ and Harper SJ (2002) Small molecule Trk receptor agonists and other neurotrophic factor mimetics. Curr. Drug Targets CNS Neurol. Disord 1, 59–80 [DOI] [PubMed] [Google Scholar]
- 142.Farooqui T et al. (1997) Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J. Neurochem 68, 2348–2355 [DOI] [PubMed] [Google Scholar]
- 143.Lazareno S et al. (2000) Allosteric interactions of staurosporine and other indolocarbazoles with N-[methyl-3H]scopolamine and acetylcholine at muscarinic receptor subtypes: identification of a second allosteric site. Mol. Pharmacol 58, 194–207 [DOI] [PubMed] [Google Scholar]
- 144.Pollack S et al. (1999) The staurosporine-like compound L-753,000 (NB-506) potentiates the neurotrophic effects of neurotrophin-3 by acting selectively at the TrkA receptor. Mol. Pharmacol 56, 185–195 [DOI] [PubMed] [Google Scholar]
- 145.Padayatti PS et al. (2004) Structural insights into the regulation and the activation mechanism of mammalian guanylyl cyclases. Pharmacol. Ther 104, 83–99 [DOI] [PubMed] [Google Scholar]
- 146.Martin E et al. (2001) YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc. Natl. Acad. Sci. U. S. A 98, 12938–12942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stasch JP et al. (2001) NO-independent regulatory site on soluble guanylate cyclase. Nature 410, 212–215 [DOI] [PubMed] [Google Scholar]
- 148.Stasch JP et al. (2002) NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br. J. Pharmacol 136, 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]