

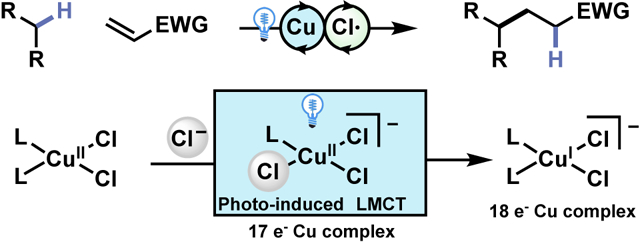
Abstract
Utilizing catalytic CuCl2 we report the functionalization of numerous feedstock chemicals via the coupling of unactivated C(sp3)–H bonds with electron deficient olefins. The active cuprate catalyst undergoes Ligand-to-Metal Charge Transfer (LMCT) to enable the generation of a chlorine radical which acts as a powerful hydrogen atom transfer reagent capable of abstracting strong electron rich C(sp3)–H bonds. Of note is that the chlorocuprate catalyst is an exceedingly mild oxidant (0.5 V vs SCE), and that a proposed protodemetallation mechanism offers a broad scope of electron deficient olefins, offering high diastereoselectivity in the case of endocyclic alkenes. The coupling of chlorine radical generation with Cu reduction through LMCT enables the generation of a highly active HAT reagent in an operationally simple and atom economical protocol.
Graphical Abstract
The catalytic functionalization of unactivated C(sp3)–H bonds has been a long-standing goal in synthetic methods development.1–2 Due to the inert and non-polarized nature of C(sp3)–H bonds, manipulation of these positions requires the formation of high energy intermediates that must differentiate comparable sites of reactivity to enable selectivity,3–4 a challenge made more profound when one also targets the desirable outcome of sustainability.5
In recent years, intermolecular hydrogen atom transfer (HAT) has evolved into a synthetically viable mechanism for the selective C–H functionalization of alkanes.6–10 (Scheme 1A) The generated alkyl radical provides a reactive intermediate poised for bond formation with myriad coupling partners including those activated via transition-metal catalysis.11–15 Selectivity of HAT is governed by the electronic and steric characteristics of both the C(sp3)–H bonds of the substrate as well as the HAT reagent.16–21 The recent expansion of photoredox catalysis in organic synthesis has enabled the formation of reactive radicals capable of HAT for myriad functional groups in a catalytic fashion.22–31
Scheme 1.
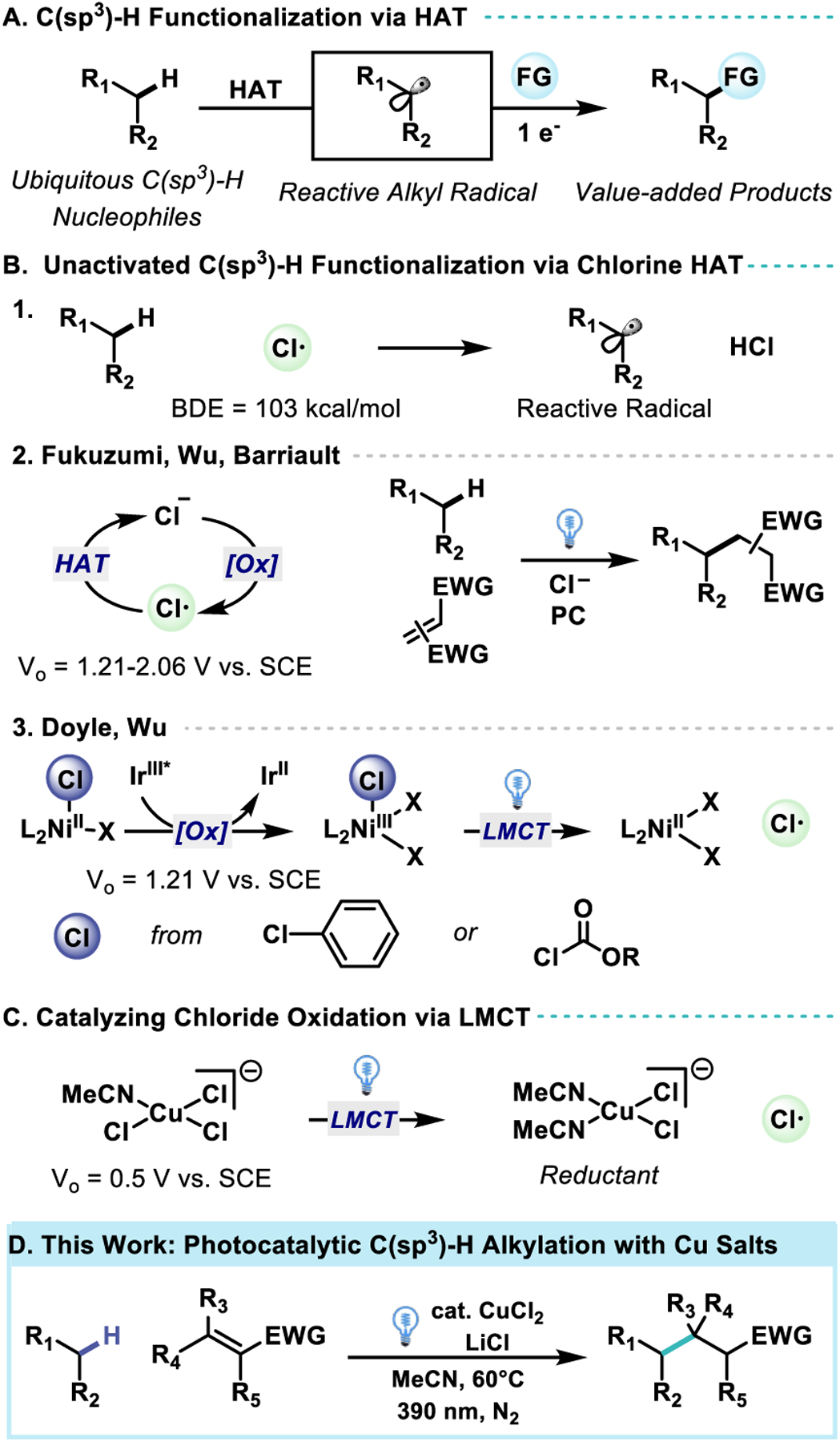
Cu catalyzed formation of chlorine radical via ligand-to-metal charge-transfer (LMCT) enables unactivated C(sp3)–H Alkylation
Due to the abundance and inexpensive nature of chloride salts, the formation of chlorine radical from chloride anion for use as a HAT reagent has long been an attractive strategy towards the functionalization of C(sp3)–H bonds. HAT with chlorine radical forms a strong polarized bond in HCl, (BDE = 103 kcal/mol) enabling the abstraction of strong electron rich C(sp3)–H bonds to form the corresponding alkyl radical. (Scheme 1B–1) While there exists a surfeit of HAT reagents capable of abstracting relatively weak C(sp3)–H bonds, there are relatively few methods capable of HAT of strong C(sp3)–H bonds.32–36 Classical methods of chlorine radical generation require the employment of reactive reagents such as chlorine gas or N–Chlorosuccinimide,37–38 but recent methods have enabled the generation of chlorine radical via direct oxidation of dissolved chloride through photoredox catalysis.39–42 This direct oxidation requires potentials exceeding +1.21 V vs. SCE,42 limiting synthetic utility due to oxidation sensitivity of the substrate as well as diminishing the coupling partners available for the generated alkyl radical due to the limited reduction potentials available to highly oxidizing photocatalysts. (Scheme 1B–2)
Scheme 2. Representative examples of alkane nucleophiles.
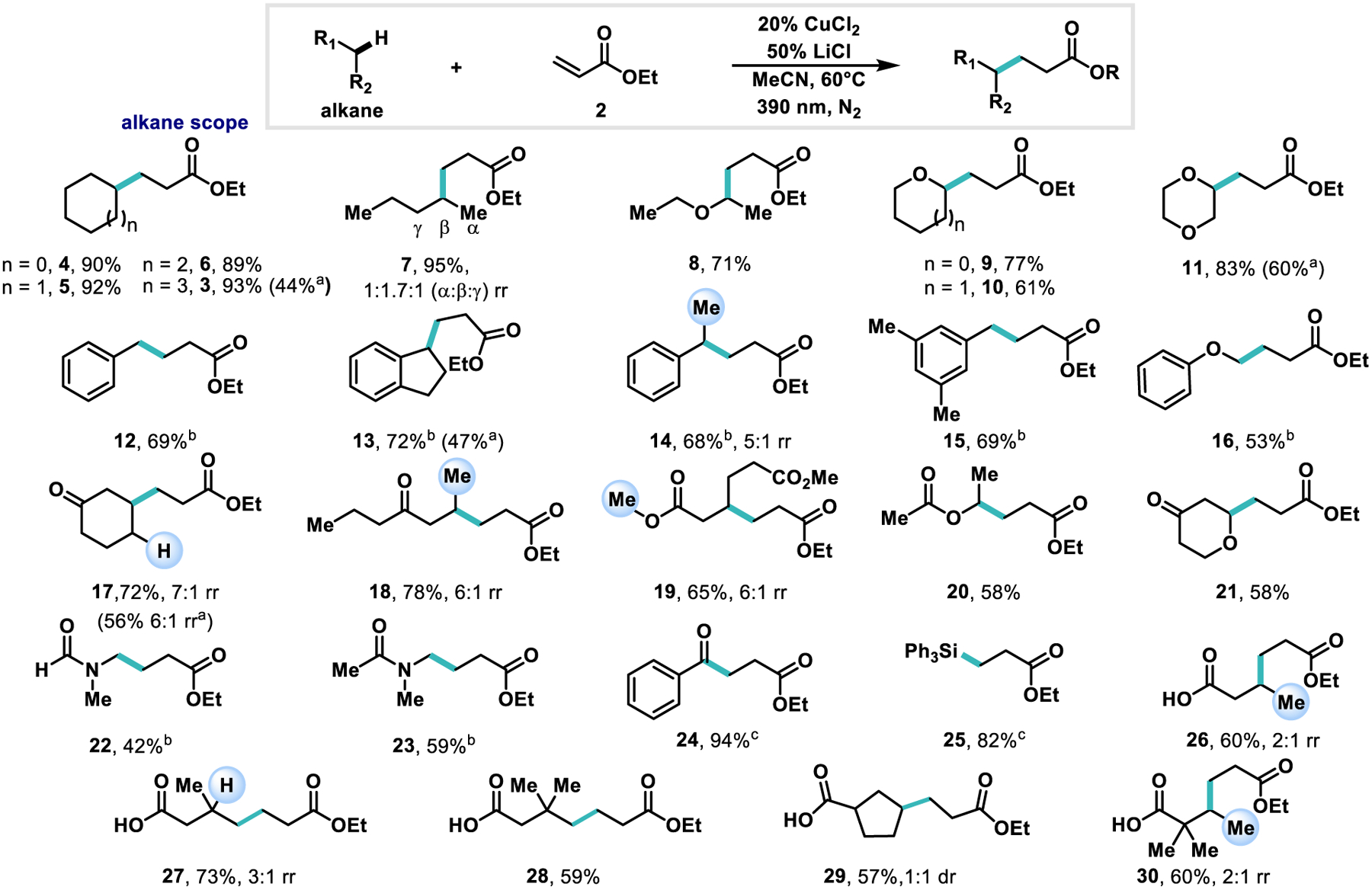
Reactions were run on 0.3 mmol scale with 5 equiv. of alkane nucleophile, 20 mol % CuCl2, 50 mol % LiCl, in 1.0 mL of MeCN for 36 h. For inseparable regioisomeric mixtures, the C(sp3)-H bond corresponding to the minor product site of functionalization is highlighted. a1 equiv. of alkane nucleophile, 1.1 equiv. of ethyl acrylate b 50 mol % CuCl2, 1.25 equiv. LiCl. c 3 equiv. of alkane nucleophile.
Photoinduced ligand-to-metal charge-transfer (LMCT) has proven an effective means towards the formation of HAT reagents in organic synthesis.43 Coordination of the reactive functional group with the metal complex enables chemoselective oxidation via direct excitation of the metal-ligand complex. Through the coupling of the ligand oxidation with metal reduction, the reactive radical may be generated under low electrochemical potentials, complementing strategies enabled by SET. Pioneering work by the Doyle, Wu, and Zuo labs has shown LMCT as an effective strategy to catalyze the formation of chlorine and alkoxy radicals to functionalize strong C(sp3)–H bonds.43–47 (Scheme 1B–3) Previous work in our group demonstrated the ability of CoII–acetylides to undergo LMCT towards CoI catalyzed [2+2+2] cycloaddition of alkynes.48 Hypothesizing that other air stable metal salts could complement these HAT centered protocols in terms of substrate scope, selectivity, and reactivity, we explored the competency of various metal halides to undergo LMCT to enable novel reactivity in HAT catalysis. Stimulated by early work from Kochi who proved the competence of cupric chloride in the stoichiometric chlorination of alkanes in acetonitrile,49–54 we noted Tarnovsky’s follow-up work characterizing the various photoactive species capable of LMCT for CuII chlorocomplexes in acetonitrile.55 Recent reports have utilized this mechanism towards the chlorination of unactivated olefins and oxidation of diarylalkynes with air as the terminal oxidant to enable catalytic loadings of Cu.56–57 (Scheme 1C)
Scheme 3. Representative examples of electrophiles.
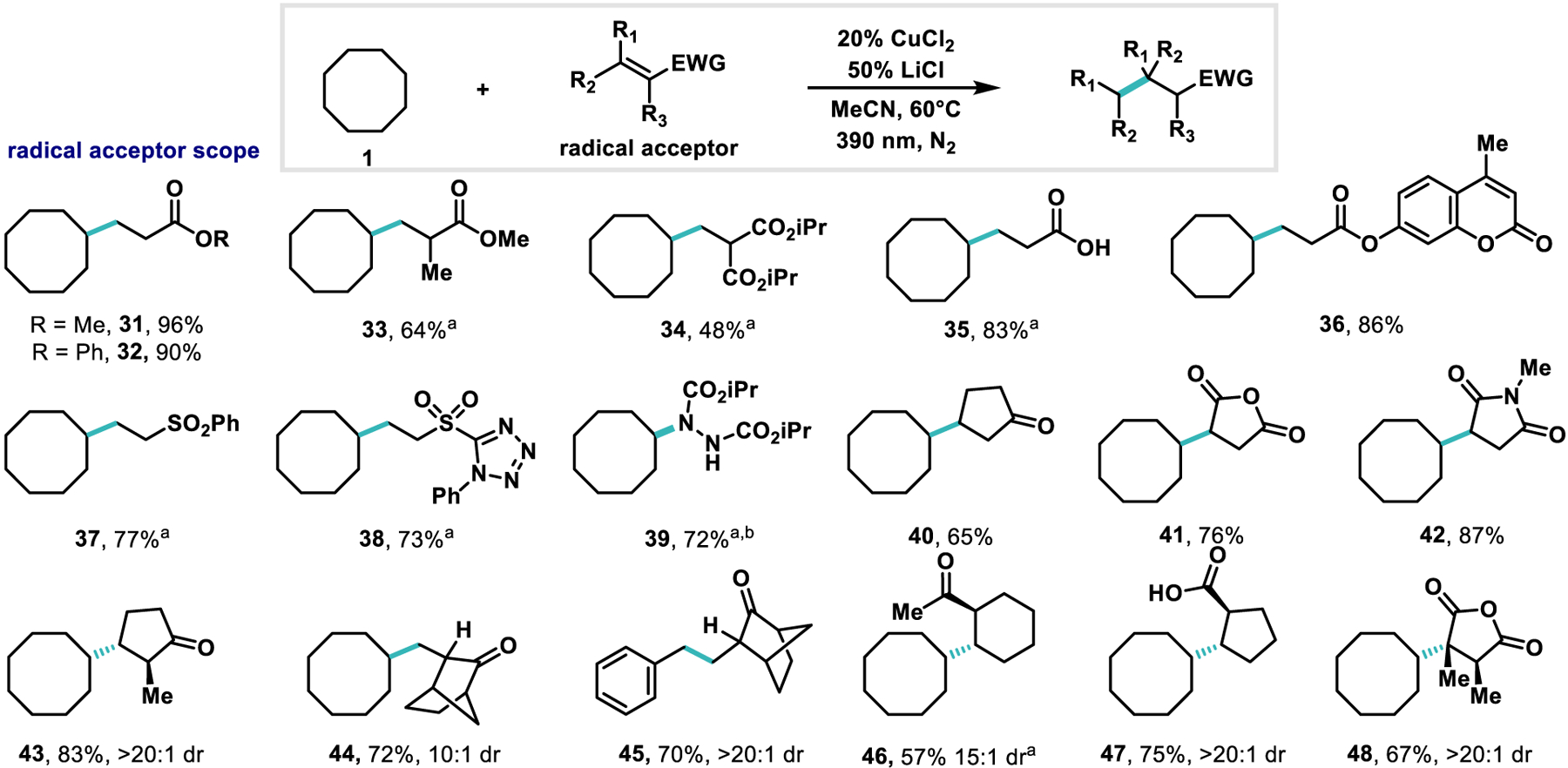
Reactions were run on 0.3 mmol scale with 5 equiv. of alkane nucleophile, 20 mol % CuCl2, 50 mol % LiCl, in 1.0 mL of MeCN for 36 h. a 50 mol % CuCl2, 1.25 equiv. LiCl.
Herein we report a catalytic method of alkane functionalization using a common base-metal salt –CuCl2– in which photoirradiation enables LMCT of a CuII species to generate a highly reactive chlorine radical. This permits the selective C(sp3)–H alkylation and amination of feedstock alkanes with abundant commodity chemicals such as acrylates and vinyl sulfones to synthesize value-added materials. (Scheme 1D) The protocol operates effectively on a number of alkane nucleophiles, including the remote functionalization of aliphatic carboxylic acids which has been enabled by the low oxidation potential of the Cu catalyst (+0.5 V vs. SCE).58–59 For endocyclic electron deficient alkenes we also observe high diastereoselectivity which we propose stems from facially selective protonation of an intermediate Cu-enolate.
Initial reaction optimization focused on the coupling of cyclooctane with ethyl acrylate, an abundant and inexpensive electrophile.60 Initial investigations revealed moderate yield with 20% loading of the CuCl2 catalyst and ethyl acrylate as the limiting component. The addition of exogenous chloride was found to increase turnover numbers. UV-Vis spectra of CuCl2 in MeCN with and without exogenous chloride reveal the active CuCl3− catalyst to be present as a greater mole fraction of photoactive species in solutions with added LiCl as well as demonstrating higher solubility (See Supplemental Figure S6). Due to the propensity of the product to undergo alkylation when in competitive concentration with the starting material, improved yields are observed when an excess of up to five equivalents of C–H nucleophile is used. Yields increase at elevated temperatures, with the conditions shown corresponding to the radiant heat generated by the 390 nm lamps. 440 nm irradiation also furnished product albeit in lower yield. Control studies revealed the necessity of light and CuCl2 for the reaction to occur. While anhydrous CuCl2 produces higher yields of 3, the air and moisture stable CuCl2 dihydrate also performs the coupling effectively, giving 3 in 83% yield.
With optimized conditions, we sought to explore the scope of nucleophiles capable of selective C–H functionalization. (Scheme 2) Unfunctionalized cyclic alkanes proved to be effective coupling partners (3–6). In the case of cyclooctane (3) the protocol scales well up to at least 3 mmol, delivering a yield of 86%. n-Pentane (7) shows a regioselectivity profile of 1:1.7:1 with preferential alkylation at the 2-C position and high overall reactivity. This selectivity matches previous reports of Giese alkylation via chlorine radical formation in MeCN.40 Ethers (8–11, 16) show selectivity for the α-position to oxygen which is expected due to the stabilization granted to the resulting radical by the proximal O atom. Benzylic positions (12–15) prove reactive towards alkylation selectively in the presence of other aliphatic positions, giving 5:1 rr in the case of 14. Ketones and esters drive selectivity towards more electron rich positions enabling the selective β-alkylation of cyclohexanone and 4-heptanone with good regioselectivity (17–18). Amides also proved to be effective nucleophiles with high selectivity for the hydrogen atoms at the α-position to the N atom (22–23). The product of the alkylation of dimethyladipate (19) demonstrates the unfavorability of functionalization proximal to carbonyls, showing reactivity both at the β-position of the carbonyl and at the α-position of oxygen of the ester (6:1 rr). Both triphenyl silane (25) and benzaldehyde (24) show high reactivity demonstrating the propensity of electron rich C–H and Si–H bonds to undergo the transformation effectively even at lower equivalents of alkane nucleophile (3 equiv.). Butanoic acid’s (26) ability to undergo alkylation with moderate regioselectivity motivated the employment of other alkyl acids (27–30) for which coupling proceeded at the β- and γ- positions with moderate to high regioselectivity. Alkyl carboxylic acids are commonly employed as alkyl radical precursors via oxidation-decarboxylation,61–64 and we observe no products corresponding to this mechanism. Finally, we have synthesized 5, 11, 13, and 15 utilizing the alkane as the limiting reagent with 1.1 equiv. ethyl acrylate to furnish products in synthetically useful isolated yields (44–60%).
We next sought to evaluate the scope of electrophiles capable of coupling with cyclooctane. A number of electron deficient unsaturated systems are competent for the reaction including various acrylates (3, 31–34, 36), vinyl sulfones (37–38), as well as diisopropyl azodicarboxylate (39). The ability to append the functional group 1-phenyl-5-(sulfonyl)-1H-tetrazole (PT-sulfone) found in 38 onto alkanes provides a useful functional handle for further derivatization.65–70 Vinyl ketones, carboxylic acids, and acid anhydrides (40–41, 43–48) all proved to be effective coupling partners. Maleic anhydride and its derivatives are prone to reduction (−0.98 V vs SCE59) illustrating the mild reduction potentials generated by the CuCl2− reductant. Substrates bearing endocyclic alkenes (43, 46–48) provide high diastereoselectivity which we propose is enabled by the inter-mediacy of a CuII-enolate which undergoes stereoselective protonation to regenerate the oxidized copper catalyst. This compares favorably to decatungstate based systems in which 45 is reported to be formed in 29% yield with 2:1 dr.61,71
Giese reactions of alkanes have been conducted previously, but typically require multiple electron withdrawing substituents on the olefin acceptor to promote electrophilicity, stabilize the resulting radical, and facilitate reduction in the case of a SET mechanism.41,47 A notable exception lies in decatungstate anion catalysis, which permits the alkylation of cyclohexane with methyl acrylate to proceed in moderate yield (65%) under 310 nm irradiation.72 Previous work in radical Giese reactions display low diastereoselectivity except for in the case of highly strained enolate intermediates (see Scheme 3, 44, 45).9,47,73–75 In the presented system, all endocyclic alkene coupling partners demonstrate high (>10:1) dr, augmenting previously reported radical Giese reactions.
We next sought to interrogate the mechanism of this reaction through a series of probes. Based on previous reports,55 the concentration of chloride in cupric-acetonitrile solutions produces various CuClx (x−2)− complexes, of which only conditions known to produce CuCl3− proved effective for this reaction. This was further confirmed via UV-Vis spectroscopy of the catalyst mixture. (See Supplemental Figure S1) A competition experiment between cyclohexane and cyclohexane-d12 at low conversion (6 hours) revealed a secondary kinetic isotope effect, alluding that the (H/D)AT event is not a turnover limiting step of the catalytic cycle (Scheme 4A). The addition of catalytic base, sufficient to neutralize the acid produced via HAT, results in depressed yield with complete consumption of acrylate through polymerization (Table 1, entry 9). Adding exogenous D2SO4 to the normal reaction conditions, as shown in Scheme 4B, permits the incorporation of deuterium into the product. The limited incorporation (17%), despite much higher initial concentrations of deutero acid as compared to proteo acid reveals that protonation must occur faster than deuteration, which is in accordance with an expected kinetic isotope effect in a protodemetallation mechanism. In the isotopically inverted experiment in which cyclohexane-d12 is used as the nucleophile with added H2SO4, deuterium incorporation is even lower (7%) in agreement with the proposed mechanism (see Supporting Information). To ensure that D incorporation does not occur after product formation, 3 was subjected to alkylation conditions and reisolated to show that D incorporation must occur in an intermediate en route to product (Scheme 4C). Previous investigations into the Cu catalyzed amidation of alkyl halides through a photoexcited intermediate presented the possibility that an in-situ generation of an alkyl chloride could precede and enable alkylation through Cu(I) halide abstraction.76–77 To ensure that the reaction did not proceed via an alkyl chloride intermediate, chlorocyclohexane was subjected to the reaction conditions to reveal no production of the alkylated cyclohexane. Instead various regio- and stereo-isomers of alkylated chlorocyclohexane were detected by GC-MS (Scheme 4D).
Scheme 4.
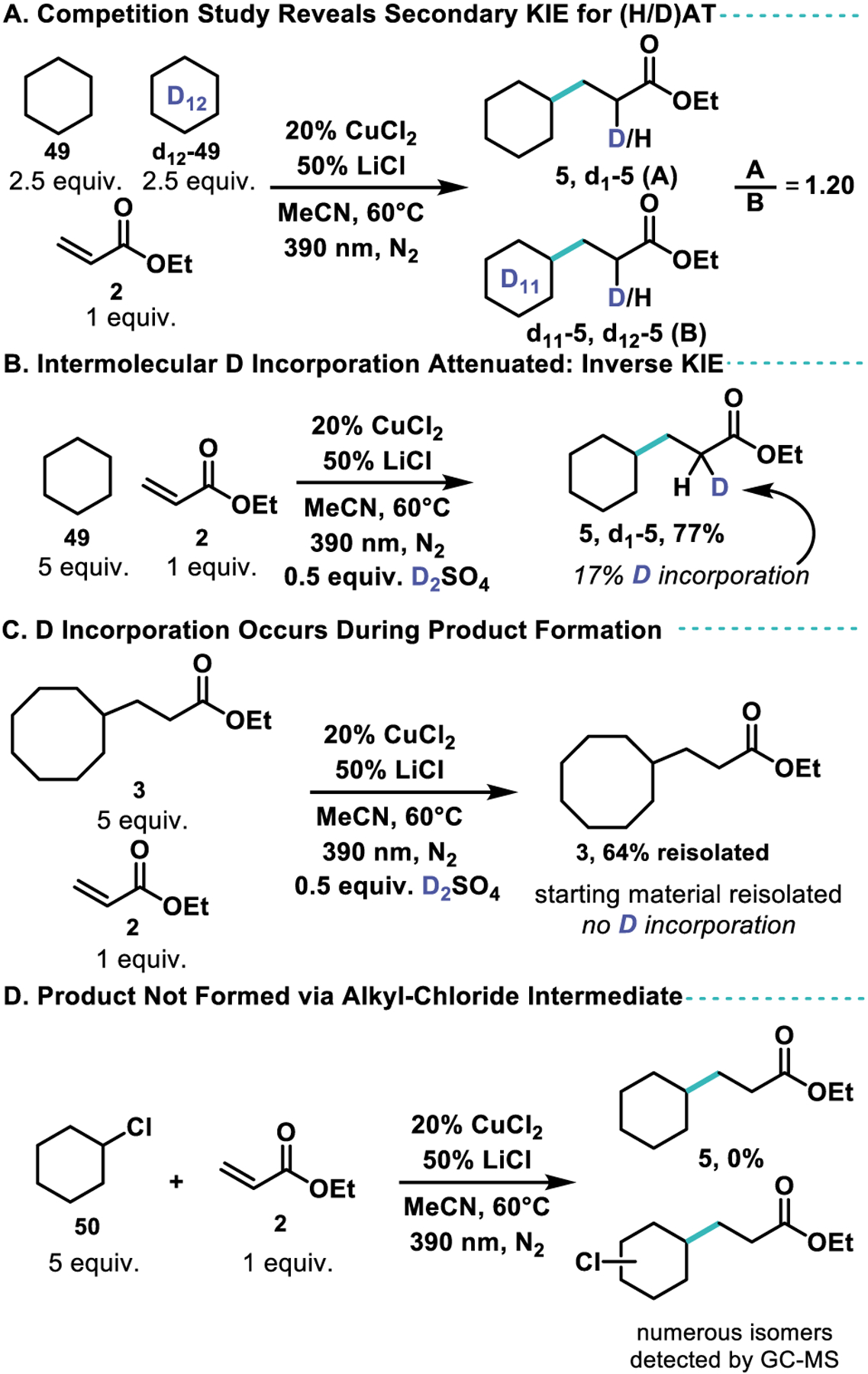
Mechanistic Investigations.
Table 1:
Optimization and Control Studies
| ||
|---|---|---|
| Entry | Deviation from Standard Conditions | Yield 3 (%) |
| 1 | none | 93 |
| 2 | 3 equiv. cyclooctane | 73 |
| 3 | 0% CuCl2 | 0 |
| 4 | 0% LiCl | 79 |
| 5 | in the dark | 0 |
| 6 | 440 nm LED | 66 |
| 7 | 30°C | 45 |
| 8 | under air | 55 |
| 9 | with 10% Cs2CO3 | 22 |
| 10 | Irradiated 1 hour, then in the dark | 14 |
Optimizations were performed on a 0.3 mmol scale using 5 equiv. of 1 and 1 equiv. of 2.
Based on our mechanistic experiments and previous photochemical studies of copper-chloro complexes in acetonitrile,55 we propose the mechanism shown in Scheme 5. First, coordination of chloride to dissolved CuCl2 produces the photo-active CuCl3−. Then, irradiation enables LMCT to generate a reactive chlorine radical. This radical undergoes HAT with the alkane nucleophile to form HCl and a reactive alkyl radical. The alkyl radical attacks ethyl acrylate to form a more stable radical α to an ester. This electron deficient radical recombines with CuCl2− to form a stable CuII-enolate. Protodemetallation with HCl forms the product and regenerates the photo-active CuCl3−.
Scheme 5.
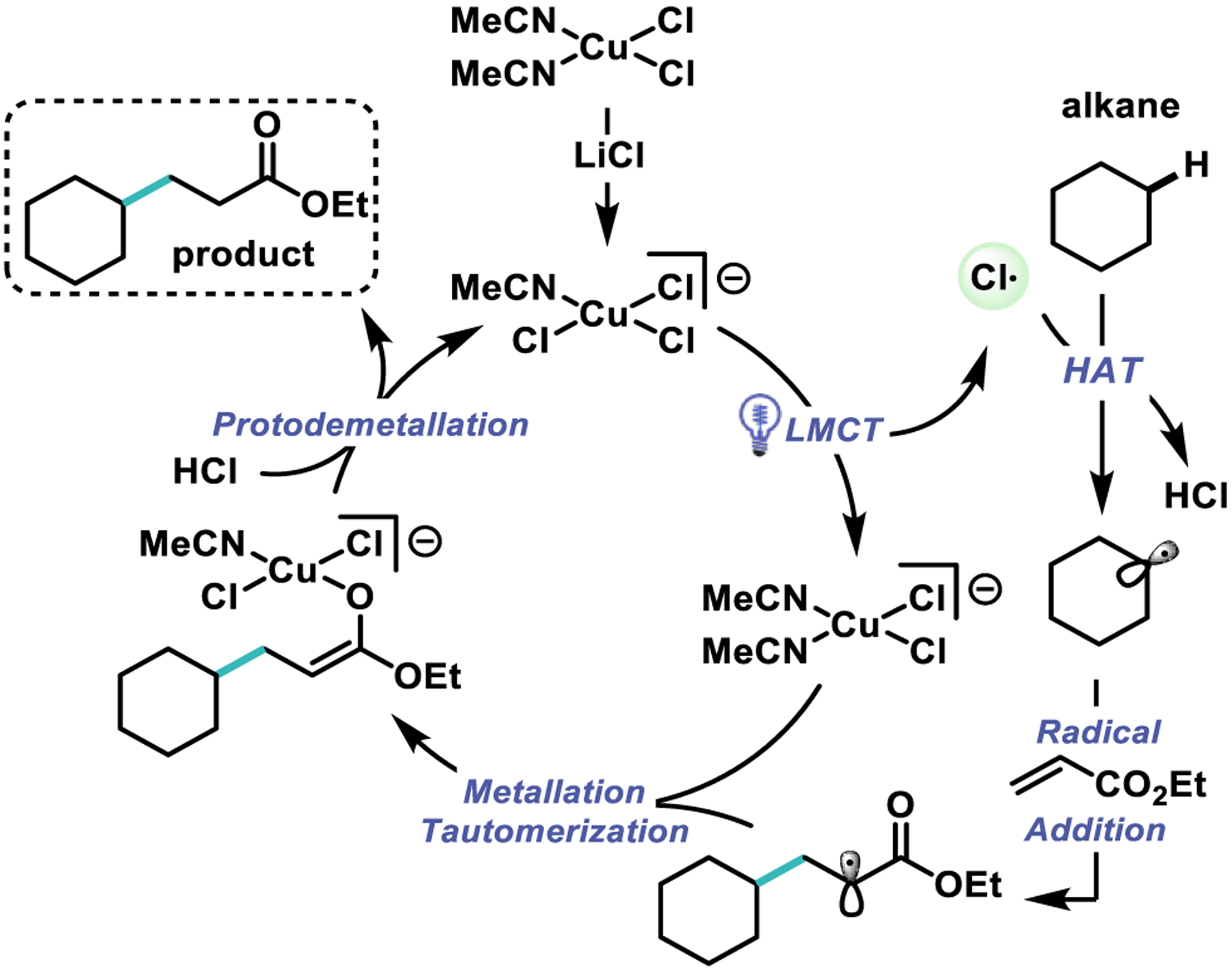
Proposed Mechanism.
In conclusion, we report an effective protocol for the alkylation of alkanes with electron deficient-olefins via the photosensitization of CuII salts. This work offers a synthetically useful and operationally simple method to couple feedstock chemicals to create value-added products. Further efforts are underway to apply LMCT for the functionalization of more selective and powerful HAT reagents under mild conditions.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS (GM125206) for support.
Footnotes
Experimental procedures and characterization data, including supplemental figures S1–S7. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Hartwig JF; Larsen MA Undirected, Homogeneous C-H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci 2016, 2 (5), 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bergman RG Organometallic Chemistry: C-H Activation. Nature 2007, 446, 391. [DOI] [PubMed] [Google Scholar]
- (3).Newhouse T; Baran PS If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem. Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chu JCK; Rovis T Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed 2018, 57, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chirik P; Morris R Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res 2015, 48, 2495–2495. [DOI] [PubMed] [Google Scholar]
- (6).Ludwig R Hydrogen-Transfer Reactions.Edited by Hynes JT, Klinman JP, Limbach H-H and Schowen RL. Chem-PhysChem 2007, 8, 2539–2539. [Google Scholar]
- (7).Hu A; Guo JJ; Pan H; Zuo Z Selective Functionalization of Methane, Ethane, and Higher Alkanes by Cerium Photocatalysis. Science 2018, 361, 668–672. [DOI] [PubMed] [Google Scholar]
- (8).Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Laudadio G; Deng Y; van der Wal K; Ravelli D; Nuño M; Fagnoni M; Guthrie D; Sun Y; Noël T C(Sp3)-H Functionalizations of Light Hydrocarbons Using Decatungstate Photocatalysis in Flow. Science 2020, 369, 92–96. [DOI] [PubMed] [Google Scholar]
- (10).Yi H; Zhang G; Wang H; Huang Z; Wang J; Singh AK; Lei A, Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chemical Reviews 2017, 117 (13), 9016–9085. [DOI] [PubMed] [Google Scholar]
- (11).Twilton J; Le C. (Chip); Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nature Reviews Chemistry 2017, 1, 52. [Google Scholar]
- (12).De Abreu M; Belmont P; Brachet E Synergistic Photoredox/Transition-Metal Catalysis for Carbon-Carbon Bond Formation Reactions. Eur. J. Org. Chem 2020, 10, 1327–1378. [Google Scholar]
- (13).Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl Sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jeffrey JL; Terrett JA; MacMillan DWC O-H Hydrogen Bonding Promotes H-Atom Transfer from α C-H Bonds for C-Alkylation of Alcohols. Science 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective Sp 3 C-H Alkylation via Polarity-Match-Based Cross-Coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Roberts BP Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev 1999, 28, 25–35. [Google Scholar]
- (20).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bietti M Activation and Deactivation Strategies Promoted by Medium Effects for Selective Aliphatic C–H Bond Functionalization. Angew. Chemie Int. Ed 2018, 57, 16618–16637. [DOI] [PubMed] [Google Scholar]
- (22).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166 [DOI] [PubMed] [Google Scholar]
- (24).Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev, 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- (25).Hu XQ; Chen JR; Xiao WJ Controllable Remote C–H Bond Functionalization by Visible-Light Photocatalysis. Angew. Chemie - Int. Ed 2017, 56, 1960–1962. [DOI] [PubMed] [Google Scholar]
- (26).Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C-H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chu JCK; Rovis T Amide-Directed Photoredox-Catalysed C-C Bond Formation at Unactivated Sp 3 C-H Bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen DF; Chu JCK; Rovis T Directed γ-C(sp3)-H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(sp3)-H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200–16203. [DOI] [PubMed] [Google Scholar]
- (30).Cuthbertson JD; MacMillan DWC The Direct Arylation of Allylic sp3 C-H Bonds via Organic and Photoredox Catalysis. Nature 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tanaka H; Sakai K; Kawamura A; Oisaki K; Kanai M Sulfonamides as New Hydrogen Atom Transfer (HAT) Catalysts for Photoredox Allylic and Benzylic C-H Arylations. Chem. Commun 2018, 54, 3215–3218. [DOI] [PubMed] [Google Scholar]
- (32).Shen Y; Gu Y; Martin R sp3 C-H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc 2018, 140, 12200–12209. [DOI] [PubMed] [Google Scholar]
- (33).Protti S; Fagnoni M; Ravelli D Photocatalytic C-H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. [Google Scholar]
- (34).Okada M; Fukuyama T; Yamada K; Ryu I; Ravelli D; Fagnoni M Sunlight Photocatalyzed Regioselective β-Alkylation and Acylation of Cyclopentanones. Chem. Sci 2014, 5, 2893–2898. [Google Scholar]
- (35).Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ Site-Selective Aliphatic C-H Bromination Using N - Bromoamides and Visible Light. J. Am. Chem. Soc 2014, 136, 14389–14392. [DOI] [PubMed] [Google Scholar]
- (36).Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C-H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fokin AA; Schreiner PR Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev 2002, 102, 1551–1594. [DOI] [PubMed] [Google Scholar]
- (38).Carey FA; Sundberg RJ Advanced Organic Chemistry Part A: Structure and Mechanisms; Springer: New York, 2007; 965–1063 [Google Scholar]
- (39).Ohkubo K; Fujimoto A; Fukuzumi S Metal-Free Oxygenation of Cyclohexane with Oxygen Catalyzed by 9-Mesityl-10-Methylacridinium and Hydrogen Chloride under Visible Light Irradiation. Chem. Commun 2011, 47, 8515–8517. [DOI] [PubMed] [Google Scholar]
- (40).Ohkubo K; Mizushima K; Fukuzumi S Oxygenation and Chlorination of Aromatic Hydrocarbons with Hydrochloric Acid Photosensitized by 9-Mesityl-10-Methylacridinium under Visible Light Irradiation. Res. Chem. Intermed 2013, 39, 205–220. [Google Scholar]
- (41).Deng H-P; Zhou Q; Wu J Microtubing-Reactor-Assisted Aliphatic C–H Functionalization with HCl as a Hydrogen-Atom-Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chemie Int. Ed 2018, 57, 12661–12665. [DOI] [PubMed] [Google Scholar]
- (42).Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chemie Int. Ed 2018, 57, 15664–15669. [DOI] [PubMed] [Google Scholar]
- (43).Hu A; Guo JJ; Pan H; Tang H; Gao Z; Zuo Z δ-Selective Functionalization of Alkanols Enabled by Visible-Light-Induced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2018, 140, 1612–1616. [DOI] [PubMed] [Google Scholar]
- (44).Shields BJ; Doyle AG Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Deng HP; Fan XZ; Chen ZH; Xu QH; Wu J Photoinduced Nickel-Catalyzed Chemo- and Regioselective Hydroalkylation of Internal Alkynes with Ether and Amide α-Hetero C(sp3)-H Bonds. J. Am. Chem. Soc 2017, 139, 13579–13584. [DOI] [PubMed] [Google Scholar]
- (46).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C-C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium-Catalyzed C-H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]
- (48).Ravetz BD; Wang JY; Ruhl KE; Rovis T Photoinduced Ligand-to-Metal Charge Transfer Enables Photocatalyst-Independent Light-Gated Activation of Co(II). ACS Catal. 2019, 9, 200–204. [Google Scholar]
- (49).Kochi JK, Photolyses of Metal Compounds: Cupric Chloride in Organic Media. J. Am. Chem. Soc 1962, 84, 2121–2127. [Google Scholar]
- (50).Shul’pin GB; Kats MM Ferric Chloride Catalyzed Photooxidation of Alkanes by Air in Organic Solvents. React. Kinet. Catal. Lett 1990, 41, 239. [Google Scholar]
- (51).Shul’pin GB; Nizova GV Photo-oxidation of Cyclohexane by Atmospheric Oxygen in Acetonitrile Catalysed by Chloride Complexes of Iron, Copper, and Gold. Petrol. Chem 1993, 33, 107. [Google Scholar]
- (52).Shul’pin GB; Nizova GV Kozlov YN Photochemical Aerobic Oxidation of Alkanes Promoted by Iron Complexes. New J. Chem 1996, 20, 1243. [Google Scholar]
- (53).Takaki K; Yamamoto J; Matsushita Y; Morii H; Shishido T; Takehira K Oxidation of Alkanes with Dioxygen Induced by Visible Light and Cu(II) and Fe(III) Chlorides Oxidation of Alkanes by Visible Light. Bull. Chem. Soc. Jpn 2003, 76, 393–398. [Google Scholar]
- (54).Takaki K; Yamamoto J; Komeyama K; Kawabata T; Takehira K Photocatalytic Oxidation of Alkanes with Dioxygen by Visible Light and Copper(II) and Iron(III) Chlorides: Preference Oxidation of Alkanes over Alcohols and Ketones. Bull. Chem. Soc. Jpn 2004, 77, 2251–2255. [Google Scholar]
- (55).Mereshchenko AS; Olshin PK; Karimov AM; Skripkin MY; Burkov KA; Tveryanovich YS; Tarnovsky AN Photochemistry of Copper(II) Chlorocomplexes in Acetonitrile: Trapping the Ligand-to-Metal Charge Transfer Excited State Relaxations Pathways. Chem. Phys. Lett 2014, 615, 105–110. [Google Scholar]
- (56).Charpe VP; Sagadevan A; Hwang KC, Visible light-induced aerobic oxidation of diarylalkynes to α-diketones catalyzed by copper-superoxo at room temperature. Green Chemistry 2020, 22, 4426–4432. [Google Scholar]
- (57).Lian P; Long W; Li J; Zheng Y; Wan X, Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2. Angewandte Chemie International Edition 2020, 59, 23603–23608. [DOI] [PubMed] [Google Scholar]
- (58).Manahan SE; Iwamoto RT Chloro Complexes of Copper(II) and Copper(I) in Acetonitrile. Inorg. Chem 1965, 4, 1409–1413. [Google Scholar]
- (59).Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (60).As of the date of this submission (08/2020), the price per gram of ethyl acrylate from Fisher Scientific is $0.02–0.03 / gram.
- (61).Chu L; Ohta C; Zuo Z; MacMillan DWC, Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (±)-Pregabalin. J. Am. Chem. Soc 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Griffin JD; Zeller MA; Nicewicz DA, Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc 2015, 137, 11340–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Johnston CP; Smith RT; Allmendinger S; MacMillan DWC, Metallaphotoredox-catalysed sp3–sp3 cross-coupling of carboxylic acids with alkyl halides. Nature 2016, 536, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Sun X; Chen J; Ritter T, Catalytic dehydrogenative decarboxyolefination of carboxylic acids. Nature Chemistry 2018, 10, 1229–1233. [DOI] [PubMed] [Google Scholar]
- (65).Blakemore PR; Cole WJ; Kocieński PJ; Morley A A Stereoselective Synthesis of Trans -1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1 H -Tetrazol-5-Yl Sulfones. Synlett 1998, 1, 26–28. [Google Scholar]
- (66).Blakemore PR The Modified Julia Olefination: Alkene Synthesis via the Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. J. Chem. Soc. Perkin 1 2002, 2, 2563–2585. [Google Scholar]
- (67).Aïssa C Mechanistic Manifold and New Developments of the Julia-Kocienski Reaction. European J. Org. Chem 2009, 12, 1831–1844. [Google Scholar]
- (68).Rodrigo E; Morales S; Duce S; Ruano JLG; Cid MB Enantioselective Organocatalytic Formal Allylation of α-Branched Aldehydes. Chem. Commun 2011, 47, 11267–11269. [DOI] [PubMed] [Google Scholar]
- (69).Rodrigo E; Alonso I; García Ruano JL; Cid MB Expanding the Potential of Heteroaryl Vinyl Sulfones. J. Org. Chem 2016, 81, 10887–10899. [DOI] [PubMed] [Google Scholar]
- (70).For a recent innovative development involving the use of PT-sulfones as effective substrates for Ni catalyzed cross coupling see:; Merchant RR; Edwards JT; Qin T; Kruszyk MM; Bi C; Che G; Bao DH; Qiao W; Sun L; Collins MR; Fadeyi OO; Gallego GM; Mousseau JJ; Nuhant P; Baran PS Modular Radical Cross-Coupling with Sulfones Enables Access to sp3-Rich (Fluoro)Alkylated Scaffolds. Science 2018, 360, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).We also synthesized 43 using 2% tetrabutylammonium decatungstate in 78% yield and 1:1 dr under otherwise identical reaction conditions to our copper catalyzed method.
- (72).Dondi D; Fagnoni M; Albini A Tetrabutylammonium Decatungstate-Photosensitized Alkylation of Electrophilic Alkenes: Convenient Functionalization of Aliphatic C-H Bonds. Chem. - A Eur. J 2006, 12, 4153–4163. [DOI] [PubMed] [Google Scholar]
- (73).Qrareya H; Ravelli D; Fagnoni M; Albini A, Decatungstate Photocatalyzed Benzylation of Alkenes with Alkylaromatics. Advanced Synthesis & Catalysis 2013, 355, 2891–2899. [Google Scholar]
- (74).Dondi D; Cardarelli AM; Fagnoni M; Albini A, Photomediated synthesis of β-alkylketones from cycloalkanes. Tetrahedron 2006, 62 (23), 5527–5535. [Google Scholar]
- (75).Capaldo L; Merli D; Fagnoni M; Ravelli D, Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion. ACS Catalysis 2019, 9 (4), 3054–3058. [Google Scholar]
- (76).Do HQ; Bachman S; Bissember AC; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Alkylation of Amides with Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc 2014, 136, 2162–2167. [DOI] [PubMed] [Google Scholar]
- (77).Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C-N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.