

Abstract
The diuretic drug amiloride and its analogues were found previously to be allosteric modulators of antagonist binding to A2A adenosine receptors. In this study, the possibility of the allosteric modulation by amiloride analogues of antagonist binding at A1 and A3 receptors, as well as agonist binding at A1, A2A, and A3 receptors, was explored. Amiloride analogues increased the dissociation rates of two antagonist radioligands, [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX) and [3H]8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one ([3H]PSB-11), from A1 and A3 receptors, respectively. Amiloride and 5-(N,N-dimethyl)amiloride (DMA) were more potent at A1 receptors than at A3 receptors, while 5-(N,N-hexamethylene)amiloride (HMA) was more potent at A3 receptors. Thus, amiloride analogues are allosteric inhibitors of antagonist binding at A1, A2A, and A3 adenosine receptor subtypes. In contrast to their effects on antagonist-occupied receptors, amiloride analogues did not affect the dissociation rates of the A1 agonist [3H]N6-[(R)-phenylisopropyl]adenosine ([3H]R-PIA) from A1 receptors or the A2A agonist [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine ([3H]CGS21680) from A2A receptors. The dissociation rate of the A3 agonist radioligand [125I]N6-(4-amino-3-iodobenzyl)-adenosine-5′-N-methyluronamide ([125I]I-AB-MECA) from A3 receptors was decreased significantly by amiloride analogues. The binding modes of amiloride analogues at agonist-occupied and antagonist-occupied receptors differed markedly, which was demonstrated in all three subtypes of adenosine receptors tested in this study. The effects of the amiloride analogues on the action of the A3 receptor agonist were explored further using a cyclic AMP functional assay in intact CHO cells expressing the human A3 receptor. Both binding and functional assays support the allosteric interactions of amiloride analogues with A3 receptors.
Keywords: Adenosine receptors, Allosteric modulation, Amiloride, Sodium ions, Cyclic AMP, Receptor binding
1. Introduction
Four subtypes of adenosine receptors have been cloned, termed A1, A2A, A2B, and A3. Activation of A1 and A3 adenosine receptors induces the inhibition of the enzyme adenylate cyclase, whereas activation of A2A and A2B receptors leads to the stimulation of this enzyme. All four subtypes belong to the superfamily of GPCRs, which possess seven membrane-spanning α-helices [1].
A number of GPCRs are modulated by certain agents that bind to an allosteric site, which is distinct from the primary ligand binding site [2–4]. Allosteric modulation of GPCRs has been characterized most extensively for muscarinic receptors [5]. The allosteric modulation of a number of other GPCRs has also been well characterized recently using dissociation kinetic and functional assays. These include adenosine receptors [2,6,7], adrenergic receptors [8,9], dopamine receptors [10,11], metabotropic glutamate receptors [12], and others. The flexible nature of the interactions between the receptors and various allosteric modulators, together with the potential for subtype selectivity, makes allosteric sites attractive for therapeutic intervention [2,13,14]. In the case of the GABAA receptor [15], which is a transmitter-gated ion channel, the benzodiazepines acting on an allosteric site on the receptor showed substantial therapeutic effects and acceptable side-effects. By contrast, direct-acting GABAA agonists have not been used clinically due to the side-effects. Recently, positive allosteric modulators for GABAB receptors, which potentiate the effects of agonists, have also been identified and characterized [4] and might open new routes for the development of drugs in the field of metabotropic glutamate receptors [16]. The presence of an allosteric site on the A3 adenosine receptor [7,17] suggested that it might be possible to develop novel allosteric modulators that potentiate the effects of endogenous adenosine in the same way that benzodiazepines potentiate GABA.
It has been shown that both amiloride and sodium are allosteric modulators for a number of GPCRs [8–11,18–20]. In the case of the α2-adrenergic receptors, the residue responsible for the sodium modulation is found on the second transmembrane (TM) domain [8]. The diuretic drug amiloride, an inhibitor of the Na+/H+ antiporter, interacts with a number of cation-binding proteins [21]. Amiloride and its analogues have been shown to allosterically increase the antagonist dissociation rate from A2A adenosine receptors [19]. However, it was not known whether this effect was A2A-selective and/or antagonist-selective. Modulation by amiloride analogues of antagonist binding at A1 and A3 receptors and agonist binding at A1, A2A, and A3 receptors has not been explored previously.
In this study, the possibility of allosteric modulation of antagonist binding at A1 and A3 receptors by amiloride analogues (Fig. 1) was tested by detecting their effects on the dissociation rates of [3H]DPCPX and [3H]PSB-11 from A1 and A3 receptors, respectively. The possibility of allosteric modulation of agonist binding at A1, A2A, and A3 adenosine receptors was also studied by examining their effects on the dissociation of the agonist radioligands, [3H]R-PIA, [3H]CGS21680, and [125I]I-AB-MECA, from A1, A2A, and A3 receptors, respectively. The effects of amiloride analogues on the agonist actions at A3 receptors were explored further using a cyclic AMP functional assay.
Fig. 1.
Chemical structures of the amiloride analogues used in the present study.
2. Materials and methods
2.1. Materials
[125I]I-AB-MECA (2000 Ci/mmol), [3H]PSB-11 (53 Ci/mmol), [3H]R-PIA (34 Ci/mmol), [3H]DPCPX (120 Ci/mmol), [3H]CGS21680 (47 Ci/mmol), and [3H]cyclic AMP (40 Ci/mmol) were obtained from Amersham Pharmacia Biotech. CGS15943 was a gift from Novartis. CPA, NECA, Cl-IB-MECA, amiloride, DMA, HMA, and MIBA were purchased from the Sigma Chemical Co. All other chemicals were from standard commercial sources and of analytical grade.
2.2. Cell culture and membrane preparation
Chinese hamster ovary (CHO) cells expressing recombinant human A3 receptors were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, 100 μg/mL of streptomycin, 2 μmol/mL of glutamine, and 800 μg/mL of geneticin. RBL-2H3 (rat basophilic leukemia) cells expressing endogenous rat A3 receptors were cultured in the same medium without geneticin. Cells were harvested by trypsinization immediately followed by dilution of trypsin with medium; they were then centrifuged at 100 g for 5 min at room temperature, and the pellet was resuspended in 50 mM Tris·HCl buffer (pH 7.4). The suspension was homogenized and recentrifuged at 20,000 g for 20 min at 4°. The resultant pellets were resuspended in buffer in the presence of 3 units/mL of adenosine deaminase, and the suspension was stored at −80° prior to the binding experiments. The preparation of rat forebrain and rat striatal membranes was as described previously [7]. The protein concentration was measured using the Bradford assay [22].
2.3. Dissociation kinetics of the agonist [125I]I-AB-MECA and the antagonist [3H]PSB-11 from A3 adenosine receptors
The dissociation of [125I]I-AB-MECA was measured as follows. Membranes (20 μg protein) were preincubated at 25° with 1.0 nM [125I]I-AB-MECA (Kd = 0.99 nM), in a total volume of 200 μL Tris·HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2, and 1 mM EDTA for 60 min. The dissociation was then initiated by the addition of 3 μM Cl-IB-MECA with or without allosteric modulators. Amiloride analogues were dissolved in DMSO; the final DMSO concentration was ≤1.0%, and the appropriate vehicle was added in control experiments. The time course of dissociation of total binding was measured by rapid filtration at appropriate time intervals. Nonspecific binding was measured after a 60-min incubation in the presence of 3 μM Cl-IB-MECA. Binding reactions were terminated by filtration through Whatman GF/B glass-fiber filters under reduced pressure using an MT-24 cell harvester (Brandel), and radioactivity was determined using a Beckman 5500B γ-counter.
For the dissociation of the antagonist radioligand [3H]PSB-11 (Kd = 4.9 nM) [7,23] from A3 adenosine receptors, membranes (80 μg) were preincubated with [3H]PSB-11 (5 nM) in a total assay volume of 200 μL for 120 min. The dissociation was initiated by the addition of 3 μM Cl-IB-MECA in the presence and absence of the tested compounds. The time course of dissociation was measured by rapid filtration at appropriate time intervals.
2.4. Dissociation of the agonist [3H]R-PIA and the antagonist [3H]DPCPX from A1 adenosine receptors
Binding of 1 nM [3H]R-PIA to A1 adenosine receptors in rat forebrain membranes (80 μg protein) was carried out at 37° for 90 min in 50 mM Tris·HCl buffer (pH 7.7) containing 10 mM MgCl2 in a total assay volume of 400 μL. Binding of [3H]DPCPX to A1 adenosine receptors in rat forebrain membranes (60 μg protein) was carried out at 25° for 90 min in 50 mM Tris·HCl buffer (pH 7.4) in a total assay volume of 400 μL. The dissociation was begun by the addition of 10 μM CPA with or without the tested compounds. Samples were filtered after incubation at the time points indicated.
2.5. Dissociation of [3H]CGS21680 from A2A adenosine receptors
Rat striatal membranes (80 μg protein) were incubated with 15 nM [3H]CGS21680 at 25° for 90 min in 400 μL of 50 mM Tris·HCl, pH 7.7, containing 10 mM MgCl2. Dissociation was started by the addition of 10 μM NECA in the presence and absence of tested compounds.
2.6. Cyclic AMP accumulation assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method [24]. CHO cells that expressed recombinant human A3 adenosine receptors were harvested by trypsinization. After centrifugation (100 g for 5 min at room temperature) and resuspension in medium, cells were plated in 24-well dishes in 1.0 mL of medium. After 24 hr, the medium was removed, and the cells were washed three times with 1 mL DMEM containing 50 mM HEPES, pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min, forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL of 0.1 M HCl or 50 μL of cyclic AMP solution (0–16 pmol/200 μL for the standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
2.7. Statistical analysis
Binding parameters were estimated by GraphPAD Prism software (GraphPAD). Data are expressed as means ± SEM.
3. Results
3.1. Effects of amiloride analogues on the dissociation kinetics of the antagonist [3H]DPCPX and the agonist [3H]R-PIA binding to A1 adenosine receptors
We have described previously the allosteric modulation of A2A receptors by amiloride analogues through examining the effects of amiloride analogues on the dissociation kinetics of an A2A receptor antagonist, [3H]ZM241385, from A2A receptors [19]. In this study, we further tested the possibility of allosteric modulation of A1 receptors by amiloride analogues, which was achieved by examining the effects of these compounds on the dissociation of the A1 receptor antagonist [3H]DPCPX from A1 receptors. Similar to the effects of amiloride analogues on A2A receptors, these compounds also increased the dissociation of the A1 antagonist radioligand, [3H]DPCPX, from A1 receptors (Fig. 2A). The order of potencies of amiloride analogues to increase the dissociation rates was DMA > HMA > amiloride. We next examined the possible modulation of agonist-occupied A1 receptors by amiloride analogues. In contrast to the effects on the dissociation of antagonist, amiloride and its analogues had no effects on the dissociation of the A1 receptor agonist [3H]R-PIA from A1 receptors (Fig. 2B). This contrasted with the effects of PD81723 on A1 receptors, which enhanced agonist binding but did not affect antagonist binding [6]. The dissociation rates of [3H]DPCPX and [3H]R-PIA in the absence and presence of amiloride analogues are summarized in Table 1.
Fig. 2.
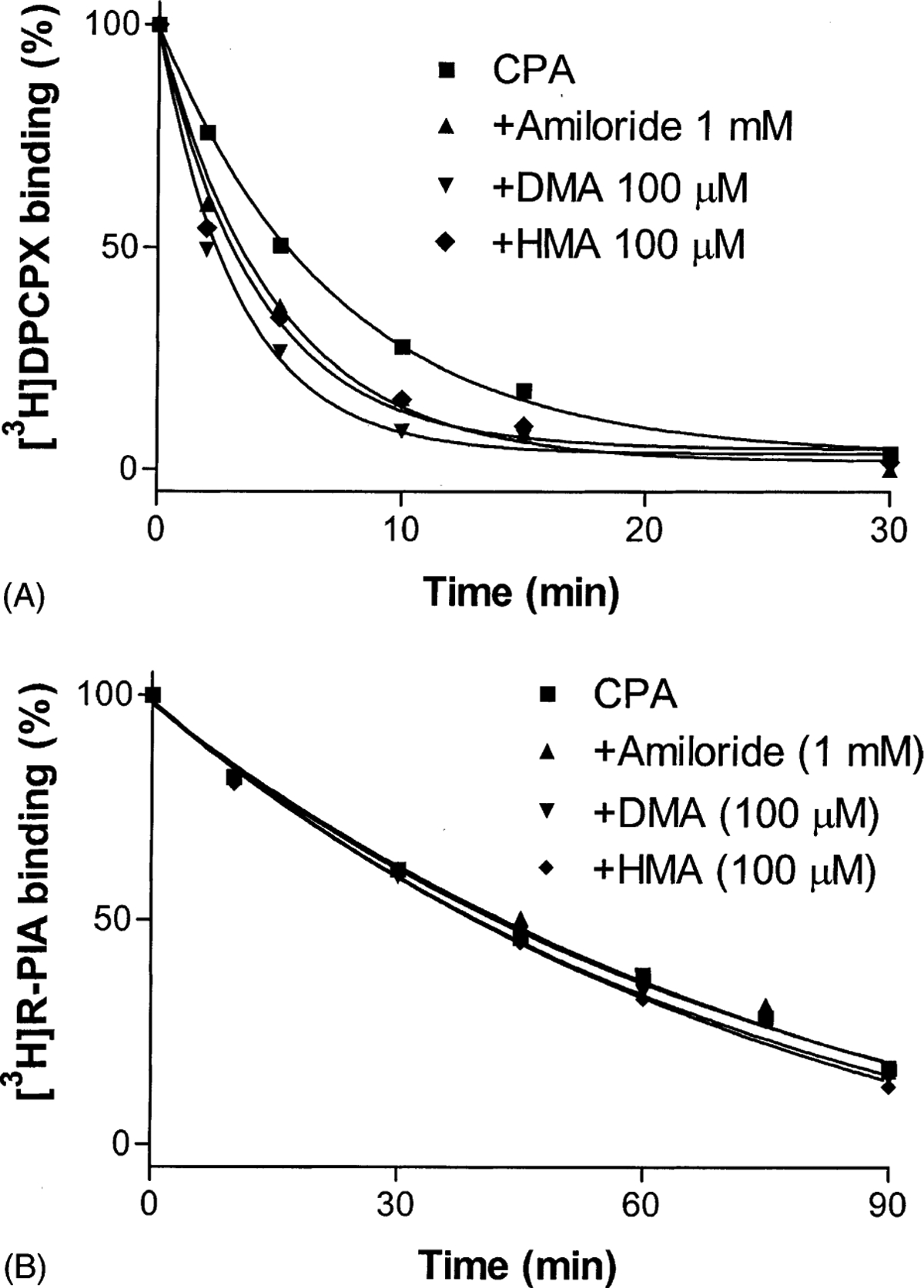
Time course of the dissociation of [3H]DPCPX (A) and [3H]R-PIA (B) from rat brain A1 adenosine receptors. Binding of [3H]DPCPX to A1 adenosine receptors in rat forebrain membranes (60 μg protein) was carried out at 25° for 90 min in 50 mM Tris·HCl buffer (pH 7.4) in a total assay volume of 400 μL. Binding of 1 nM [3H]R-PIA to A1 adenosine receptors in rat forebrain membranes (80 μg protein) was carried out at 37° for 90 min in 50 mM Tris·HCl buffer (pH 7.7) containing 10 mM MgCl2 in a total assay volume of 400 μL. The dissociation was begun by the addition of 10 μM CPA with or without the tested compounds. Samples were filtered after incubation at the time points indicated. The data shown were derived from one experiment performed in duplicate and are typical of three independent experiments giving similar results. The k−1 values were calculated from three independent experiments and are listed in Table 1.
Table 1.
Dissociation rates (k−1) of radiolabeled agonists and antagonists in the absence or presence of amiloride analogues
| Derivative | k−1 (min−1) | |||||
|---|---|---|---|---|---|---|
| A1 receptor | A2A receptor | A3 receptor | ||||
| [3H]R-PIA (agonist) | [3H]DPCPX (antagonist) | [3H]CGS21680 (agonist) | [3H]ZM241385a (antagonist) | [125I]I-AB-MECA (agonist) | [3H]PSB-11 (antagonist) | |
| Control | 0.012 ± 0.003 | 0.13 ± 0.02 | 0.066 ± 0.009 | 0.007 ± 0.002 | 0.059 ± 0.009 | 0.007 ± 0.001 |
| +Amiloride (1 mM) | 0.012 ± 0.002 | 0.19 ± 0.02* | 0.087 ± 0.022 | 0.008 ± 0.002 | 0.058 ± 0.006 | 0.008 ± 0.002 |
| +DMA (0.1 mM) | 0.013 ± 0.003 | 0.25 ± 0.04* | 0.084 ± 0.017 | NDb | 0.047 ± 0.009* | 0.009 ± 0.001* |
| +HMA (0.1 mM) | 0.013 ± 0.003 | 0.22 ± 0.02* | ND | 0.080 ± 0 010* | 0.031 ± 0.006* | 0.016 ± 0 003* |
| +MIBA(0.1mM) | ND | ND | ND | 0.040 ± 0005* | 0.035 ± 0.007* | 0.011 ±0002* |
Human A3 receptors were expressed in CHO cells, and the rat A3 receptor of RBL-2H3 cells was used. A1 and A2A receptors of rat brain (forebrain and striatum, respectively) were used. The k−1 values for [3H]PSB-11 were from an experiment performed at 4°; similar results from an experiment performed at 25° are shown in Fig. 4A. Results are expressed as mean ± SEM and are from at least three independent experiments performed in duplicate.
Data from Ref. [19].
ND = not determined.
P < 0.05, compared with the control.
3.2. Effects of amiloride analogues on the dissociation of the agonist [3H]CGS21680 from A2A adenosine receptors
As described above, it has been demonstrated that the dissociation of the antagonist radioligand [3H]ZM241385 from A2A receptors was modulated by amiloride analogues [19]. In this study, we further examined if the agonist-occupied A2A receptor can be modulated by amiloride analogues. Similar to their effects on the dissociation of the A1 agonist [3H]R-PIA from A1 receptors, amiloride analogues did not enhance the dissociation rate of the agonist [3H]CGS21680 from A2A receptors (Fig. 3). Their respective k−1 values in the absence and presence of amiloride and its analogues are summarized in Table 1.
Fig. 3.
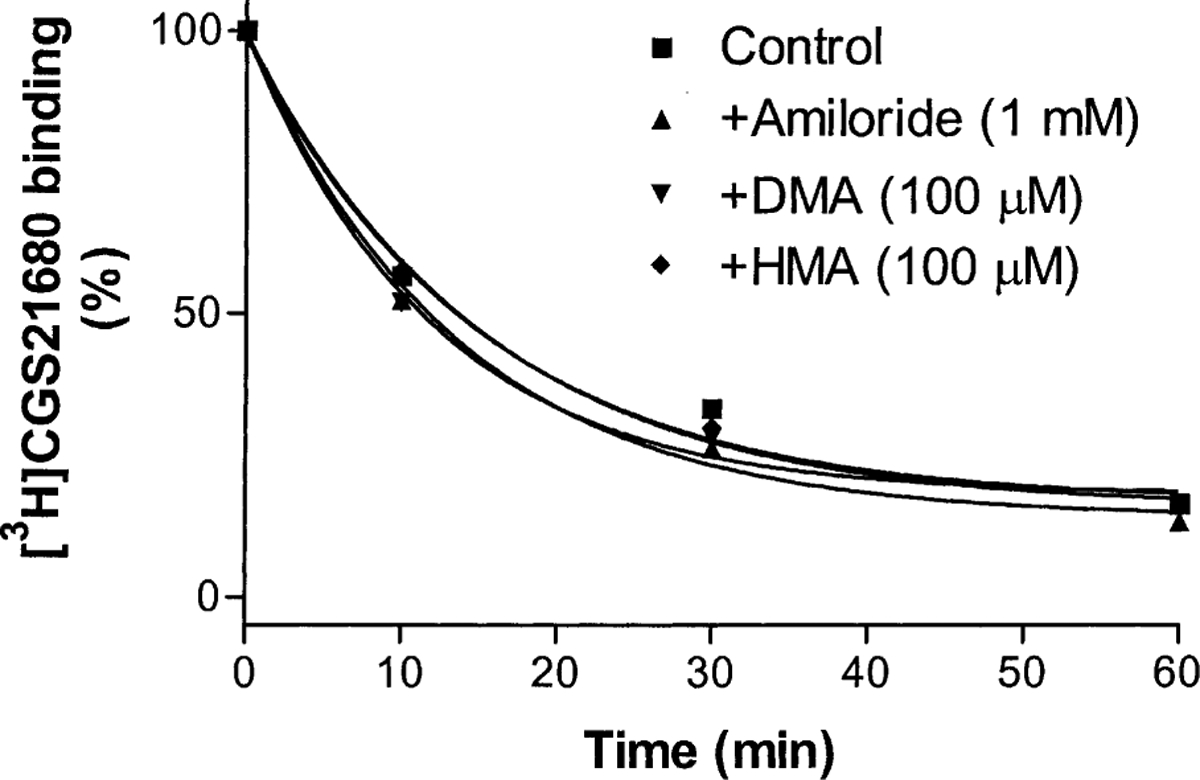
Time course of the dissociation of [3H]CGS21680 from A2A adenosine receptors. Rat striatal membranes (80 μg protein) were incubated with 15 nM [3H]CGS21680 at 25° for 90 min in 400 μL of 50 mM Tris·HCl, pH 7.7, containing 10 mM MgCl2. NECA (10 μM) was used to define nonspecific binding. Dissociation was started by the addition of 10 μM NECA in the presence and absence of the tested compounds. The data shown were from one experiment performed in duplicate. The k−1 values were calculated from three independent experiments and are listed in Table 1.
3.3. Effects of amiloride analogues on the dissociation of the antagonist radioligand [3H]PSB-11 and the agonist radioligand [125I]I-AB-MECA from A3 receptors
The dissociation rate of the antagonist [3H]PSB-11 was first measured at 25°. Amiloride (1 mM), which significantly increased the dissociation rate of [3H]DPCPX from A1 receptors and of [3H]ZM241385 from A2A receptors, only slightly affected the dissociation rate of [3H]PSB-11 from A3 receptors (Fig. 4A). The amiloride analogue HMA was found to be more potent than amiloride itself. HMA significantly increased the dissociation rate at 100 μM. However, another analogue, DMA (100 μM), which was more potent than HMA in increasing the dissociation rate of [3H]DPCPX from A1 receptors, only slightly increased the dissociation rate (Fig. 4A). In contrast to the amiloride analogues, sodium ions decreased the dissociation rate (Fig. 4A). The dissociation rates (k−1) in the absence and presence of NaCl (100 mM) were 0.27 ± 0.04 and 0.16 ± 0.02 min−1, respectively, which were significantly different (P < 0.05).
Fig. 4.
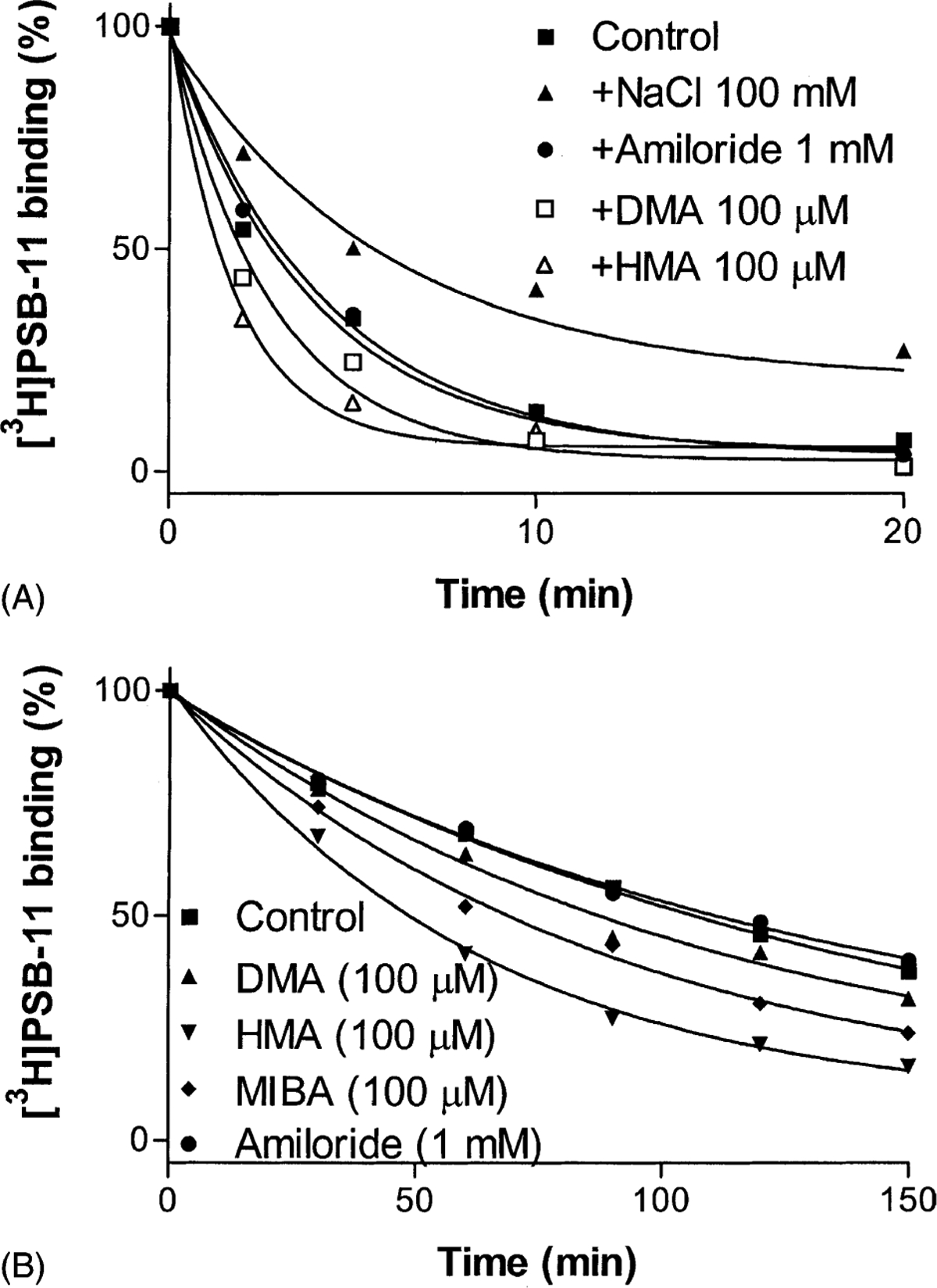
Time course of the dissociation of [3H]PSB-11 from human A3 adenosine receptors expressed in CHO cells. Membranes (80 μg) were preincubated with 5 nM [3H]PSB-11 in a total assay volume of 200 μL for 60 min at 25° (A) or for 120 min at 4° (B). The dissociation of [3H]PSB-11 from the A3 adenosine receptor was started by the addition of 3 μM Cl-IB-MECA in the absence or presence of the tested compounds. The reaction was measured by rapid filtration at appropriate time intervals. Data are from one experiment performed in duplicate. The k−1 values were calculated from three independent experiments and are listed in Table 1.
The dissociation of [3H]PSB-11 at 25° was very fast, particularly in the presence of amiloride analogues. Hence, in order to measure the dissociation rates more precisely, we also measured the dissociation rates at 4° (Fig. 4B). The dissociation rates (k−1) in the presence and absence of the amiloride analogues are summarized in Table 1.
The dissociation of the agonist [125I]I-AB-MECA from human A3 receptors was measured in the absence and presence of amiloride (Fig. 5A). The rate of dissociation was modified only slightly at 1 mM amiloride. At a higher concentration (3 mM), amiloride induced a modest but significant decrease of the dissociation. The dissociation rates in the absence and presence of 3 mM amiloride were 0.059 ± 0.009 and 0.046 ± 0.005 min−1, respectively. Amiloride analogues, alkyl-substituted at the 5-amino group (HMA), were found to be more potent than amiloride itself in decreasing the rate of [125I]I-AB-MECA dissociation (Fig. 5B). By comparison, the A1 receptor enhancer of agonist action, PD81723 (20 μM), did not affect the dissociation rate (Fig. 5B). The dissociation rates of [125I]I-AB-MECA in the absence and presence of amiloride analogues are summarized in Table 1.
Fig. 5.
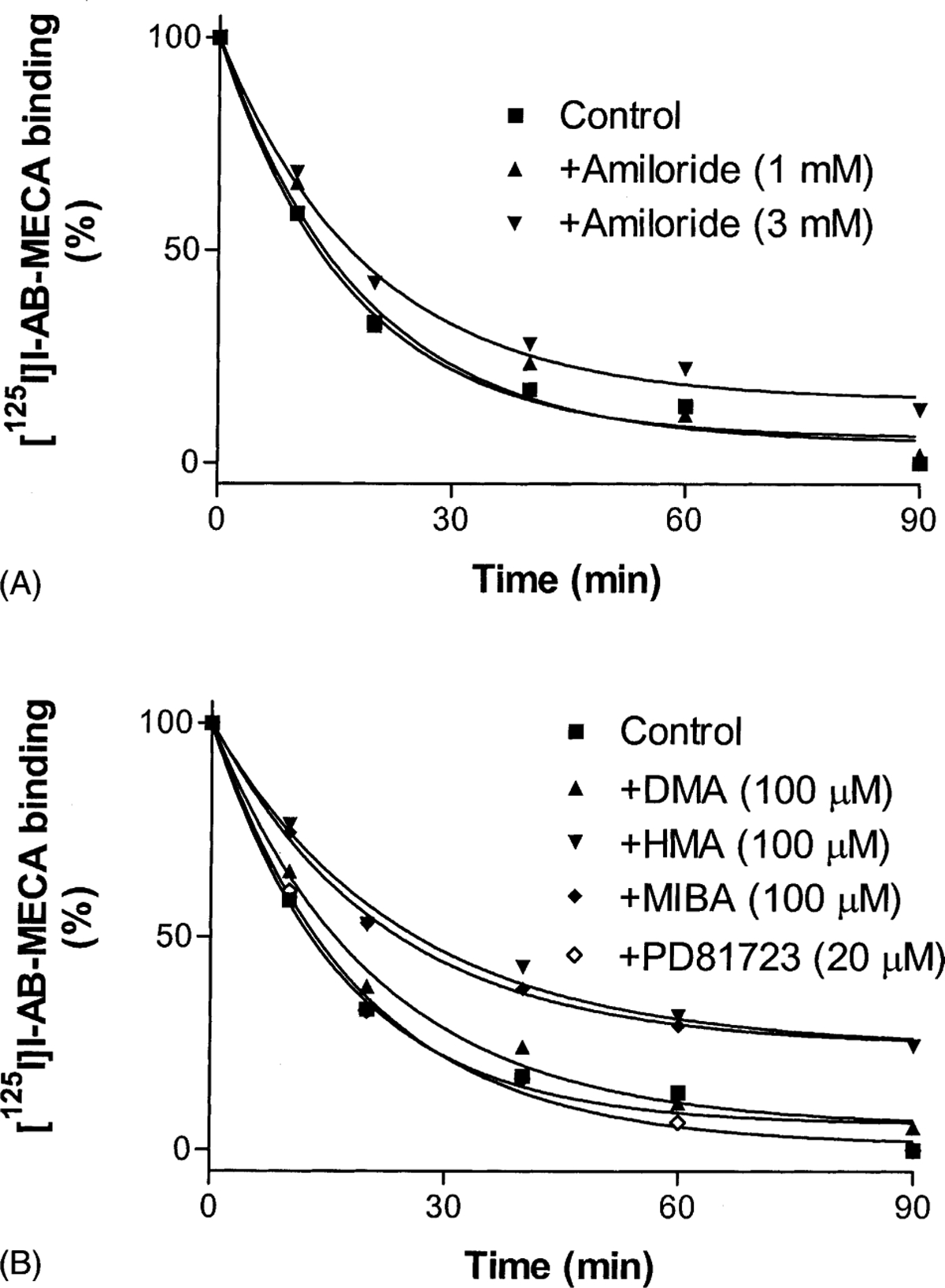
Effects of amiloride (A) and amiloride analogues (B) on the dissociation of [125I]I-AB-MECA from human A3 adenosine receptors expressed in CHO cells. [125I]I-AB-MECA (1.0 nM) was pre-associated with the A3 adenosine receptor membranes for 1 hr at 25° in a total assay volume of 200 μL. Nonspecific binding was determined in parallel by the addition of 3 μM Cl-IB-MECA before the pre-association. After 1 hr pre-association, dissociation was initiated by the addition of Cl-IB-MECA (final concentration, 3 μM) alone or with the amiloride analogues. The data shown were derived from one experiment performed in duplicate and are typical of three independent experiments giving similar results. The k−1 values were calculated from three independent experiments and are listed in Table 1.
The allosteric modulation of agonist binding was also demonstrated in rat A3 adenosine receptors in RBL-2H3 cells. The potency of these compounds on the binding and dissociation of [125I]I-AB-MECA from the rat A3 receptor was very similar to their effects on the human A3 receptor (Fig. 6, Table 2). No obvious species difference was observed for the amiloride analogues at the A3 receptor using the agonist radioligand. Unfortunately, the use of an antagonist radioligand at the A3 receptor is limited to humans, since there is no antagonist of suitable affinity at the rat A3 receptor.
Fig. 6.
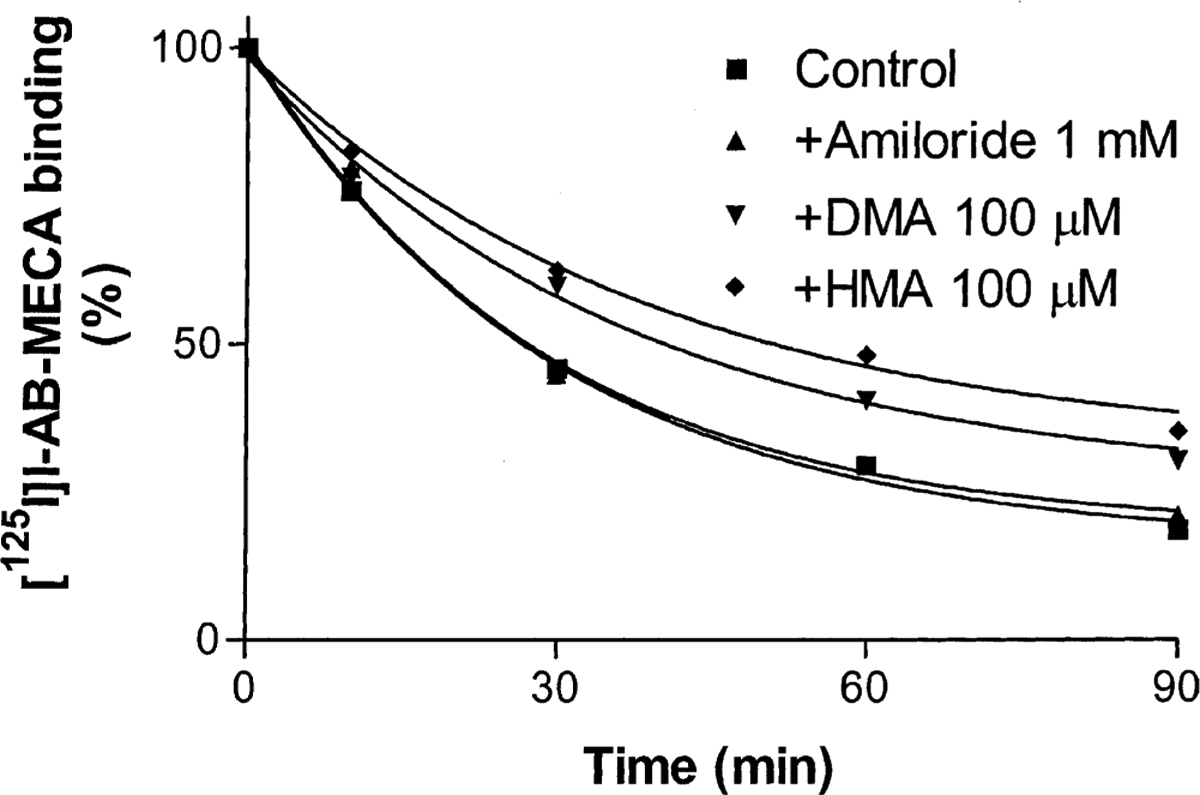
Time course of the dissociation of [125I]I-AB-MECA from rat A3 adenosine receptors. [125I]I-AB-MECA (1.0 nM) was pre-associated with rat A3 adenosine receptors from RBL-2H3 cell membranes (80 μg protein) at 25° for 1 hr. Nonspecific binding was determined in parallel by the addition of 3 μM Cl-IB-MECA before the pre-association. After 1 hr of pre-association, dissociation was initiated by the addition of Cl-IB-MECA (final concentration, 3 μM) and the tested agents. The data shown were derived from one experiment performed in duplicate and are typical of three independent experiments giving similar results.
Table 2.
Displacement by amiloride analogues of binding at A1, A2A, and A3 adenosine receptors
| Radioligand and receptor | ic50 (μM) | |||
|---|---|---|---|---|
| Amiloride | DMA | HMA | MIBA | |
| Antagonist [3H]DPCPX, 1 nM Rat A1 (slope) | 197 ± 23 (1.3 ± 0.1) | 7.9 ± 1.7 (1.2 ± 0.1) | 21.5 ± 3.7 (1.4 ± 0.2) | 12.6 ± 1.4 (1.4 ± 0.1) |
| Antagonist [3H]ZM241385, 1 nM Rat A2A | 9.7 ± 1.1a | NDb | 3.3 ± 0.5a | 3.0 ± 0.2a |
| Agonist [125I]I-AB-MECA, 1 nM Rat A3 (slope) | >100c | 19.7 ± 3.2 (1.1 ± 0.1) | 7.0 ± 1.4 (1.0 ± 0.2) | 7.1 ± 1.5 (1.2 ± 0.1) |
| Antagonist [3H]PSB-11, 5 nM Human A3 | 82.3 ± 7.2 | 12.8 ± 2.1 | 5.7 ± 0.9 | 8.2 ± 1.3 |
Concentrations of the radioligands used were close to their Kd values. Human A3 receptors were expressed in CHO cells, and the rat A3 receptor of RBL-2H3 cells was used. A1 and A2A receptors of rat brain (forebrain and striatum, respectively) were used. Results are expressed as mean ± SEM and are from at least three independent experiments performed in duplicate.
Ki values. Data from Ref. [19].
ND = not determined.
Displaced less than 15% of [125I]I-AB-MECA binding at 100 μM.
3.4. Concentration-dependent effect of amiloride analogues on the dissociation of the agonist radioligand [125I]I-AB-MECA and the antagonist radioligand [3H]PSB-11 from the A3 receptor
To further demonstrate the allosteric effects of these compounds, we observed their concentration-dependent effects on the dissociation of [125I]I-AB-MECA and [3H]PSB-11 from the human A3 receptor. To be comparable, 10 mM MgCl2 was present in both experiments. Figure 7A shows the influence of increasing concentrations of amiloride analogues on the dissociation of [125I]I-AB-MECA in the presence of 3 μM Cl-IB-MECA. Amiloride analogues decreased the dissociation rate in a concentration-dependent manner. The amiloride analogues were more potent than amiloride itself in decreasing the dissociation. The order of potencies was HMA ≥ MIBA > DMA > amiloride. In contrast to their effects on the dissociation of the agonist radioligand, the amiloride analogues accelerated the dissociation of the antagonist radioligand ([3H]PSB-11) from the receptor in a concentration-dependent manner (Fig. 7B).
Fig. 7.
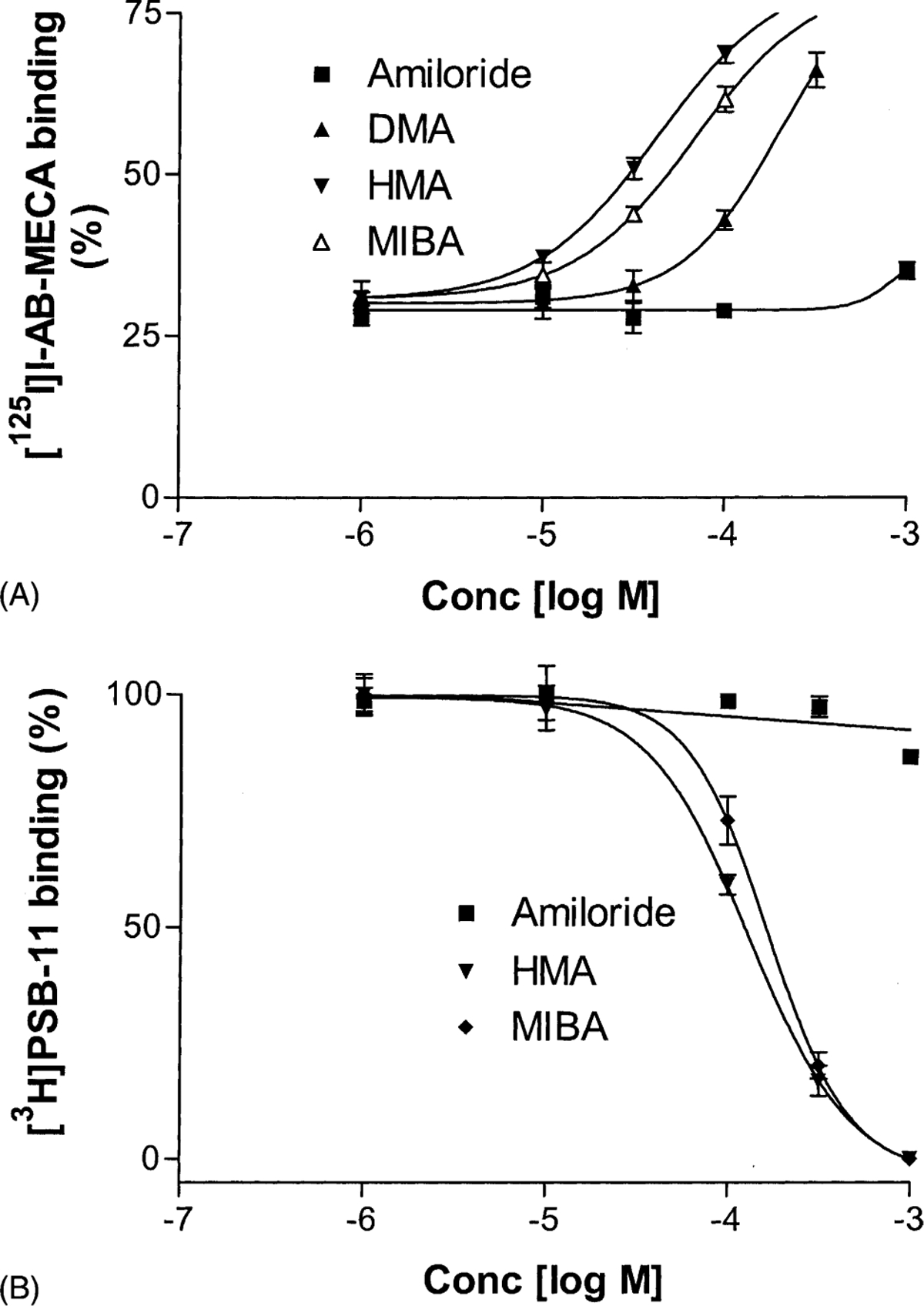
Concentration–response curves for slowing the dissociation of the agonist [125I]I-AB-MECA (A) and for increasing the dissociation of the antagonist [3H]PSB-11 (B) from human A3 receptors by the amiloride analogues. [125I]I-AB-MECA (1.0 nM) was pre-associated with CHO cell membranes (20 μg protein) for 60 min at 25° without additions (total binding) or in the presence of 3 μM Cl-IB-MECA (nonspecific binding). At the end of the incubation period, 3 μM Cl-IB-MECA was added simultaneously with vehicle or various concentrations of the test compounds. The incubation was terminated after an additional 45 min. Control specific binding at the end of the 60-min dissociation period was approximately 25% of the total binding. Data are mean values from three independent experiments performed in duplicate. The procedure for the experiments regarding [3H]PSB-11 was similar to that of [125I]I-AB-MECA except that it was performed at 4°. MgCl2 was present in both experiments. One hundred percent refers to the specific binding remaining after 45-min dissociation in the absence of added amiloride or amiloride analogues.
3.5. Displacement by amiloride analogues of ligand binding to A1, A2A, and A3 receptors
The amiloride analogues inhibited radioligand binding in all cases, including the binding of [125I]I-AB-MECA to rat A3 receptors. The displacement curves for rat A1 and A3 receptors are shown in Fig. 8. The affinities of these compounds for A1, A2A, and A3 receptors are summarized in Table 2. In general, the presence of an N5-alkyl substitution increased affinity; however, selectivity among the subtypes was not pronounced except that amiloride showed moderate selectivity for A2A receptors.
Fig. 8.
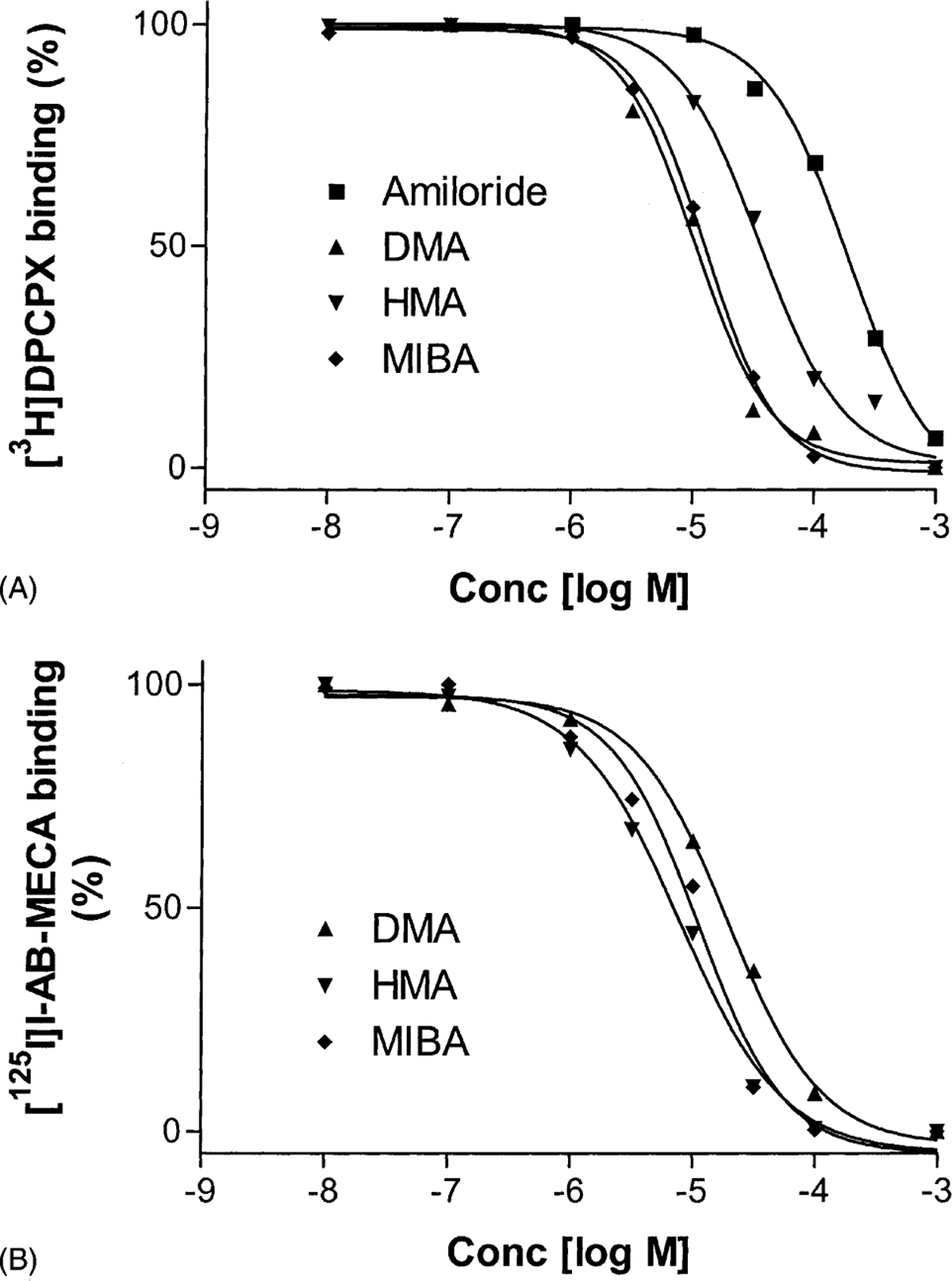
Displacement of radioligand binding to rat A1 ([3H]DPCPX) and A3 ([125I]I-AB-MECA) receptors by amiloride analogues. The procedures were described in “Section 2.” The data points are from a representative experiment performed in duplicate. The mean Ki values calculated from three independent experiments are listed in the text.
3.6. Effects of the amiloride analogues on A3 agonist-induced inhibition of forskolin-stimulated cyclic AMP production in intact CHO cells expressing human A3 receptor
The A3 adenosine receptor is a Gi-coupled receptor. The potent and selective A3 receptor agonist Cl-IB-MECA inhibited forskolin-stimulated cyclic AMP production in a concentration-dependent manner, while amiloride and HMA had no direct effects on forskolin-stimulated cyclic AMP production in the absence of agonists (Fig. 9). The agonist concentration–response curve was shifted to the right by the competitive A3 antagonist MRS1220 [25] in a concentration-dependent manner (Fig. 9).
Fig. 9.
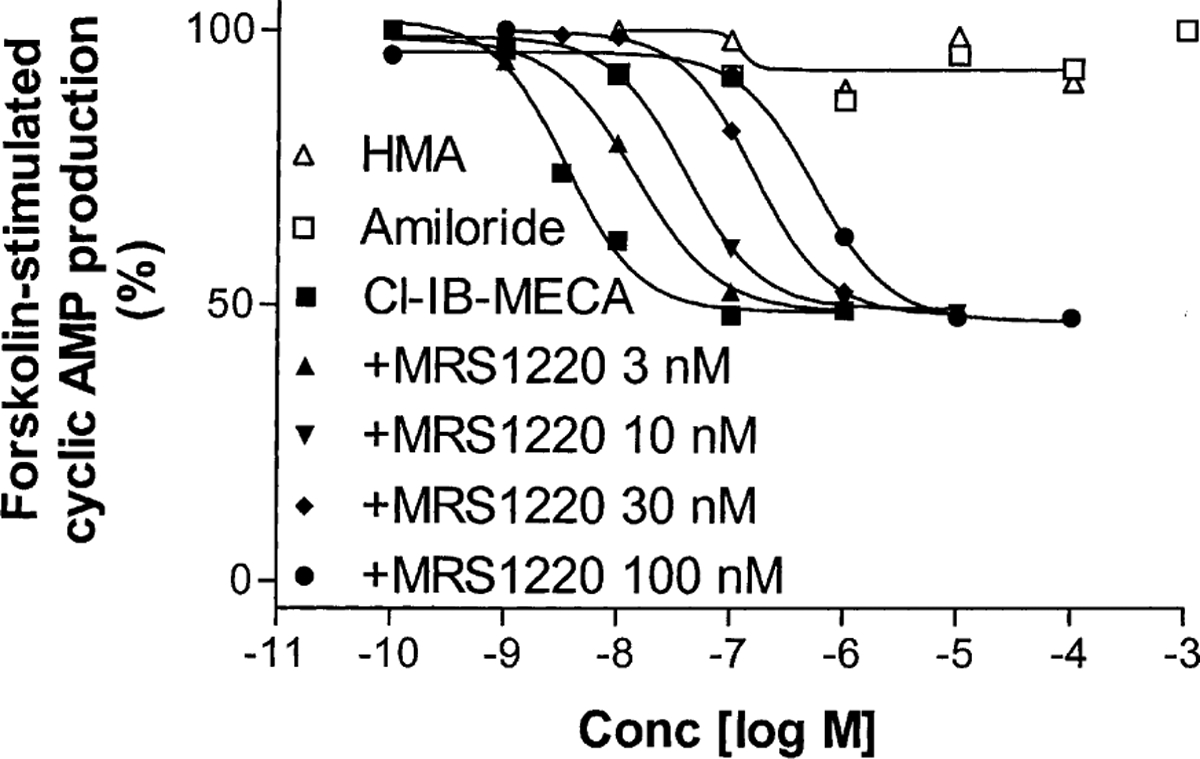
Agonist-induced inhibition of forskolin-stimulated cyclic AMP production in CHO cells stably transfected with human A3 receptors. All experiments were performed in the presence of 10 μM rolipram and 3 units/mL of adenosine deaminase. Forskolin (10 μM) was used to stimulate cyclic AMP levels. The level of cyclic AMP corresponding to 100% was 210 ± 32 pmol/mL. The data shown are from one experiment performed in duplicate and are typical of three independent experiments giving similar results.
As amiloride analogues decreased the dissociation rate of the agonist [125I]I-AB-MECA from A3 receptors, we further examined their effects on agonist-induced inhibition of forskolin-stimulated cyclic AMP production in intact CHO cells expressing human A3 receptors. Both amiloride and amiloride analogues caused a rightward shift of the Cl-IB-MECA concentration–response curve (Fig. 10). However, the pattern of the shift by the amiloride analogues was different from that by the competitive A3 antagonist MRS1220 (Fig. 9), e.g. 1.0 mM amiloride (ic50 = 82 μM) and 100 μM MIBA (ic50 = 8.2 μM) only induced a 2- and 3-fold rightward shift, respectively, while 10 nM MRS1220 (approx. 10 times its Ki value) induced an 11-fold shift of the Cl-IB-MECA concentration–response curve. In the case of HMA (ic50 = 5.7 μM), it induced a relatively larger rightward shift of the agonist concentration–response curve and in a parallel manner (8-fold at 100 μM). To examine the mechanisms of HMA action in the cyclic AMP assay, Schild analysis [26] was used to examine the rightward shift produced by the modulator (Fig. 10; only concentrations of less than 30 μM were used). This analysis gave a Schild slope of 0.58 ± 0.11, which was significantly different from unity (P < 0.05). MRS1220, a competitive antagonist, yielded a slope of 0.97 ± 0.08 in a similar experiment. At a higher concentration (100 μM), both HMA and MIBA also caused a significant decrease of the maximal effect of Cl-IB-MECA (Fig. 10). The cyclic AMP levels in the presence of 100 μM HMA and MIBA were 139 ± 11 and 128 ± 8 pmol/mL, respectively, which were significantly different from that in the absence of the amiloride analogues (107 ± 10 pmol/mL) (P < 0.05). Due to the fact that only a limited number of concentrations of amiloride and MIBA could be used, Schild analysis of the rightward shift of the Cl-IB-MECA concentration–response curves in the presence of amiloride and MIBA was not feasible.
Fig. 10.
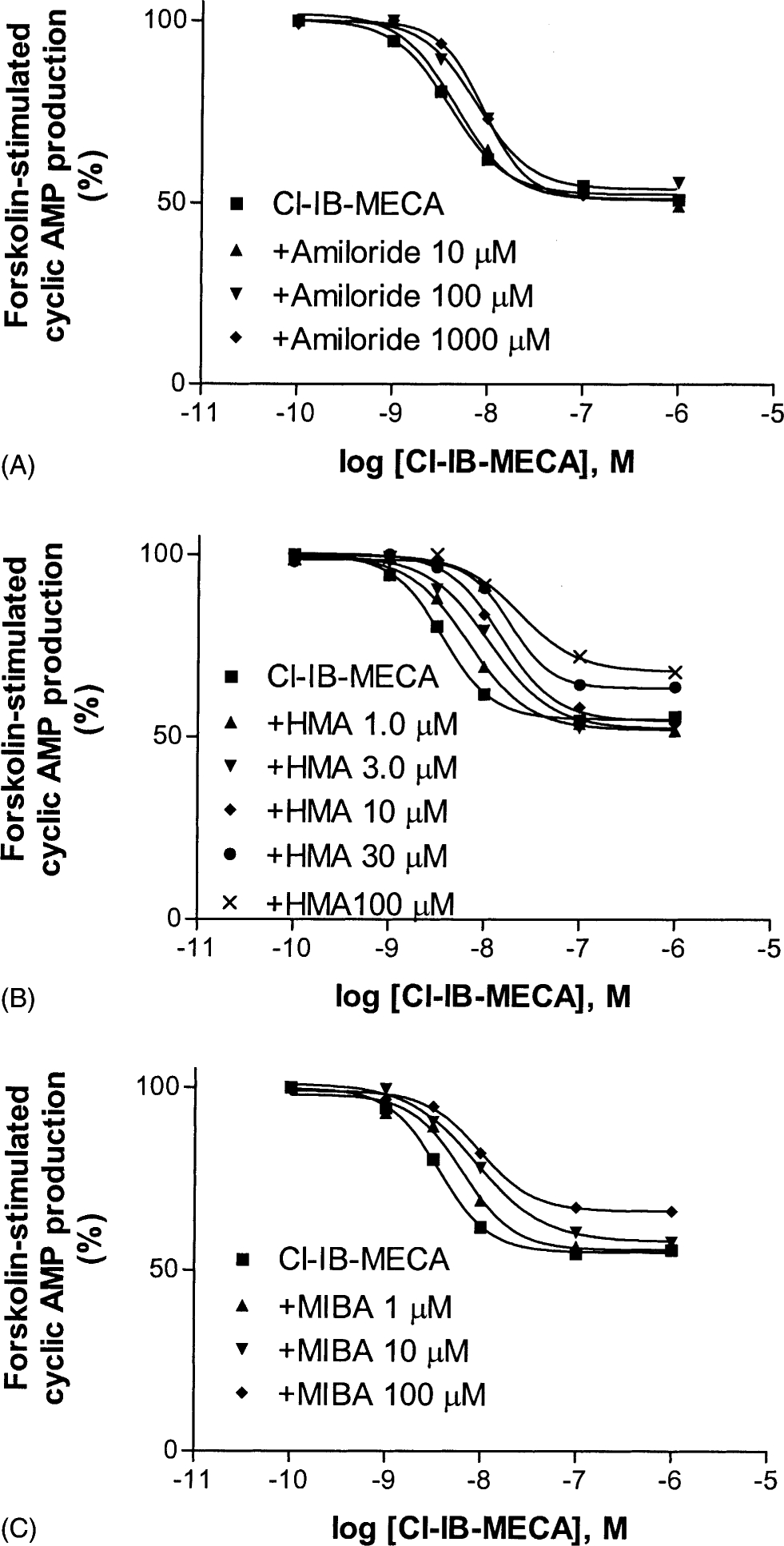
Effect of amiloride analogues on the A3 agonist-induced inhibition of forskolin-stimulated cyclic AMP production in CHO cells expressing human A3 receptors. The experiments were performed in the presence of 10 μM rolipram and 3 units/mL of adenosine deaminase. Forskolin (10 μM) was used to stimulate cyclic AMP levels. The level of cyclic AMP corresponding to 100% was 210 ± 32 pmol/mL. The data are from a single experiment representative of three independent experiments.
3.7. Effect of amiloride on the potency of the A1 receptor antagonist to block A1 receptors
Since it was determined in the dissociation kinetic experiment that amiloride increased the antagonist dissociation rate, we further examined if amiloride can affect the potency of an A1 receptor antagonist to block A1 receptors in the functional cyclic AMP assay. Figure 11 shows that amiloride (1 mM) induced a modest but significant decrease of the potency of the antagonist DPCPX. The ec50 values of DPCPX in the absence and presence of amiloride were 45 ± 12 and 86 ± 13 nM, respectively, which were significantly different (P < 0.05).
Fig. 11.
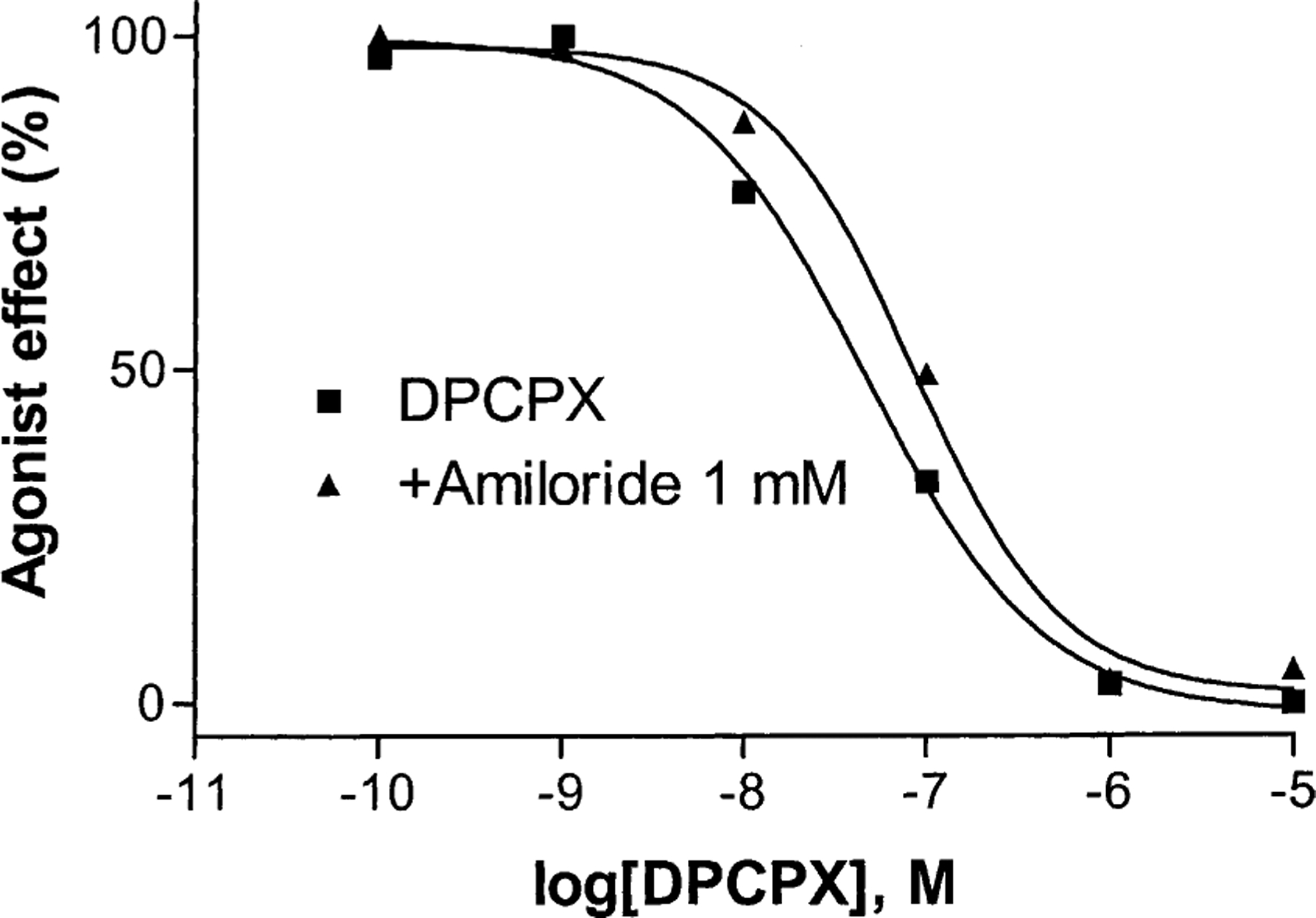
Effect of amiloride on the potency of the A1 receptor antagonist (DPCPX) to block A1 receptors expressed in CHO cells. The experiments were performed in the presence of 10 μM rolipram and 3 units/mL of adenosine deaminase. The concentration of the A1 receptor agonist CPA was 100 nM. Forskolin (10 μM) was used to stimulate cyclic AMP levels. The level of cyclic AMP corresponding to 100% agonist effect (100 nM CPA) was 97 ± 23 pmol/mL, and 0% corresponded to 248 ± 42 pmol/mL. The data are from a single experiment representative of three independent experiments. The ec50 values from three independent experiments are listed in the text.
4. Discussion
The potassium sparing diuretic amiloride has been shown to act as both an antagonist and an allosteric modulator for a number of GPCRs [9,11,18,27–29]. Recently, we also showed that amiloride and amiloride analogues are allosteric modulators for A2A receptors by increasing the antagonist dissociation rate [19]. The present study further demonstrated that amiloride analogues are allosteric modulators for agonist binding at A3 but not A1 and A2A receptors, and that they are allosteric inhibitors for antagonist binding to A1, A2A, and A3 receptors. The binding modes of the amiloride analogues at the agonist-occupied and antagonist-occupied receptors were markedly different.
It has been suggested that amiloride and amiloride analogues are competitive antagonists for A1 receptors [30], as these compounds did not affect the dissociation of the antagonist radioligand [3H]DPCPX from A1 receptors in calf brain. However, only relatively low concentrations of amiloride and amiloride analogues were used in that study. The present result does not necessarily contradict those earlier results [30]. It is possible that the amiloride analogues can compete for the antagonist binding site at low concentrations and bind at the allosteric site at higher concentrations.
It has been reported that the A1 adenosine receptor was modulated allosterically by a series of aminobenzoylthiophenes, including PD81723 [6]. However, in contrast to the present results, PD81723 only affected the agonist-occupied A1 receptors without influencing antagonist-occupied receptors, suggesting different mechanisms of action of these two series of modulators for A1 receptors.
We recently identified two other chemical classes of compounds, including the imidazoquinoline DU124183 and the pyridinylisoquinoline VUF5455, as allosteric modulators of A3 receptors [7,17]. However, in contrast to the amiloride analogues, DU124183 and VUF5455 only selectively affected the agonist-occupied A3 receptor without any effect on antagonist-occupied A1, A2A, and A3 receptors, as well as the agonist-occupied A1 and A2A receptors, suggesting that these compounds utilize different mechanisms of action in modulating A3 receptors.
Compared with the effect of HMA on the antagonist-occupied A2A receptors, which increased by more than 10-fold the [3H]ZM241385 dissociation rate [19], the effects of HMA on antagonist-occupied A1 and A3 receptors, as well as agonist-occupied A3 receptors were relatively small (approximately a 2-fold increase or decrease of the dissociation rates). This suggested that HMA is relatively selective for A2A receptors.
The most potent analogues examined were HMA and MIBA, suggesting that the binding site for amiloride analogues can accommodate large steric bulk in the region of the N5-amine. Similar results have also been demonstrated in α2-adrenergic and D2 dopamine receptors [11,31].
The effect of allosteric modulators on agonist action at A3 receptors was assessed in an experiment using Cl-IB-MECA-induced inhibition of forskolin-stimulated cyclic AMP production in intact CHO cells expressing human A3 receptors. Amiloride analogues induced a rightward shift of the agonist concentration–response curve and also elicited a marked reduction of the maximum efficacy of the agonist. Amiloride (1.0 mM, ic50 = 82 μM) and MIBA (100 μM, ic50 = 8.2 μM) only induced 2- and 3-fold rightward shifts, respectively, possibly suggesting a noncompetitive interaction. Schild analysis of the effects of HMA on the Cl-IB-MECA concentration–response curves gave a Schild slope significantly different from unity, whereas in an analogous experiment with a competitive antagonist, MRS1220, the Schild slope was close to 1. The inhibitory effect on the maximum response was indicative of negative cooperativity in the functional activation of the receptor [32].
In conclusion, the present study demonstrated that the agonist-occupied and antagonist-occupied adenosine receptors were differentially modulated by amiloride analogues. Amiloride analogues are both antagonists and allosteric inhibitors of antagonist binding at A1 A2A, and A3 receptors; however, only the agonist binding at A3, but not A1 and A2A receptors was allosterically modulated by these compounds. Further characterization of the interaction between amiloride analogues and the allosteric site on the receptor may provide useful information for the rational design of subtype-selective drugs.
Acknowledgments
We thank Drs. Gary Stiles (Duke University) and Joel Linden (University of Virginia) for the gifts of CHO cells expressing the human A3 receptor and of RBL-2H3 cells expressing the rat A3 receptor, respectively. Z.-G.G. thanks Gilead Sciences for financial support.
Abbreviations:
- CPA
N6-cyclopentyladenosine
- DMA
5-(N,N-dimethyl)-amiloride
- GABA
γ-aminobutyric acid
- GPCR
G protein-coupled receptor
- HMA
5-(N,N-hexamethylene)amiloride
- MIBA
5-(N-methyl-N-isobutyl)amiloride
- NECA
5′-N-ethylcarboxamidoadenosine
- AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide
- CGS15943
5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline
- CGS21680
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- PD81723
2-amino-4,5-dimethyl-3-thienyl-[3-(trifluoromethyl)phenyl]-methanone
- R-PIA
N6-[(R)-phenylisopropyl]adenosine
- PSB-11
8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-5-one
- ZM241385
4-{2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethyl}phenol
References
- [1].Olah ME, Stiles GL. The role of receptor structure in determining adenosine receptor activity. Pharmacol Ther 2000;85:55–75. [DOI] [PubMed] [Google Scholar]
- [2].Linden J Allosteric enhancement of adenosine receptors. In: Jacobson KA, Jarvis MF, editors. Purinergic approaches in experimental therapeutics. New York: Wiley-Liss; 1997. p. 85–97. [Google Scholar]
- [3].Kobilka B Allosteric activation of CaR by l-amino acids. Proc Natl Acad Sci USA 2000;97:4419–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, Bettler B, Kaupmann K. Positive allosteric modulation of native and recombinant γ-aminobutyric acidB receptors by 2,6-di-tert-butyl-4-(3-hydroxy-2,2-dimethyl-propyl)-phenol (CGP7930) and its aldehyde analog CGP13501. Mol Pharmacol 2001;60:963–71. [PubMed] [Google Scholar]
- [5].Holzgrabe U, Mohr K. Allosteric modulators of ligand binding to muscarinic acetylcholine receptors. Drug Discov Today 1998;3:214–22. [Google Scholar]
- [6].Bruns RF, Fergus JH. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol 1990;38:939–49. [PubMed] [Google Scholar]
- [7].Gao Z-G, Van Muijlwijk-Koezen JE, Chen A, Müller CE, IJzerman AP, Jacobson KA. Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol 2001;60:1057–63. [PMC free article] [PubMed] [Google Scholar]
- [8].Hortstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ Jr, Limbird LE. An aspartate conserved among G-protein receptors confers allosteric regulation of α2-adrenergic receptors by sodium. J Biol Chem 1990;265:21590–5. [PubMed] [Google Scholar]
- [9].Leppik RA, Birdsall NJM. Agonist binding and function at the human α2A-adrenoceptor: allosteric modulation by amilorides. Mol Pharmacol 2000;58:1091–9. [DOI] [PubMed] [Google Scholar]
- [10].Neve KA. Regulation of dopamine D2 receptors by sodium and pH. Mol Pharmacol 1991;39:570–8. [PubMed] [Google Scholar]
- [11].Hoare SRJ, Strange PG. Regulation of D2 dopamine receptors by amiloride and amiloride analogs. Mol Pharmacol 1996;50:1295–308. [PubMed] [Google Scholar]
- [12].Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C, Kuhn R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol 1999;55:453–61. [PubMed] [Google Scholar]
- [13].Birdsall NJM, Cohen F, Lazareno S, Matsui H. Allosteric regulation of G-protein-linked receptors. Biochem Soc Trans 1995;23:108–11. [DOI] [PubMed] [Google Scholar]
- [14].Bhattacharya S, Linden J. Effects of long-term treatment with the allosteric enhancer, PD81,723, on Chinese hamster ovary cells expressing recombinant human A1 adenosine receptors. Mol Pharmacol 1996;50:104–11. [PubMed] [Google Scholar]
- [15].Barker JL, Harrison NL, Mariani AP. Benzodiazepine pharmacology of cultured mammalian CNS neurons. Life Sci 1986;39:1959–68. [DOI] [PubMed] [Google Scholar]
- [16].Pin JP, Parmentier ML, Prezeau L. Positive allosteric modulators for γ-aminobutyric acidB receptors open new routes for the development of drugs targeting family 3 G-protein-coupled receptors. Mol Pharmacol 2001;60:881–4. [DOI] [PubMed] [Google Scholar]
- [17].Gao ZG, Kim SG, Soltysiak KA, Melman N, IJzerman AP, Jacobson KA. Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol Pharmacol 2002;62:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Howard MJ, Hughes RJ, Motulsky HJ, Mullen MD, Insel PA. Interactions of amiloride with α- and β-adrenergic receptors: amiloride reveals an allosteric site on α2-adrenergic receptors. Mol Pharmacol 1987;32:53–8. [PubMed] [Google Scholar]
- [19].Gao Z-G, IJzerman AP. Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol 2000;60:669–76. [DOI] [PubMed] [Google Scholar]
- [20].Neve KA, Cumbay MG, Thompson KR, Yang R, Buck DC, Watts VJ, DuRand CJ, Teeter MM. Modeling and mutational analysis of a putative sodium-binding pocket on the dopamine D2 receptor. Mol Pharmacol 2001;60:373–81. [DOI] [PubMed] [Google Scholar]
- [21].Kleyman TR, Cragoe EJ Jr. Cation transport probes: the amiloride series. Methods Enzymol 1990;191:739–55. [DOI] [PubMed] [Google Scholar]
- [22].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- [23].Müller CE, Diekmann M, Thorand M, Ozola V. [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg Med Chem Lett 2002;12:501–3. [DOI] [PubMed] [Google Scholar]
- [24].Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem 1990;189:231–4. [DOI] [PubMed] [Google Scholar]
- [25].Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 1997;36:1157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother 1959;14:48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Insel PA, Snavely MD, Healy DP, Munzel PA, Potenza CL, Nord EP. Radioligand binding and functional assays demonstrate postsynaptic α2-receptors on proximal tubules of rat and rabbit kidney. J Cardiovasc Pharmacol 1985;8:S9–17. [PubMed] [Google Scholar]
- [28].Blankesteijn WM, Siero HL, Rodrigues de Miranda JF, van Megen YJ, Russel FG. Characterization of muscarinic receptors in rat kidney. Eur J Pharmacol 1993;244:21–7. [DOI] [PubMed] [Google Scholar]
- [29].Hoare SRJ, Coldwell MC, Armstrong D, Strange PG. Regulation of human D1, D2(long), D2(short), D3 and D4 dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol 2000;130:1045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Garritsen A, Beukers MW, IJzerman AP, Cragoe EJ Jr, Soudijn W. The mode of interaction of amiloride and some of its analogues with the adenosine A1 receptor. Neurochem Int 1992;20:207–13. [DOI] [PubMed] [Google Scholar]
- [31].Leppik RA, Lazareno S, Mynett A, Birdsall NJM. Characterization of the allosteric interactions between antagonists and amiloride analogues at the human α2A-adrenergic receptor. Mol Pharmacol 1998;53:916–25. [PubMed] [Google Scholar]
- [32].Hall DA. Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol 2000;58:1412–23. [DOI] [PubMed] [Google Scholar]