

Abstract
We develop and employ the Fluorescence-Activating and absorption-Shifting Tag (FAST) system for super-resolution (SR) imaging and single-molecule tracking based on single-molecule localizations. The fast off rate of fluorogen binding combined with its spatially well-separated labeling of the densely expressed FAST fusion proteins allowed single molecule measurements to be performed in both living and fixed cells. The well-separated fluorescence localization density was achieved by either reversibly controlling the fluorogen concentration or by irreversibly photobleaching the FAST-fluorogen complex. The experimentally determined resolution of 28 nm allowed us to resolve Ensconsin-labeled microtubules and to track single molecules in mitochondria. Our results demonstrate that FAST is well suited for single-molecule localization microscopy (SMLM). The small size and the availability of spectrally distinct fluorogens present unique advantages of the FAST system as a potential orthogonal labeling strategy that could be applied in conjunction with existing super-resolution dyes and photo-activatable proteins in versatile imaging applications.
Graphical Abstract
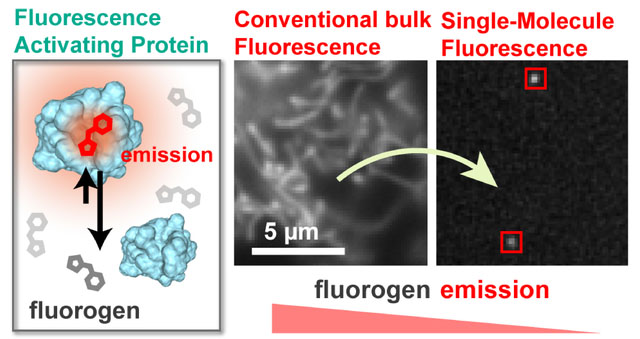
The complex biological functions of cells emerge though specific and spatiotemporally organized interactions of biomolecules. Methods to measure and quantify the organization, interactions, and dynamics of the involved biomolecules are the key to understanding specific biological events. The introduction of traditional fluorescence microscopy to visualize specific biomolecules in living and fixed cells presented a breakthrough in our understanding of biological processes.1,2 However, the limited resolution of 200–250 nm caused by the diffraction of light results in loss of valuable information about the location, interactions, and nanoscopic clustering of biomolecules.3 The development of super-resolution fluorescence microscopy overcomes these limitations by allowing us to resolve nanoscopic structures in cells with 20–40 nm resolution4– 8 and to quantify the biomolecules they contain.9–11 Single-molecule localization microscopy (SMLM) techniques such as stochastic optical reconstruction microscopy (STORM)4 and photoactivated localization microscopy (PALM)5 are based on activating only a sparse subset of fluorophores, which enables the determination of their individual centers with high precision. A super-resolution (SR) image is then built up by combining the single-molecule localizations from numerous snapshots of different fluorophores in time.
An alternative approach for spreading the signals of fluorophores in time is to continuously target structures under investigation with transiently binding fluorescent reporters.12–14 DNA-PAINT, for instance achieves < 30 nm resolution via the reversible and specific binding of dye-labeled “imager” DNA strands to complementary “docking” DNA strands, which are bound to the biological structure.15,16 These techniques require TIRF or light sheet excitation to suppress the background fluorescence of the freely diffusing fluorophores. Non-covalent fluorogen-based reporters overcome this limitation by exhibiting a dramatic increase in brightness when transiently bound to a genetically encodable fluorogen-activating protein (FAP)17 or RNA aptamers.18 By adjusting the concentration of the fluorogen, sparse stochastic binding/unbinding of the fluorogen to the FAP can be achieved, which results in blinking that is suitable for SMLM.19,20
In this work we demonstrate for the first time the application of the recently developed Fluorescence-Activating and absorption-Shifting Tag (FAST)21,22 for SMLM. FAST is a variant of the Photoactive Yellow Protein (PYP), which was engineered to specifically bind and activate the fluorescence of the fluorogenic analogs of 4-hydroxybenzylidene-rhodanine (HBR). FAST is characterized by a rapid and reversible binding of the fluorogenic HBR analogs (FAST:HBR, KD = 0.62 μM, fluorogen residence time = 60ms)21, which enables the bulk fluorescence to be rapidly switched on and off by the addition and washout of the fluorogen. The availability of fluorogen variants of three different colors makes FAST a unique and ideal system for orthogonal labeling. Furthermore, the size of FAST (14 kDa) is smaller than fluorescent proteins (26–30 kDa) and any known FAP, which minimizes interference with the function of a tagged protein. Here, we establish the SMLM capability of FAST primarily using the red variant, HBR-3,5DOM (4-hydroxy-3,5-dimethoxybenzylidene rhodamine, see Figure 1). The highly specific activation of bright fluorescence compared to unbound fluorogens allows FAST:fluorogen emitters to be precisely fit and localized over thousands of frames (Movie S1). Our results demonstrate that FAST is a viable and likely orthogonal labeling strategy for live and fixed-cell SMLM.
Figure 1.
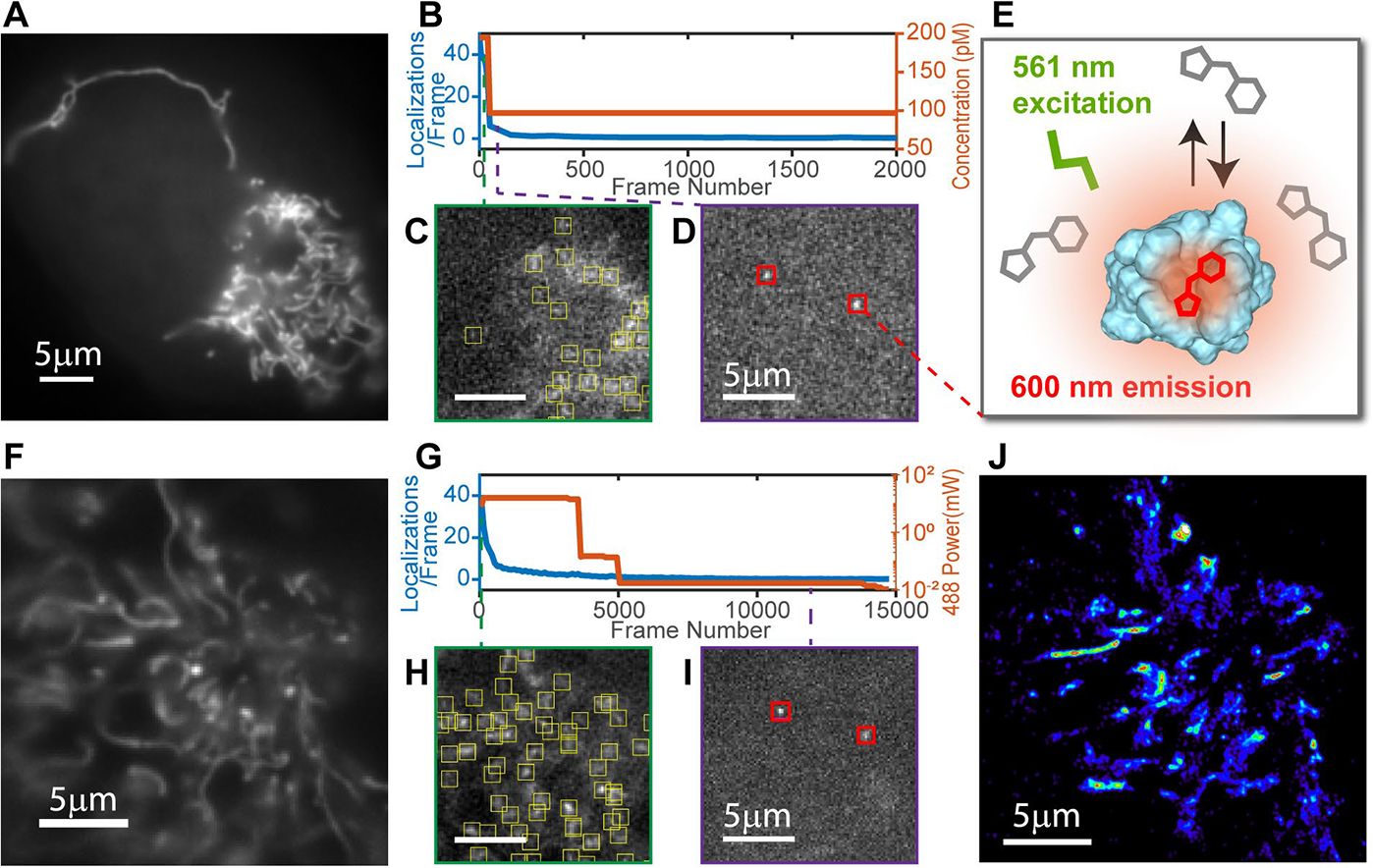
Developing SMLM capability of FAST in living U2OS cells. (A) Conventional fluorescence image of a mitochondria (25 nM HBR-3,5DOM, average 50 frames) confirms specific labeling and proper expression levels of MTS-FAST. (B) The fluorogen concentration (orange) is lowered to control the fluorescence localization density (blue). (C) A single frame at 195 pM fluorogen shows a localization density too high for identification of a single fluorophore. (D) A single frame at 97 pM HBR-3,5DOM exhibits fluorescence of single fluorophores suitable for SMLM imaging. (E) When bound to a FAST protein, HBR-3,5DOM is excited at 561 nm. (F) Conventional fluorescence image of mitochondria (100 nM fluorogen, average 50 frames) prior to photobleaching. (G) The fluorescence localization density (blue) is adjusted via photobleaching with a 488 nm laser (orange). (H) After 40 bleaching frames the localization density is still high. (I) After 2500 bleaching frames HBR-3,5DOM labeling exhibits single detectable fluorophores. (J) Rendered SR image (from frames 2,500–15,000) shows viability of FAST for living cell SMLM.
The reversible binding and external control of the fluorogen concentration makes it possible to tune the density of bound fluorogens independently from the expression level of FAST-tagged proteins.
Adjusting the concentration of fluorogen used for labeling FAST bearing a mitochondria targeting sequence (MTS-FAST) in U2OS cells allowed us to transition from conventional bulk fluorescence imaging (Figure 1A) to sparse single-molecule fluorescence imaging (Figure 1D). While these conditions allowed us to reconstruct a SR image of mitochondria (Figure S1) the localization density significantly decreased over the data acquisition time (Figure 1B, blue). This photo-damage to the FAST:fluorogen complex provided an alternative approach to decrease the localization density. Since the excitation spectrum of the FAST:HBR-3,5DOM complex ranges from 450 nm to 600 nm we used a combination of 488 nm bleaching (Figure 1G, orange) to decrease the localization density and 561 nm excitation for single molecule imaging. Applying this strategy to cells expressing MTS-FAST labeled with 10 μM HBR-3,5DOM (Figure 1F) reduced the localization density to less than 10 localizations/frame within the first ~1000 frames, allowing precise single molecule localization (Figure 1I) for SR image reconstruction (Figure 1J).
In a live cell environment, single particle tracking (SPT) offers valuable and complementary information about the dynamics of biomolecules.23 To evaluate the FAST system for SPT (Figure 2), we first verified that the fluorescence of single emitters lasted longer than just a single frame in fixed cells. The localizations from different FAST:fluorogen complexes were spatially well separated, such that localizations that appeared in subsequent frames within a distance of 150 nm could be reliably grouped to single-molecule traces. The on-time histogram of these traces confirmed that the fluorescence emission from 34% of FAST:fluorogen complexes could be reliably tracked beyond a single frame (Figure 2C). A fit of the on-time histogram revealed a characteristic time of 77 ms, which corresponds well with the fluorogen residence times from bulk measurements.21 Additionally, the characteristic on-time remained constant over a wide range of 561 nm laser powers (5mW-60mW, Figure S2A), indicating that fluorogen bleaching is not detected on the short timescale of fluorescent bursts. To study the diffusion of MTS-FAST:HBR-3,5DOM in the mitochondria, we performed SPT in live cells and used fixed cells as a control. While the parameters used to link localizations between frames were the same, we observed vastly different diffusion behaviors for each condition. The traces in fixed cells were restricted to small radii around immobile proteins (Figure 2A) and their averaged mean-square displacement (MSD) exhibited immobile behavior as expected (Figure 2C, red). In contrast the live cell traces were protracted paths along the mitochondria, indicating rapid diffusion (Figure 2B). The fit of the MSD with a linear 2D diffusion model resulted in a diffusion coefficient of D=0.4 μm2/s (Figure 2C, blue), which is consistent with but at the slow end of reported values for protein diffusion in the mitochondria (0.3–20 μm2/s)24–26 due to our sampling rate. Since proteins in the mitochondria exhibit a broad distribution of diffusion coefficients, our sampling rate of 20Hz will miss the fastest species and shift the average diffusion coefficient to slower values. Alternatively, in a two channel (red/green) experimental setup, the FAST spectral variants provide a way to image and drift-correct cells of interest with one color and to simultaneously perform SPT with another color without need for additional dilutions (Figure 2E).
Figure 2.
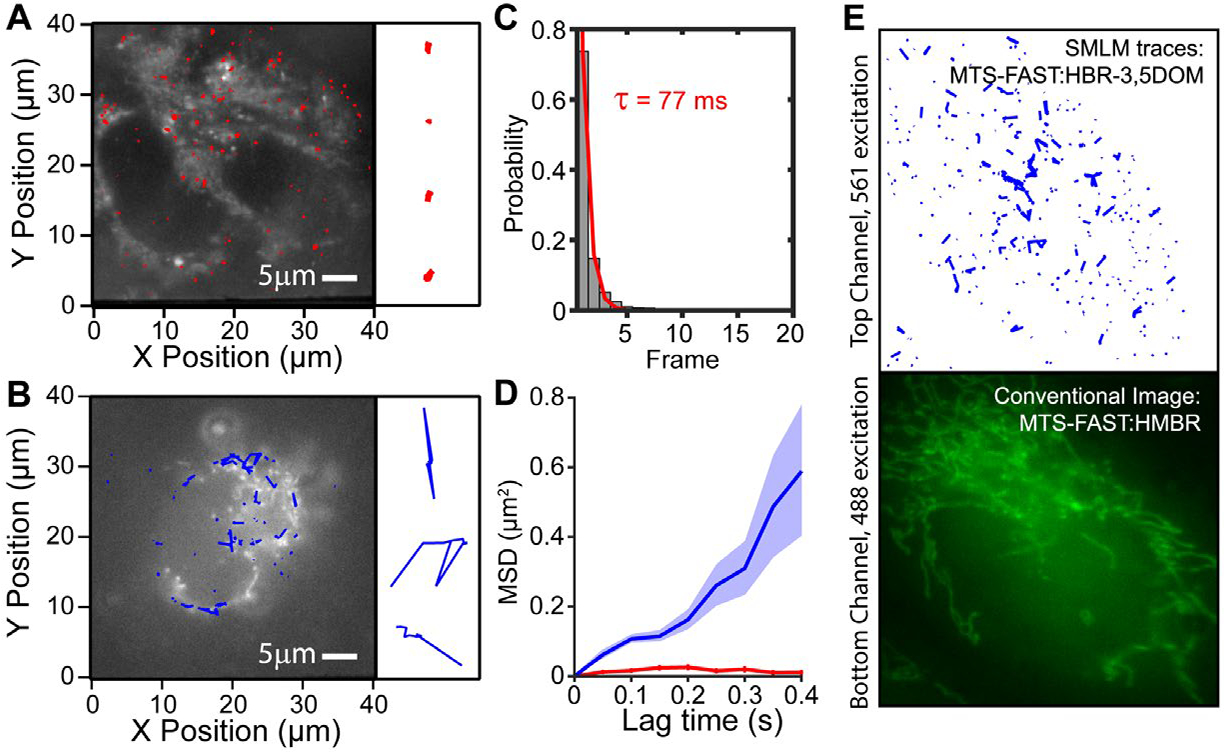
Single particle tracking analysis of MTS FAST:HBR 3–5DOM in mitochondria. (A,B) Fixed cell trajectories (red) indicating immobile proteins and live cell trajectories (blue) indicating mobile proteins superimposed on a bulk fluorescence image of mitochondria (grey). (C) On-time histogram in fixed cells; the fluorogen-FAST binding lasts for more than a single frame. (D) The average mean square displacement versus time showed immobile behavior in fixed cells (red, n=194) and mobile behavior with a diffusion coefficient of 0.4 μm2/s in live cells (blue, n=155). (E) Multicolor FAST with HMBR (25nM) and HBR-3,5DOM (100pM). Conventional fluorescence from HMBR revealed specific labeling of MTS-FAST (bottom). SPT analysis of MTS-FAST:HBR-3,5DOM resulted in extended traces (blue) indicating rapid diffusion (top).
The quality of super-resolution fluorescence images is based on the ability to accurately localize the labeled biomolecules of interest. A higher photon budget in combination with a lower background signal will increase the localization precision and subsequently lead to a higher image resolution. Thus, we further characterized the FAST system by measuring the signal properties of single FAST:fluorogen complexes. The mean photon output of single emitters was 550 photons/frame at 1.2 kW/cm2 (Figure S2B, S2C), which is slightly higher than the reported value for the MG-FAP fluorogen complex20 and similar to commonly used photoactivatable fluorescent proteins.27–29. The experimentally determined precision was 28 nm (see Figure 3F), which is comparable to other fluorogens and PALM/STORM probes.19,20,27,28,31
Figure 3.
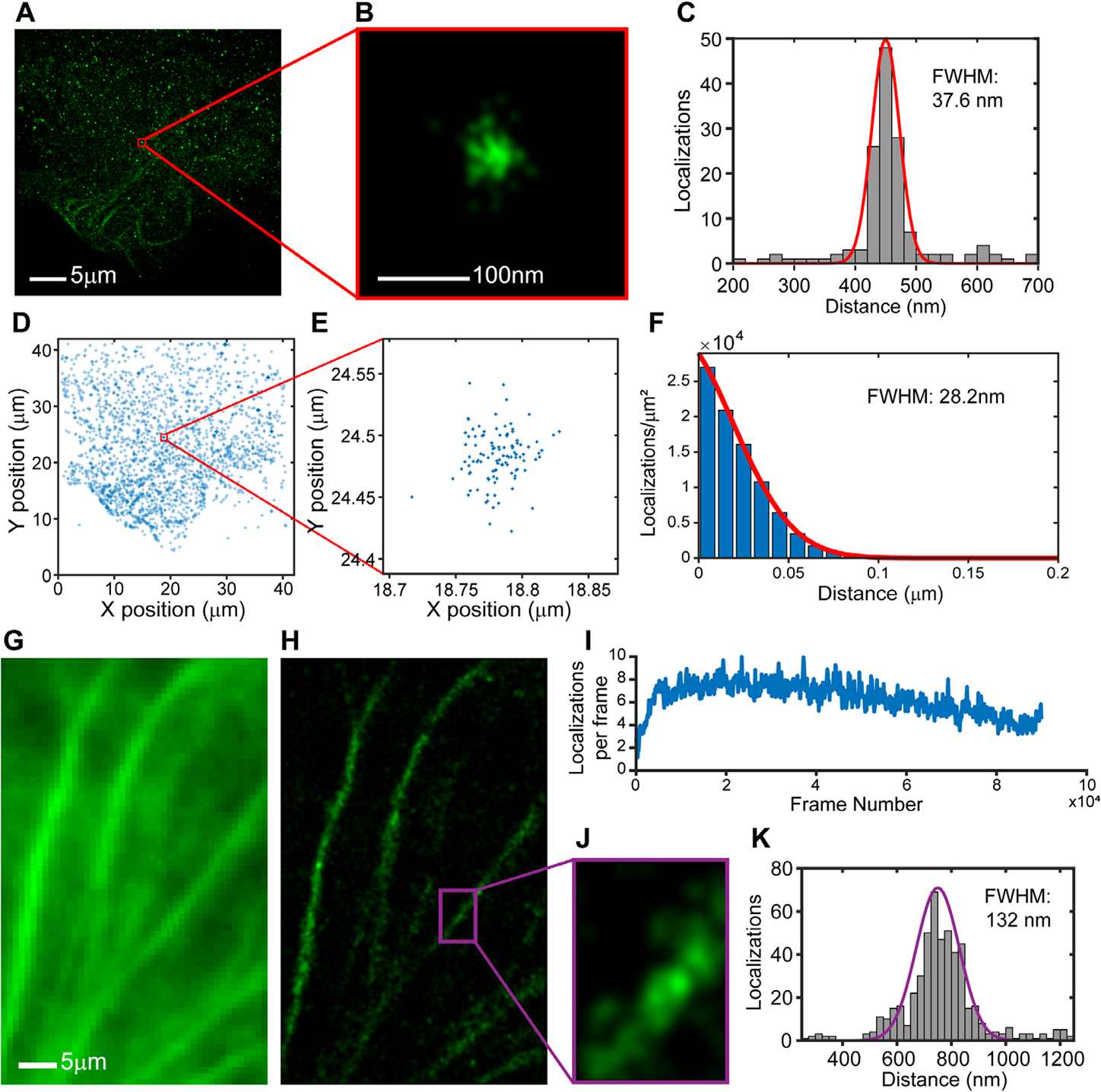
Ensconsin-FAST:HBR-3,5DOM in fixed U2OS cells. (A) A sub-set of localizations (n=4000) from consecutive frames (PBS). (B) The spread in localizations for a single protein rendered with uniform Gaussians (σ=6 nm). (C) X-projection of localizations has a FWHM of 37.6 nm (D) The same localizations (n=4000) are used to calculate the radial distribution function for the FAST:fluorogen complex. (E) Enlarged section of localizations, marked by the red box. (F) Fit of the radial distribution function (PCF, see material and methods) returns an experimental resolution of 28.2 nm. (G) Conventional fluorescence image of microtubules (320 nM fluorogen, average 50 frames) confirms specific labeling of Ensconsin-FAST. (H) Rendered SR fluorescence image (1.25 nM fluorogen, PBS+O2 scavenger, 90,000 frames) shows viability of FAST for fixed cell SMLM (I) 5–10 localization/frame maintained for 90 minutes of imaging. (J) Enlarged regions of interest (ROI) from Projection of localization from the SR ROI, along the microtuble axis, has a FWHM 132 nm.
To demonstrate the versatility of FAST for SMLM, we imaged the microtubule-binding protein Ensconsin fused to FAST in fixed U2OS cells. In PBS, the rapid photodestruction, which initially resulted in sparse super-resolution images (Figure S3), allowed us to characterize the experimental resolution with the few remaining and spatially well separated FAST:fluorogen complexes (Figure 3A). The x-projection of the spread in localizations for a single protein (Figure 3B) resulted in a full width at half maximum of 37.6 nm (Figure 3C). The radial distribution function, which quantified the spread of all localizations around individual and well separated FAST:fluorogen complexes from 4000 localizations (Figure 3D,E) exhibited a full width at half-maximum of 28.2 nm (Figure 3F). This size represents the experimental precision of super-resolution images. The addition of oxygen scavengers to the buffer prevented photodamage and allowed a rate of 5–10 localizations/frame to be maintained for over one hour of imaging (Figure 3I). The large number of localizations from each FAST:fluorogen complex together with dense incorporation of Ensconsin-FAST into microtubules resulted in super-resolution images reconstructed from 90,000 frames (Figure 3H,J).
In this paper, we showed that the Fluorescence-Activating and absorption-Shifting Tag (FAST) is well suited for single-molecule based imaging and tracking methods. The fast off rate of the FAST:fluorogen complex combined with the specific activation of detectable fluorophores allows single molecule measurements to be performed in both living and fixed cells. We evaluated the signal properties of the FAST system and showed that the photon budget and experimental resolution (28 nm) of the FAST system is comparable to other fluorogenic systems and PALM/STORM labels. We successfully performed SPT measurements of FAST in mitochondria and obtained a diffusion coefficient, which matched accepted values. In fixed U2OS cells, we resolved ensconsin associated microtubules and imaged them for over one hour, thus showing that the FAST:fluorogen system is a viable option to complement existing super-resolution dyes and photo-activatable proteins. Preventing photodamage of the FAST:fluorogen complex with oxygen scavengers resulted in an almost constant localization rate over more than one hour of imaging. This result presents a unique advantage over traditional PALM/STORM labels and indicates that future mutations at the fluorogen binding site of the FAST protein could lead to an improved localization rate in long term measurements without the need of oxygen scavengers. Since no fluorogen bleaching is detected over the used range of excitation powers, future mutations or changes in the fluorogen could also result in longer on-times and improve the quality of SPT data. The FAST system, with its highly specific activation of fluorescence and seamless change of color through spectrally distinct fluorogen analogs, provides a potentially orthogonal labeling option that could easily be used in conjunction with photoactivatable fluorescent proteins, STORM dyes, and/or covalent fluorogenic labeling tags (SNAP-tag, CLIP-tag, Halo-tag).
METHODS
Sample Preparation.
U2OS cells maintained in DMEM (Gibco) medium with 10% fetal bovine serum were subcultered in eight-well coverglass chamber slides (Nunc, ThermoFischer) 12 hours before transfection. Transient transfections were carried out using GeneJET (ThemoFisher) 12–24 hours prior to the measurements. The growth medium was replaced with Dulbecco’s phosphate-buffered saline (PBS) with calcium and magnesium (Giboco) for measurements. For fixation, cells were treated with 4% (vol/vol) formaldehyde for 15 minutes. (SI Material and Methods gives details). The cloning of the Ensconsin-FAST (microtubule-binding protein) and MTS-FAST (mitochondria targeting sequence) plasmids for mammalian cell expression have been previously described.21,22 Fluorogens (HBR-3,5 DOM and HMBR) were added to well at various concentrations prior to measurement (SI Material and Methods). For the experiment shown in Figure 3, glucose oxidase and catalase (GOC) was added to the well just prior to super-resolution imaging (SI Material and Methods).
Superresolution microscopy.
Super-resolution experiments were performed on a custom-built microscope as previously described (SI Material and Methods give details).9 The 561-nm laser powers at the objective ranged from 0.1–1 mW (~6–62W/cm2) for conventional fluorescence imaging and from 15–18 mW (power density ~1kW/cm2) for super-resolution imaging. Imaging sequences and powers were varied based on the fluorogen concentration, the cell preparation conditions (fixed versus live), and the experiment (SI Material and Methods give details). A typical SR image was generated from a sequence of 50,000–100,000 image frames, recorded at 20 Hz. Fluorescent intensity profiles from single fluorogens were fit with Gaussians and drift corrected (SI Material and Methods).
Basic Image Analysis.
Localizations belonging to the same FAST protein will create a spatial signature (radial distribution function, RDF) that defines the uncertainty in position determination and thus the underlying experimental resolution of the system.9,32–34 We determine the RDF by determining the distribution of distances between any pair of two localization (SI Materials and Methods). For single molecule diffusion analysis, traces were generated by linking localizations in consecutive frames that appear within a distance of 2 μm. For each single-molecule trajectory (n>150) longer than three consecutive frames, the mean-square displacement (MSD) was calculated by averaging the displacement of all time intervals with length delta t in a custom-written Igor-Pro program (SI Material and Methods).
Supplementary Material
ACKNOWLEDGMENT
We would like to thank R. Stefansson for developing the MSD analysis and PCF code in MATLAB and J. Mueller for the U2OS cells. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R21GM127965.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Sample preparation, experimental setup, data acquisition, image analysis, Figures S1–S4, and Movie S1. This material is available free of charge on the ACS Publications website.
REFERENCES
- (1).Combs CA (2010) Fluorescence Microscopy: A Concise Guide to Current Imaging Methods. Curr. Protoc. Neurosci. Editor. Board Jacqueline N Crawley Al 0 2, Unit2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lichtman JW, and Conchello J-A (2005) Fluorescence microscopy. Nat. Methods 2, 910–919. [DOI] [PubMed] [Google Scholar]
- (3).Sydor AM, Czymmek KJ, Puchner EM, and Mennella V (2015) Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol. 25, 730–748. [DOI] [PubMed] [Google Scholar]
- (4).Rust MJ, Bates M, and Zhuang X (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, and Hess HF (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- (6).Moerner WE, and Kador L (1989) Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 62, 2535–2538. [DOI] [PubMed] [Google Scholar]
- (7).Klar TA, Jakobs S, Dyba M, Egner A, and Hell SW (2000) Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. U. S. A. 97, 8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hell SW, and Wichmann J (1994) Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782. [DOI] [PubMed] [Google Scholar]
- (9).Puchner EM, Walter JM, Kasper R, Huang B, and Lim WA (2013) Counting molecules in single organelles with superresolution microscopy allows tracking of the endosome maturation trajectory. Proc. Natl. Acad. Sci. U. S. A. 110, 16015–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Annibale P, Vanni S, Scarselli M, Rothlisberger U, and Radenovic A (2011) Quantitative Photo Activated Localization Microscopy: Unraveling the Effects of Photoblinking. PLoS ONE 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zanacchi FC, Manzo C, Alvarez AS, Derr ND, Parajo MG, and Lakadamyali M (2017) DNA Origami offers a versatile method for quantifying protein copy-number in super-resolution. Nat. Methods 14, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sharonov A, and Hochstrasser RM (2006) Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. U. S. A. 103, 18911–18916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schoen I, Ries J, Klotzsch E, Ewers H, and Vogel V (2011) Binding-activated localization microscopy of DNA structures. Nano Lett. 11, 4008–4011. [DOI] [PubMed] [Google Scholar]
- (14).Kiuchi T, Higuchi M, Takamura A, Maruoka M, and Watanabe N (2015) Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat. Methods 12, 743–746. [DOI] [PubMed] [Google Scholar]
- (15).Jungmann R, Steinhauer C, Scheible M, Kuzyk A, Tinnefeld P, and Simmel FC (2010) Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett. 10, 4756–4761. [DOI] [PubMed] [Google Scholar]
- (16).Jungmann R, Avendaño MS, Dai M, Woehrstein JB, Agasti SS, Feiger Z, Rodal A, and Yin P (2016) Quantitative super-resolution imaging with qPAINT. Nat. Methods 13, 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Szent-Gyorgyi C, Schmidt BF, Schmidt BA, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JAJ, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, and Waggoner A (2008) Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235–240. [DOI] [PubMed] [Google Scholar]
- (18).Kolpashchikov DM (2005) Binary malachite green aptamer for fluorescent detection of nucleic acids. J. Am. Chem. Soc. 127, 12442–12443. [DOI] [PubMed] [Google Scholar]
- (19).Yan Q, Schwartz SL, Maji S, Huang F, Szent-Gyorgyi C, Lidke DS, Lidke KA, and Bruchez MP (2014) Localization microscopy using noncovalent fluorogen activation by genetically encoded fluorogen-activating proteins. Chemphyschem Eur. J. Chem. Phys. Phys. Chem. 15, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schwartz SL, Yan Q, Telmer CA, Lidke KA, Bruchez MP, and Lidke DS (2015) Fluorogen-Activating Proteins Provide Tunable Labeling Densities for Tracking FcεRI Independent of IgE. ACS Chem. Biol. 10, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Plamont M-A, Billon-Denis E, Maurin S, Gauron C, Pimenta FM, Specht CG, Shi J, Quérard J, Pan B, Rossignol J, Moncoq K, Morellet N, Volovitch M, Lescop E, Chen Y, Triller A, Vriz S, Le Saux T, Jullien L, and Gautier A (2016) Small fluorescence-activating and absorption-shifting tag for tunable protein imaging in vivo. Proc. Natl. Acad. Sci. U. S. A. 113, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li C, Plamont M-A, Sladitschek HL, Rodrigues V, Aujard I, Neveu P, Le Saux T, Jullien L, and Gautier A (2017) Dynamic multicolor protein labeling in living cells. Chem. Sci. 8, 5598–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Qian H, Sheetz MP, and Elson EL (1991) Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dieteren CEJ, Gielen SCAM, Nijtmans LGJ, Smeitink JAM, Swarts HG, Brock R, Willems PHGM, and Koopman WJH (2011) Solute diffusion is hindered in the mitochondrial matrix. Proc. Natl. Acad. Sci. U. S. A. 108, 8657–8662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Partikian A, Ölveczky B, Swaminathan R, Li Y, and Verkman AS (1998) Rapid Diffusion of Green Fluorescent Protein in the Mitochondrial Matrix. J. Cell Biol. 140, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sukhorukov VM, Dikov D, Busch K, Strecker V, Wittig I, and Bereiter-Hahn J (2010) Determination of protein mobility in mitochondrial membranes of living cells. Biochim. Biophys. Acta 1798, 2022–2032. [DOI] [PubMed] [Google Scholar]
- (27).Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, and Betzig E (2007) Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. U. S. A. 104, 20308–20313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, and Looger LL (2009) A bright and photostable photoconvertible fluorescent protein for fusion tags. Nat. Methods 6, 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang S, Moffitt JR, Dempsey GT, Xie XS, and Zhuang X (2014) Characterization and development of photoactivatable fluorescent proteins for single-molecule–based superresolution imaging. Proc. Natl. Acad. Sci. U. S. A. 111, 8452–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Thompson RE, Larson DR, and Webb WW (2002) Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dempsey GT, Vaughan JC, Chen KH, Bates M, and Zhuang X (2011) Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, and Baird BA (2012) Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PloS One 7, e31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, and Lippincott-Schwartz J (2011) Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 8, 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sengupta P, Jovanovic-Talisman T, and Lippincott-Schwartz J (2013) Quantifying spatial organization in point-localization superresolution images using pair correlation analysis. Nat. Protoc. 8, 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.