

Abstract
Acute myeloid leukaemia (AML) carrying nucleophosmin (NPM1) mutations has been defined as a distinct entity of acute leukaemia. Despite remarkable improvements in diagnosis and treatment, the long‐term outcomes for this entity remain unsatisfactory. Emerging evidence suggests that leukaemia, similar to other malignant diseases, employs various mechanisms to evade killing by immune cells. However, the mechanism of immune escape in NPM1‐mutated AML remains unknown. In this study, both serum and leukemic cells from patients with NPM1‐mutated AML impaired the immune function of CD8+ T cells in a co‐culture system. Mechanistically, leukemic cells secreted miR‐19a‐3p into the tumour microenvironment (TME) via small extracellular vesicles (sEVs), which was controlled by the NPM1‐mutated protein/CCCTC‐binding factor (CTCF)/poly (A)‐binding protein cytoplasmic 1 (PABPC1) signalling axis. sEV‐related miR‐19a‐3p was internalized by CD8+ T cells and directly repressed the expression of solute‐carrier family 6 member 8 (SLC6A8; a creatine‐specific transporter) to inhibit creatine import. Decreased creatine levels can reduce ATP production and impair CD8+ T cell immune function, leading to immune escape by leukemic cells. In summary, leukemic cell‐derived sEV‐related miR‐19a‐3p confers immunosuppression to CD8+ T cells by targeting SLC6A8‐mediated creatine import, indicating that sEV‐related miR‐19a‐3p might be a promising therapeutic target for NPM1‐mutated AML.
Keywords: AML, CD8+ T cells, creatine, extracellular vesicles, nucleophosmin
Abbreviations
- AGAT
Glycine amidinotransferase
- AML
Acute myeloid leukaemia
- CAR‐Ts
Chimeric antigen receptor‐engineered T cells
- CFSE
Carboxyfluorescein succinimidyl ester
- ChIP
Chromatin immunoprecipitation
- CK
Creatine kinase
- CM
Conditioned medium
- Cr
Creatine
- Crn
Creatinine
- CTCF
CCCTC binding factor
- FCM
Flow cytometry
- GAMT
Guaidinoacetate methyltransferase
- GSEA
Gene set enrichment analysis
- ICIs
Immune checkpoint inhibitors
- IF
Immunofluorescence
- IHC
Immunohistochemistry
- KHSRP
KH‐type splicing regulatory protein
- MMPs
Matrix metalloproteases
- NES
Nuclear export signal
- NLS
Nuclear localization signal
- NPM1
Nucleophosmin 1
- NTA
Nanoparticle tracking analysis
- PABPC1
Poly (A) binding protein cytoplasmic 1
- PCr
Phospho‐creatine
- RBPDB
RNA‐Binding Protein DataBase
- RBPs
RNA‐binding proteins
- RIP
RNA immunoprecipitation
- sEVs
small extracellular vesicles
- SLC6A8
Solute carrier family 6 member 8
- TEM
Transmission electron microscopy
- TME
Tumour microenvironment
- WHO
World Health Organization
1. INTRODUCTION
Acute myeloid leukaemia (AML) is a heterogeneous disease characterized by multiple cytogenetic and molecular abnormalities, with a very poor prognosis (Coombs et al., 2016). Nucleophosmin (NPM1) gene mutation, especially the type A NPM1 mutation (NPM1mA, the most frequent in NPM1 mutation) (Zou et al., 2017), is one of the most frequent and clinically relevant genetic alterations, accounting for approximately 30% of all AML cases (Papaioannou & Petri, 2019). NPM1 mutation, results in a functionally stronger nuclear‐export signal than a nuclear‐localization signal, giving rise to the cytoplasmic delocalization of NPM1 mutant protein, which is thought to play a key role in leukemogenesis (Papaemmanuil et al., 2016). Currently, NPM1‐mutated AML (NPM1mut AML) treatment still relies on standard chemotherapy plus an FLT3 inhibitor, with or without allogeneic hematopoietic stem cell transplantation (Falini et al., 2021). More recently, combination treatment with venetoclax plus hypomethylating agents or a low‐dose cytarabine has demonstrated promising clinical activity (Dinardo & Wei, 2020; Falini et al., 2021). Nevertheless, some patients with NPM1mut AML, especially older patients, eventually relapse and die from progressive disease (Gionfriddo et al., 2021), indicating that new and effective therapeutic strategies are needed for this AML entity.
Immunotherapy for cancers first entered into clinical practice in the late 1990s and has opened a new era of cancer therapy in recent years (Goebeler & Bargou, 2020). T cells play central roles in mediating and orchestrating immune responses against cancers (Dumauthioz et al., 2020). Many strategies are aimed at exploiting the potential of T cells to recognize and kill cancer cells in a targeted manner (Goebeler & Bargou, 2020). However, the most widely used types of T cell‐based immunotherapies, immune‐checkpoint inhibitors and chimeric antigen receptor‐engineered T cells (Ferrara et al., 2018; Goebeler & Bargou, 2020), are not effective in AML (C. Chen et al., 2020; Qasim, 2019). Recently, it was reported that CLAVEEVSL, a mutated NPM1‐derived neoantigen, could be efficiently targeted by transferring TCR retroviral transduced T cells and might be a relevant target for immunotherapy of AML (Van Der Lee et al., 2019). However, the T cells specific for CLAVEEVSL were not present at detectable levels in AML patients (Van Der Lee et al., 2019), indicating that in vivo responses against CLAVEEVSL may be suppressed by the tumour microenvironment (TME). Therefore, there is an urgent need to better understand T cell biology in the TME in order to expand the repertoire of therapeutic agents for NPM1mut AML.
Small extracellular vesicles (sEVs) are extracellular membranous vesicles with a diameter of approximately 30–200 nm that are secreted by most organisms and cell types (Abreu et al., 2021). It is well known that sEVs (including exosomes, microvesicles and other types of membrane particles) can mediate cross‐talk between cells in the TME by delivering cargo (such as proteins, lipids, and nucleic acids) to target cells (Mathieu et al., 2019). More recently, sEV trafficking is identified as a novel mechanism that mediates the anticancer immune response (Marar et al., 2021). sEVs from cancer cells may directly activate immune cells, such as T cells and natural killer cells, resulting in robust tumour clearance (Moroishi et al., 2016). In addition, tumour‐derived sEVs can act as immune suppressors. For example, exosomes from cancer cells played critical roles in T‐mediated immune escape through the miR‐21/PTEN/PD‐L1 regulatory axis in oral squamous cell carcinoma (Li et al., 2019). Glioblastoma‐derived EVs contributed to T‐cell inhibition by inducing immunosuppressive monocytes (Himes et al., 2020). However, the functions and mechanisms of tumour‐derived sEVs in T cell‐mediated immune responses in NPM1mut AML remain unknown.
It is crucial to meet the bioenergetic demand for T cell activation, effector and memory function (Siska & Rathmell, 2015). T cells face the special challenge of competing with fast‐growing tumour cells for metabolic fuels such as glucose, amino acids, and lipids, which can lead to the suppression of T cell function (Mccarthy et al., 2013). Creatine, as a non‐canonical energy fuel, can satisfy the cellular energy requirement in an acute manner via the creatine‐phosphocreatine cycle, which is coupled with the ATP–ADP transition in the brain and muscles (Ji et al., 2019). Recently, the biology of creatine in the systems other than brain and muscles have been gradually revealed, such as a role in orchestrating macrophage polarization (Ji et al., 2019). However, little is known regarding the effect of creatine metabolism on T cell function in AML, especially in NPM1mut AML.
In this study, we discovered that tumour sEV‐related miR‐19a‐3p enhanced the immune escape of NPM1mut leukaemia. The sEV‐related microRNA, miR‐19a‐3p (the expression of which is regulated by NPM1mA) could be transferred from leukemic cells to CD8+ T cells, which directly downregulated SLC6A8 expression. Furthermore, reduced SLC6A8 expression resulted in decreased creatine import and ATP production, which impaired CD8+ T cell function. Collectively, our findings suggest that the sEV‐related miR‐19a‐3p may represent a potential therapeutic target for NPM1mut AML.
2. MATERIALS AND METHODS
2.1. Patient samples and serum preparation
The peripheral blood and bone marrow of patients with AML or benign hematologic disease (BHD) (e.g., iron‐deficiency anaemia) were obtained from the First Affiliated Hospital and the Third Affiliated Hospital of Chongqing Medical University. Patients were newly diagnosed with AML through cytomorphology, cytogenetic, and molecular genetic analyses. The samples were matched in terms of the sex and age of the patients. All patients were informed and consented to participate in the study. The experiments were approved by the Ethics Committee of Chongqing Medical University. The patients’ clinical characteristics are presented in detail in Table 1 and Supplementary list 1.
TABLE 1.
Clinical characteristics of newly diagnosed patients
| Characteristics | Median (range) | All cases |
|---|---|---|
| Sex | ||
| Female | 50 | |
| Male | 31 | |
| Total | 81 | |
| Median age, y | 53.3 (26–77) | |
| Younger than 40 y | 19 | |
| 40–60 y | 35 | |
| Older than 60 y | 27 | |
| Median WBC, 109/L | 61.8 (0.5–285) | |
| Median platelets, 109/L | 122.4 (23–602) | |
| iron deficiency anemia | 27 | |
| AML | 54 | |
| AML FAB subtype | ||
| AML without maturation:M1 | 6 | |
| AML with maturation:M2 | 8 | |
| Acute promyelocytic leukaemia:M3 | 8 | |
| Acute myelomonocytic leukaemia:M4 | 17 | |
| Acute monoblastic or monocytic leukaemia:M5 | 15 | |
| unclassified | 0 | |
| Karyotype | ||
| Normal | 27 | |
| t (8;21) | 7 | |
| t (15;17) | 6 | |
| inv (16) | 14 | |
| Unknown | 0 | |
| Gene mutations | ||
| NPM1 | 27 | |
| FLT3/ITD | 14 | |
| WT1 | 8 | |
| DNMT3A | 12 |
Abbreviations: AML, acute myeloid leukaemia; FAB classification, French‐American‐British classification, a classification of acute leukaemia produced by three‐nation joint collaboration.; WBC, white blood cell; y, year old.
Serum was prepared as previously described (Hamam et al., 2016). Briefly, blood samples were collected from a peripheral vein with vacutainer tubes containing a clot‐activation additive and a barrier gel. The samples were allowed to clot for 60 min at room temperature, followed by centrifugation at 1500g for 15 min to isolate serum samples. The serum samples were collected and subjected to a second centrifugation at 2000g for 10 min to remove contaminating cells for further use.
2.2. Immunohistochemistry (IHC) analysis
Bone marrow aspirates from patients enrolled in this study were transferred to slides using a cytospin, treated with 3% H2O2, and blocked with 5% normal goat serum. Next, the slides were incubated with primary antibodies against CD3 (1:200, Abcam, United Kingdom), CD4 (1:200, Abcam), and CD8 (1:200, Abcam) at 4°C, followed by incubation with horseradish peroxidase (HRP)‐conjugated secondary antibody for 30 min and then staining with diaminobenzidine. The images were captured and evaluated using Image‐Pro 6.0 software (Media Cybernetics, USA).
2.3. Cell culture
CD8+ T cells were obtained from the peripheral blood mononuclear cells (PBMCs) of volunteers, as described previously (Yang et al., 2019). PBMCs were isolated using HISTOPAQUE‐1077 solution (Sigma‐Aldrich, USA), according to the manufacturer's instructions. Briefly, fresh peripheral blood was collected in ethylenediaminetetraacetic acid anticoagulant tubes and subsequently layered onto the HISTOPAQUE‐1077 solution. After centrifugation at 650g for 30 min, the PBMCs remained at the plasma–HISTOPAQUE‐1077 interface and were carefully transferred to a new tube, then washed twice with 1× phosphate‐buffered saline (PBS; Invitrogen, USA). CD8+ T cells were purified by positive selection with CD8 microbeads (Miltenyi Biotec, USA) from the above PBMCs and resuspended in PBS. The purity of the CD8+ T cells was >90%, as determined by flow cytometry (FCM). CD8+ T cells were seeded at 1 × 106 cells/well in 48‐well plates and stimulated with anti‐CD3 (clone UCHT1, 10 mg/ml) and anti‐CD28 (clone UCHT1, 12 mg/ml) antibodies (BD Bioscience) to maintain the proliferation ability for 3–5 days. At the indicated time points, CD8+ T cells were collected for further analysis.
To obtain leukemic blast cells from patients with AML, we isolated mononuclear cells from bone marrow aspirates via density‐gradient centrifugation and purified the cells with the CD45‐Positive Selection Kit (Miltenyi Biotec, UK). The identity of the leukemic blast cells was confirmed by microscopy and FCM. All leukemic blast cells were grown in StemSpan™ SFEM (STEMCELL Technologies, Canada) with 2% fetal bovine serum (FBS, STEMCELL Technologies) supplemented with rhIL3 (R&D Systems, Minneapolis, USA), rhTPO (R&D Systems), rhFLT3‐ligand (R&D Systems), and rhSCF (R&D Systems).
The human myeloid leukemic cell lines (NB4, THP1, and KG‐1a) and the human embryonic kidney cell line HEK 293T were obtained from the American Type Culture Collection (ATCC, USA). The human myeloid leukemic cell lines, OCI/AML3 (harboring NPM1mA) and OCI/AML2, were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Germany). All cell lines were tested free of mycoplasma contamination and authenticated by the short tandem repeat (STR)‐based method. Myeloid leukaemia cell lines (NB4, THP1, KG‐1a, and OCI/AML3) and CD8+ T cells were maintained in RPMI‐1640 medium (Gibco, Australia) supplemented with 10% FBS (Gibco) and 1% penicillin–streptomycin (Sangon Biotech, China). OCI/AML2 cells were cultured in MEM Alpha medium (Gibco) supplemented with 10% FBS and 1% penicillin–streptomycin. HEK293T cells were cultured in DMEM medium (Gibco) with 10% FBS and 1% penicillin‐streptomycin. All cell lines were incubated in a humidity chamber (Thermo Fisher Scientific, USA) containing 5% CO2 at 37°C.
2.4. Preparing conditioned medium (CM) from leukemic cells
When the leukemic cell growth reached approximately 80% confluency, they were washed three times with PBS, then an appropriate volume of FBS‐free medium was added to the cells, with or without 15 mM GW4869 (Selleck, USA). After culturing for 48 h, the CM was collected and centrifuged at 4000g for 10 min to remove cell debris for further study. To further obtain sEV‐free CM, the CM was spun down successively at 300g for 20 min, 2000g for 20 min, 10,000g for 30 min, and 120,000g for 70 min to deplete sEVs from the CM.
2.5. FCM analysis
To measure the proliferation ability of CD8+ T cells, a carboxyfluorescein succinimidyl ester (CFSE) proliferation assay was performed using CellTrace™ CFSE Cell Proliferation Kit (Life Technologies, USA) according to the manufacturer's instructions. Briefly, CD8+ T cells were resuspended in CFSE (5 mM) buffer, incubated at 37°C with 5% CO2 for 20 min, and washed twice in culture medium. The labelled CD8+ T cells were seeded in a 96‐well round‐bottom plate at 1 × 106 cells/well and then stimulated with anti‐human CD3/CD28 beads. Cells, with or without indicated treatment, were collected and detected by FCM using a FACSCalibur instrument (Becton Dickinson, USA).
Expression of CD25, a surface activation marker on CD8+ T cells, was measured by FCM after surface staining with fluorochrome‐labelled antibodies against CD25 (PE, Thermo Fisher, USA). The expression levels of effector molecules (Granzyme B, IL‐2, and IFN‐γ) in CD8+ T cells were measured by FCM after intracellular staining. Briefly, CD8+ T cells (1 × 106/well in 48‐well plates) were stimulated with 50 ng/ml phorbol 12‐myristate 13‐acetate and 500 ng/ml ionomycin (Sigma‐Aldrich, USA) in the presence of GolgiStop solution (Becton Dickinson) for 3.5 h. Then, the cells were stained with anti‐granzyme B (APC, Thermo Fisher), anti‐IL‐2 (APC, BioLegend, USA), or anti‐IFN‐γ (APC, Thermo Fisher) after fixation and permeabilization. Finally, the stained cells were detected by FCM.
2.6. Enzyme‐linked immunosorbent assay (ELISA) analysis
ELISAs were performed to detect the levels of IFN‐γ and IL‐2 secreted into supernatants from CD8+ T cells. Briefly, the supernatants were collected and centrifuged at 2000g for 5 min to clear the cells for further study. The levels of IFN‐γ and IL‐2 were comparatively analyzed using an IFN Gamma Human ELISA Kit (Thermo Fisher) and an IL‐2 Human ELISA Kit (Thermo Fisher), respectively following the manufacturers’ instructions.
2.7. sEV isolation and characterization
sEVs were prepared via the differential‐centrifugation method, as previously reported (Yan et al., 2018). Briefly, CM was prepared by incubating cells grown at sub‐confluence in FBS‐free media for 48 h and then pre‐clearing the culture medium by centrifugation at 2000g for 20 min and then at 10,000g for 30 min. sEVs were isolated by ultracentrifugation at 120,000g for 70 min (XE‐100, Beckman Coulter, USA) and washed in PBS using the same ultracentrifugation conditions. The pelleted sEVs were resuspended in ∼100 μl of PBS and subjected to several experiments, including nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM) analysis, western blotting analysis, and quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis, or CD8+ T cell treatment.
NTA was used to assess the size distribution and concentration of sEVs. Briefly, sEVs were resuspended in PBS and injected into the sample chamber of a ZetaView PMX 110 instrument (Particle Metrix, Germany), and the particles were measured based on Brownian motion and the diffusion coefficient. Data analysis was performed using the manufacturer's software (ZetaView 8.02.28).
TEM was performed to visualize the morphologies of sEVs. The isolated sEVs were loaded onto a grid (ProSciTech, Australia), fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), and stained with a drop of 2% uranyl acetate (Sigma, USA) for 10 min at room temperature. The morphologies of the sEVs were visualized using a JEM‐1011 transmission electron microscope (Hitachi, Japan).
At the designated CM‐collection time, CM was collected for sEV isolation. The protein contents of the isolated sEV samples were assessed using a BCA Protein Assay Kit (P0012, Beyotime, China). For CD8+ T cell‐treatment experiments in vitro, 2 μg of sEVs (equivalent to that collected from ∼5 × 106 producer cells) was added to 1 × 106 recipient cells at an sEV concentration of 10 μg/ml.
2.8. Reverse transcription PCR and qRT‐PCR
Total RNA samples were isolated using the TRIzol reagent (Takara, Japan), and then reverse‐transcribed using the Prime‐Script™ RT Reagent Kit (Takara). qRT‐PCR analysis was performed in an MJ Mini™ Gradient Thermal Cycler Real‐Time PCR machine (Bio‐Rad, USA). β‐actin expression was detected as an internal control for the indicated genes. U6 expression was detected as an internal control for the intracellular miRNA levels. As a spike‐in control for sEV‐related miRNA levels, 20 fmol of synthetic cellular miR‐19a‐3p (cel‐miR‐19a‐3p), cel‐miR‐19b‐3p, and cel‐miR‐29b‐3p were added to sEVs from an equal number of cells during RNA extraction, and their levels were subsequently used for data normalization after performing miScript miRNA RT‐qPCR assays (Qiagen, Germany). The sequences of the primers used are shown in Table 2.
TABLE 2.
Sequences of primers used in qRT‐PCR
| Target gene | Primer sequences | |
|---|---|---|
| SLC6A8 | Forward | 5′‐CCTTATTCCCTACGTCCTGATC‐3′ |
| Reverse | 5′‐GGTAAAGGACTTGACCAGGTAA‐3′ | |
| AGAT | Forward | 5′‐TGGCTGATGAGCTTTATAACCA‐3′ |
| Reverse | 5′‐TGCCTAGGTAGTTTGTAACCTG‐3′ | |
| GAMT | Forward | 5′‐TCACTTTGATGGGATCCTGTAC‐3′ |
| Reverse | 5′‐AACATGATGGTGATGTCTGAGT‐3′ | |
| CKB | Forward | 5′‐CGACTTCAGAAGCGAGGCACAG‐3′ |
| Reverse | 5′‐TCACTCCGTCCACCACCATCTG‐3′ | |
| miR17HG | Forward | 5′‐TTTGCCACGTGGATGTGAAGA‐3′ |
| Reverse | 5′‐CAGTGTGTCTTCAAACTACAGGAG‐3′ | |
| b‐actin | Forward | 5′‐TGACGTGGACATCCGCAAAG‐3′ |
| Reverse | 5′‐CTGGAAGGTGGACAGCGAGG‐3′ | |
| miR‐19a‐3p | Forward | 5′‐GCGTGTGCAAATCTATGCAA‐3′ |
| Reverse | 5′‐AGTGCAGGGTCCGAGGTATT‐3′ | |
| miR‐19b‐3p | Forward | 5′‐CGTGTGCAAATCCATGCAA‐3′ |
| Reverse | 5′‐AGTGCAGGGTCCGAGGTATT‐3′ | |
| miR‐29b‐3p | Forward | 5′‐CGCGTAGCACCATTTGAAATC‐3′ |
| Reverse | 5′‐AGTGCAGGGTCCGAGGTATT‐3′ | |
| U6 | Forward | 5′‐CGGGTTCTCCAAAAGAAAGCA‐3′ |
| Reverse | 5′‐CAGCCACAAAAGAGCACAAT‐3′ | |
| PABPC1 | Forward | 5′‐AGCAAATGTTGGGTGAACGG‐3′ |
| Reverse | 5′‐ACCGGTGGCACTGTTAACTG‐3′ | |
| CTCF E1 for ChIP | Forward | 5′‐CCGCCCGACCTGCGCCTTC‐3′ |
| Reverse | 5′‐GCTGCGGTAGTCGTCCCCAC‐3′ | |
| CTCF E2 for ChIP | Forward | 5′‐GCGCTCCGCGAGGCTG‐3′ |
| Reverse | 5′‐CCACGGCGGCTCGTTCTTG‐3′ | |
| CTCF E3 for ChIP | Forward | 5′‐CAGGAAGCCCCGGGAAGCGT‐3′ |
| Reverse | 5′‐GCCCTCCGTTAACAAGCTCT‐3′ | |
| H3K27ac for ChIP | Forward | 5′‐CCGCGCAGCCAGTTGGTGTCA‐3′ |
| Reverse | 5′‐TCCTACCCGGACCGCACTACGAG‐3′ |
2.9. Western blot assays
Western blot assays were performed as previously described (Jin et al., 2018). Briefly, total cellular or sEV proteins were obtained using RIPA lysis buffer (P0013B, Beyotime) and quantified with a BCA Protein Assay Kit (P0012, Beyotime). The proteins were electrophoresed by 6%–12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and subsequently incubated with primary antibodies against TSG101 (1:1000, Abcam), HSP70 (1:1000, Abcam), CD9 (1:1000, Abcam), CD63 (1:500, Bimake, China), calnexin (1:1000, Abcam), SLC6A8 (1:1000, SAB, USA), ATAG (1:1000, Abcam), GAMT (1:1000, Abcam), CKB (1:1000, Abcam), NPM1mA (1:1000, Thermo Fisher Scientific), PABPC1 (1:1000, SAB), KHSRP (1:1000, Abcam), β‐actin (1:1000, ZSGBBIO, China). β‐actin was detected as a loading control. The appropriate HRP‐conjugated anti‐mouse or anti‐rabbit IgG (ZSGBBIO) was used as a secondary antibody. Protein signals were visualized using an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Japan).
2.10. Creatine and ATP quantification
Creatine and ATP were quantified using the Creatine Assay Kit (ab65339; Abcam) and Luminescent ATP Detection Assay Kit (ab113849; Abcam), respectively, according to the manufacturers’ instructions. All experiments were performed at least three times, and the data were normalized to the cell numbers or protein contents.
2.11. Plasmid constructs, siRNA interference, cell transfection and infection
An expression vector encoding SLC6A8 was constructed by inserting SLC6A8 cDNA sequence into the pBABE‐puro vector at the BamHI and EcoRI sites. An miR‐19a‐3p mimic, a small‐interfering RNA (siRNA) targeting SLC6A8, and a lentivirus‐mediated short‐hairpin RNA (shRNA) against NPM1 were purchased from GenePharma (Shanghai, China). The shRNA specifically targeting miR‐19a‐3p was reconstructed by subcloning the corresponding DNA sequence into the lentiviral vector pLVX‐shRNA (Clontech, USA) at the BamHI and EcoRI sites. All the sequences described above are shown in Table 3.
TABLE 3.
Core sequences of siRNA against target genes
| Target gene | siRNA sequences |
|---|---|
| NC | 5′‐TTCTCCGAACGTGTCACGT‐3′ |
| SLC6A8 | 1# 5′‐CAGAUGGACUUCAUCAUGUTT‐3′ |
| 2# 5′‐CCAUCAUCCUGGCUCUCAUTT‐3′ | |
| 3# 5′‐CCGCUUCAUGGACGACAUUTT‐3′ | |
| NPM1 | 1# 5′‐GGTACATTGTTGATTGTTATG‐3′ |
| 2# 5′‐GGAACAGGAGAGTAGACTAGG‐3′ | |
| 3# 5′‐CAGTAGTCAGATTATAGAATC‐3′ | |
| miR‐19a‐3p | 5′‐GCTCAAACTGTTTATCTTCCATGCGAGTTTG‐3′ |
| PABPC1 | 1# 5′‐ TCACTGGCATGTTGTTGGA ‐3′ |
| 2# 5′‐ GAAAGGAGCTCAATGGAAA ‐3′ | |
| 3# 5′‐GCAAACATAATGCTAGTCC‐3′ | |
| KHSRP | 1# 5′‐ TACTACTCACACTACTACTCGAGTAGTAGTGTGAGTAG ‐3′ |
| 2# 5′‐ AGAAGATTGCTCATATACTCGAGTATATGAGCAATCTTC ‐3′ | |
| 3# 5′‐GAGTGAAGATGATCTTACTCGAGTAAGATCATCTTCACT‐3′ |
Plasmid transfections were performed using a transfection reagent (Invitrogen, USA), according to the manufacturer's instructions. The cells were transiently transfected with siRNA or an miRNA mimic using RFectSP siRNA/miRNA Transfection Reagent (BaiDai, China) according to the manufacturer's instructions. After 48 h of transfection, the cells were collected for qRT‐PCR or western blot analysis.
The cells were infected with a lentivirus encoding an shRNA in the presence of 5 μg/ml polybrene (Millipore Sigma, USA), and then selected in the presence of 2 μg/ml puromycin (Millipore Sigma) for 7 days. The puromycin‐resistant cells were collected for further analysis.
2.12. Luciferase reporter assays
To explore the regulatory effect of miR‐19a‐3p on SLC6A8 expression, luciferase reporter assays were performed. First, the pMIR‐SLC6A8‐3′UTR‐wt and pMIR‐SLC6A8‐3′UTR‐mut plasmids were designed by cloning the 3′‐untranslated regions (UTRs) of SLC6A8 wild‐type (WT) and an SLC6A8 mutant (MUT) into the 3′‐UTR of the firefly luciferase reporter gene in the pMIR‐REPORT vector (Promega, USA) at the SpeI and HindIII sites. Next, CD8+ T cells were seeded at a density of 1 × 106 cells in 96‐well plates and co‐transfected with either pMIR‐SLC6A8‐3′UTR‐wt or pMIR‐SLC6A8‐3′UTR‐mut along with an miR‐19a‐3p mimic or scrambled mimic. Luciferase activities were measured using the Dual‐Luciferase Reporter Assay System (Promega, USA) after 48 h of transfection. Renilla luciferase activity was normalized to firefly luciferase activity.
Similarly, to determine the transcriptional activity of CTCF on MIR17HG, pGL3‐MIR17HG WT reporter and pGL3‐MIR17HG mutant reporters (E1 MUT, E2 MUT, and E3 MUT) were designed by cloning the MIR17HG‐WT‐promoter and MIR17HG‐MUT‐promoters (E1 MUT, E2 MUT, or E3 MUT) separately into the pGL3‐basic luciferase reporter plasmid at the KpnI and BglII sites, upstream of the luciferase gene. Next, HEK293T cells (1 × 106 cells/well) were co‐transfected with the above pGL3‐MIR17HG vectors and the CTCF expression vector. Renilla luciferase activity was normalized to firefly luciferase activity.
2.13. Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed to detect CTCF binding to the MIR17HG promoter or H3K27ac enrichment on the PABPC1 promoter. Briefly, leukemic cells (with or without NPM1 silencing) were collected from each group at a density of 1 × 108 cells, treated with 37% formaldehyde, and incubated for 15 min at room temperature to generate DNA–protein cross‐links. The chromatin was sheared into small fragments of approximately 200–300 base pairs using sonication. An anti‐CTCF or anti‐H3K27ac antibody and protein G beads were used to pull down the target proteins. The target proteins were digested with proteinase K at 45°C for 50 min. After collection and purification, the target protein‐bound DNA was quantitated by qRT‐PCR analysis using the primers indicated in Table 2. All qRT‐RCR products were subjected to electrophoresis on a 2% agarose gel.
2.14. Biotin–miRNA pulldown assays
To detect the binding between miR‐19a‐3p and PABPC1, biotin–miRNA pulldown assays were performed. Briefly, lysates of leukemic cells were incubated with 100 pmol of synthetic biotin‐labeled miR‐19a‐3p overnight at 4°C. Streptavidin beads (Invitrogen) were added to each binding reaction, after which each mixture was washed and eluted. The eluted solutions were analyzed by SDS‐PAGE, followed by western blotting to detect PABPC1 expression.
2.15. RNA immunoprecipitation (RIP) assays
To detect binding between miR‐19a‐3p and PABPC1, RIP experiments were performed using the Magna RIP™ RNA‐Binding Protein Immunoprecipitation Kit (Millipore, USA), according to the manufacturer's instructions. Briefly, OCI/AML3 cells were collected and lysed in RIP lysis buffer supplemented with protease and RNase inhibitors. The protein extract was incubated overnight at 4°C with an anti‐PABPC1 antibody or a control IgG. After washing the beads, the bead‐bound immunoprecipitate was digested with proteinase K. RNA were purified using a phenol–chloroform–isoamyl alcohol mixture and then subjected to qRT‐PCR. RNA levels were normalized to that of the input control. The fold‐enrichment of miR‐19a‐3p was determined as a percentage of the input RNA and compared with the IgG isotypic control.
2.16. Immunofluorescence (IF) assays
To define the transfer of sEVs from leukemic cells to CD8+ T cells, sEVs were labelled with the red fluorescent membrane dye PKH26 (Sigma‐Aldrich). Briefly, after an initial pelleting of the sEVs by ultracentrifugation, they were resuspended in 50 μl of PBS and then mixed with 50 μl of PKH26 staining solution (1:50, diluted in diluent). CD8+ T cells were incubated with labelled sEVs at a concentration of 10 μg/ml for 12 h in a 48‐well plate. Nuclei were stained with 4′,6′‐diamidino‐2‐phenylindole (DAPI, 1:50; Beyotime). The sEVs internalized in CD8+ T cells was observed using a Leica TCS SP8 confocal microscope (Leica, Wetzlar, Germany).
Cy3‐labelled miR‐19a‐3p was transfected into leukemic cells to study the transfer of sEV‐related miR‐19a‐3p from leukemic cells to CD8+ T cells. Cy3‐miR‐19a‐3p‐expressing leukemic cells were then co‐cultured with CD8+ T cells using a transwell chamber in a 24‐well format. Cells were prepared for immunofluorescence analysis. Briefly, the CD8+ T cells were washed, spun onto slides, and then treated with 4% paraformaldehyde, and stained. The cell nuclei were stained with DAPI. Finally, the internalization of sEV‐related miR‐19a‐3p in CD8+ T cells was measured by confocal microscopy.
2.17. Animal experiments
The animal experiments were approved by the Animal Care Ethics Committee of Chongqing Medical University. Humanized NOD/SCID/IL‐2Rγ−/− (huHSC‐NSG) mice (Charles River Laboratories, China) were generated by transplanting human hematopoietic stem cells (hHSCs) into NSG mice to study the development and function of the human immune system (Lang et al., 2011). Twelve‐week‐old female huHSC‐NSG mice were randomly divided into four groups (n = 6/group). Stably engineered OCI‐AML3 cells (OCI/AML3‐NC cells or OCI/AML3‐miR‐19a‐3p KD cells) were generated by infecting a lentivirus encoding an shNC or shmiR‐19a‐3p into OCI/AML3 cells. Then, OCI/AML3 cells or engineered OCI/AML3 cells (5 × 106) were injected into the mice via the tail vein. After the day of cell transplantation, the mice in the OCI/AML3‐miR‐19a‐3p KD group were injected every 3 days with OCI/AML3‐sEVs (10 μg/mouse) via the tail vein, until the mice were sacrificed. The body weights and the general appearance of the mice were observed every other day. The mice were sacrificed after 100 days by cervical dislocation. The Kaplan–Meier method was used to analyze the survival curves of the mice in each group. Human CD45+ cells from peripheral blood were analyzed using FCM. Immature cells from bone marrow cells were evaluated using Wright's staining. Livers and spleens were excised and serially sectioned into 4 μm‐thick sections, and then infiltration of leukemic cells in the liver and spleen was analyzed by hematoxylin and eosin (H&E) staining and IF staining for the NPM1mA protein.
2.18. Statistical analysis
Statistical significance was determined using SPSS software (version 17.0). All data were derived from three independent experiments and are presented as the mean ± SD. The Kruskal‐Wallis test and Wilcoxon rank‐sum test were used to compare multiple groups and two groups, respectively. Statistical significance was set at p < 0.05.
3. RESULTS
3.1. NPM1mut AML cells impair CD8+ T cell function
To explore T cell function in AML, we first performed IHC to quantify T cells in the bone marrow aspirates of AML patients and individuals with BHD. IHC results showed that the bone marrow aspirates from patients with NPM1mut AML (NPM1mut AMLs) contained significantly fewer CD8+ T cells, and had a lower CD8+ T cell: total T cell (CD3+CD8+: CD3+) ratio than those from patients with NPM1‐wildtype AML (NPM1wt AMLs) or BHD (Figure S1A–C). Next, we examined CD8+ T cell proliferation and effector cytokine secretion (e.g., IL‐2 and IFN‐γ) in these patients. Less CD8+ T cell proliferation and IL‐2 and IFN‐γ secretion were observed in NPM1mut AMLs than in NPM1wt AMLs or patients with BHD (Figure S1D‐‐F), indicating that the CD8+ T cell population was lower in NPM1mut AML and had abnormal function.
To further explore the mechanism whereby CD8+ T cell are suppressed in NPM1mut AMLs, we treated primary CD8+ T cells with serum derived from patients with AML or BHD. Interestingly, the serum derived from NPM1mut AMLs significantly inhibited CD8+ T cell function, including proliferation (Figure 1a), effector cytokine production (e.g., IL‐2 and IFN‐γ; Figure 1b,c), surface activation marker expression (e.g., CD25; Figure 1d), and cytotoxic molecule production (e.g., Granzyme B; Figure 1e), by comparing these functions to those in serum samples from patients with NPM1wt AMLs or BHD. Moreover, CM from the NPM1mut cell line (OCI/AML3‐CM) inhibited CD8+ T cell proliferation (Figure 1f), IL‐2 and IFN‐γ secretion (Figure 1g,h), CD25 expression (Figure 1i), and granzyme B production (Figure 1j), when compared with CM from NPM1wt cell lines (OCI/AML2‐CM and NB4‐CM). To expand these findings, we isolated leukemic primary blast cells from patients with AML and found that CM from NPM1mut blasts (Blasts/mut‐CM) inhibited CD8+ T cell function, when compared with the CM from NPM1wt blasts (Blasts/wt‐CM) (Figure S2A–E). Taken together, the above data indicate that NPM1mut leukemic cells play an essential role in functionally impaired CD8+ T cells.
FIGURE 1.
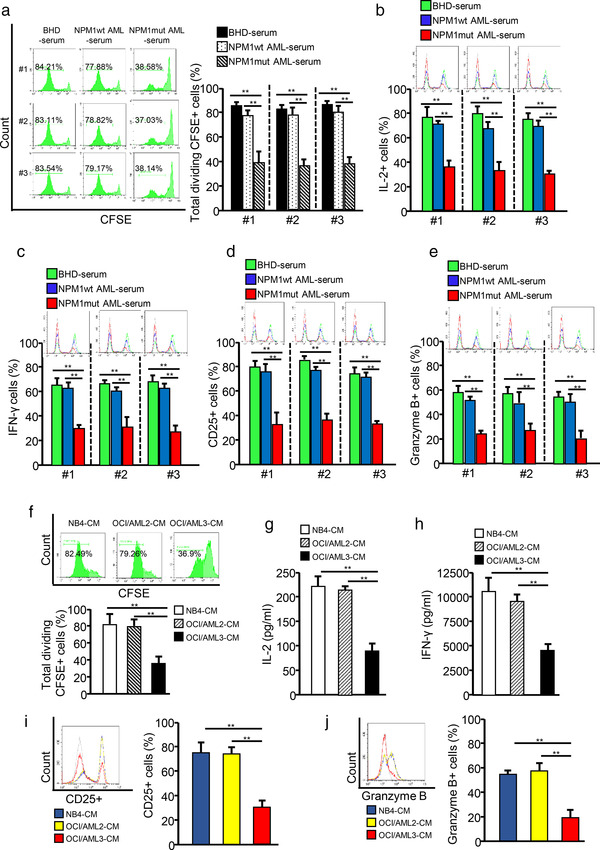
NPM1mut AML cells impair CD8+ T cell function. CD8+ T cells were purified from blood samples from volunteers and stimulated with anti‐CD3 and anti‐CD28 antibodies to maintain their proliferative ability. (a) CD8+ T cells were pre‐labelled with CFSE and incubated with serum samples from patients with BHD, NPM1wt AML, or NPM1mut AML. CD8+ T cell proliferation was evaluated by FCM, and quantitative diagrams are shown. (b–e) CD8+ T cells were incubated with serum from patients with BHD, NPM1wt AML, and NPM1mut AML. Representative FCM analysis of IL‐2 (B), IFN‐γ (C), CD25 (d), and granzyme B (e) expression levels are presented. (f–j) CD8+ T cells were incubated with NB4‐CM, OCI/AML2‐CM, or OCI/AML3‐CM. (f) The proliferation of CD8+ T cells was evaluated by FCM. (g and h) IL‐2 (g) and IFN‐γ (h) levels in cell culture supernatants were analyzed for using ELISA kits. (i and j) The expression of CD25 (i) and granzyme B (j) was detected by FCM (*p < 0.05; **p < 0.01; ***p < 0.001; n.s, not significant)
FIGURE 2.
Leukemic cells impair the immune function of CD8+ T cells via sEV release. (a–e) CD8+ T cells were pre‐labelled with CFSE and incubated with CM control (CM Ctrl), OCI/ALM3‐DMSO‐CM (CM from DMSO‐treated OCI/AML3 cells), OCIAML3‐GW4869‐CM (CM from GW4869‐treated OCI/AML3 cells), OCI/AML3‐CM, or OCI/AML3‐CMsEVs‐/– (sEVs‐depleted OCI/AML3‐CM). (a) CD8+ T cell proliferation was evaluated by FCM analysis. (b and c) IL‐2 (b) and IFN‐γ (c) levels in supernatants from cell cultures were analyzed using ELISA kits. (d and e) CD25 (d) and granzyme B (e) expression of CD8+ T cells were detected by FCM analysis. (F–J) CD8+ T cells were incubated with NB4‐sEVs, OCI/AML2‐sEVs, or OCI/AML3‐sEVs. (f) The proliferation of CD8+ T cells was evaluated by FCM analysis. (G and H) IL‐2 (g) and IFN‐γ (h) levels in supernatants from cell cultures were analyzed using ELISA kits. (i and j) CD25 (i) and granzyme B (j) expression of CD8+ T cells were determined by FCM (*p < 0.05; **p < 0.01; ***p < 0.001; n.s, not significant)
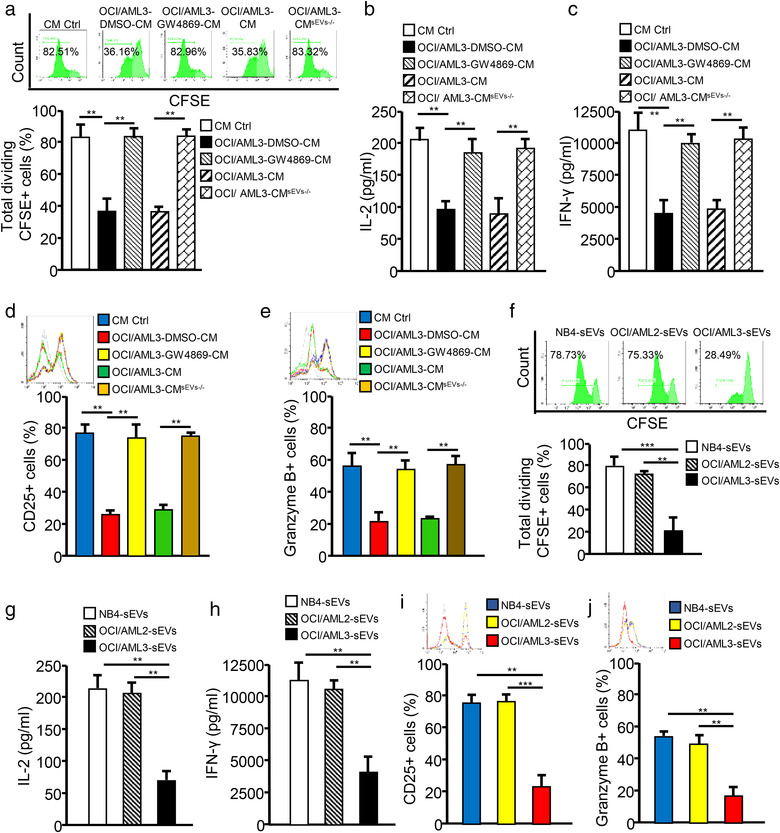
3.2. Leukemic cells impair the immune function of CD8+ T cells via sEV release
We performed bioinformatics analysis of array data (accession numbers: GSE14468, GSE15434, and GSE34860) deposited in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geoprofiles/) with R software and the DAVID database (https://david.ncifcrf.gov/conversion.jsp) to reveal the roles of differentially expressed genes between NPM1mut AMLs and NPM1wt AMLs, as indicated by their biological processes and the corresponding pathways. As shown in Figure S3A,B, the differentially expressed genes in NPM1mut AMLs were found to be involved in various biological processes, including the immune response and extracellular exosomes (a main component of sEVs). sEVs, which are nanometric membrane vesicles secreted by almost all cell types, are becoming increasingly appreciated as important regulators of cellular communications in the TME (Flemming et al., 2020). Thus, we studied whether leukemic cell‐derived sEVs play roles in affecting CD8+ T cell function. GW4869, an inhibitor of sEV secretion (Ge et al., 2021), decreased sEV secretion from leukemic cells (Figure S3C) and showed almost no effect on leukemic cell and CD8+ T cell viability (Figure S3D). Interestingly, blocking sEV secretion by leukemic cells with GW4869 rescued CD8+ T cell proliferation (Figures 2a and S4A), IL‐2 and IFN‐γ secretion (Figures 2b,c and S4B,C), CD25 expression (Figures 2d and S4D), and granzyme B production (Figures 2e and S4E). In addition, physically removing sEVs from leukemic cell‐CM by ultracentrifugation also partially restored CD8+ T cell function (Figures 2a–e and S4A–E). These data support the possibility that sEVs derived from NPM1mut AML cells may participate in CD8+ T cell dysfunction.
We then purified sEVs from the CM of leukemic cells through ultracentrifugation and characterized them by TEM, NTA, and western blotting. The results showed that sEVs from leukemic cells (NB4, OCI/AML2, and OCI/AML3) were approximately 70 nm in diameter (Figure S5A) with a double‐layer membrane structure (Figure S5B), and that the sEVs expressed sEV‐specific protein markers (TSG101, HSP70, CD9, and CD63), but not calnexin, an endoplasmic reticulum protein (Figure S5C). By incubating CD8+ T cells with leukaemia cell‐derived sEVs labelled with PKH26 (a lipophilic dye with red fluorescence), we observed that PKH26‐labelled sEVs could be internalized by CD8+ T cells, with no significant difference in the efficiency of internalization of NB4‐sEVs, OCI/AML2‐sEVs, and OCI/AML3‐sEVs (Figure S5D) (the data for sEVs derived from leukaemia primary blasts not shown). Next, we cultured CD8+ T cells with leukemic sEVs, and found that the sEVs from NPM1mut leukemic cells (OCI/AML3‐sEVs, Blasts/mut‐sEVs) inhibited CD8+ T cell proliferation (Figures 2f and S6A), IL‐2 and IFN‐γ secretion (Figures 2g,h and S6B,C), CD25 expression (Figures 2i and S6D), and granzyme B production (Figures 2j and S6E), compared with the corresponding findings from NPM1wt leukemic cells. Collectively, these data confirm that leukemic cells impair CD8+ T cell function via sEV release.
3.3. Leukemic cell‐derived sEVs inhibit SLC6A8‐mediated creatine import to impair CD8+ T cell immune function
Creatine metabolism, a non‐canonical pathway of energy production, plays a critical role in T cell activation and survival in the nutrient‐deficient TME through the ATP–ADP transition as shown in Figure 3a (Di Biase et al., 2019). Interestingly, both OCI/AML3‐sEV and Blasts/mut‐sEV treatment decreased total creatine (Figures 3b and S7A) and ATP (Figures 3c and S7B) production in CD8+ T cells, while increasing extracellular creatine levels (Figures 3d and S7C). In addition, the mRNA‐ and protein‐expression levels of the creatine metabolism‐related enzymes were also assessed. The expression of SLC6A8, a specific creatine‐transporter, sharply decreased in CD8+ T cells treated with OCI/AML3‐sEVs or Blasts/mut‐sEVs (Figures 3e,f and S7D,E), indicating that leukemic cell‐derived sEVs may downregulate SLC6A8‐mediated creatine import.
FIGURE 3.
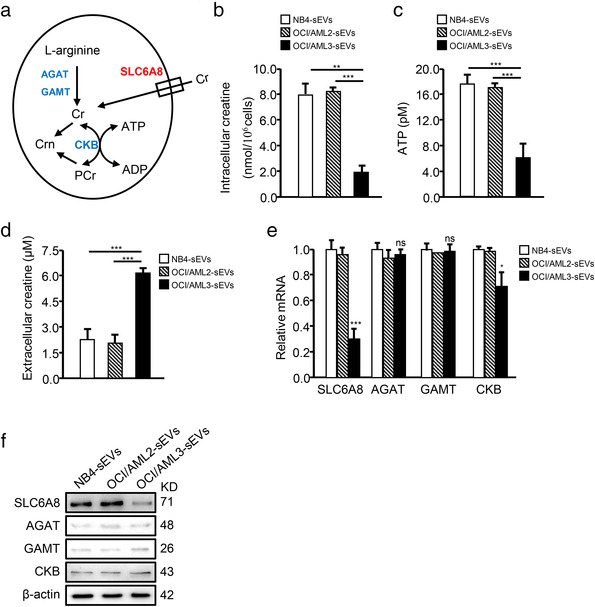
Leukemic cell‐derived sEVs inhibit creatine import by CD8+ T cells. (a) Diagram showing creatine uptake and creatine‐mediated bioenergy buffering in cells. SLC6A8, solute carrier family six member eight; AGAT, glycine amidinotransferase; GAMT, guanidinoacetate methyltransferase; Cr, creatine; PCr, phospho‐creatine; Crn, creatinine; CK, creatine kinase. (b–f) CD8+ T cells were treated with NB4‐sEV, OCI/AML2‐sEV, or OCI/AML3‐sEV, and the levels of intracellular creatine (b), ATP (c), and extracellular creatine (d) are shown. The mRNA‐expression (e) and protein‐expression (f) levels of the creatine transporter SLC6A8, two enzymes controlling the de novo creatine synthesis (AGAT and GAMT), and creatine kinase (CK) were measured via qRT‐PCR and western blotting, respectively (*p < 0.05; **p < 0.01; ***p < 0.001; n.s, not significant)
To further explore whether leukemic cell‐derived sEVs could impair CD8+ T cell function by inhibiting SLC6A8‐mediated creatine import in NPM1mut AML, we silenced SLC6A8 expression using a matching siRNA (Figure S8A) and observed that SLC6A8 knockdown (KD) significantly decreased creatine (Figure S8B) and ATP (Figure S8C) production, while inhibiting CD8+ T function (Figure S8D–H). Importantly, overexpressing SLC6A8 (Figures 4a and S9A) in the CD8+ T cells treated with OCI/AML3‐sEVs or Blasts/mut‐sEVs rescued cellular creatine levels (Figures 4b and S9B), ATP production (Figures 4c and S9C) and CD8+ T cell function (Figures 4d–h and S9D–H). These data suggest that leukemic cell‐derived sEVs can suppress creatine import and the immune function of CD8+ T cells by inhibiting SLC6A8 expression.
FIGURE 4.
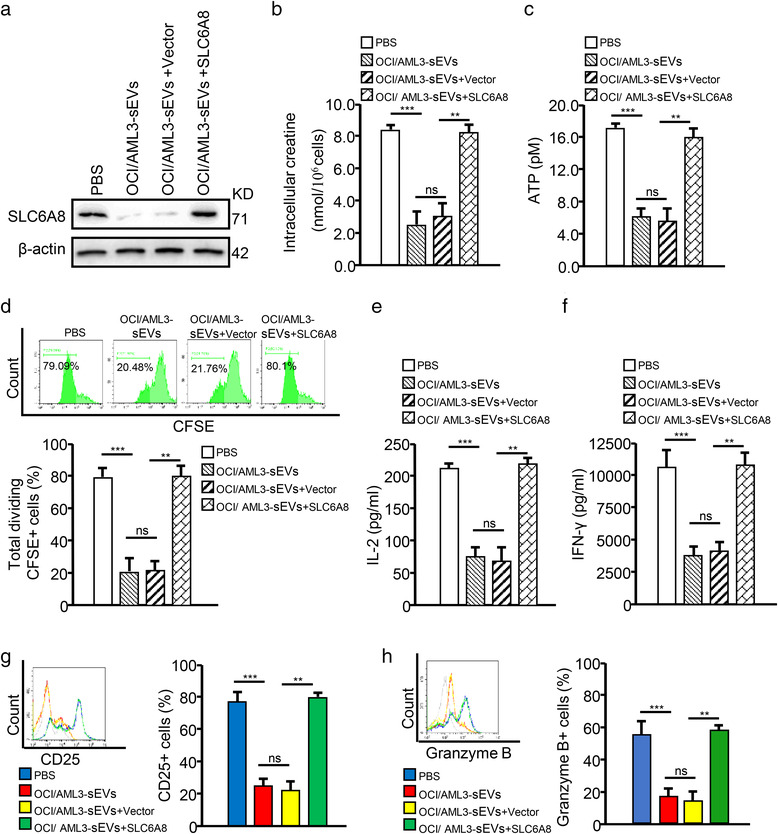
Leukemic cell‐derived sEVs impair CD8+ T cell function by decreasing SLC6A8 expression. (a–h) CD8+ T cells transfected with an SLC6A6‐overexpression vector (SLC6A8) or negative control vector (vector) were cultured with OCI/AML3‐sEVs. (a) SLC6A8 proteins were detected by western blotting. (b and c) The intracellular levels of creatine (b) and ATP (c) in CD8+ T cells are shown. (d) CD8+ T cell proliferation was evaluated by FCM. (e and f) IL‐2 (e) and IFN‐γ (f) levels in supernatants from cell cultures were analyzed for using commercially available ELISA kits. (g and h) CD25 (g) and Granzyme B (h) expression of CD8+ T cells were detected by FCM (*p < 0.05; **p < 0.01; ***p < 0.001; n.s, not significant)
3.4. Leukemic cell‐derived sEVs transfer miR‐19a‐3p to inhibit SLC6A8 expression in CD8+ T cells
It has been reported that RNAs, especially miRNAs, are responsible for tumour cell‐derived sEVs that reprogram immune cell function (Que et al., 2016). Therefore, we wondered whether sEV‐related miRNAs could regulate SLC6A8 expression and further affect CD8+ T cell function. First, we focused on the upregulated miRNAs in NPM1mut AMLs from a previous study, in which a unique miRNA‐signature associated with NPM1mut AMLs was identified, using an oligonucleotide microchip for genome‐wide microRNA profiling (Garzon et al., 2008). Simultaneously, we predicted the potential upstream miRNAs targeting SLC6A8 using three miRNA prediction algorithms (miRBase (https://www.mirbase.org/), miRDB (http://mirdb.org/), and TargetScan (http://www.targetscan.org/vert_71/)). By comparing the upregulated miRNAs in NPM1mut AMLs and the potential upstream miRNAs of SLC6A8, we screened out three potential miRNAs (miR‐19a‐3p, miR‐19b‐3p, and miR‐29b‐3p) that might be involved in regulating SLC6A8 expression in NPM1mut AMLs (Figure S10A). qRT‐PCR analysis revealed that miR‐19a‐3p was more abundantly expressed compared with miR‐19b‐3p and miR‐29b‐3p in NPM1mut leukemic cell‐derived sEVs (OCI/AML3‐sEVs and Blasts/mut‐sEVs) (Figure S10B). Importantly, miR‐19a‐3p, but not miR‐19b‐3p and miR‐29b‐3p, was highly increased in CD8+ T cells when treated with sEVs from NPM1mut leukemic cells, as compared to those from NPM1wt leukemic cells (Figure S10C–E). In addition, we co‐cultured CD8+ T cells with NPM1mut leukemic cells transfected with Cy3‐tagged miR‐19a‐3p and found that miR‐19a‐3p could be detected in CD8+ T cells (Figure 5a). The transfer of miR‐19a‐3p from leukemic cells to CD8+ T cells decreased when the leukemic cells were pre‐treated with GW4869 (Figure 5b). These data demonstrate that miR‐19a‐3p can be transported from leukemic cells into CD8+ T cells via sEV release.
FIGURE 5.
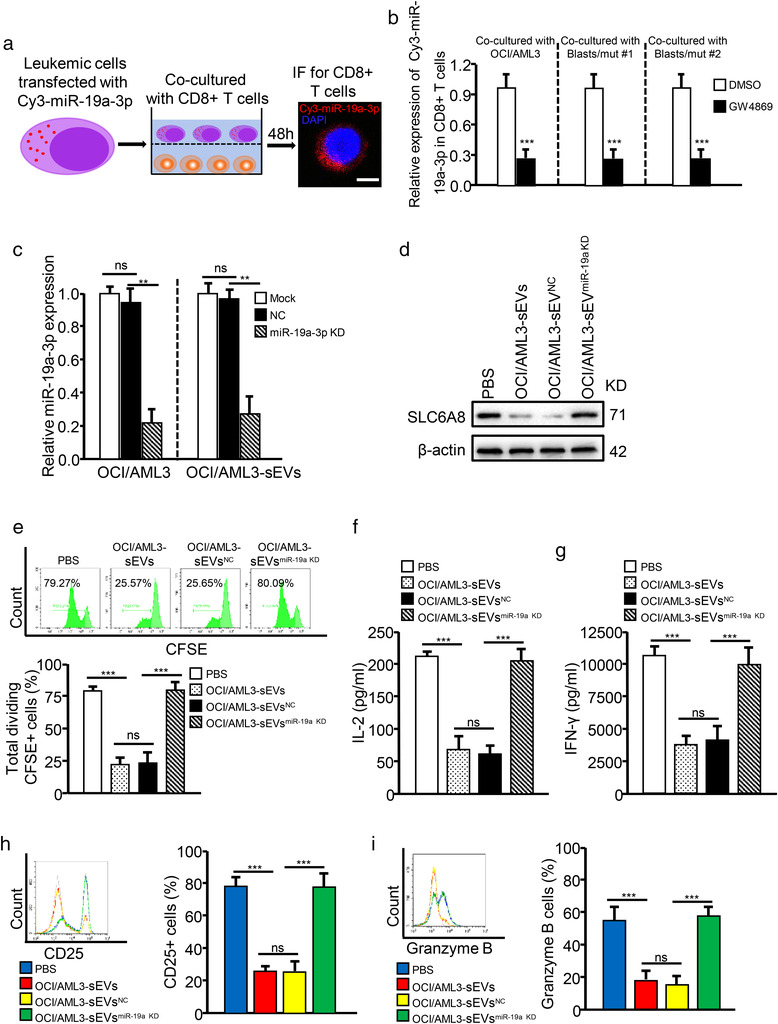
Leukaemia‐derived sEVs transfer miR‐19a‐3p to inhibit SLC6A8 expression in CD8+ T cells. (a and b) Leukemic cells transfected with Cy3‐tagged miR‐19a‐3p (Cy3‐miR‐19a‐3p) were co‐cultured with CD8+ T cells for 48 h, with or without GW4869. Fluorescence microscopy was used to detect the related red fluorescent signals in CD8+ T cells. (c) The interference efficiency of miR‐19a‐3p in OCI/AML3 cells and OCI/AML3‐sEVs was detected by qRT‐PCR. U6 expression was detected as an internal control. (d–i) CD8+ T cells incubated with OCI/AML3‐sEVs, with or without miR‐19a‐3p KD. OCI/AML3‐sEVsNC, OCI/AML3 cells transfected with negative control vector; OCI/AML3‐sEVsmiR‐19a KD, OCI/AML3 cells transfected with shRNA targeting miR‐19a‐3p. (d) SLC6A8 expression was analyzed by western blotting. (e) CD8+ T cell proliferation was evaluated by FCM. (f and g) IL‐2 (f) and IFN‐γ (g) levels in supernatants from cell cultures were analyzed using ELISA kits. (h and i) CD25 (h) and Granzyme B (i) expression by CD8+ T cells were determined by FCM
We then explored whether SLC6A8 expression in CD8+ T cells is directly regulated by miR‐19a‐3p. Bioinformatics analysis identified a potential miR‐19a‐3p‐binding fragment in the 3′‐UTR of the SLC6A8 gene (Figure S11A). A luciferase‐reporter assay showed that the miR‐19a‐3p mimic significantly inhibited the transcriptional activity of SLC6A8 (Figure S11B). Additionally, gene set‐enrichment analysis (GSEA) confirmed that high miR‐19a‐3p expression correlated negatively with creatine metabolism in AML (Figure S11C). The miR‐19a‐3p mimic treatment (Figure S11D) reduced endogenous SLC6A8 expression (Figure S11E), creatine production (Figure S11F), ATP levels (Figure S11G), and cell function (Figure S11H–L) of CD8+T cells. Notably, overexpressing SLC6A8 in CD8+ T cells treated with the miR‐19a‐3p mimic rescued miR‐19a‐3p mimic‐induced reductions in cellular creatine levels, ATP production and CD8+ T cell function (Figure S11E–L). Conversely, shRNA‐mediated KD of miR‐19a‐3p in leukemic cells markedly decreased miR‐19a‐3p levels in sEVs (Figures 5c and S12A). Furthermore, treating CD8+ T cells with those sEVs (OCI/AML3‐sEVsmiR‐19a‐3p KD or Blasts/mut‐sEVsmiR‐19a‐3p KD) partially rescued SLC6A8 expression (Figures 5d and S12B) and enhanced CD8+ T cell function (Figures 5e–i and S12C–G). Additionally, the levels of serum sEV‐related miR‐19a‐3p increased significantly in NPM1mut AMLs compared with those in patients with BHD or those in NPM1wt AMLs (Figure 6a) and correlated negatively with SLC6A8 mRNA expression (Figure 6b) and the immune function of CD8+ T cells (Figure 6c–e). Collectively, these data confirm that leukemic cell‐derived sEVs transfer miR‐19a‐3p to CD8+ T cells to inhibit SLC6A8 expression and affect CD8+ T cell function.
FIGURE 6.
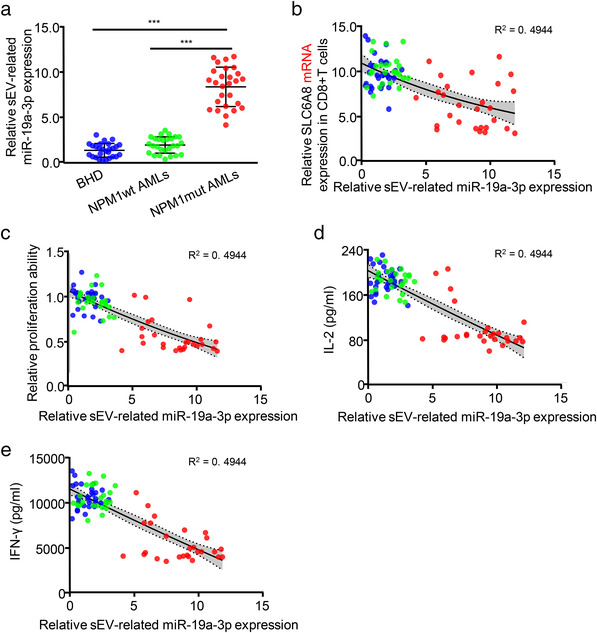
Serum sEV‐related miR‐19a‐3p correlates negatively with CD8+ T cell function. (a–e) The blue, green, and red circles represent data from patients with BHD, NPM1wt AML, and NPM1mut AML, respectively. (a) Expression of the sEV‐related miR‐19a‐3p was analyzed via qRT‐PCR. (b) The correlation between sEV‐related miR‐19a‐3p expression and SLC6A8 mRNA expression is shown. (C–E) Correlations between sEV‐related miR‐19a‐3p expression and peripheral CD8+ T cell proliferation (c), IL‐2 secretion (d), or IFN‐γ secretion (e) are shown. The grey areas mark 95% confidence intervals
3.5. NPM1mA mediates miR‐19a‐3p expression and packaging into sEVs
Based on the observation that serum sEV‐related miR‐19a‐3p levels were significantly higher in NPM1mut AMLs than in other AMLs (Figure 6a), we investigated whether sEV‐related miR‐19a‐3p was specifically released from NPM1mut leukemic cells. By assessing miR‐19a‐3p levels in AML cells and cell‐derived sEVs, we found that miR‐19a‐3p was higher in both NPM1mut leukemic cells (OCI/AML3 and Blasts/mut) and their sEVs, when compared with those in the indicated NPM1wt leukemic cells (Figure S13A). In addition, NPM1 KD using a lentivirus‐mediated shNPM1 strategy (Figure S13B) significantly attenuated miR‐19a‐3p levels in NPM1mut leukemic cells (OCI/AML3 and Blasts/mut) and their corresponding sEVs (Figure 7a). Thus, the above data suggest that sEV‐related miR‐19a‐3p secretion might mainly occur in NPM1mut AML and that NPM1mA is required for the miR‐19a‐3p expression and/or its packaging into sEVs.
FIGURE 7.
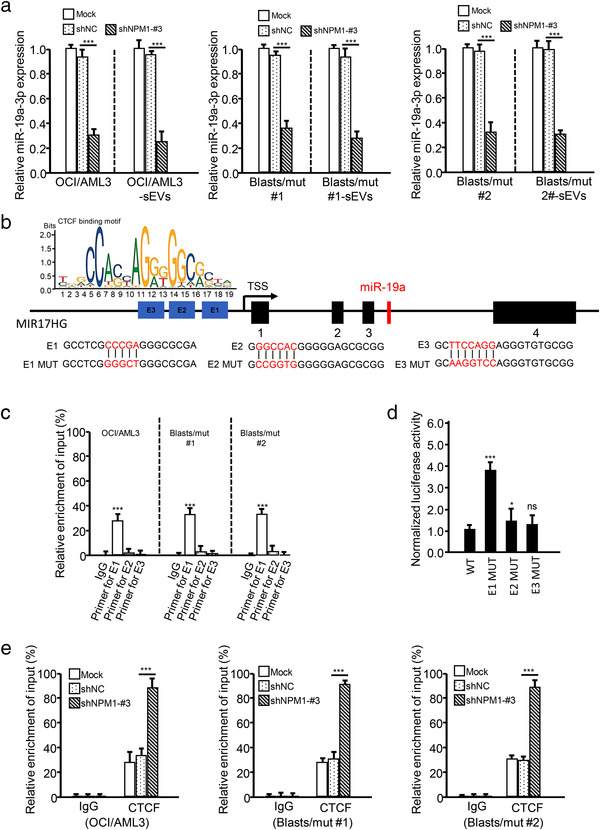
NPM1mA induces miR‐19a‐3p expression by CTCF‐mediated transcription inhibition. (a) Leukemic cells were transfected with shRNA against NPM1 (shNPM1‐#3), and the expression of miR‐19a‐3p in the indicated cells and cell‐derived sEVs was detected by qRT‐PCR. (b) Illustration of the genomic location of miR‐19a in the host gene, MIR17HG. The CTCF‐binding motifs in the promoter region of MIR17HG are indicated. (c) CTCF binding to the promoter of MIR17HG was determined by performing ChIP assays. miR‐19a‐3p levels in immunoprecipitated samples were determined by qRT‐PCR and are shown as percentages with respect to the input sample (% input). (d) Transcriptional inhibition of MIR17HG by CTCF was determined by performing dual‐luciferase assays. (e) The enrichment of CTCF at the MIR17HG promoter in leukemic cells (treated with or without NPM1 shRNA) was detected by ChIP
To determine how NPM1mA regulates miR‐19a‐3p expression, we searched for the host gene of miR‐19a‐3p using the University of California Santa Cruz (UCSC) (http://genome.ucsc.edu/) database (Liang et al., 2018). As shown in Figure 7b, miR‐19a was derived from MIR17HG. Additionally, NPM1 KD led to a significant decrease in MIR17HG expression at the transcriptional level in leukemic cells (Figure S13C). After screening for potential transcription factors of MIR17HG using the hTFtarget database (http://bioinfo.life.hust.edu.cn/hTFtarget#!/) (H. Zhang, Xu, et al., 2020), the master regulator CCCTC binding factor (CTCF) caught our attention (Figure S13D). ChIP assays showed that CTCF could bind to the MIR17HG promoter at CCCGA (E1) (Figure 7c), and dual‐luciferase assays showed that the CTCF inhibited MIR17HG transcription (Figure 7d). It was reported that CTCF, as a transcription inhibition factor, could be transported from the nucleus to the cytoplasm in conjunction with NPM1mA, which would reduce nuclear CTCF levels, and increase the transcription of CTCF‐mediated target genes in NPM1mut AML (Wang et al., 2020). Here, we verified that CTCF interacted with NPM1mA in NPM1mut leukemic cells (Figure S13E) and that NPM1 KD enhanced CTCF binding to the MIR17HG promoter (Figure 7e). Taken together, these data demonstrate that NPM1mA upregulates miR‐19a‐3p expression by reducing CTCF‐mediated inhibition of miR‐19a‐3p host gene transcription.
We next investigated whether NPM1mA can regulate the specific packaging of miR‐19a‐3p into sEVs. Given the critical role of RNA‐binding proteins (RBPs) in packaging miRNAs into exosomes (Qin et al., 2019), we first analyzed the RBPs of miR‐19a‐3p using the RNA‐Binding Protein DataBase (RBPDB; http://rbpdb.ccbr.utoronto.ca/) and found that poly (A)‐binding protein cytoplasmic 1 (PABPC1) and KH‐type splicing regulatory protein (KHSRP) potentially served as RBPs that packaging miR‐19a‐3p into sEVs (Figure S14A). Further investigations revealed that knocking down PABPC1 expression (Figure S14B, left blot image), but not KHSRP expression (Figure S14B, right blot image), significantly decreased miR‐19a‐3p levels in OCI/AML3‐sEVs and Blasts/mut‐sEVs (Figure 8a), whereas miR‐19a‐3p levels in OCI/AML3 cells and Blasts/mut cells were nearly unchanged (Figure S14C). Additionally, the interaction between PABPC1 and miR‐19a‐3p in OCI/AML3 and Blasts/mut cells was verified by performing miRNA pull‐down assays (Figure 8b) and RIP assays (Figure 8c). These data indicate that PABPC1, as an RBP, plays a critical role in miR‐19a‐3p packaging into sEVs. Notably, knocking down NPM1 expression decreased PABPC1 expression (Figure S14D,E) and miR‐19a‐3p binding with PABPC1 in NPM1mut leukemic cells (Figure S14F). We further explored the probable mechanisms whereby NPM1mA regulates PABPC1 expression using the UCSC database and found that the promoter region of PABPC1 may be highly enriched for H3K27ac (an important histone marker associated with active chromatin) (Figure 8d). As reported previously, NPM1mA can modulate H3K27 acetylation at the HOX promoter to enhance its expression (Brunetti et al., 2018). Similarly, we found that NPM1mA suppression decreased H3K27ac enrichment in the PABPC1 gene promoter in OCI/AML3 and Blasts/mut cells (Figure 8e), indicating that NPM1mA regulated PABPC1 expression by modulating H3K27 acetylation.
FIGURE 8.
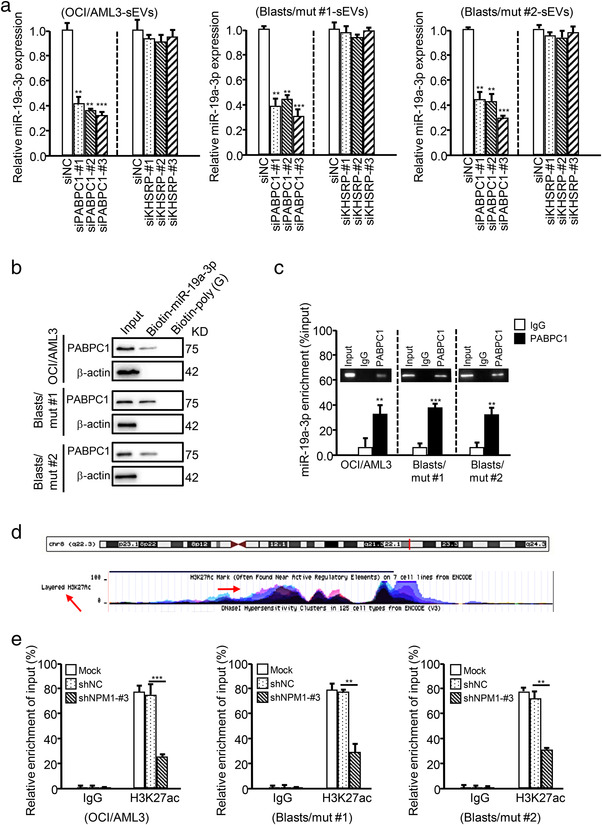
NPM1mA accelerates the production of sEV‐related miR‐19a‐3p by upregulating cellular miR‐19a‐3p and PABPC1 expression. (a) The expression of miR‐19a‐3p in the sEVs derived from leukemic cells (with or without PABPC1 or KHSRP KD) was detected by qRT‐PCR. (b) PABPC1 expression in leukemic cell lysates (recovered by miRNA pulldown) was determined by western blotting. Biotin‐poly (g) was used as a negative control. (c) RIP assays with an anti‐PABPC1 antibody (or IgG as a control) were performed with leukemic cells. MiR‐19a‐3p levels in immunoprecipitated samples were determined by qRT‐PCR and are reported as percentages with respect to the input sample (% input). (d) The H3K27ac‐enriched area in the promoter of PABPC1 was predicted using the UCSC Genome Browser. (e) The enrichment of H3K27ac in the PABPC1 promoter in OCI/AML3 cells (treated with or without shNPM1‐#3) was detected by ChIP. H3K27ac levels in immunoprecipitated samples were determined by qRT‐PCR and are reported as percentages with respect to the input sample (% input)
Collectively, these findings show that NPM1mA accelerates the production of sEV‐related miR‐19a‐3p by upregulating cellular miR‐19a‐3p expression and its packaging into sEVs.
3.6. Leukemia‐derived sEVs impair CD8+ T cell antitumour function in vivo
To expand our findings, OCI/AML3 cells and engineered OCI‐AML3 cells were injected into huHSC‐NSG mice to test their effects on leukemogenesis and CD8+ T cell function in vivo. The mice injected with OCI/AML3 miR‐19a‐3p KD cells showed longer survival time than those injected with OCI/AML3 mock cells or OCI/AML3 NC cells, whereas OCI/AML3‐sEV supplementation shortened the survival time (Figure 9a). Next, leukemic cell infiltration was assessed. As shown in Figure 9b–e, miR‐19a‐3p KD reduced human‐CD45+ cells in the peripheral blood (Figure 9b) and leukemic cells in the bone marrow (Figure 9c), and curbed liver and splenic infiltration as identified by H&E staining (Figure 9d) and IF staining (for the leukaemia biomarker NPM1mA) (Figure 9e). In addition, sEV‐related miR‐19a‐3p expression decreased in the bone marrow aspirates (Figure S15A). Administering OCI/AML3‐sEVs to miR‐19a‐3p KD mice could partially rescue leukemic cell infiltration (Figure 9b–e). Finally, CD8+ T cell levels in the bone marrow were investigated, and the results showed that miR‐19a‐3p KD led to a higher CD8+ T cell contents (Figure 9f) and increased IL‐2 and IFN‐γ secretion (Figure 9g–i), with OCI/AML3‐sEVs being able to partially reverse these effects (Figure 9f–i). Additionally, miR‐19a‐3p KD increased SLC6A8 expression (Figure 9j) and creatine production (Figure 9k) in CD8+ T cells, and OCI/AML3‐sEV supplementation attenuated these effects (Figure 9j,k). These data suggest that leukemic cells impair CD8+ T cell antitumour function by transferring sEV‐related miR‐19a‐3p, which contributes to the immune evasion of leukemic cells in vivo.
FIGURE 9.
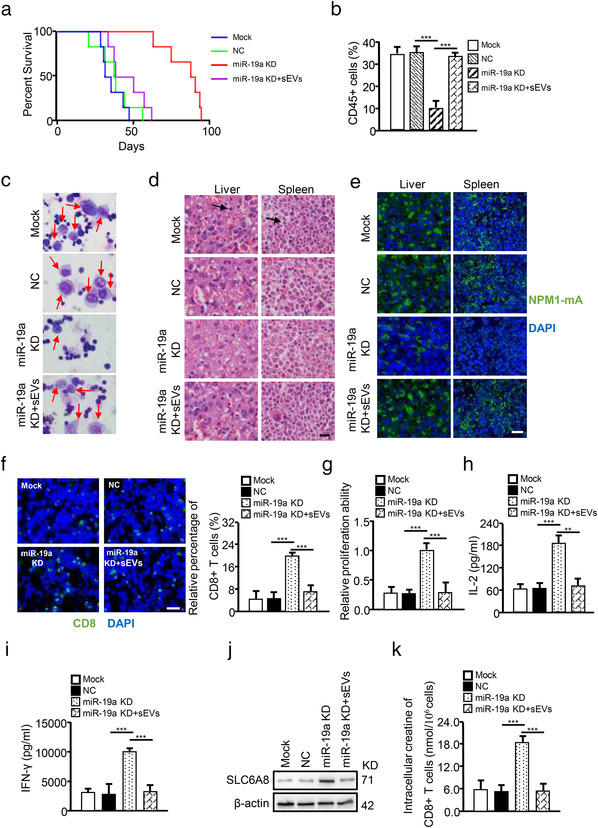
Leukaemia‐derived sEV‐related miR‐19a‐3p impairs CD8+ T cell antitumour function in vivo. The indicated OCI/AML3 cells (OCI/AML3, OCI/AML3‐NC, or OCI/AML3‐miR‐19a‐3p KD) were injected into HuHSC‐NSG mice. OCI/AML3, mock (untreated cells); OCI/AML3‐NC, NC (OCI/AML3 cells transfected with negative control vector); OCI/AML3‐miR‐19a‐3p KD, miR‐19a KD (OCI/AML3 cells transfected with shRNA targeting miR‐19a‐3p); OCI/AML3‐sEVs, sEVs (sEVs‐derived from OCI/AML3 cells). (a) Kaplan–Meier analysis of the survival curves of the mice in each group (n = 6). (b) The number of CD45+ human leukemic cells were determined by FCM. (c) Immature cells from the bone marrow were checked using Wright's stain. (d and e) Spleen and liver infiltration were analyzed by H&E staining (d) and IF for NPM1mA (e). (f) CD8+ T cells in bone marrow detected by IF for CD8. Proliferation (g), IL‐2 secretion (H), and IFN‐γ secretion (i) of bone marrow CD8+ T cells are shown. (j) SLC6A8 protein expression in CD8+ T cells was detected by western blotting. (k) Intracellular creatine in CD8+ T cells was determined using the Creatine Assay Kit (*p < 0.05; **p < 0.01; ***p < 0.001; n.s, not significant)
Taken together, our findings confirm that sEV‐related miR‐19a‐3p is enhanced by NPM1mA and can be transferred from leukemic cells to CD8+ T cells, and thereby suppresses the immune function of CD8+ T cells by inhibiting SLC6A8‐mediated creatine import (Figure 10).
FIGURE 10.
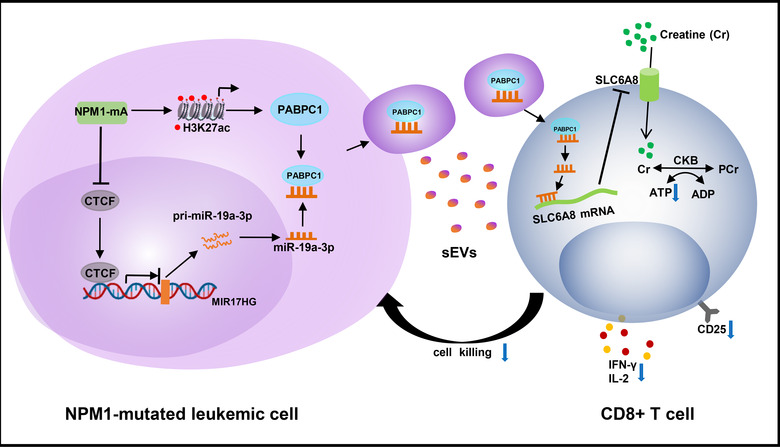
Proposed model for how miR‐19a‐3p suppresses the immune function of CD8+ T cells. sEV‐related miR‐19a‐3p (regulated by NPM1mA) shuttles from leukemic cells to CD8+ T cells to inhibit SLC6A8‐mediated creatine import, thereby impairing the immune function of CD8+ T cells in AML
4. DISCUSSION
The aim of cancer immunotherapy is to improve antitumour immune responses by activating or boosting the immune system to attack cancer cells. This approach has recently shifted the paradigm for cancer treatment and has fewer off‐target effects than chemotherapies and other treatments (Rosenberg, 2014). Immunotherapy is recognized as a promising strategy to treat and cure certain types of cancers (Berraondo et al., 2019), but this benefit remains limited to patients with NPM1mut AML. Therefore, there is an urgent need to improve our understanding of immune regulation in NPM1mut AML. Herein, our data demonstrated that leukemic cells impaired the immune function of CD8+ T cells by inhibiting creatine import.
Metabolic reprogramming of immune cells plays a critical role in cancer progression. Enhanced glycolysis, fatty acid metabolism, and amino acid metabolism promote the immune effects of related cells, such as T cells and macrophages (D.‐ P. Chen et al., 2019; Makowski et al., 2020). Recently, growing evidence has shown that the TME can alter the metabolism of immune‐related cells, thus impairing antitumour immunity. LDHA‐associated acid accumulation in melanomas inhibits the immune function of natural killer and T cells, and leads to tumour immune escape (Brand et al., 2016). Tumour cells disrupt methionine metabolism in CD8+ T cells, thereby lowering the intracellular levels of methionine and the methyl donor S‐adenosylmethionine, which impairs T cell immunity (Bian et al., 2020). Here, we discovered that leukemic cells downregulated creatine import in CD8+ T cells, leading to the repression of immune function. Creatine is broadly used by bodybuilders and athletes to increase muscle mass and improve performance (Wyss & Kaddurah‐Daouk, 2000). A few recent reports have shown that creatine is involved in maintaining T cell function. For example, Di Biase et al. revealed that tumor‐infiltrating T cells used glycolysis and/or the tricarboxylic acid cycle to convert nutrients (e.g., glucose, amino acids, and lipids) into bioenergy in the form of ATP, while using creatine as a “molecular battery” to store bioenergy and buffer the intracellular ATP level, which supported T cell antitumour activities (Di Biase et al., 2019). Our research shows that leukemic cells inhibit creatine import in CD8+ T cells, which is pivotal to further efforts to understand the effect of creatine metabolism on immune cell function.
Tumour‐derived exosomes can shuttle from cancer cells to immune cells and participate in tumour immune escape (Whiteside, 2016). Accumulating evidence suggests that exosomal miRNAs contribute to reprogramming the function of immune target cells (Li et al., 2019). For example, exosomal miR‐23a from endoplasmic reticulum‐stressed hepatocellular carcinoma cells was found to upregulate PD‐L1 expression in macrophages via the PTEN/AKT pathway and inhibit T cell antitumour function (Sun, in press). Exosomal miR‐125b‐5p from cancer cells can be delivered to macrophages and induce the observed tumour‐promoting macrophage phenotype in melanoma (Gerloff & Lützkendorf, 2020). However, few studies have focused on leukemic cell‐derived sEVs. Here, we revealed that sEV‐related miR‐19a‐3p from NPM1mut leukemic cells could be transferred to CD8+ T cells and impair CD8+ T cell function. It has been reported that the miR‐17–19 cluster is a potent and pleiotropic regulator of T cell responses and miR‐19a is mainly involved in promoting Th1 and Th2 cell differentiation (Jiang et al., 2011; Simpson et al., 2014). Additionally, recent findings have revealed that miR‐19a‐3p can be secreted into the microenvironment via sEVs. For example, miR‐19a‐3p contained in angiogenic exosomes can induce angiogenesis and decrease myocardial ischemia (Gollmann‐Tepeköylü et al., 2020). Exosomal miR‐19a from adipose‐derived stem cells can suppress the differentiation of rabbit corneal keratocytes into myofibroblasts by inhibiting HIPK2 expression (Shen et al., 2020). In this study, we found that sEV‐related miR‐19a‐3p from NPM1mut leukemic cells impaired CD8+ T cell immune function.
SLC6A8 belongs to the solute carrier six family, specifically acting to transport creatine into cells, and is mainly expressed in organs such as the brain and skeletal muscles that have high demands for creatine (Stockebrand et al., 2018). Recently, it was reported that SLC6A8 was highly expressed in immune cells, such as macrophages (Ji et al., 2019) and tumour‐infiltrating T cells (Di Biase et al., 2019). Here, we showed that SLC6A8 was highly expressed in activated CD8+ T cells and that leukaemia‐derived sEV‐related miR‐19a‐3p directly inhibited SLC6A8 expression in CD8+ T cells. Previous findings have suggested that SLC6A8 activity/expression may be regulated by extracellular and cytosolic creatine levels (Alfieri et al., 2006), hormones (Omerovic et al., 2003), and kinase activity (such as AMP‐activated kinase and glycogen synthase kinase) (Darrabie et al., 2011; Fezai et al., 2016). Herein, we revealed that miR‐19a‐3p directly inhibited SLC6A8 expression at the transcriptional level.
The targeted export of miRNAs to sEVs may require specific mechanisms. Recently, some reports have indicated that RBPs participate in the specific loading of miRNAs into exosomes (Santangelo et al., 2016). For example, hnRNPA2B1 was shown to enhance exosomal miRNA export from T cells (Villarroya‐Beltri et al., 2013). In addition, relevant functions in miRNA trafficking were demonstrated for AGO2 and Y‐box in colon cancer cells and HEK293T cells, respectively (Mckenzie et al., 2016; Shurtleff et al., 2016). However, the effects of RBPs on sEV‐related miRNA secretion in AML, especially in NPM1mut AML, remain poorly understood. PABPC1 is a cytosolic protein that binds to the indicated miRNAs and regulates miRNA‐mediated gene function (Lemay et al., 2010). For example, PABPC1 interacts with AGO2 or GW182 to enhance miRNA‐mediated gene silencing in high‐grade hepatocellular carcinoma (Huntzinger et al., 2010; H. Zhang et al., 2015). Recently, proteomic profiling of two distinct populations of extracellular vesicles isolated from human seminal plasma showed that PABPC1 may facilitate the RNA loading of extracellular vesicles (Q. Zhang, Liu, et al., 2020). Here, we found that PABPC1 interacted with miR‐19a‐3p and regulates miR‐19a‐3p packaging into sEVs.
NPM1mA participates in various cellular processes in leukaemia progression, such as leukemic cell survival (Jin et al., 2018; W. Zhang et al., 2018), invasion (Xian et al., 2016), and autophagy (Zou et al., 2017). Here, we revealed that NPM1mA enhanced the immune escape of leukemic cells by promoting sEV‐related miR‐19a‐3p secretion. NPM1mA enhanced cellular miR‐19a‐3p expression by regulating CTCF. NPM1mA was previously reported to interact with CTCF and mediate CTCF translocation from the nucleus to the cytoplasm, which repressed the function of CTCF‐mediated transcription inhibition (Wang et al., 2020). In this study, we found that decreased CTCF expression in the nucleus attenuated its transcriptional inhibition of MIR17HG, the host gene of miR‐19a‐3p, and thus upregulated miR‐19a‐3p levels. In addition, NPM1mA promoted miR‐19a‐3p packaging into sEVs by upregulating H3K27 acetylation‐activated PABPC1. As reported previously, NPM1mA modulated H3K27 acetylation at the HOX promoter to enhance HOX expression (Brunetti et al., 2018). Additionally, H3K27 acetylation of PABPC1 was enhanced by the long non‐coding RNA SNHG14 in hepatocellular carcinoma and breast cancer cells (Dong et al., 2018; X. Zhang, Vos, et al., 2020). This evidence supports our finding that NPM1mA modulated H3K27 acetylation at the PABPC1 promoter to enhance its expression.
5. CONCLUSION
In summary, our findings revealed that leukemic cells can impair CD8+ T cell antitumour immunity by repressing creatine import. NPM1mA enhanced the secretion of sEV‐related miR‐19a‐3p from leukemic cells by upregulating cellular miR‐19a‐3p expression and packaging miR‐19a‐3p into sEVs. The sEV‐related miR‐19a‐3p was internalized by CD8+ T cells and further inhibited SLC6A8‐mediated creatine import, which impaired immune function. Collectively, our present findings provide a new perspective on leukaemia immune escape mechanisms, and suggest that sEV‐related miR‐19a‐3p, especially the signalling that leads to preferential packaging of miR‐19a‐3p into sEVs, may be an important therapeutic target for NPM1mut AML.
DISCLOSURE OF INTEREST
The authors report no conflict of interest.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval was given by the Medical Ethics Committee of Chongqing Medical University. Animal experiments were permitted by the Animal Ethics Committees of Chongqing Medical University. All animal work was conducted according to the approved protocols and the institutional animal welfare guidelines.
AUTHORS’ CONTRIBUTIONS
MP and LZ designed this study; MP, JR, JH, YP, and LL performed the experiments; XJ, QX and XW analyzed the data; MP, ZY, ZY, QZ and CL drew the figures and tables; MP and LZ drafted the initial manuscript; MP, LZ, GJ and XZ revised this manuscript; all authors read and approved the final manuscript.
CONSENT FOR PUBLICATION
All authors are aware of and agree to the content of the paper and their being listed as a co‐author of the paper.
Supporting information
Supplementary Figure 1. Detecting T cell function in AML. (A) Representative IHC staining for CD3, CD4 and CD8 in patients with BHD, NPM1wt AML, and NPM1mut AML are shown. Scale bar, 50 mm. (B) The dot plots show the relative numbers of CD3+, CD4+, and CD8+ cells found by IHC staining. (C) The pie charts show the relative numbers of CD3+ T cells, CD4+ T cells, CD8+ T cells, and other T cells found by IHC staining. (D‐F) Proliferation (D), IL‐2 secretion (E) and IFN‐γ secretion (F) of peripheral blood CD8+ T cells in patients with BHD (n = 27), NPM1wt AMLs (n = 27) and NPM1mut AMLs (n = 27) are shown. Supplementary Figure 2. Effects of leukemic blast cells on CD8+ T cell function. (A‐E) CD8+ T cells were incubated with Blasts/wt‐CM or Blasts/mut‐CM. (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression levels were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 3. Bioinformatics analysis of sequence data from NPM1wt AMLs and NPM1wt AMLs. (A) Geometric mean‐centred, hierarchical cluster heat map from microarray data (accession numbers: GSE14468, GSE15434, and GSE34860) of NPM1wt AMLs and NPM1mut AMLs in the Gene Expression Omnibus (GEO) database. (B) Significantly aberrant canonical pathways in NPM1mut AML were identified using the DAVID database. (C) The sEV concentrations from leukemic cells treated with or without GW4869 were detected by performing NTA assays. (D) The cells were treated with GW4869. Cell viability was determined using an Annexin V‐FITC Kit and FCM, and the percentages of viable cells were calculated. Supplementary Figure 4. Related to Figure 2. (a‐e) CD8+ T cells were incubated with CM Ctrl, Blasts/mut‐DMSO‐CM (CM from DMSO‐treated Blasts/mut), Blasts/mut‐GW4869‐CM (CM from GW4869‐treated Blasts/mut), Blasts/mut‐CM, or Blasts/mut‐CMsEVs‐/– (sEV‐depleted Blasts/mut‐CM). (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression of CD8+ T cells were detected by FCM. Supplementary Figure 5. Small EV identification. (A) The size distribution and concentrations of sEVs from leukemic cells (NB4, OCI/AML2, and OCI/AML3) were detected by nanoparticle tracking analysis (NTA). (B) The morphologies of sEVs were visualized by transmission electron microscopic analysis (TEM). (C) Specific sEV marker proteins (TSG101, HSP70, CD9, and CD63) and the negative sEV marker protein (calnexin) in sEVs and parental cell lysates were detected by western blotting. (D) Representative confocal microscopy image showing the internalization of PKH26‐labeled leukemic cell sEVs (red) by CD8+ T cells. Supplementary Figure 6. Related to Figure 2. (a‐e) CD8+ T cells were incubated with Blasts/wt‐sEV or Blasts/mut‐sEV. (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression of CD8+ T cells were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 7. Related to Figure 3. (a‐f) CD8+ T cells were treated with Blasts/wt‐sEVs or Blasts/mut‐sEVs. Intracellular creatine (A) and ATP (B) levels, as well as extracellular levels of creatine (C) are shown. The mRNA‐expression (D) and protein‐expression (E) levels of SLC6A8, AGAT, GAMT, and CK were measured via qRT‐PCR and western blotting, respectively. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 8. Related to Figure 4. (a‐h) CD8+ T cells were treated with or without siRNA targeting SLC6A8. (A) The efficiency of SLC6A8 silencing was determined by western blotting. (B‐C) The intracellular creatine (B) and ATP (C) levels in CD8+ T cells are shown. (D) CD8+ T cell proliferation was evaluated by FCM. (E‐F) IL‐2 (E) and IFN‐γ (F) levels in supernatants were analyzed using ELISA kits. (G‐H) CD25 (G) and Granzyme B (H) expression of CD8+ T cells were detected by FCM. Supplementary Figure 9. Related to Figure 4. (a‐i) CD8+ T cells transfected with an SLC6A6‐overexpression vector (SLC6A8) or negative control vector (vector) were cultured with Blasts/mut‐sEVs. (A) SLC6A8 proteins were detected by western blotting. (B‐C) The intracellular levels of creatine (B) and ATP (C) in CD8+ T cells are shown. (D) CD8+ T cell proliferation was evaluated by FCM. (E‐F) IL‐2 (E) and IFN‐γ (F) levels in supernatants from cell cultures were analyzed using ELISA kits. (G‐H) CD25 (G) and Granzyme B (H) expression of CD8+ T cells were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 10. Prediction of sEV‐related miRNAs. (A) Venn diagram showing that miR‐19a‐3p, miR‐19b‐3p, and miR‐29b‐3p putatively target SLC6A8 mRNA in NPM1mut AML. The upstream miRNAs of SLC6A8 were predicted using the miRanda, miRDB, and TargetScan databases. The upregulated miRNAs in patients with NPM1mut AML were obtained from The Cancer Genome Atlas database. (B) The miRNA contents in sEVs derived from leukemic cells were detected by qRT‐PCR. (C‐E) The levels of miR‐19a‐3p (C), miR‐19b‐3p (D), and miR‐29b‐3p (E) in CD8+ T cells (treated with or without leukemia‐sEVs) were assessed by qRT‐PCR. Supplementary Figure 11. Related to Figure 5. (a) Base pairing between miR‐19a‐3p and the 3′‐UTR of SLC6A8 was predicted using TargetScan, and DNA sequences for SLC6A8 3′‐UTR‐WT and SLC6A8 3′‐UTR‐MUT were inserted into dual‐luciferase vectors, as shown. (B) Dual‐luciferase analysis of CD8+ T cells co‐transfected with an miR‐19a‐3p mimic or a scrambled mimic, and either pGL3‐MIR17HG WT or pGL3‐MIR17HG MUT, is shown. (C) GSEA analysis of miR‐19a‐3p in AML is shown. (D) The efficiency of miR‐19a‐3p overexpression in CD8+ T cells was determined by qRT‐PCR. (E‐L) CD8+ T cells were transfected with an miR‐19a‐3p mimic or a scrambled mimic or/and ectopic SLC6A8. (E) SLC6A8 proteins expression was detected by western blotting. (F‐G) The intracellular levels of creatine (F) and ATP (G) in CD8+ T cells are shown. (H) CD8+ T cell proliferation was evaluated by FCM. (I‐J) IL‐2 (I) and IFN‐γ (J) levels in supernatants from cell cultures were analyzed using commercially available ELISA kits. (K‐L) CD25 (K) and Granzyme B (L) expression of CD8+ T cells were detected by FCM. Supplementary Figure 12. Knockdown of the sEV‐related miR‐19a‐3p rescues CD8+ T cell function. (A) The interference efficiency of miR‐19a‐3p in Blasts/mut cells and Blasts/mut‐sEVs was detected by qRT‐PCR. U6 expression was detected as an internal control. (B‐G) CD8+ T cells incubated with Blasts/mut‐sEVs, with or without miR‐19a‐3p KD. Blasts/mut‐sEVsNC, Blasts/mut cells transfected with negative control vector; Blasts/mut‐sEVsmiR‐19a KD, Blasts/mut cells transfected with shRNA targeting miR‐19a‐3p. (B) SLC6A8 protein expression was analyzed by western blotting. (C) CD8+ T cell proliferation was evaluated by FCM. (D‐E) IL‐2 (D) and IFN‐γ (E) levels in cell culture supernatants were analyzed using ELISA kits. (F‐G) CD25 (F) and Granzyme B (G) expression of CD8+ T cells were determined by FCM. Supplementary Figure 13. Related to Figure 7. (a) The expression levels of miR‐19a‐3p in leukemic cells and cell‐derived sEVs were detected by qRT‐PCR. (B) The efficiency of NPM1 silencing was determined by western blotting. (C) MIR17HG mRNA expression in Blasts/mut cells was detected by qRT‐PCR. (D) The transcription factors for MIR17HG were predicted using the hTFtargets database. (E) Total lysates from leukemic cells were immunoprecipitated with anti‐NPM1mA or control rabbit IgG antibodies (upper panel) or anti‐CTCF or control mouse IgG antibodies (lower panel), followed by immunoblotting for CTCF and NPM1mA, respectively. Supplementary Figure 14. Related to Figure 8. (a) A specific interaction between the miR‐19a‐3p sequence and RBP motifs was predicted through RBPDB analysis. (B) The efficiencies of PABPC1 and KHSRP silencing in leukemic cells were determined by western blotting. (C) The expression levels of miR‐19a‐3p in leukemic cells (with or without PABPC1 KD) were detected by qRT‐PCR. (D) PABPC1 RNA expression in leukemic cells (treated with or without shNPM1) was determined by qRT‐PCR. (E) Leukemic cells were treated with or without shNPM1‐#3, and PABPC1 expression in the indicated cells was detected by western blotting. (F) RIP assays with anti‐PABPC1 antibody (or IgG as a control) were performed using cell lysates from leukemic cells, treated with or without shNPM1. miR‐19a‐3p levels in immunoprecipitated samples were determined by qRT‐PCR and are reported as percentages with respect to the input sample (% input). Supplementary Figure 15. Related to Figure 9. (A) Expression levels of the sEV‐related miR‐19a‐3p in mouse bone marrow aspirates were detected by qRT‐PCR.
Supporting information.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China under Grant NSFC81873973; National Natural Science Foundation of China under Grant NSFC82072353; outstanding Postgraduate Fund of Chongqing Medical University under Grant BJRC202009.
Peng, M. , Ren, J. , Jing, Y. , Jiang, X. , Xiao, Q. , Huang, J. , Tao, Y. , Lei, L. , Wang, X. , Yang, Z. , Yang, Z. , Zhan, Q. , Lin, C. , Jin, G. , Zhang, X. , & Zhang, L. (2021). Tumor‐derived small extracellular vesicles suppress CD8+ T cell immune function by inhibiting SLC6A8‐mediated creatine import in NPM1‐mutated acute myeloid leukemia. Journal of Extracellular Vesicles, 10,e12168. 10.1002/jev2.12168
REFERENCES
- Abreu, R. C. , Ramos, C. V. , Becher, C. , Lino, M. , Jesus, C. , Costa Martins, P. A. , Martins, P. A. T. , Moreno, M. J. , Fernandes, H. , & Ferreira, L. (2021).Exogenous loading of miRNAs into small extracellular vesicles. Journal of Extracellular Vesicles, 10(10), e12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri, R. R. , Bonelli, M. A. , Cavazzoni, A. , Brigotti, M. , Fumarola, C. , Sestili, P. , Mozzoni, P. , De Palma, G. , Mutti, A. , Carnicelli, D. , Vacondio, F. , Silva, C. , Borghetti, A. F. , Wheeler, K. P. , & Petronini, P. G. (2006).Creatine as a compatible osmolyte in muscle cells exposed to hypertonic stress. The Journal of Physiology, 576(Pt 2), 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berraondo, P. , Sanmamed, M. F. , Ochoa, M. C. , Etxeberria, I. , Aznar, M. A. , Pérez‐Gracia, J. L. , Rodríguez‐Ruiz, M. E. , Ponz‐Sarvise, M. , Castañón, E. , & Melero, I. (2019).Cytokines in clinical cancer immunotherapy. British Journal of Cancer, 120(1), 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, Y. , Li, W. , Kremer, D. M. , Sajjakulnukit, P. , Li, S. , Crespo, J. , Nwosu, Z. C. , Zhang, Li , Czerwonka, A. , Pawłowska, A. , Xia, H. , Li, J. , Liao, P. , Yu, J. , Vatan, L. , Szeliga, W. , Wei, S. , Grove, S. , Liu, J. R. , … Zou, W. (2020).Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature, 585(7824), 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, A. , Singer, K. , Koehl, G E. , Kolitzus, M. , Schoenhammer, G. , Thiel, A. , Matos, C. , Bruss, C. , Klobuch, S. , Peter, K. , Kastenberger, M. , Bogdan, C. , Schleicher, U. , Mackensen, A. , Ullrich, E. , Fichtner‐Feigl, S. , Kesselring, R. , Mack, M. , Ritter, U. , … Kreutz, M. (2016).LDHA‐associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell metabolism, 24(5), 657–671. [DOI] [PubMed] [Google Scholar]
- Brunetti, L. , Gundry, M. C. , Sorcini, D. , Guzman, A. G. , Huang, Y. ‐. H. , Ramabadran, R. , Gionfriddo, I. , Mezzasoma, F. , Milano, F. , Nabet, B. , Buckley, D. L. , Kornblau, S. M. , Lin, C. Y. , Sportoletti, P. , Martelli, M. P. , Falini, B. , & Goodell, M. A. (2018).Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell, 34(3), 499–512.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Liang, C. , Wang, S. , Chio, C. , Zhang, Y. , Zeng, C. , et al. (2020). Expression patterns of immune checkpoints in acute myeloid leukemia. Journal of Hematology & Oncology, 13(1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D.‐ P. , Ning, W‐Ru , Jiang, Ze‐Z. , Peng, Z. ‐. P. , Zhu, L. ‐. Y. , Zhuang, S. ‐. M. , Kuang, D. ‐. M. , Zheng, L. , & Wu, Y. (2019).Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3‐PD‐L1 axis in human hepatocellular carcinoma. Journal of Hepatology, 71(2), 333–343. [DOI] [PubMed] [Google Scholar]
- Coombs, C. C. , Tallman, M. S. , & Levine, R. L. (2016).Molecular therapy for acute myeloid leukaemia. Nature Reviews Clinical Oncology, 13(5), 305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrabie, M. D. , Arciniegas, A. J. L. , Mishra, R. , Bowles, D. E. , Jacobs, D. O. , & Santacruz, L. (2011).AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. American Journal of Physiology Endocrinology and Metabolism, 300(5), E870–E876. [DOI] [PubMed] [Google Scholar]
- Di Biase, S. , Ma, X. , & Wang, X. (2019). Creatine uptake regulates CD8 T cell antitumor immunity, Journal of Experimental Medicine 216(12), 2869–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinardo, C. D. , & Wei, A. H. (2020).How I treat acute myeloid leukemia in the era of new drugs. Blood, 135(2), 85–96. [DOI] [PubMed] [Google Scholar]
- Dong, H. , Wang, W. , Mo, S. , Liu, Q. , Chen, X. , Chen, Ru , Zhang, Yu , Zou, K. , Ye, M. , He, X. , Zhang, F. , Han, J. , & Hu, J. (2018).Long non‐coding RNA SNHG14 induces trastuzumab resistance of breast cancer via regulating PABPC1 expression through H3K27 acetylation. Journal of Cellular and Molecular Medicine, 22(10), 4935–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumauthioz, N. , Tschumi, B. , Wenes, M. , Marti, B. , Wang, H. , Franco, F. , Li , W., Lopez‐Mejia , I. C., Fajas , L., Ho , P.‐C., Donda , A., Romero , P., & Zhang , L. (2020). Enforced PGC‐1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cellular & Molecular Immunology, 18(7):1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini, B. , Brunetti, L. , & Martelli, M. P. (2021).How I diagnose and treat NPM1‐mutated AML. Blood, 137(5), 589–599. [DOI] [PubMed] [Google Scholar]
- Ferrara, R. , Mezquita, L. , Texier, M. , Lahmar, J. , Audigier‐Valette, C. , Tessonnier, L. , Mazieres, J. , Zalcman, G. , Brosseau, S. , Le Moulec, S. , Leroy, L. , Duchemann, B. , Lefebvre, C. , Veillon, R. , Westeel, V. , Koscielny, S. , Champiat, S. , Ferté, C. , Planchard, D. , … Caramella, C. (2018).Hyperprogressive disease in patients with advanced non‐small cell lung cancer treated with PD‐1/PD‐L1 inhibitors or with single‐agent chemotherapy. JAMA Oncology, 4(11), 1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fezai, M. , Jemaà, M. , Fakhri, H. , Chen, H. , Elsir, B. , Pelzl, L. , & Lang, F. (2016).Down‐regulation of the Na+,Cl‐ coupled creatine transporter CreaT (SLC6A8) by glycogen synthase kinase GSK3ß. Cellular Physiology and Biochemistry : International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology, 40(5), 1231–1238. [DOI] [PubMed] [Google Scholar]
- Flemming, J. P. , Hill, B. L. , Haque, M. W. , Raad, J. , Bonder, C. S. , Harshyne, L. A. , Rodeck, U. , Luginbuhl, A. , Wahl, J. K. , Tsai, K. Y. , Wermuth, P. J. , Overmiller, A. M. , & Mahoney, Mỹ G. (2020).miRNA‐ and cytokine‐associated extracellular vesicles mediate squamous cell carcinomas. Journal of Extracellular Vesicles, 9(1), 1790159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon, R. , Garofalo, M. , Martelli, M. P. , Briesewitz, R. , Wang, L. , Fernandez‐Cymering, C. , Volinia, S. , Liu, C. ‐ G. , Schnittger, S. , Haferlach, T. , Liso, A. , Diverio, D. , Mancini, M. , Meloni, G. , Foa, R. , Martelli, M. F. , Mecucci, C. , Croce, C. M. , & Falini, B. (2008). Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proceedings of the National Academy of Sciences of the United States of America, 105(10), 3945–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X. , Meng, Q. , Wei, Lu , Liu, J. , Li, M. , Liang, X. , Lin, F. , Zhang, Y. , Li, Y. , Liu, Z. , Fan, H. , & Zhou, X. (2021).Myocardial ischemia‐reperfusion induced cardiac extracellular vesicles harbour proinflammatory features and aggravate heart injury. Journal of Extracellular Vesicles, 10(4), e12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerloff, D. , & Lützkendorf, J. (2020). Melanoma‐derived exosomal miR‐125b‐5p educates tumor associated macrophages (TAMs) by targeting lysosomal acid lipase A (LIPA), Cancers 12(2) 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gionfriddo, I. , Brunetti, L. , Mezzasoma, F. , Milano, F. , Cardinali, V. , Ranieri, R. , Venanzi, A. , Pierangeli, S. , Vetro, C. , Spinozzi, G. , Dorillo, E. , Wu, H. C. , Berthier, C. , Ciurnelli, R. , Griffin, M. J. , Jennings, C. E. , Tiacci, E. , Sportoletti, P. , Falzetti, F. , … Falini, B. (2021).Dactinomycin induces complete remission associated with nucleolar stress response in relapsed/refractory NPM1‐mutated AML. Leukemia, 35, 2552–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebeler, M.‐ E. , & Bargou, R. C. (2020).T cell‐engaging therapies ‐ BiTEs and beyond. Nature Reviews Clinical Oncology, 17(7), 418–434. [DOI] [PubMed] [Google Scholar]
- Gollmann‐Tepeköylü, C. , Pölzl, L. , Graber, M. , Hirsch, J. , Nägele, F. , Lobenwein, D. , Hess, M. W. , Blumer, M. J. , Kirchmair, E. , Zipperle, J. , Hromada, C. , Mühleder, S. , Hackl, H. , Hermann, M. , Al Khamisi, H. , Förster, M. , Lichtenauer, M. , Mittermayr, R. , Paulus, P. , … Holfeld, J. (2020).miR‐19a‐3p containing exosomes improve function of ischaemic myocardium upon shock wave therapy. Cardiovascular Research, 116(6), 1226–1236. [DOI] [PubMed] [Google Scholar]
- Hamam, D. , Abdouh, M. , Gao, Z. H. , Arena, V. , Arena, M. , & Arena, G. O. (2016).Transfer of malignant trait to BRCA1 deficient human fibroblasts following exposure to serum of cancer patients. Journal of Experimental & Clinical Cancer Research : CR, 35, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himes, B. T. , Peterson, T. E. , De Mooij, T. , Garcia, L. M. C. , Jung, Mi‐Y. , Uhm, S. , Yan, D. , Tyson, J. , Jin‐Lee, H. J. , Parney, D. , Abukhadra, Y. , Gustafson, M. P. , Dietz, A. B. , Johnson, A. J. , Dong, H. , Maus, R. L. , Markovic, S. , Lucien, F. , & Parney, I. F. (2020).The role of extracellular vesicles and PD‐L1 in glioblastoma‐mediated immunosuppressive monocyte induction. Neuro‐oncology, 22(7), 967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger, E. , Braun, J. E. , Heimstädt, S. , Zekri, L. , & Izaurralde, E. (2010).Two PABPC1‐binding sites in GW182 proteins promote miRNA‐mediated gene silencing. The EMBO Journal, 29(24), 4146–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, L. , Zhao, X. , Zhang, B. , Kang, L. , Song, W. , Zhao, B. , Xie, W. , Chen, L. , & Hu, X. (2019).Slc6a8‐mediated creatine uptake and accumulation reprogram macrophage polarization via regulating cytokine responses. Immunity, 51(2), 272–284.e7. [DOI] [PubMed] [Google Scholar]
- Jiang, S. , Li, C. , Olive, V. , Lykken, E. , Feng, F. , Sevilla, J. , Wan, Y. , He, L. , & Li, Qi‐J. (2011).Molecular dissection of the miR‐17‐92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood, 118(20), 5487–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, H. , Yang, L. , Wang, L. , Yang, Z. , Zhan, Q. , & Tao, Y. et al. (2018).INPP4B promotes cell survival via SGK3 activation in NPM1‐mutated leukemia. Journal of Experimental & Clinical Cancer Research : CR, 37(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, J. , Weiss, N. , Freed, B. M. , Torres, R. M. , & Pelanda, R. (2011).Generation of hematopoietic humanized mice in the newborn BALB/c‐Rag2null Il2rγnull mouse model: A multivariable optimization approach. Clinical Immunology (Orlando, Fla), 140(1), 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay, J. ‐. F. , Lemieux, C. , St‐André, O. , & Bachand, F. (2010).Crossing the borders: Poly(A)‐binding proteins working on both sides of the fence. RNA Biology, 7(3), 291–295. [DOI] [PubMed] [Google Scholar]
- Li, L. , Cao, B. , Liang, X. , Lu, S. , Luo, H. , Wang, Z. , Wang, S. , Jiang, J. , Lang, J. , Zhu, G. (2019). Microenvironmental oxygen pressure orchestrates an anti‐ and pro‐tumoral γδ T cell equilibrium via tumor‐derived exosomes, Oncogene 38(15), 2830–2843. [DOI] [PubMed] [Google Scholar]
- Liang, H. , Xiao, Ji , Zhou, Z. , Wu, J. , Ge, F. , Li, Z. , Zhang, H. , Sun, J. , Li, F. , Liu, R. , & Chen, C. (2018).Hypoxia induces miR‐153 through the IRE1α‐XBP1 pathway to fine tune the HIF1α/VEGFA axis in breast cancer angiogenesis. Oncogene, 37(15), 1961–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski, L. , Chaib, M. , & Rathmell, J. C. (2020). From basic mechanisms to translation. Immunological Reviews, 295(1), 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marar, C. , Starich, B. , & Wirtz, D. (2021).Extracellular vesicles in immunomodulation and tumor progression. Nature Immunology, 22(5), 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Théry, C. (2019).Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology, 21(1), 9–17. [DOI] [PubMed] [Google Scholar]
- Mccarthy, S. A. , Mufson, R. A. , Pearce, E. J. , Rathmell, J. C. , & Howcroft, T. K. (2013).Metabolic reprogramming of the immune response in the tumor microenvironment. Cancer Biology & Therapy, 14(4), 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenzie, A. J. , Hoshino, D. , Hong, N. H. , Cha, D. J. , Franklin, J. L. , Coffey, R. J. , Patton, J. G. , & Weaver, A. M. (2016).KRAS‐MEK signaling controls Ago2 sorting into exosomes. Cell Reports, 15(5), 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi, T. , Hayashi, T. , Pan, W. ‐. W. , Fujita, Yu , Holt, M V. , Qin, J. , Carson, D A. , & Guan, K.‐L. (2016). The hippo pathway kinases LATS1/2 suppress cancer immunity. Cell, 167(6):1525–1539.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omerovic, E. , Bollano, E. , Lorentzon, M. , Walser, M. , Mattsson‐Hultén, L. , & Isgaard, J. (2003).Growth hormone induces myocardial expression of creatine transporter and decreases plasma levels of IL‐1beta in rats during early postinfarct cardiac remodeling. Growth Hormone & IGF Research : Official Journal of the Growth Hormone Research Society and the International IGF Research Society, 13(5), 239–245. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil, E. , Gerstung, M. , Bullinger, L. , Gaidzik, V. I. , Paschka, P. , Roberts, N. D. , Potter, N. E. , Heuser, M. , Thol, F. , Bolli, N. , Gundem, G. , Van Loo, P. , Martincorena, I. , Ganly, P. , Mudie, L. , Mclaren, S. , O'meara, S. , Raine, K. , Jones, D. R. , … Campbell, P. J. (2016).Genomic classification and prognosis in acute myeloid leukemia. The New England Journal of Medicine, 374(23), 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaioannou, D. , & Petri, A. (2019). The long non‐coding RNA HOXB‐AS3 regulates ribosomal RNA transcription in NPM1‐mutated acute myeloid leukemia. Nature Communications 10(1):5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasim, W. (2019).Allogeneic CAR T cell therapies for leukemia. American Journal of Hematology, 94, S50–S54. [DOI] [PubMed] [Google Scholar]
- Qin, X. , Guo, H. , Wang, X. , Zhu, X. , Yan, M. , Wang, Xu , Xu, Q. , Shi, J. , Lu, E. , Chen, W. , & Zhang, J. (2019).Exosomal miR‐196a derived from cancer‐associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biology, 20(1), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que, Ri‐S. , Lin, C. , Ding, G. ‐. P. , Wu, Z. ‐. R. , & Cao, L.‐P. (2016).Increasing the immune activity of exosomes: The effect of miRNA‐depleted exosome proteins on activating dendritic cell/cytokine‐induced killer cells against pancreatic cancer. Journal of Zhejiang University Science B, 17(5), 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, S. A. (2014).IL‐2: The first effective immunotherapy for human cancer. Journal of Immunology;192(12), 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo, L. , Giurato, G. , Cicchini, C. , Montaldo, C. , Mancone, C. , Tarallo, R. , Battistelli, C. , Alonzi, T. , Weisz, A. , & Tripodi, M. (2016).The RNA‐binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling microRNA sorting. Cell Reports, 17(3), 799–808. [DOI] [PubMed] [Google Scholar]
- Shen, T. , Zheng, Q. , Luo, H. , Li, X. , Chen, Z. , Song, Z. , Zhou, G. , & Hong, C. (2020).Exosomal miR‐19a from adipose‐derived stem cells suppresses differentiation of corneal keratocytes into myofibroblasts. Aging, 12(5), 4093–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff, M. J. , Temoche‐Diaz, M. M. , Karfilis, K. V. , Ri, S. , & Schekman, R. (2016). Y‐box protein 1 is required to sort microRNAs into exosomes in cells and in a cell‐free reaction. Elife 5, e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, L. J. , Patel, S. , Bhakta, N. R. , Choy, D. F. , Brightbill, H. D. , Ren, X. , Wang, Y. , Pua, H. H. , Baumjohann, D. , Montoya, M. M. , Panduro, M. , Remedios, K. A. , Huang, X. , Fahy, J. V. , Arron, J. R. , Woodruff, P. G. , & Anse, K. M. (2014). A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nature Immunology 15(12), 1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska, P. J. , & Rathmell, J. C. (2015).T cell metabolic fitness in antitumor immunity. Trends in Immunology, 36(4), 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockebrand, M. , Sasani, A. , Das, D. , Hornig, S. , Hermans‐Borgmeyer, I. , Lake, H. A. , Isbrandt, D. , Lygate, C. A. , Heerschap, A. , Neu, A. , & Choe, C.‐U. (2018).A mouse model of creatine transporter deficiency reveals impaired motor function and muscle energy metabolism. Frontiers in Physiology, 9, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, G. Hepatology (Baltimore, Md). [Google Scholar]
- Van Der Lee, D. I. , Reijmers, R. M. , Honders, M. W. , Hagedoorn, R. S. , De Jong, R. C.M. , Kester, M. G.D. , Van Der Steen, D. M. , De Ru, A. H. , Kweekel, C. , Bijen, H. M. , Jedema, I. , Veelken, H. , Van Veelen, P. A. , Heemskerk, M. H.M. , Falkenburg, J. H. F. , & Griffioen, M. (2019).Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. The Journal of Clinical Investigation, 129(2), 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroya‐Beltri, C. , Gutiérrez‐Vázquez, C. , Sánchez‐Cabo, F. , Pérez‐Hernández, D. , Vázquez, J. , Martin‐Cofreces, N. , Martinez‐Herrera, D. J. , Pascual‐Montano, A. , Mittelbrunn, M. , Sánchez‐Madrid, F. (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nature Communications, 4:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A. J. , Han, Y. , Jia, N. , Chen, P. , & Minden, M. D. (2020).NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia, 34(5), 1278–1290. [DOI] [PubMed] [Google Scholar]
- Whiteside, T. L. (2016).Exosomes and tumor‐mediated immune suppression. The Journal of Clinical Investigation, 126(4), 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss, M. , & Kaddurah‐Daouk, R. (2000).Creatine and creatinine metabolism. Physiological reviews, 80(3), 1107–1213. [DOI] [PubMed] [Google Scholar]
- Xian, J. , Shao, H. , Chen, X. , Zhang, S. , Quan, J. , Zou, Q. , Jin, H. , & Zhang, L. (2016).Nucleophosmin mutants promote adhesion, migration and invasion of human leukemia THP‐1 cells through MMPs up‐regulation via Ras/ERK MAPK Signaling. International Journal of Biological Sciences, 12(2), 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W. , Wu, X. , Zhou, W. , Fong, M. Y. , Cao, M. , Liu, J. Chen, C.‐H. , Fadare, O. , Pizzo, D. P. , Wu, J. , Liu, L. , Liu, X. , Chin, A. R. , Ren, X. , Chen, Y. , Locasale, J. W. , & Wang, S. E. (2018). Cancer‐cell‐secreted exosomal miR‐105 promotes tumour growth through the MYC‐dependent metabolic reprogramming of stromal cells. Nature Cell Biology 20(5), 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, R. , Cheng, S. , Luo, N. , Gao, R. , Yu, K. , Kang, B. Wang, L. , Zhang, Q. , Fang, Q. , Zhang, L. , Li, C. , He, A. , Hu, X. , Peng, J. , Ren, X. & Zhang, Z. (2019). Distinct epigenetic features of tumor‐reactive CD8+ T cells in colorectal cancer patients revealed by genome‐wide DNA methylation analysis. Genome Biology 21(1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Sheng, C. , Yin, Y. , Wen, S. , Yang, G. , Cheng, Z. , & Zhu, Q. (2015).PABPC1 interacts with AGO2 and is responsible for the microRNA mediated gene silencing in high grade hepatocellular carcinoma. Cancer Letters, 367(1), 49–57. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Xu, H. , Kurban, E. , & Luo, H. (2020).LncRNA SNHG14 promotes hepatocellular carcinoma progression via H3K27 acetylation activated PABPC1 by PTEN signaling. Cell Death & Disease, 11(8), 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Liu, W. , Zhang, H. , Xie, G. , Miao, Y. , & Xia, M. , Guo, A.‐Y. (2020).hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genomics, Proteomics & Bioinformatics, 18(2), 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Zhao, C. , Zhao, J. , Zhu, Y. , Weng, X. , Chen, Q. , Sun, H. , Mi, J. ‐. Q. , Li, J. , Zhu, J. , Chen, Z. , Pandolfi, P. P. , Chen, S. , Yan, X. , & Xu, J. (2018).Inactivation of PBX3 and HOXA9 by down‐regulating H3K79 methylation represses NPM1‐mutated leukemic cell survival. Theranostics, 8(16), 4359–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Vos, H. R. , Tao, W. , & Stoorvogel, W. (2020).Proteomic profiling of two distinct populations of extracellular vesicles isolated from human seminal plasma. International Journal of Molecular Sciences, 21(21), 7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Q. , Tan, S. , Yang, Z. , Zhan, Q. , Jin, H. , Xian, J. , Zhang, S. , Yang, L. , Wang, Lu , & Zhang, L. (2017).NPM1 mutant mediated PML delocalization and stabilization enhances autophagy and cell survival in leukemic cells. Theranostics, 7(8), 2289–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Detecting T cell function in AML. (A) Representative IHC staining for CD3, CD4 and CD8 in patients with BHD, NPM1wt AML, and NPM1mut AML are shown. Scale bar, 50 mm. (B) The dot plots show the relative numbers of CD3+, CD4+, and CD8+ cells found by IHC staining. (C) The pie charts show the relative numbers of CD3+ T cells, CD4+ T cells, CD8+ T cells, and other T cells found by IHC staining. (D‐F) Proliferation (D), IL‐2 secretion (E) and IFN‐γ secretion (F) of peripheral blood CD8+ T cells in patients with BHD (n = 27), NPM1wt AMLs (n = 27) and NPM1mut AMLs (n = 27) are shown. Supplementary Figure 2. Effects of leukemic blast cells on CD8+ T cell function. (A‐E) CD8+ T cells were incubated with Blasts/wt‐CM or Blasts/mut‐CM. (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression levels were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 3. Bioinformatics analysis of sequence data from NPM1wt AMLs and NPM1wt AMLs. (A) Geometric mean‐centred, hierarchical cluster heat map from microarray data (accession numbers: GSE14468, GSE15434, and GSE34860) of NPM1wt AMLs and NPM1mut AMLs in the Gene Expression Omnibus (GEO) database. (B) Significantly aberrant canonical pathways in NPM1mut AML were identified using the DAVID database. (C) The sEV concentrations from leukemic cells treated with or without GW4869 were detected by performing NTA assays. (D) The cells were treated with GW4869. Cell viability was determined using an Annexin V‐FITC Kit and FCM, and the percentages of viable cells were calculated. Supplementary Figure 4. Related to Figure 2. (a‐e) CD8+ T cells were incubated with CM Ctrl, Blasts/mut‐DMSO‐CM (CM from DMSO‐treated Blasts/mut), Blasts/mut‐GW4869‐CM (CM from GW4869‐treated Blasts/mut), Blasts/mut‐CM, or Blasts/mut‐CMsEVs‐/– (sEV‐depleted Blasts/mut‐CM). (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression of CD8+ T cells were detected by FCM. Supplementary Figure 5. Small EV identification. (A) The size distribution and concentrations of sEVs from leukemic cells (NB4, OCI/AML2, and OCI/AML3) were detected by nanoparticle tracking analysis (NTA). (B) The morphologies of sEVs were visualized by transmission electron microscopic analysis (TEM). (C) Specific sEV marker proteins (TSG101, HSP70, CD9, and CD63) and the negative sEV marker protein (calnexin) in sEVs and parental cell lysates were detected by western blotting. (D) Representative confocal microscopy image showing the internalization of PKH26‐labeled leukemic cell sEVs (red) by CD8+ T cells. Supplementary Figure 6. Related to Figure 2. (a‐e) CD8+ T cells were incubated with Blasts/wt‐sEV or Blasts/mut‐sEV. (A) CD8+ T cell proliferation was evaluated by FCM. (B‐C) IL‐2 (B) and IFN‐γ (C) levels in supernatants from cell cultures were analyzed using ELISA kits. (D‐E) CD25 (D) and granzyme B (E) expression of CD8+ T cells were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 7. Related to Figure 3. (a‐f) CD8+ T cells were treated with Blasts/wt‐sEVs or Blasts/mut‐sEVs. Intracellular creatine (A) and ATP (B) levels, as well as extracellular levels of creatine (C) are shown. The mRNA‐expression (D) and protein‐expression (E) levels of SLC6A8, AGAT, GAMT, and CK were measured via qRT‐PCR and western blotting, respectively. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 8. Related to Figure 4. (a‐h) CD8+ T cells were treated with or without siRNA targeting SLC6A8. (A) The efficiency of SLC6A8 silencing was determined by western blotting. (B‐C) The intracellular creatine (B) and ATP (C) levels in CD8+ T cells are shown. (D) CD8+ T cell proliferation was evaluated by FCM. (E‐F) IL‐2 (E) and IFN‐γ (F) levels in supernatants were analyzed using ELISA kits. (G‐H) CD25 (G) and Granzyme B (H) expression of CD8+ T cells were detected by FCM. Supplementary Figure 9. Related to Figure 4. (a‐i) CD8+ T cells transfected with an SLC6A6‐overexpression vector (SLC6A8) or negative control vector (vector) were cultured with Blasts/mut‐sEVs. (A) SLC6A8 proteins were detected by western blotting. (B‐C) The intracellular levels of creatine (B) and ATP (C) in CD8+ T cells are shown. (D) CD8+ T cell proliferation was evaluated by FCM. (E‐F) IL‐2 (E) and IFN‐γ (F) levels in supernatants from cell cultures were analyzed using ELISA kits. (G‐H) CD25 (G) and Granzyme B (H) expression of CD8+ T cells were determined by FCM. (* p < 0.05; ** p < 0.01; *** p < 0.001; n.s, not significant). Supplementary Figure 10. Prediction of sEV‐related miRNAs. (A) Venn diagram showing that miR‐19a‐3p, miR‐19b‐3p, and miR‐29b‐3p putatively target SLC6A8 mRNA in NPM1mut AML. The upstream miRNAs of SLC6A8 were predicted using the miRanda, miRDB, and TargetScan databases. The upregulated miRNAs in patients with NPM1mut AML were obtained from The Cancer Genome Atlas database. (B) The miRNA contents in sEVs derived from leukemic cells were detected by qRT‐PCR. (C‐E) The levels of miR‐19a‐3p (C), miR‐19b‐3p (D), and miR‐29b‐3p (E) in CD8+ T cells (treated with or without leukemia‐sEVs) were assessed by qRT‐PCR. Supplementary Figure 11. Related to Figure 5. (a) Base pairing between miR‐19a‐3p and the 3′‐UTR of SLC6A8 was predicted using TargetScan, and DNA sequences for SLC6A8 3′‐UTR‐WT and SLC6A8 3′‐UTR‐MUT were inserted into dual‐luciferase vectors, as shown. (B) Dual‐luciferase analysis of CD8+ T cells co‐transfected with an miR‐19a‐3p mimic or a scrambled mimic, and either pGL3‐MIR17HG WT or pGL3‐MIR17HG MUT, is shown. (C) GSEA analysis of miR‐19a‐3p in AML is shown. (D) The efficiency of miR‐19a‐3p overexpression in CD8+ T cells was determined by qRT‐PCR. (E‐L) CD8+ T cells were transfected with an miR‐19a‐3p mimic or a scrambled mimic or/and ectopic SLC6A8. (E) SLC6A8 proteins expression was detected by western blotting. (F‐G) The intracellular levels of creatine (F) and ATP (G) in CD8+ T cells are shown. (H) CD8+ T cell proliferation was evaluated by FCM. (I‐J) IL‐2 (I) and IFN‐γ (J) levels in supernatants from cell cultures were analyzed using commercially available ELISA kits. (K‐L) CD25 (K) and Granzyme B (L) expression of CD8+ T cells were detected by FCM. Supplementary Figure 12. Knockdown of the sEV‐related miR‐19a‐3p rescues CD8+ T cell function. (A) The interference efficiency of miR‐19a‐3p in Blasts/mut cells and Blasts/mut‐sEVs was detected by qRT‐PCR. U6 expression was detected as an internal control. (B‐G) CD8+ T cells incubated with Blasts/mut‐sEVs, with or without miR‐19a‐3p KD. Blasts/mut‐sEVsNC, Blasts/mut cells transfected with negative control vector; Blasts/mut‐sEVsmiR‐19a KD, Blasts/mut cells transfected with shRNA targeting miR‐19a‐3p. (B) SLC6A8 protein expression was analyzed by western blotting. (C) CD8+ T cell proliferation was evaluated by FCM. (D‐E) IL‐2 (D) and IFN‐γ (E) levels in cell culture supernatants were analyzed using ELISA kits. (F‐G) CD25 (F) and Granzyme B (G) expression of CD8+ T cells were determined by FCM. Supplementary Figure 13. Related to Figure 7. (a) The expression levels of miR‐19a‐3p in leukemic cells and cell‐derived sEVs were detected by qRT‐PCR. (B) The efficiency of NPM1 silencing was determined by western blotting. (C) MIR17HG mRNA expression in Blasts/mut cells was detected by qRT‐PCR. (D) The transcription factors for MIR17HG were predicted using the hTFtargets database. (E) Total lysates from leukemic cells were immunoprecipitated with anti‐NPM1mA or control rabbit IgG antibodies (upper panel) or anti‐CTCF or control mouse IgG antibodies (lower panel), followed by immunoblotting for CTCF and NPM1mA, respectively. Supplementary Figure 14. Related to Figure 8. (a) A specific interaction between the miR‐19a‐3p sequence and RBP motifs was predicted through RBPDB analysis. (B) The efficiencies of PABPC1 and KHSRP silencing in leukemic cells were determined by western blotting. (C) The expression levels of miR‐19a‐3p in leukemic cells (with or without PABPC1 KD) were detected by qRT‐PCR. (D) PABPC1 RNA expression in leukemic cells (treated with or without shNPM1) was determined by qRT‐PCR. (E) Leukemic cells were treated with or without shNPM1‐#3, and PABPC1 expression in the indicated cells was detected by western blotting. (F) RIP assays with anti‐PABPC1 antibody (or IgG as a control) were performed using cell lysates from leukemic cells, treated with or without shNPM1. miR‐19a‐3p levels in immunoprecipitated samples were determined by qRT‐PCR and are reported as percentages with respect to the input sample (% input). Supplementary Figure 15. Related to Figure 9. (A) Expression levels of the sEV‐related miR‐19a‐3p in mouse bone marrow aspirates were detected by qRT‐PCR.
Supporting information.