

PURPOSE
Somatic KRAS mutations occur in approximately half of the patients with metastatic colorectal cancer (mCRC). Biologic tumor characteristics differ on the basis of the KRAS mutation variant. KRAS mutations are known to influence patient prognosis and are used as predictive biomarker for treatment decisions. This study examined clinical features of patients with mCRC with a somatic mutation in KRAS G12, G13, Q61, K117, or A146.
METHODS
A total of 419 patients with colorectal cancer with initially unresectable liver-limited metastases, who participated in a multicenter prospective trial, were evaluated for tumor tissue KRAS mutation status. For the subgroup of patients who carried a KRAS mutation and were treated with bevacizumab and doublet or triplet chemotherapy (N = 156), pretreatment circulating tumor DNA levels were analyzed, and total tumor volume (TTV) was quantified on the pretreatment computed tomography images.
RESULTS
Most patients carried a KRAS G12 mutation (N = 112), followed by mutations in G13 (N = 15), A146 (N = 12), Q61 (N = 9), and K117 (N = 5). High plasma circulating tumor DNA levels were observed for patients carrying a KRAS A146 mutation versus those with a KRAS G12 mutation, with median mutant allele frequencies of 48% versus 19%, respectively. Radiologic TTV revealed this difference to be associated with a higher tumor load in patients harboring a KRAS A146 mutation (median TTV 672 cm3 [A146] v 74 cm3 [G12], P = .036). Moreover, KRAS A146 mutation carriers showed inferior overall survival compared with patients with mutations in KRAS G12 (median 10.7 v 26.4 months; hazard ratio = 2.5; P = .003).
CONCLUSION
Patients with mCRC with a KRAS A146 mutation represent a distinct molecular subgroup of patients with higher tumor burden and worse clinical outcomes, who might benefit from more intensive treatments. These results highlight the importance of testing colorectal cancer for all KRAS mutations in routine clinical care.
INTRODUCTION
Oncogenic KRAS mutations are highly prevalent in multiple cancers and drive cell differentiation and proliferation.1 KRAS mutations stimulate KRAS to stay in its active state, thereby triggering the oncogenic signaling pathway.2 Around 40%-50% of the patients with metastatic colorectal cancer (mCRC) harbor a somatic KRAS mutation.3-5 In general, patients with a KRAS wild-type tumor have a better prognosis than patients carrying a KRAS-mutated tumor.6,7 Moreover, KRAS mutation status is a predictive marker for poor response to anti–epidermal growth factor receptor (EGFR) monoclonal antibody therapy,8 one of the options for systemic treatment for patients with mCRC.9 Therefore, analysis of KRAS mutation status has been widely adopted in routine clinical practice.10
CONTEXT
Key Objective
The distribution of KRAS mutation variants across tumor types is not uniform. The KRAS A146 mutation is predominantly seen in patients with colorectal cancer. Here, we evaluated how clinical features like tumor load and overall survival differ between patients with metastatic colorectal cancer (mCRC) carrying distinct somatic KRAS G12, G13, Q61, K117, or A146 mutations.
Knowledge Generated
This study revealed that within patients with mCRC, A146 is the third most common KRAS mutation variant. Patients with mCRC with a KRAS A146–mutated tumor represent a distinct molecular subgroup of patients with higher tumor burden that is associated with worse clinical outcomes.
Relevance
These results highlight the clinical importance of testing colorectal cancer for all KRAS mutations in routine diagnostics. The distinct clinical implications of KRAS A146 mutations in patients with mCRC warrant further investigation regarding therapeutic strategies to target and treat KRAS A146 mutant tumors.
It is known that the biologic characteristics of tumors, like cellular phenotypes and metabolomic characteristics, differ on the basis of the KRAS mutation variant and amino acid substitution.11-13 In a substantial part of routine diagnostic KRAS tissue panels, only the most common driver mutations in KRAS codons G12 and G13 are tested, which are affected in 28% and 8% of all patients with mCRC, respectively. However, mutations are also commonly present in KRAS Q61 (2%), K117 (1%), and A146 (4%).4,5,14 Here, we investigated clinical features like tumor load and overall survival of patients with mCRC with a somatic mutation in KRAS G12, G13, Q61, K117, or A146.
METHODS
Patient Characteristics
Liquid biopsies of patients with histologically proven colorectal cancer (CRC) with isolated, previously untreated, initially unresectable colorectal liver metastases (CRLM) were collected in the ongoing multicenter phase III CAIRO5 trial (NCT02162563).15 A total of 419 patients with CRLM, enrolled between November 2014 and July 2019, were evaluated in this study. For all patients, tissue KRAS mutation analyses were performed in the participating hospitals before randomization following routine clinical practice. Only those patients who were randomized for treatment with bevacizumab and chemotherapy consisting of 5-fluorouracil, leucovorin, oxaliplatin, and/or irinotecan were selected for the current study. Clinical follow-up was performed according to standard of care, including clinical review every three months as well as computed tomography (CT) imaging every six months, for patients with resectable disease and every two months for patients with unresectable disease, using the RECIST 1.1 for reporting. Follow-up was recorded until March 23, 2020. The study was performed in accordance with the Declaration of Helsinki and a medical ethical committee approved the trial, and all patients signed written informed consent for study participation and liquid biopsy collection.
Liquid Biopsy Collection
Liquid biopsies were collected before study treatment using a cell-stabilizing BCT tube (Streck, La Vista, NE) in the participating hospitals and shipped to the Netherlands Cancer Institute. Here, cell-free plasma was collected in a two-step centrifugation process: 10 minutes at 1.700 g followed by 10 minutes at 20.000 g, and stored at –80°C until further processing. Cell-free DNA (cfDNA) was isolated using the QIAsymphony (Qiagen, Hilden, Germany) with an elution volume set to 60 µL. The concentration of cfDNA was measured using the Qubit dsDNA High-Sensitivity Assay (TFS, Waltham, MA).
Liquid Biopsy Mutation Analyses
For patients with an established KRAS mutation on the basis of tumor analysis, liquid biopsy mutation analyses were performed using four droplet digital polymerase chain reaction (ddPCR; Bio-Rad, Hercules, CA) screening kits, namely ddPCR KRAS G12/G13 (#1863506), ddPCR KRAS Q61 (#12001626), ddPCR KRAS K117N (#10049047), and ddPCR KRAS A146T (#10049550). Table 1 in the Data Supplement shows the different amino acid variants detected by these assays. The ddPCR assays were performed according to the manufacturer's instruction, making use of 1 µL of the multiplex assay, 11 µL of the ddPCR supermix for probes (no dUTP), 9 µL of sample, and 1 µL H2O. All measurements were performed in duplicate and included a blank (nuclease-free water) and a positive control. Data were analyzed using the QuantaSoft software version 1.6.6 (Bio-Rad, Hercules, CA). The number of mutant copies per mL plasma (MTc/mL) and mutant allele frequency (MAF) were used as outcome measures. For the cfDNA samples with a KRAS A146 mutation, orthogonal validation was performed using targeted deep sequencing, as described previously.16 In brief, genomic libraries were prepared from 125 ng of cfDNA, following normalization, end-repair, A-tailing, adapter ligation, and PCR amplification. Target capture was performed using a panel consisting of 58 genes, covering 81 kb. Candidate somatic alterations across the region of interest were identified using VariantDx (Personal Genome Diagnostics, Baltimore, MD).
Radiologic Total Tumor Volume Quantification
For patients with an identified KRAS mutation on tumor tissue, pretreatment contrast-enhanced abdominal CT images were used for semiautomatic segmentation in the Tumor Tracking Modality of IntelliSpace Portal 9.0 (Philips, Eindhoven, the Netherlands). The liver itself and all metastases were segmented by two trained members of the research team and subsequently adjusted and verified by a radiologist specialized in abdominal pathology. All segmentations and related CT images were processed and analyzed with the SAS Viya analytical platform (SAS Institute Inc, Cary, NC) for volume quantification using the quantifyBioMed Images action.17 This action calculates the total tumor volume (TTV) directly out of the segmentation from all tumors presented in the liver by determining the volume of one voxel and multiplying this volume with the number of voxels included in the tumor segmentation. A CT scan is built up by voxels, the three-dimensional equivalent of a pixel, and a voxel's volume depends on the pixel spacing and slice thickness attributes of the CT scan. The volume of the liver was calculated similarly, on the basis of the three-dimensional liver segmentation. In addition, the percentage TTV of the total liver volume, including TTV, was calculated. Furthermore, the radiologist registered the number of liver lesions.
Statistical Analyses
A Brown–Forsythe analysis of variance test using Dunnett's multiple comparisons was used for the liquid biopsy analyses. A one-way analysis of variance corrected for multiple comparisons using Tukey's multiple comparisons test was used for the volumetric analyses. A two-sided P-value of .05 was used as a cutoff for significance. A Mantel–Cox log-rank test using a Bonferroni-corrected threshold of P < .005 for significance was performed for the survival analyses. To determine the equivalence between ddPCR and sequencing circulating tumor DNA (ctDNA) levels, a Pearson correlation was used. Clinical patient characteristics were compared between carriers of different KRAS mutant variants using Fisher's exact tests. Univariate and multivariate Cox proportional hazards regression analyses were performed to analyze prognostic factors for overall survival, adjusted for potential confounders. Analyses were performed with Prism version 8 (GraphPad Software, Inc, San Diego, CA) and SPSS software version 27 (IBM, New York, NY).
RESULTS
Patient Characteristics
Of the 419 patients evaluated, 178 patients (42%) met the selection criteria and carried a tumor tissue KRAS mutation. Three patients who did not receive bevacizumab and 19 patients unavailable for follow-up were excluded, leaving 156 patients for analyses (Fig 1). The majority of these patients carried a KRAS G12 mutation (N = 112, 71.8%), followed by mutations in G13 (N = 15, 9.6%), A146 (N = 12, 7.7%), Q61 (N = 9, 5.8%), K117 (N = 5, 3.2%), and A59 (N = 1, 0.6%). The codon affected was unknown for two patients (1.3%; Fig 2). Clinical patient characteristics per KRAS mutation and per KRAS most frequent G12 residues (G12A, G12C, G12D, and G12V) are shown in Table 1 and Table 2 in the Data Supplement, respectively.
FIG 1.

Flowchart of patient selection. KRAS, NRAS, and BRAF mutation status was determined on tumor tissue for a total of 419 patients with CRC with isolated and initially unresectable liver metastases enrolled in the CAIRO5 clinical trial. Patients without a KRAS-mutated tumor were excluded from the current study. Next, patients not treated with bevacizumab and chemotherapy were excluded to ensure a homogenous patient group. Last, patients without clinical follow-up were excluded, resulting in 156 patients with mCRC for analyses. CRC, colorectal cancer; ctDNA, circulating tumor DNA; CT, computed tomography; mCRC; metastatic colorectal cancer.
FIG 2.
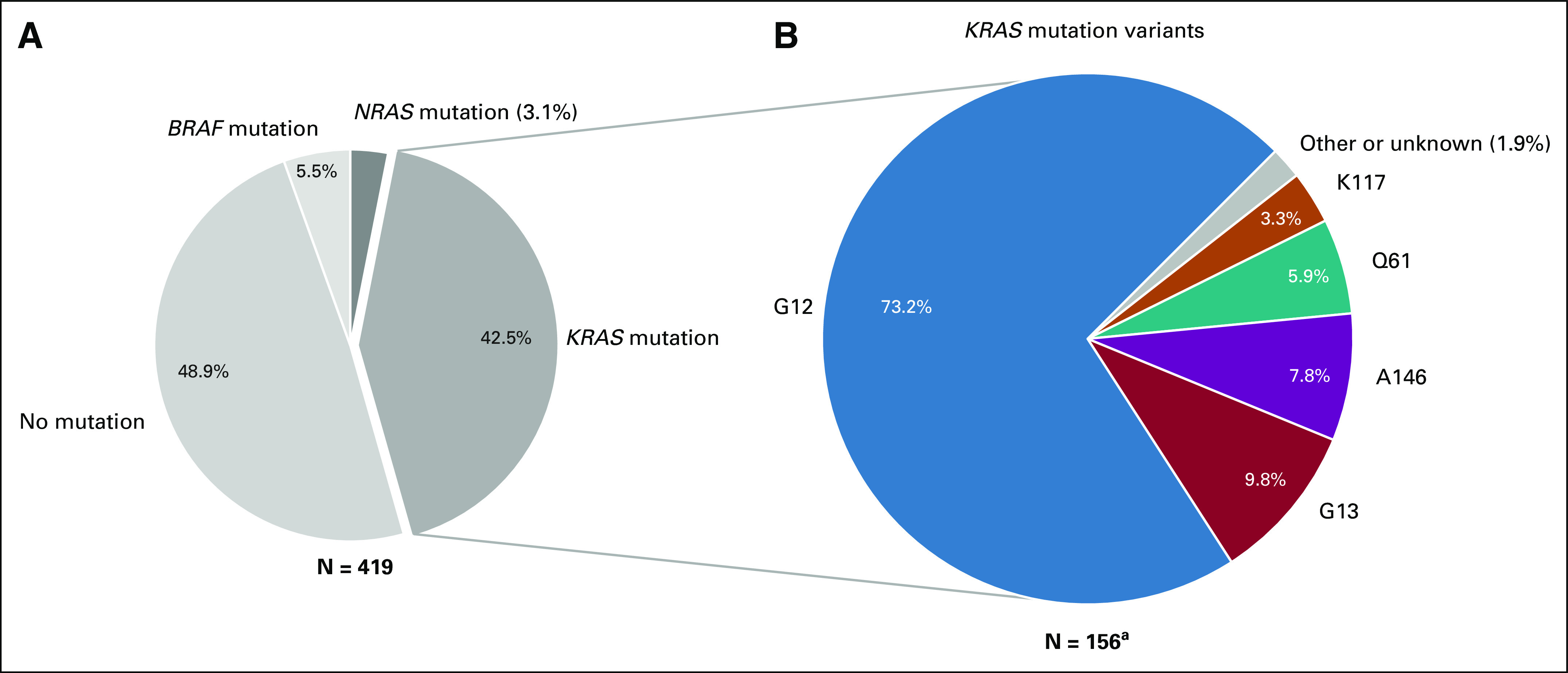
(A) The distribution of KRAS/NRAS/BRAF mutations among the 419 patients with CRLM evaluated in this study and (B) the relative distribution of KRAS codon variants among 156 KRAS mutation carriers. aOf note, 42.5% of the 419 patients (N = 178) carried a tumor tissue KRAS mutation, of whom 156 patients were used for further analyses (see Fig 1). CRLM, colorectal liver metastases.
TABLE 1.
Clinical Patient Characteristics per KRAS Mutation Variant

High Plasma ctDNA Levels in Patients With KRAS A146 Mutant Tumors
We previously measured plasma ctDNA levels in 100 patients with CRLM and noticed remarkably high plasma ctDNA levels in patients harboring a KRAS A146–mutated tumor, an observation that warranted further investigation.18 The current study investigated the liquid biopsy ctDNA levels for all 156 patients included. Patients without a pretreatment liquid biopsy (N = 32) and patients carrying a tumor with a KRAS mutation that could not be detected by the ddPCR kits (N = 2) were excluded, leaving 122 ctDNA samples for liquid biopsy analyses (Data Supplement Figure 1). Liquid biopsy ddPCR analyses showed more MTc/mL plasma and a higher MAF for patients with KRAS A146–mutated tumors (N = 10, median MTc/mL = 35,338, median MAF = 48%) compared with patients carrying a different KRAS variant, for example, a KRAS G12 mutation (N = 92, median MTc/mL = 700, median MAF = 19%), see Figure 3A (MTc/mL) and Figure 3B (MAF). To ensure that these high plasma ctDNA levels were not because of the KRAS codon 146 ddPCR assay's test characteristics, we performed orthogonal testing using a targeted deep-sequencing approach. A strong confirmation of the high KRAS A146 ctDNA levels was observed, with a Pearson correlation (R2) of 0.98 (95% CI, 0.96 to 1.00; P < .0001) between the ddPCR and sequencing MAF results (Figure 2 in Data Supplement). The high plasma ctDNA levels in KRAS A146 mutation carriers were not caused by DNA copy-number gains or focal amplification of the KRAS locus (see methods in Data Supplement). Moreover, the high plasma ctDNA levels in patients harboring a KRAS A146–mutated tumor were accompanied by high plasma ctDNA levels for other genes like TP53, TERT, and PIK3CA (Figure 3 in Data Supplement), implying that high plasma ctDNA levels for KRAS A146–mutated tumors are associated with tumor burden.
FIG 3.
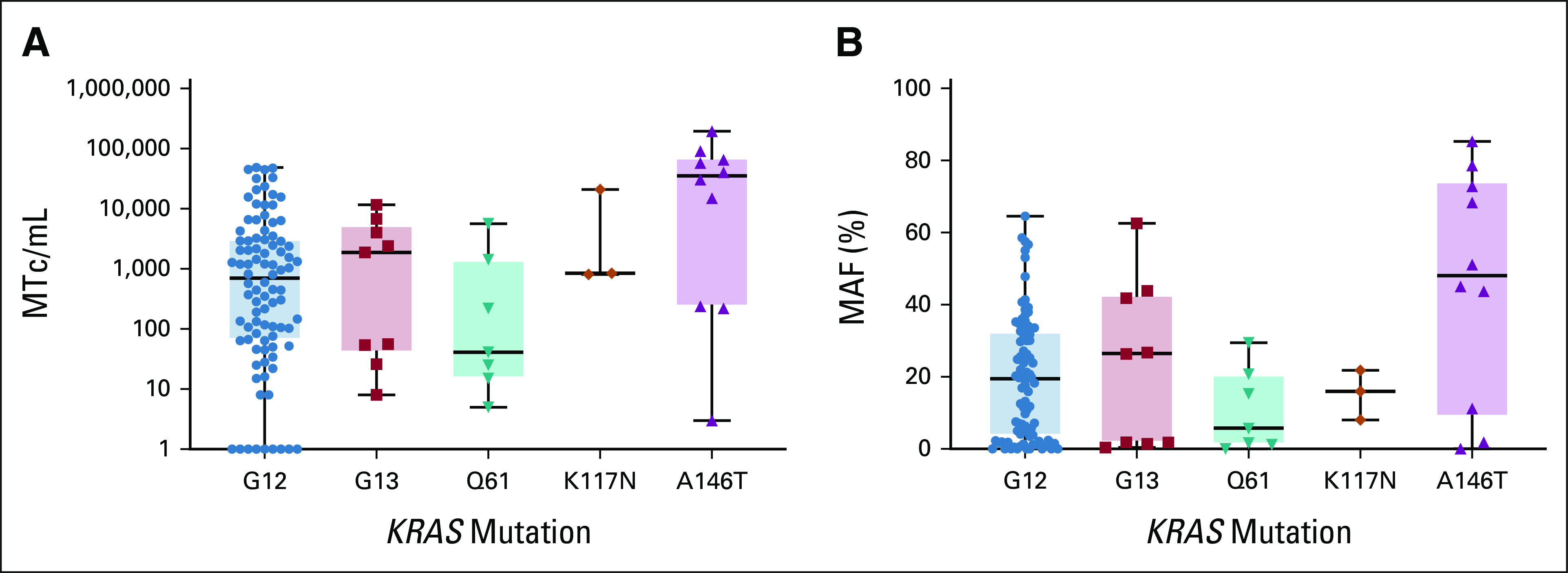
(A) MTc/mL plasma and (B) MAF detected by ddPCR analyses of pretreatment liquid biopsies stratified per KRAS mutation variant. No significant differences were observed upon Brown-Forsythe ANOVA and Dunnett's multiple comparison testing. ANOVA, analysis of variance; ddPCR, droplet digital polymerase chain reaction; MAF, mutant allele frequency; MTc/mL, mutant copies per mL.
Patients With KRAS A146–Mutated Tumors Have High TTV
As all patients in this study had liver-only metastases, total tumor burden could be assessed by measuring the pretreatment TTV. Since abdominal contrast-enhanced CT images could be used for segmentation, patients with a magnetic resonance imaging (N = 17) and positron emission tomography-CT (PET-CT) or non–contrast-enhanced scans (N = 4) were excluded from the volumetric analysis. Other reasons for exclusion were technical errors in the segmentation software (N = 3), missing scans (N = 2), and incomplete scans (N = 4), leaving 126 patients for volumetric analysis. Figure 4A shows the absolute TTV and Figure 4B shows the relative TTV as percentage of the liver volume. Patients with a KRAS A146–mutated tumor have a significantly higher absolute and relative TTV (median TTV of 672 cm3 and 24.5% of total liver volume) compared with patients with a KRAS G12 mutation (median TTV of 74 cm3 and 4.1% of total liver volume; P = .036 and P = .053, respectively) and G13 mutation (median TTV of 55 cm3 and 3.5% of total liver volume; P = .021 and P = .026, respectively). In addition, the median number of lesions tended to be higher for patients with KRAS A146 mutant tumors (median = 25) compared with patients with one of the other KRAS variants (median number of lesions KRAS G12 = 11, G13 = 10.5, Q61 = 10, K117 = 14; see Figure 4 in the Data Supplement). High TTV was also observed in patients with the less prevalent KRAS K117 mutation (median absolute TTV = 592 cm3, relative TTV = 24.1%). The volumetric results of the four most frequent G12-mutated residues (G12A, G12C, G12D, and G12V) did not differ significantly (Figure 5 in Data Supplement).
FIG 4.
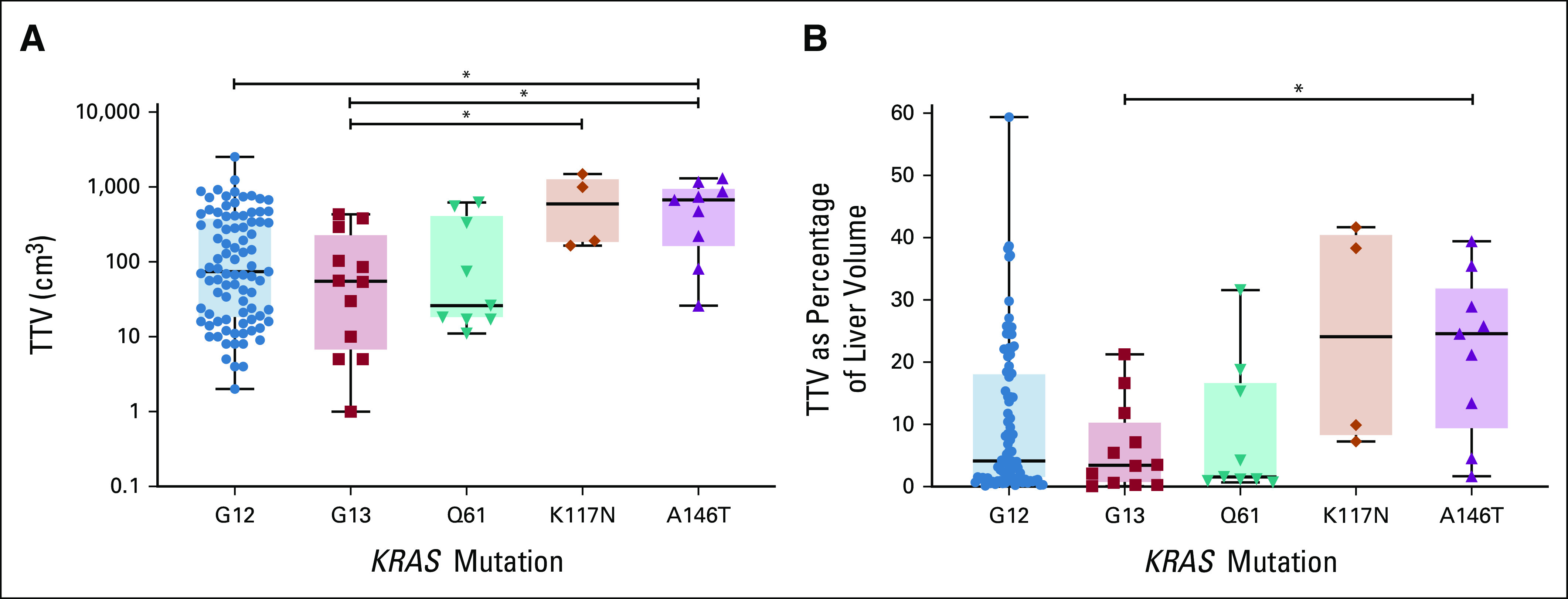
(A) The absolute TTV in cm3 and (B) the relative TTV as a percentage of the liver volume per KRAS mutation variant assessed using volumetric analyses of pretreatment CT imaging. *P < .05; one-way ANOVA; Tukey's multiple comparisons test. ANOVA, analysis of variance; CT, computed tomography; TTV, total tumor volume.
KRAS A146–Mutated Tumors Are Associated With Poor Overall Survival
Patients with mCRC with a KRAS A146–mutated tumor showed a worse prognosis than patients with another KRAS mutation variant (Fig 5). The overall survival of patients with a KRAS A146–mutated tumor was significantly shorter compared with patients with a KRAS G12–mutated tumor (median 10.7 v 26.4 months). Also, patients with the less common KRAS K117 mutation progressed faster, whereas patients with a tumor with a mutation in KRAS G13 had the most favorable prognosis. Univariable Cox regression analyses showed that age, sex, sidedness of the primary tumor, and WHO performance status were not associated with overall survival (Table 3 in Data Supplement). After adjusting for these clinical characteristics (age, sex, sidedness, and performance status), the multivariable Cox regression analysis showed that only the KRAS alteration was an independent prognostic factor for worse overall survival. The reported contrast between the KRAS mutation variants showed that KRAS A146 was the only significant feature behind this observation (hazard ratio = 2.5; 95% CI, 1.4 to 4.6; log-rank P = .003). No indications of an association between the baseline patient characteristics and the KRAS mutation variants were found (Table 1). No significant differences were seen in overall survival between the four most frequently mutated G12 residues (G12A, G12C, G12D, and G12V; see Figure 6 in the Data Supplement). Furthermore, the location of disease progression showed similar patterns for the different KRAS mutation variants (Figure 7 in Data Supplement).
FIG 5.

Overall survival of patients with mCRC carrying a tumor with a KRAS G12, G13, Q61, K117, or A146 mutation. Median survival in months was 26.4 (KRAS G12), undefined (KRAS G13), 27.9 (KRAS Q61), 20.6 (KRAS K117), and 10.7 (KRAS A146). aA significant difference was observed between KRAS G12 and A146 (log-rank P = .0045), by the Mantel-Cox log-rank test using a Bonferroni-corrected threshold for every combination of P < .005 for significance. mCRC, metastatic colorectal cancer.
DISCUSSION
Oncogenic KRAS mutations occur in approximately 50% of patients with mCRC and are known to be predictive for treatment response and to affect patient prognosis.10,11,19 The level of KRAS oncogene activation can vary depending on the amino acid change, resulting in different biologic and clinical behavior.11,20 Here, we studied the clinical impact of KRAS mutation variants in a homogenous group of patients with CRLM and demonstrated that patients with CRLM with a KRAS A146–mutated tumor have a high tumor load, which was associated with inferior survival compared with patients with other KRAS mutations.
The observations made in this study cannot directly be translated to other cancer types, since the behavior of RAS mutation variants is dependent on the location and cell type of the tumor.11 For example, the large number of samples analyzed and reported in cBioPortal21 show that KRAS A146 mutations are rarely reported in other cancer types except for CRC.21-23 Although the prevalence of A146 mutations among KRAS mutation carriers in CRC is approximately 8%, similar to our observation in this study, it is only 0%-0.5% in both lung24-26 and pancreas cancer.27,28
Biologically, KRAS mutation variants display distinct metabolic profiles. Oncogenic KRAS can dysregulate cell metabolism via glycolysis and the following tricarboxylic acid cycle. Enhanced glycolysis of cancer cells generating lactate even when exposed to abundant oxygen (Warburg effect)29 is shown to be upregulated via oncogenic KRAS.30-32 This Warburg effect is marked by low levels of ATP. However, a human CRC cell line study revealed distinct metabolic profiles of different KRAS mutation variants. Where for most KRAS variants the nucleotide imbalance is shifted toward a decrease in ATP and other nucleotides like guanosine triphosphate (GTP), cell lines harboring the KRAS A146 mutation displayed increased levels of these nucleotides.12 The distinct metabolic profiles among KRAS mutation variants do not directly explain the observed differences in clinical outcome. Future research is needed to examine whether KRAS A146–mutated tumors are a distinct metabolic subgroup for which other therapeutic targets might be beneficial.
The differences in biology and consequently clinical outcome between KRAS mutation variants observed in this study might originate from the molecular mechanism of oncogenic activation. KRAS changes between two nucleotide-binding states, the inactive form (guanosine diphosphate–bound) and the active (GTP-bound) form, with the help of guanine nucleotide exchange factors (GEF) and GTPase-activating proteins (GAP). Somatic mutations stimulate KRAS to be in the active GTP-bound form,33 but the impairment of GTP hydrolysis occurs via different mechanisms.11,34 KRAS G12 and Q61 mutations mainly affect GAP-driven GTP hydrolysis, whereas mutations in G13 and K117 influence both GEF and GAP.35,36 By contrast, mutations in KRAS A146 cause an increase in GEF-mediated nucleotide exchange without affecting GAP activity,37 suggesting that tumors with a KRAS A146 mutation may be prone to respond to GEF inhibitors. Inhibition of the GEF Son Of Sevenless protein 1 (SOS1) reduces KRAS activation, especially when combined with an MEK inhibitor.38 Likewise, RAS activation via GEFs was reduced by inhibition of the protein tyrosine phosphatase SHP2,39 which was more effective in cells harboring KRAS G12C compared with cells harboring KRAS G12D.40 Recently, AMG 510 (sotorasib), an antitumor agent targeting KRAS G12C mutant advanced solid tumors, has shown to improve the efficacy of (targeted) treatments in vivo.41 AMG 510 is currently under investigation in a clinical trial (NCT03600883), including patients with CRC and non–small-cell lung cancer.42 Another potential treatment strategy could be dual phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibition. Overexpression of the PI3K/Akt/mTOR signaling pathway is common in (m)CRC, resulting in enhanced tumor growth. Dual PI3K/mTOR inhibitors have shown to reduce cell proliferation of PIK3CA mutant tumors in mice43 and phase I clinical studies.44 However, this effect was not seen in cell lines where KRAS and PIK3CA mutations co-occurred.45 When combining the dual PI3K/mTOR inhibitor with an MEK inhibitor, significant tumor reduction was seen in KRAS mutant tumors.46,47 No data are available on combined PI3K/mTOR and MEK inhibition for patients with KRAS A146 mCRC specifically. The high tumor burden observed in our study makes the PI3K/mTOR signaling pathway an interesting potential druggable target for patients with KRAS A146–mutated tumors. Future research is needed to find out whether PI3K/mTOR inhibition combined with an MEK inhibitor has potential for KRAS A146–mutated tumor and if the poor long-term tolerability found in other advanced solid tumors48 is also pertinent in patients with KRAS A146 mutant tumors. Taken together, these results show promising potential for therapeutic targeting of KRAS mutation variants and warrant further investigation regarding therapeutic strategies to specifically target tumors with a KRAS A146 mutation.
A better insight into the KRAS mutation status can help guide and personalize the treatment approach of patients with mCRC. Previous studies in patients with early-stage CRC and CRLM showed worse outcomes for patients with a KRAS G12V,49-53 G12C,50-53 or G12S49 tumor mutation compared with other frequently occurring G12 variants, like G12A and G12D. In our study, patients with KRAS G12C and G12V mutations tended to have inferior survival compared with KRAS G12D and G12A mutations. Interestingly, in contrast to the current study investigating patients with CRC in the metastatic setting, patients with nonmetastatic CRC with a KRAS A146–mutated tumor showed better survival compared with patients with mutations in other KRAS codons.4,54 Whereas the patients with early-stage CRC carrying a KRAS A146 mutation in the study of Janakiraman et al had more frequent KRAS copy-number gains, we did not observe such copy-number aberrations in the patients with KRAS A146 mutant mCRC. The location and extent of metastases might also influence survival differences. The focus on unresectable liver-only metastases is specific for this study. Furthermore, the biologic characteristics of tumors might differ on the basis of the specific KRAS A146 amino acid substitution, similar to the differences observed between KRAS G12 variants.
In current clinical practice, no distinction is made based between KRAS mutation variants with regards to anti-EGFR treatment. However, data on the effect of anti-EGFR treatment in patients with CRC with a KRAS A146 tumor mutation are conflicting. Some studies describe a more favorable clinical outcome in patients with CRC with a KRAS A146-mutated tumor upon anti-EGFR treatment compared with patients with tumors carrying another KRAS mutation.54-57 Other studies show that tumors with a KRAS A146 mutation, like other KRAS mutations, are responsible for anti-EGFR resistance.58-61In this study, the homogenous population of initially unresectable liver-only metastatic CRC patients all receiving the same treatment regimen allowed for an unbiased comparison of the clinical features of patients harboring different KRAS tumor mutations. Another strength of this study is the objective assessment of tumor burden by TTV quantification on the basis of semiautomatic segmentations of the tumor. However, the assessment of TTV is not implemented in clinical practice, as it remains time-consuming and advanced volumetric software is not yet widely available in every radiology department. Since KRAS A146 mutations occur in roughly 4% of patients with mCRC, only a limited number of patients were available with a KRAS A146–mutated tumor despite the large number of patients included in the clinical trial. The percentage of patients with a KRAS A146–mutated tumor might be even higher than depicted in this study because routine molecular diagnostics is sometimes limited to the most common KRAS driver mutations, that is, the G12 and G13 variants. Although KRAS Q61, K117, and A146 mutations occur less frequently, this study indicates it is important to implement KRAS mutation testing for all variants (G12, G13, Q61, K117, and A146) in routine diagnostics.8,59,62
In conclusion, patients with mCRC with a KRAS A146 mutation represent a distinct molecular subtype of patients with poor survival who might benefit from more intensive treatments. Therefore, KRAS A146 mutation testing should be adopted in routine diagnostic testing.
ACKNOWLEDGMENT
The authors thank Mirthe Lanfermeijer, Dorothé Linders, Kalpana Ramkisoensing, Pien Delis-van Diemen, Margriet Lemmens, Anne Bolijn, and Marianne Tijssen for laboratory assistance. The authors would like to acknowledge the NKI-AVL Core Facility Molecular Pathology & Biobanking (CFMPB) for supplying NKI-AVL Biobank material and the NKI-AVL Genomics Core Facility (GCF) for shallow sequencing of FFPE tumor tissue DNA.
Alessandro Leal
Employment: Delfi Diagnostics
Stock and Other Ownership Interests: Delfi Diagnostics
Honoraria: Roche, Amgen, AstraZeneca, Libbs
Consulting or Advisory Role: AstraZeneca, Amgen, Delfi Diagnostics
Speakers' Bureau: Oncologia Brasil
Patents, Royalties, Other Intellectual Property: Delfi Diagnostics
Daan van den Broek
Consulting or Advisory Role: Roche Molecular Diagnostics (Inst)
Victor E. Velculescu
Leadership: Personal Genome Diagnostics, Delfi Diagnostics
Stock and Other Ownership Interests: Personal Genome Diagnostics, Delfi Diagnostics
Consulting or Advisory Role: Ignyta, Personal Genome Diagnostics, Takeda, Delfi Diagnostics, Danaher, Genentech, Bristol Meyers Squibb, Merck
Research Funding: Delfi Diagnostics
Patents, Royalties, Other Intellectual Property: Receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Qiagen, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Myriad, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Personal Genome Diagnostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Genzyme, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Roche, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Sysmex-Inostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Delfi Diagnostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Agios.
Travel, Accommodations, Expenses: Delfi Diagnostics, Bristol Myers Squibb, Genentech
Geert Kazemier
Travel, Accommodations, Expenses: SAS Analytics (Inst)
Cornelis J. A. Punt
Consulting or Advisory Role: Nordic Bioscience (Inst)
Gerrit A. Meijer
Stock and Other Ownership Interests: crcBioscreen BV
Research Funding: Exact Sciences (Inst), Sysmex (Inst), Sentinel Diagnostics (Inst), Personal Genome Diagnostics (Inst), Hartwig Medical Foundation (Inst)
Patents, Royalties, Other Intellectual Property: Several Patents Pending (Inst)
Remond J. A. Fijneman
Research Funding: Merck BV (Inst), Personal Genome Diagnostics (Inst), Delfi Diagnostics (Inst), Cergentis (Inst)
Patents, Royalties, Other Intellectual Property: Several Patents pending (Inst)
No other potential conflicts of interest were reported.
DISCLAIMER
The funders had no role in the design, conduct and submission of the study, nor the decision to submit the manuscript for publication.
SUPPORT
Supported by the Dutch Cancer Society (grant number 10438), the Stand Up To Cancer–Dutch Cancer Society International Translational Cancer Research Dream Team Grant (SU2C-AACR-DT1415)—Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research, and the Mark Foundation for Cancer Research. C.J.A.P. received scientific grants from Roche and Amgen for the CAIRO5 trial. This funding is not related to the current research.
AUTHOR CONTRIBUTIONS
Conception and design: Iris van 't Erve, Remond J. A. Fijneman
Financial support: Victor E. Velculescu, Geert Kazemier, Cornelis J. A. Punt, Gerrit A. Meijer, Remond J. A. Fijneman
Provision of study materials or patients: Karen Bolhuis
Collection and assembly of data: Iris van 't Erve, Nina J. Wesdorp, Jamie E. Medina, Leonardo Ferreira, Alessandro Leal, Joost Huiskens, Karen Bolhuis, Jan-Hein T. M. van Waesberghe
Data analysis and interpretation: Iris van 't Erve, Nina J. Wesdorp, Jamie E. Medina, Leonardo Ferreira, Jan-Hein T. M. van Waesberghe, Victor E. Velculescu, Geert Kazemier, Cornelis J. A. Punt, Gerrit A. Meijer, Remond J. A. Fijneman
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Alessandro Leal
Employment: Delfi Diagnostics
Stock and Other Ownership Interests: Delfi Diagnostics
Honoraria: Roche, Amgen, AstraZeneca, Libbs
Consulting or Advisory Role: AstraZeneca, Amgen, Delfi Diagnostics
Speakers' Bureau: Oncologia Brasil
Patents, Royalties, Other Intellectual Property: Delfi Diagnostics
Daan van den Broek
Consulting or Advisory Role: Roche Molecular Diagnostics (Inst)
Victor E. Velculescu
Leadership: Personal Genome Diagnostics, Delfi Diagnostics
Stock and Other Ownership Interests: Personal Genome Diagnostics, Delfi Diagnostics
Consulting or Advisory Role: Ignyta, Personal Genome Diagnostics, Takeda, Delfi Diagnostics, Danaher, Genentech, Bristol Meyers Squibb, Merck
Research Funding: Delfi Diagnostics
Patents, Royalties, Other Intellectual Property: Receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Qiagen, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Myriad, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Personal Genome Diagnostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Genzyme, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Roche, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Sysmex-Inostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Delfi Diagnostics, receives royalties for discoveries our group has made at Johns Hopkins that have been licensed by Agios.
Travel, Accommodations, Expenses: Delfi Diagnostics, Bristol Myers Squibb, Genentech
Geert Kazemier
Travel, Accommodations, Expenses: SAS Analytics (Inst)
Cornelis J. A. Punt
Consulting or Advisory Role: Nordic Bioscience (Inst)
Gerrit A. Meijer
Stock and Other Ownership Interests: crcBioscreen BV
Research Funding: Exact Sciences (Inst), Sysmex (Inst), Sentinel Diagnostics (Inst), Personal Genome Diagnostics (Inst), Hartwig Medical Foundation (Inst)
Patents, Royalties, Other Intellectual Property: Several Patents Pending (Inst)
Remond J. A. Fijneman
Research Funding: Merck BV (Inst), Personal Genome Diagnostics (Inst), Delfi Diagnostics (Inst), Cergentis (Inst)
Patents, Royalties, Other Intellectual Property: Several Patents pending (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Nussinov R, Tsai CJ, Jang H: Oncogenic Ras isoforms signaling specificity at the membrane. Cancer Res 78:593-602, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simanshu DK, Nissley DV, McCormick F: RAS proteins and their regulators in human disease. Cell 170:17-33, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peeters M, Kafatos G, Taylor A, et al. : Prevalence of RAS mutations and individual variation patterns among patients with metastatic colorectal cancer: A pooled analysis of randomised controlled trials. Eur J Cancer 51:1704-1713, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Imamura Y, Lochhead P, Yamauchi M, et al. : Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: Cohort study and literature review. Mol Cancer 13:135, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaeger R, Chatila WK, Lipsyc MD, et al. : Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33:125-136.e3, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eklof V, Wikberg ML, Edin S, et al. : The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br J Cancer 108:2153-2163, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foltran L, De Maglio G, Pella N, et al. : Prognostic role of KRAS, NRAS, BRAF and PIK3CA mutations in advanced colorectal cancer. Future Oncol 11:629-640, 2015 [DOI] [PubMed] [Google Scholar]
- 8.Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. : The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 53:852-864, 2014 [DOI] [PubMed] [Google Scholar]
- 9.Vale CL, Tierney JF, Fisher D, et al. : Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat Rev 38:618-625, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Sepulveda AR, Hamilton SR, Allegra CJ, et al. : Molecular biomarkers for the evaluation of colorectal cancer: Guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. Arch Pathol Lab Med 141:625-657, 2017 [DOI] [PubMed] [Google Scholar]
- 11.Munoz-Maldonado C, Zimmer Y, Medova M: A Comparative analysis of individual RAS mutations in cancer biology. Front Oncol 9:1088, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varshavi D, Varshavi D, McCarthy N, et al. : Metabolic characterization of colorectal cancer cells harbouring different KRAS mutations in codon 12, 13, 61 and 146 using human SW48 isogenic cell lines. Metabolomics 16:51, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zafra MP, Parsons MJ, Kim J, et al. : An in vivo Kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov 10:1654-1671, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sjoblom T, Jones S, Wood LD, et al. : The consensus coding sequences of human breast and colorectal cancers. Science 314:268-274, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Huiskens J, van Gulik TM, van Lienden KP, et al. : Treatment strategies in colorectal cancer patients with initially unresectable liver-only metastases, a study protocol of the randomised phase 3 CAIRO5 study of the Dutch Colorectal Cancer Group (DCCG). BMC Cancer 15:365, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phallen J, Sausen M, Adleff V, et al. : Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 9:eaan2415, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.SAS Visual Data Mining and Machine Learning Programming Guide: The quantifyBioMedImages Action 2020. https://go.documentation.sas.com/?cdcId=pgmsascdc&cdcVersion=9.4_3.5&docsetId=casactml&docsetTarget=casactml_biomedimage_details05.htm&locale=en [Google Scholar]
- 18.Erve IV, Greuter MJE, Bolhuis K, et al. : Diagnostic strategies towards clinical implementation of liquid biopsy RAS/BRAF circulating tumor DNA analyses in patients with metastatic colorectal cancer. J Mol Diagn 22:1430-1437, 2020 [DOI] [PubMed] [Google Scholar]
- 19.Tejpar S, Celik I, Schlichting M, et al. : Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol 30:3570-3577, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Serebriiskii IG, Connelly C, Frampton G, et al. : Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun 10:3722, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.cBioPortal : www.cBioPortal.org
- 22.Cerami E, Gao J, Dogrusoz U, et al. : The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401-404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, et al. : Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell JD, Alexandrov A, Kim J, et al. : Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607-616, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Yang H, Teo ASM, et al. : Genomic landscape of lung adenocarcinoma in East Asians. Nat Genet 52:177-186, 2020 [DOI] [PubMed] [Google Scholar]
- 26.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. : Tracking the evolution of non-small-cell lung cancer. N Engl J Med 376:2109-2121, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Bailey P, Chang DK, Nones K, et al. : Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531:47-52, 2016 [DOI] [PubMed] [Google Scholar]
- 28.Witkiewicz AK, McMillan EA, Balaji U, et al. : Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6:6744, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warburg O: On respiratory impairment in cancer cells. Science 124:269-270, 1956 [PubMed] [Google Scholar]
- 30.Son J, Lyssiotis CA, Ying H, et al. : Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101-105, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu CM, Tien SC, Hsieh PK, et al. : High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab 29:1334-1349.e10, 2019 [DOI] [PubMed] [Google Scholar]
- 32.Santana-Codina N, Roeth AA, Zhang Y, et al. : Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 9:4945, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edkins S, O'Meara S, Parker A, et al. : Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther 5:928-932, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter JC, Manandhar A, Carrasco MA, et al. : Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 13:1325-1335, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Haigis KM: KRAS alleles: The devil is in the detail. Trends Cancer 3:686-697, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feig LA, Cooper GM: Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol Cell Biol 8:2472-2478, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulin EJ, Bera AK, Lu J, et al. : Tissue-specific oncogenic activity of KRASA146T. Cancer Discov 9:738-755, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofmann MH, Gmachl M, Ramharter J, et al. : BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov 11:142-157, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nichols RJ, Haderk F, Stahlhut C, et al. : RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20:1064-1073, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Mulla F, Milner-White EJ, Going JJ, et al. : Structural differences between valine-12 and aspartate-12 Ras proteins may modify carcinoma aggression. J Pathol 187:433-438, 1999 [DOI] [PubMed] [Google Scholar]
- 41.Hong DS, Fakih MG, Strickler JH, et al. : KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med 383:1207-1217, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canon J, Rex K, Saiki AY, et al. : The clinical KRASG12C inhibitor AMG 510 drives anti-tumour immunity. Nature 575:217-223, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Foley TM, Payne SN, Pasch CA, et al. : Dual PI3K/mTOR inhibition in colorectal cancers with APC and PIK3CA mutations. Mol Cancer Res 15:317-327, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janku F, Wheler JJ, Naing A, et al. : PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res 73:276-284, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim A, Lee JE, Lee SS, et al. : Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int J Cancer 133:984-996, 2013 [DOI] [PubMed] [Google Scholar]
- 46.Migliardi G, Sassi F, Torti D, et al. : Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 18:2515-2525, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Engelman JA, Chen L, Tan X, et al. : Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14:1351-1356, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schram AM, Gandhi L, Mita MM, et al. : A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer 119:1471-1476, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margonis GA, Kim Y, Spolverato G, et al. : Association between specific mutations in KRAS codon 12 and colorectal liver metastasis. JAMA Surg 150:722-729, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayama T, Hashiguchi Y, Okamoto K, et al. : G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int J Colorectal Dis 34:1491-1496, 2019 [DOI] [PubMed] [Google Scholar]
- 51.Andreyev HJ, Norman AR, Cunningham D, et al. : Kirsten ras mutations in patients with colorectal cancer: The ‘RASCAL II’ study. Br J Cancer 85:692-696, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imamura Y, Morikawa T, Liao X, et al. : Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res 18:4753-4763, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones RP, Sutton PA, Evans JP, et al. : Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer 116:923-929, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janakiraman M, Vakiani E, Zeng Z, et al. : Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 70:5901-5911, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Roock W, Claes B, Bernasconi D, et al. : Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 11:753-762, 2010 [DOI] [PubMed] [Google Scholar]
- 56.Korphaisarn K, Pongpaibul A, Roothumnong E, et al. : High frequency of KRAS codon 146 and FBXW7 mutations in Thai patients with stage II-III colon cancer. Asian Pac J Cancer Prev 20:2319-2326, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray SM, Lee J, Kim ST, et al. : Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci Rep 9:15365, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Douillard JY, Oliner KS, Siena S, et al. : Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369:1023-1034, 2013 [DOI] [PubMed] [Google Scholar]
- 59.Hecht JR, Douillard JY, Schwartzberg L, et al. : Extended RAS analysis for anti-epidermal growth factor therapy in patients with metastatic colorectal cancer. Cancer Treat Rev 41:653-659, 2015 [DOI] [PubMed] [Google Scholar]
- 60.Rebersek M, Mesti T, Boc M, et al. : Molecular biomarkers and histological parameters impact on survival and response to first-line systemic therapy of metastatic colorectal cancer patients. Radiol Oncol 53:85-95, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loupakis F, Ruzzo A, Cremolini C, et al. : KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 101:715-721, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richman SD, Fairley J, Butler R, et al. : How close are we to standardised extended RAS gene mutation testing? The UK NEQAS evaluation. J Clin Pathol 70:58-62, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]