

Abstract
Folate metabolism can be an effective target for cancer treatment. However, standard cell culture conditions utilize folic acid, a non-physiological folate source for most tissues. We find that the enzyme that couples the folate and methionine metabolic cycles, methionine synthase (MTR), is required for cancer cell proliferation and tumor growth when 5-methyl tetrahydrofolate (THF), the major folate found in circulation, is the extracellular folate source. In such physiological conditions, MTR incorporates 5-methyl THF into the folate cycle to maintain intracellular levels of folates needed for nucleotide production. 5-methyl THF can sustain intracellular folate metabolism in the absence of folic acid. Therefore, cells exposed to 5-methyl THF are more resistant to methotrexate, an antifolate drug that specifically blocks folic acid incorporation into the folate cycle. Together, these data argue that the environmental folate source has a profound effect on folate metabolism, determining how both folate cycle enzymes and antifolate drugs impact proliferation.
Folate metabolism is critical for cell proliferation. One-carbon units carried by folate cofactors are necessary for many one-carbon transfer reactions1, including those required for nucleotide synthesis2. Folate species carry distinct one-carbon units in different oxidation states to support biosynthetic reactions, with each folate species in cells derived from a common tetrahydrofolate (THF) scaffold (Fig. 1A). THF cannot be synthesized by mammalian cells de novo3; thus, mammalian cells rely on uptake of exogenous precursors which can be used to generate THF and enable the one-carbon transfer reactions that support proliferation.
Figure 1. Cells can be cultured in medium with 5-methyl THF as the folate source.
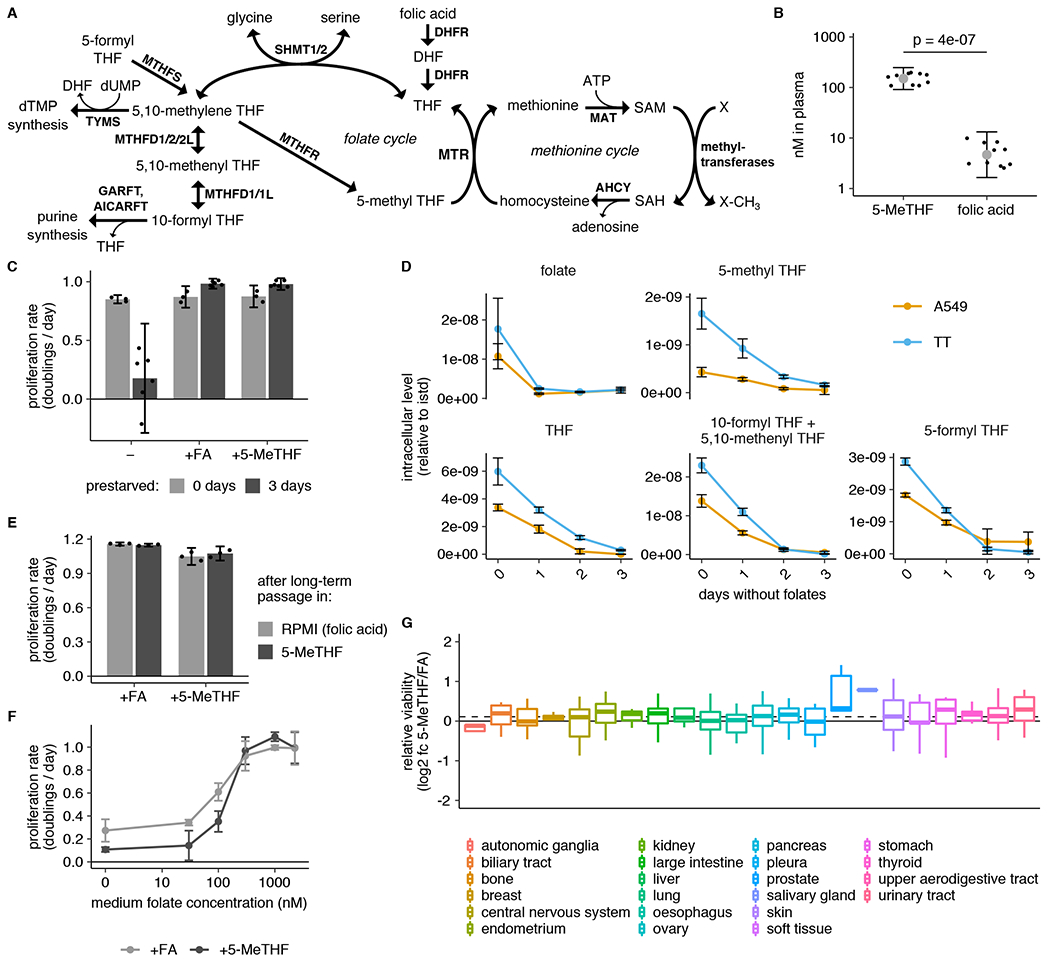
(A) Schematic showing reactions involved in folate and methionine metabolism. THF: tetrahydrofolate. SAM: S-adenosyl methionine. SAH: S-adenosyl homocysteine. dTMP: deoxythymidine monophosphate. dUMP: deoxyuridine monophosphate. MTR: methionine synthase. DHFR: dihydrofolate reductase. SHMT: serine hydroxymethyl transferase. MTHFR: methylenetetrahydrofolate reductase. MTHFD: methylenetetrahydrofolate dehydrogenase. MTHFS: methylenetetrahydrofolate synthetase. AHCY: adenosylhomocysteinase. MAT: methionine adenosyltransferase. TYMS: thymidylate synthase. AICARFT: aminoimidazole-4-carboxamide ribonucleotide (AICAR) formyltransferase (AICAR). GARFT: phosphoribosylglycinamide (GAR) formyltransferase. (B) LC/MS measurement of 5-methyl THF (5-MeTHF) and folic acid levels in plasma from NSG mice (n = 10 mice; p = 4*10−7). p-value is derived from a two-tailed, unpaired Welch’s t test. (C) Proliferation rate of A549 cells in medium containing no folate (−), folic acid (+FA), or 5-methyl THF (+5-MeTHF) before (0 days, n = 3 independent samples) or after (3 days, n = 6) of culture with no folate (“prestarved”). (D) LC/MS measurement of intracellular folic acid (folate), THF, 5-methyl THF, combined 5,10-methenyl THF/10-formyl THF, and 5-formyl THF levels in A549 or T.T cells cultured for the indicated number of days in medium lacking folates (n = 3 independent samples). Data are normalized to cell number and an internal standard. (E) Proliferation rate of A549 cells when switched to medium with the indicated folate for 4 days, after 3 weeks of continuous passaging in folic acid or 5-methyl THF (n = 3 independent samples). (F) Proliferation rate of A549 cells in medium containing the indicated amounts of either folic acid or 5-methyl THF (n = 3 independent samples). (G) Log2 fold change in relative viability for 489 barcoded cell lines grouped by tissue of origin when cultured 5-methyl THF versus folic acid (log2 fc 5-MeTHF/FA). Median (box center line), interquartile range (IQR) (box), and 1.5*IQR (whiskers) are plotted; dashed line represents the population median. (A-G) Mean +/− SD error bars are displayed.
The available precursor for THF differs in standard cell culture medium compared to circulating plasma in mammals. Standard culture medium contains folic acid, while the predominant circulating folate is 5-methyl tetrahydrofolate (5-methyl THF); folic acid and other folates from the diet comprise less than 10% of folates in circulation3,4. Further, the folate transporter SLC19A1 exhibits substantially higher affinity for 5-methyl THF compared to other folates5,6. As environmental nutrients can strongly influence cellular metabolism7,8, culturing cells in 5-methyl THF may impact intracellular metabolite levels, dependence on metabolic pathways that utilize folate species, and sensitivity to antifolate drugs.
Cells use different pathways to convert folic acid and 5-methyl THF to THF, which can accept a one-carbon unit for use in subsequent biosynthetic reactions (Fig. 1A)3. Folic acid is directly converted to THF by the enzyme dihydrofolate reductase (DHFR), which is also the target of methotrexate, an antifolate chemotherapy drug. In contrast, the conversion of 5-methyl THF to THF is coupled to synthesis of the amino acid methionine by transfer of a methyl group to homocysteine in a reaction catalyzed by the vitamin B12 (cobalamin)-dependent enzyme methionine synthase (MTR) (Fig. 1A)9,10. MTR is the only known enzyme that can utilize 5-methyl THF as a substrate and regenerate the THF scaffold. Therefore, loss of MTR activity secondary to cobalamin deficiency causes accumulation of intracellular 5-methyl THF, clinically referred to as the “methyl-folate trap”11,12. This irreversible sequestration of folates in the form of 5-methyl THF in cells results in limited THF availability to accept new one-carbon units.
Several lines of evidence suggest that MTR’s role in the folate cycle may be critical for tumor growth. Loss of MTR activity due to impairment of cobalamin binding or availability perturbs the folate cycle and slows cancer cell proliferation13,14. Further, clues from human medicine suggest that cobalamin availability is important to sustain tumor growth15–17. MTR activity also regenerates the amino acid methionine and the methyl donor cofactor S-adenosyl methionine (SAM)11,18–20 (Fig. 1A). In animals, MTR activity can affect systemic methionine and SAM levels11,21–23, and MTR inhibition has been suggested to impair cancer cell proliferation by specifically interfering with production of these metabolites18,19. However, the degree to which MTR activity is critical for cell-autonomous methionine and SAM metabolism is unclear. Further, the general metabolic consequences of perturbing MTR activity have not been examined under physiological folate conditions, and whether targeted loss of MTR expression would curtail tumor growth in animals is unknown. In addition, as MTR is required for assimilation of 5-methyl THF, but not the folic acid provided in cell culture, into the intracellular folate pool, its activity in different folate sources may alter susceptibility to antifolate chemotherapies that target environmental folate scavenging.
Results
5-methyl THF supports cancer cell proliferation
To begin to assess the impact of physiological folates on cancer cell metabolism and proliferation, we first confirmed that 5-methyl THF is the most abundant folate in mice fed a standard chow diet (Fig. 1B). Previous work has shown that leukemia cells can be cultured for short periods in 5-methyl THF13,18,19. To generalize these findings to cancer cells derived from solid tumors, we assessed the effects of culturing A549 and T.T cells in medium containing folic acid or 5-methyl THF, which exhibit average MTR expression levels relative to cancer cells included in the Cancer Cell Line Encyclopedia24 (Extended Data Fig. 1A,B). Folate deprivation did not diminish proliferation of either cell line over four days of exponential growth in culture (Fig. 1C, Extended Data Fig. 1C). This suggests that cells contain excess folates that are sufficient to sustain proliferation over this period and is consistent with previous observations in leukemia cells13. Additionally, the dialyzed fetal bovine serum used to supplement culture media contains low levels of both folic acid and 5-methyl THF (Extended Data Fig. 1D), providing a potential alternative source of some folates.
In order to study cell proliferation and metabolism in a well-defined folate source, we pre-incubated cells in media lacking any added folic acid beyond the trace levels found in dialyzed serum in an attempt to deplete intracellular folate reserves before replacing the extracellular folate source with either folic acid or 5-methyl THF. Three days of such folate “prestarvation” was sufficient to deplete intracellular levels of the folate species 5,10-methenyl THF/10-formyl THF and 5-methyl THF (Fig. 1D). This prestarvation period did not reduce intracellular levels of methionine or SAM, whose de novo synthesis is linked to folate metabolism through MTR activity (Extended Data Fig. 1E,F). Intracellular nucleotide levels were also not reduced, although the purine and dTMP synthesis intermediates directly upstream of folate-utilizing steps, glycinamide ribonucleotide (GAR), 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR), and dUMP began to accumulate after 1 day of folate deprivation (Extended Data Fig. 1G,H). Nevertheless, proliferation rate is maintained over three days in folate-depleted medium (Extended Data Fig. 1I), arguing that short-term folate deprivation allows for controlled replacement of the environmental folate source.
To examine the proliferation rates of cells cultured in different folate sources, cells were cultured for three days in folate depleted media and then cultured for four more days in media with folic acid, 5-methyl THF, or no replacement folate. Cells continued to proliferate at the same rate when cultured in media with either folic acid or 5-methyl THF as the folate source, but proliferation was impaired when deprived of folates for this extended period of time (Fig. 1C, Extended Data Fig. 1C). Importantly, cells maintain the same proliferation rate after culture in 5-methyl THF for 3 weeks, and this proliferation rate remains unchanged when cells are switched back to standard media containing folic acid as the folate source (Fig. 1E). 5-methyl THF and folic acid also sustained proliferation over a similar concentration range (Fig. 1F). Of note, the average concentration of 5-methyl THF in human plasma is 37.5 +/− 1.5 nM4 and in mouse plasma is 154.2 +/− 26.1 nM (Fig. 1B), neither of which is able to sustain maximal proliferation of A549 cells in culture (Fig. 1F). Thus, folate availability may limit rapid proliferation in some physiological settings, consistent with reports that folate supplementation accelerates the growth of some cancers25–27. Together, these results suggest that 5-methyl THF is sufficient to sustain proliferation of human cancer cells in the absence of other folate sources.
To assess whether cancer cells with varied tissues of origin and oncogenic drivers can utilize 5-methyl THF, we performed a multiplexed proliferation assay with a library of 489 barcoded adherent cancer cell lines (PRISM)28 in medium containing either folic acid or 5-methyl THF as the folate source. After 4 days of culture, we measured the relevant barcode abundance in each media condition to calculate the viability of each cell line in each condition. Most cell lines had similar viability in 5-methyl THF and folic acid media, and different cell types displayed minimal differences in viability when grown in the two folate sources, consistent with our observation that 5-methyl THF is a suitable replacement folate source for cell culture (Fig. 1G, Extended Data Fig. 1J). Of note, neither the relative viability of a cell line grown in 5-methyl THF compared to folic acid nor its proliferation rate in either medium alone correlated with its previously measured MTR mRNA expression level24 (Extended Data Fig. 1K,L), suggesting that existing variability in cancer cell MTR expression is not limiting for use of 5-methyl THF as a folate source. To determine how MTR expression changed given the provided folate source, we measured MTR protein level and found that it was slightly lower in A549 and T.T cells cultured in 5-methyl THF compared to folic acid (Extended Data Fig. 1M).
MTR is required to grow in physiological folates
Given the lack of correlation between MTR expression level and cell proliferation in 5-methyl THF, we questioned whether MTR is necessary to support proliferation when 5-methyl THF is the major folate source. To do this, we disrupted MTR expression in A549 and T.T cells using CRISPR-Cas9 genome editing (sgMTR) and then re-expressed an sgRNA-resistant version of MTR (+MTR) or an empty-vector control (+EV) to create isogenic cell lines that differ in MTR expression (Fig. 2A). In A549 and T.T cells, MTR is essential for proliferation when 5-methyl THF is the sole folate source (Fig. 2B) or when 5-methyl THF comprises 96% of the folate source with 4% folic acid, approximating the physiological ratio of reduced folates to folic acid in human plasma4 (Extended Data Fig. 2A). MTR knockout cells display >10-fold increased steady-state levels of 5-methyl THF regardless of folate source (Fig. 2C), consistent with previous results29. As expected, the 5-methyl THF pool remained stable in MTR knockout cells upon folate starvation (Extended Data Fig. 2B), consistent with an inability of these cells to utilize 5-methyl THF through MTR. 10-formyl THF and 5,10-methenyl THF, which are synthesized by adding one-carbon units to THF, were less abundant in MTR knockout cells than control cells and were further depleted in 5-methyl THF media (Fig. 2C, Extended Data Fig. 2B). This is consistent with a defect in THF regeneration from 5-methyl THF in MTR knockout cells in 5-methyl THF medium and some 5-methyl THF trapping even in folic acid medium. These species were rapidly depleted following folate starvation (Extended Data Fig. 2B); as expected, MTR knockout cells were more sensitive to a three-day folate pre-starvation period than control cells (Extended Data Fig. 2C). Interestingly, culture in 5-methyl THF also led to accumulation of 5-formyl THF (Fig. 2C), a storage form of folate that may alter the activity of other folate-dependent enzymes29, suggesting that growth in 5-methyl THF may broadly affect intracellular folate metabolism.
Figure 2. Methionine synthase is only essential for proliferation when 5-methyl THF is the folate source.
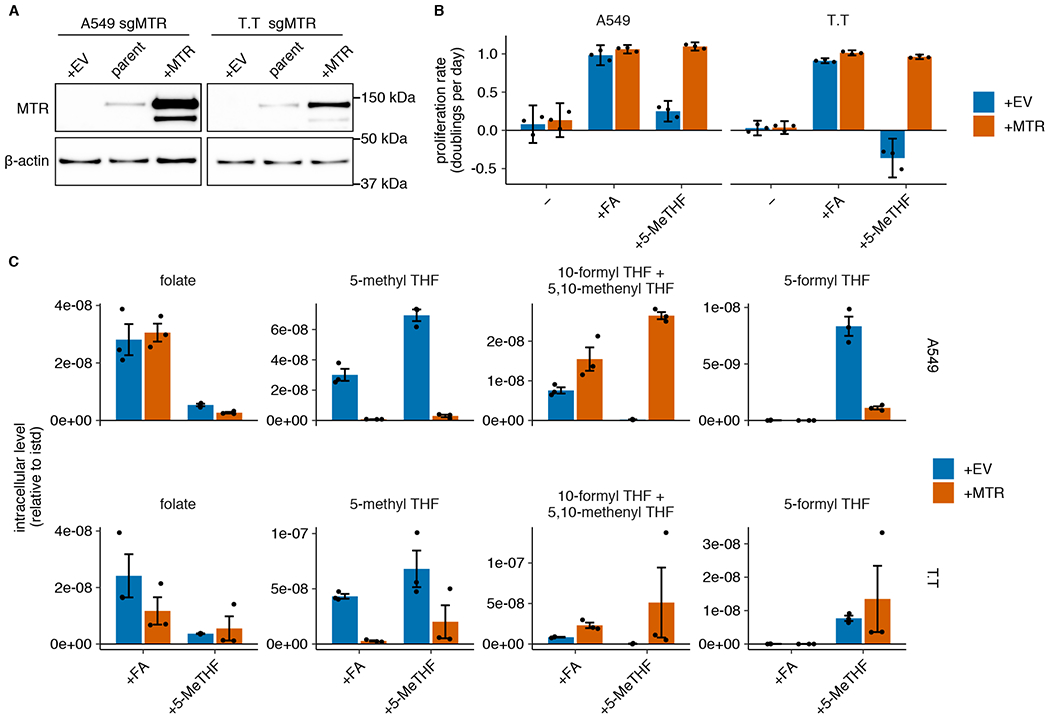
(A) Western blot analysis of MTR expression in parental A549 and T.T cells as well as MTR knockout cells expressing an empty vector (+EV) or MTR (+MTR), cultured in RPMI (representative of 2 independent experiments). (B) Proliferation rates of A549 and T.T MTR knockout cells +EV or +MTR after culture in folic acid (+FA) or 5-methyl THF (+5-MeTHF) for 4 days after a folate pre-starvation period (n = 3 independent samples). Mean +/− SD error bars are displayed. (C) LC/MS measurement of intracellular folate, 5-methyl THF, combined 5,10-methenyl THF/10-formyl THF, and 5-formyl THF levels in A549 and T.T MTR knockout cells +EV or +MTR after culturing cells in the indicated folate as in B. Data are normalized to cell number and an internal standard. Mean +/− SEM error bars are displayed.
Methionine synthase is required to supply tetrahydrofolate
We next sought to characterize why MTR activity is metabolically necessary for proliferation in physiological folate medium. MTR enables production of methionine, the downstream methyl donor SAM, and THF, which carries one-carbon units derived from the amino acid serine for purine and dTMP synthesis. Based on our observations that folate starvation affects intermediates in nucleotide synthesis without reducing methionine or SAM levels (Extended Data Fig. 1G,H), and previous work linking MTR inactivation to impaired pyrimidine synthesis in standard culture conditions29, we began by assessing the role of MTR in purine and pyrimidine synthesis. Purine synthesis requires 10-formyl THF, which is indistinguishable from 5,10-methenyl THF when measured by LC/MS due to rapid interconversion after cell lysis30,31. We find that the combined levels of 10-formyl THF and 5,10-methenyl THF are depleted in MTR knockout cells, suggesting that purine synthesis may be compromised (Fig. 2C). Levels of adenosine and guanosine nucleotides were decreased in MTR knockout cells, particularly when cultured in 5-methyl THF relative to folic acid (Fig. 3A–B, Extended Data Fig. 3A–B), correlating with the reduced proliferation rate of MTR knockout cells in 5-methyl THF (Figure 2B). The decrease in these nucleotides, even when MTR knockout cells are cultured in folic acid, may be related to the finding that some THF produced from folic acid is irreversibly trapped as 5-methyl THF (Fig. 2C, Extended Data Fig. 2B).
Figure 3. MTR is required for nucleotide synthesis in 5-methyl THF medium.
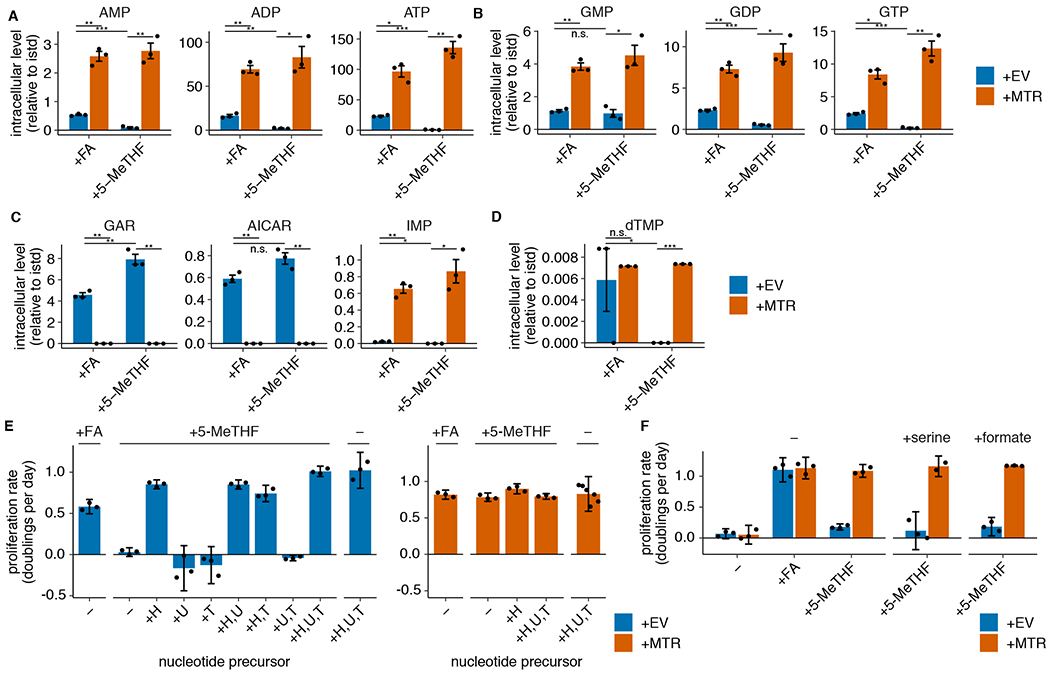
(A-D) LC/MS measurement of intracellular AMP, ADP, ATP (A), GMP, GDP, GTP (B), GAR, AICAR, IMP (C), and dTMP (D) levels in A549 MTR knockout cells without (+EV) or with MTR expression (+MTR) cultured for 4 days in folic acid (+FA) or 5-methyl THF (5-MeTHF) after a folate prestarvation period (n = 3 independent samples; +EV +FA vs +MTR +FA p-values: AMP: 0.006, ADP: 0.003, ATP: 0.015, GMP: 0.004, GDP: 0.007, GTP: 0.011, GAR: 0.002, AICAR: 0.003, IMP: 0.007, dTMP: 0.79; +EV +5-methyl THF vs +MTR +5-methyl THF p-values: AMP: 0.01, ADP: 0.022, ATP: 0.005, GMP: 0.019, GDP: 0.015, GTP: 0.009, GAR: 0.003, AICAR: 0.005, IMP: 0.025, dTMP: 0.001; +EV +FA vs +EV +5-methyl THF p-values: AMP: 0.0002, ADP: 0.008, ATP: 0.0006, GMP: 0.54, GDP: 0.0004, GTP: 0.0007, GAR: 0.01, AICAR: 0.07, IMP: 0.016, dTMP: 0.019). Data are normalized to protein concentration and an internal standard. Mean +/− SEM error bars are displayed. (E-F) Proliferation rates of A549 MTR knockout cells without +EV or +MTR cultured as in A-D in the indicated folate, with or without the addition of 100 μM each of the indicated nucleotide precursors hypoxanthine (H), uridine (U), thymidine (T) (n = 3 independent samples; except +MTR +H,U,T n = 6) or the addition of 1 mM serine or formate (n = 3 independent samples). Mean +/− SD error bars are displayed. p-values are derived from a two-tailed, unpaired Welch’s t test (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
If purine synthesis is impaired due to a lack of folate species derived from THF, intermediates prior to the steps that utilize folates should accumulate (Extended Data Fig. 1G,H)32. Consistent with this hypothesis, GAR and AICAR levels are elevated in MTR knockout cells and further increase in MTR knockout cells cultured in 5-methyl THF relative to folic acid (Fig. 3C, Extended Data Fig. 3C). Further supporting this hypothesis, inosine monophosphate (IMP), a purine precursor downstream of GAR and AICAR, is depleted in MTR knockout cells, particularly when cultured in 5-methyl THF (Fig. 3C, Extended Data Fig. 3C).
Synthesis of the pyrimidine nucleotide deoxythymidine monophosphate (dTMP) requires the folate species 5,10-methylene THF (Fig. 1A), which is unstable and not directly measurable by LC/MS30. If THF production is disrupted by MTR loss in physiological folate medium, cells would be predicted to also have low levels of 5,10-methylene THF and therefore exhibit impaired pyrimidine synthesis. Indeed, dTMP levels are depleted in MTR knockout cells cultured in 5-methyl THF (Fig. 3D, Extended Data Fig. 3D).
To assess whether purine and/or pyrimidine depletion causes proliferation arrest of MTR knockout cells in 5-methyl THF, we performed a series of metabolic rescue experiments. Addition of the pyrimidine salvage precursors uridine and thymidine and the purine salvage precursor hypoxanthine supports proliferation of MTR knockout and +MTR cells in the absence of any folate source and rescues MTR knockout cell proliferation in 5-methyl THF (Fig. 3E, Extended Data Fig. 3E), confirming that the proliferation defect is caused by nucleotide depletion. Exogenous hypoxanthine alone rescued proliferation of MTR knockout cells in 5-methyl THF over a four-day period (Fig. 3E, Extended Data Fig. 3E). However, MTR knockout cells ceased proliferation after 12 days in hypoxanthine alone (Extended Data Fig. 3F), likely because dTMP cannot be synthesized without folate precursors and is not rescued by hypoxanthine. The short-term rescue of proliferation by hypoxanthine alone is consistent with the fact that purine synthesis requires higher flux from the folate cycle than dTMP synthesis33. As a result, trace residual folate sources may sustain pyrimidine synthesis longer than purine synthesis. Addition of thymidine reduced proliferation in T.T cells (Extended Data Fig. 3E), likely due to its ability to cause cell cycle arrest34. Together, these data suggest that nucleotide synthesis is impaired by MTR knockout.
In addition to requiring THF, nucleotide synthesis can be limited by availability of the one-carbon precursor units carried by THF. As MTR transfers a one-carbon unit from 5-methyl THF to methionine, it has been proposed that methionine and nucleotide synthesis compete for one-carbon units in some contexts35. MTR knockout would thus be predicted to increase one-carbon precursor availability; therefore, one carbon units are not likely to explain the growth defect of MTR knockout cells in physiological folates. Indeed, exogenous addition of the one-carbon precursors serine or formate did not rescue growth of MTR knockout cells in physiological folate medium (Fig. 3F, Extended Data Fig. 3G), confirming that one-carbon units are not limiting for proliferation in these cells. Thus, MTR knockout limits nucleotide synthesis by preventing production of THF from 5-methyl THF.
MTR is not required to maintain intracellular methionine
MTR can support both methionine and folate metabolism9,11. Though nucleotide precursors were sufficient to fully rescue MTR knockout in physiological folate medium, and providing additional methionine did not rescue proliferation of MTR knockout cells (Extended Data Fig. 4A), high methionine levels in the medium could mask an additional requirement for MTR to synthesize methionine in physiological conditions. Standard culture medium contains 100 μM methionine, a concentration similar to that found in murine plasma36. However, human plasma methionine levels are closer to 25 μM37. Thus, we tested whether restriction of exogenous methionine to physiological levels would specifically inhibit proliferation in MTR knockout cells, and whether supplementation of nucleotide precursors would fail to fully rescue proliferation in these conditions. Proliferation was not affected by a reduction from 100 μM to 25 μM extracellular methionine, independent of the folate source (Fig. 4A, Extended Data Fig. 4B). A further reduction in extracellular methionine to 10 μM did slightly reduce proliferation, but this effect was independent of the folate source provided, and nucleotide precursors still rescued both MTR knockout and +MTR cell proliferation to the same, methionine-limited maximum rate (Fig. 4A, Extended Data Fig. 4B). Consistent with a limited role for MTR in maintaining methionine availability, methionine levels are higher in MTR knockout cells than control cells, especially in 5-methyl THF (Fig. 4B, Extended Data Fig. 4C); this accumulation of methionine may be related to reduced growth and protein synthesis, as serine and lysine are also higher in this condition. Further, a parallel study that traced the fate of labeled methionine found little to no role for MTR in new methionine synthesis in mouse tumors or tissues38. These results suggest that MTR is not required for maintenance of intracellular methionine levels.
Figure 4. MTR knockout reduces SAM but not methionine levels.
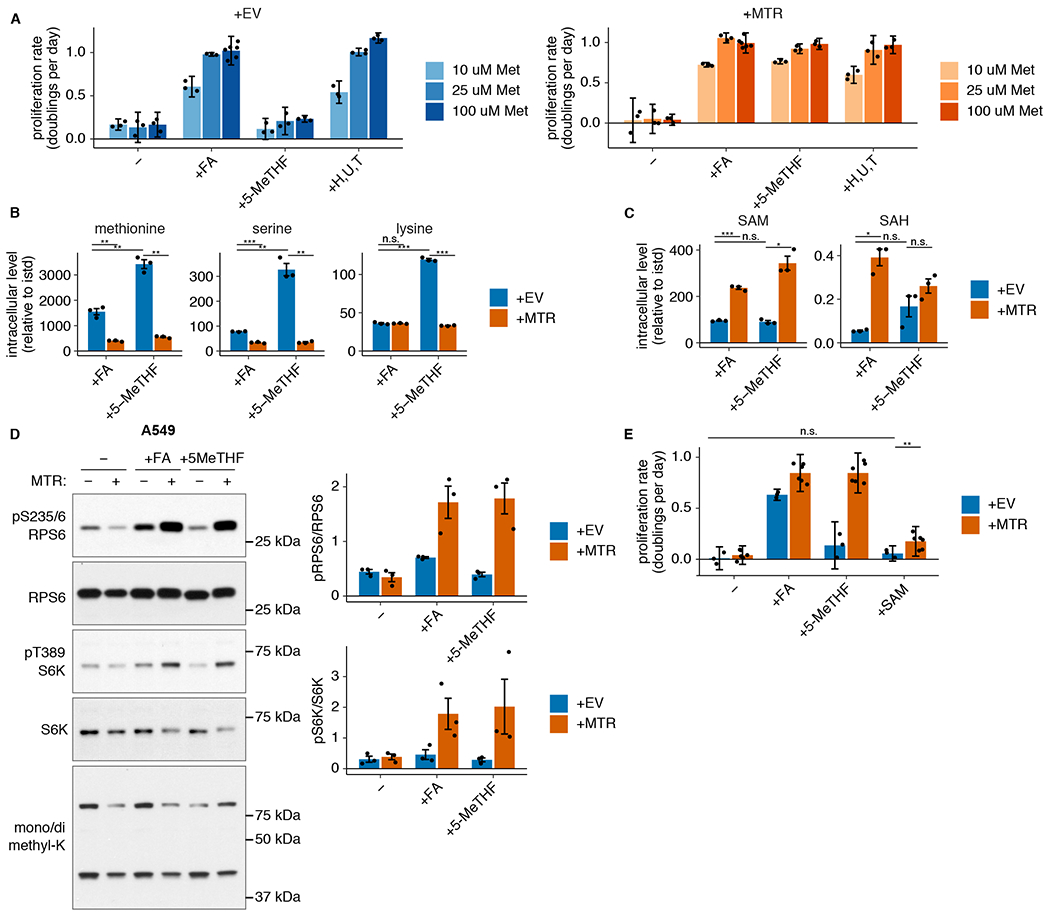
(A) Proliferation rates of A549 MTR knockout cells without (+EV) or with MTR expression (+MTR) cultured in folic acid (+FA) or 5-methyl THF (5-MeTHF) or in 100 uM each nucleotide precursor (hypoxanthine, uridine, and thymidine) for 4 days after a folate prestarvation period, at varied extracellular methionine concentrations (n = 3 independent samples, except n = 6 for +FA 100 uM methionine +EV/+MTR). (B-C) LC/MS measurement of intracellular methionine, serine, lysine (B), SAM, and SAH levels (C) in A549 MTR knockout cells +EV or +MTR cultured in the indicated folate as in A (n = 3 independent samples; +EV +FA vs +MTR +FA p-values: methionine: 0.01, serine: 0.0001, lysine: 0.936, SAM: 0.0009, SAH: 0.012; +EV +5-methyl THF vs +MTR +5-methyl THF p-values: methionine: 0.003, serine: 0.007, lysine: 0.0001, SAM: 0.012, SAH: 0.152; +EV +FA vs +EV +5-methyl THF p-values: methionine: 0.001, serine: 0.01, lysine: 9.1*10−6, SAM: 0.47, SAH: 0.1). Data are normalized to protein concentration and an internal standard. (D) Western blots to assess phosphorylation of mTORC1 targets and levels of mono- or dimethyl lysine-containing proteins in A549 MTR knockout cells +EV or +MTR cultured in the indicated folate for 16 hours. The ratio of phospho-protein to total protein signal from 3 independent replicates is shown. (E) Proliferation rates of A549 MTR knockout cells +EV or +MTR cultured in the indicated folate as in A, with or without the addition of 1 mM SAM (n = 3 independent samples for +EV, n = 6 for +MTR; +EV −folate vs +EV +SAM p = 0.084, +EV +SAM vs +MTR +SAM p = 0.001). (A,E) Mean +/− SD error bars are displayed. (B-D) Mean +/− SEM error bars are displayed. p-values are derived from a two-tailed, unpaired Welch’s t test (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
Methionine is directly converted to SAM by addition of adenosine triphosphate (ATP)39 (Fig. 1A), and defects in purine synthesis have previously been shown to decrease SAM levels40. Consistent with this, SAM and S-adenosyl homocysteine (SAH) levels are decreased in MTR knockout cells (Fig. 4C, Extended Data Fig. 4D). As SAM acts as both a methyl donor and a direct signal to mTORC1 kinase, its levels could affect proliferation by altering protein or DNA methylation patterns or mTORC1 signaling41–43. However, SAM levels are equally diminished in MTR knockout cells in both environmental folate sources, arguing that SAM depletion cannot explain why these cells do not proliferate in 5-methyl THF. Changes in methylation of cytosolic proteins did not correlate with the reduced SAM levels or proliferation caused by MTR knockout in 5-methyl THF (Fig. 4D, Extended Data Fig. 4E). Phosphorylation of the direct mTORC1 kinase target S6 kinase, as well as the S6 kinase target RPS6, largely correlated with SAM levels (Fig. 4D, Extended Data Fig. 4E); however, addition of exogenous SAM43 was unable to restore MTR knockout cell proliferation in 5-methyl THF (Fig. 4E, Extended Data Fig. 4F). Therefore, MTR expression is important to maintain levels of SAM and SAH, but the decreases in SAM and SAH levels observed upon MTR knockout are not sufficient to explain reduced cell proliferation in physiological folate conditions.
Tumor formation requires MTR for folate metabolism
Given that 5-methyl THF is the predominant folate in circulation in humans4 and mice (Fig. 1B), our results suggest that MTR may be essential for tumor formation. However, as folic acid is also present at low levels in the circulation of both humans4 and mice (Fig. 1B), circulating folic acid might be sufficient to sustain tumor growth in the absence of MTR. To examine whether MTR is required for tumor growth given the available folates in vivo, A549 cells with or without MTR expression were implanted into the flanks of immunocompromised NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice fed a diet containing folic acid at levels consistent with a typical human diet44,45. MTR expression was required for tumor formation in these conditions (Fig. 5A). To test whether MTR is essential for tumor growth due to its role in utilizing circulating 5-methyl THF, mice were supplemented with excess folic acid to provide a secondary folate source that circumvents the need for MTR-mediated conversion of 5-methyl THF to THF. Unlimited provision of folic acid in the drinking water of mice resulted in increased plasma folic acid levels in some mice (Fig. 5B), without altering 5-methyl THF levels in circulation (Fig. 5C). The degree of increase in plasma folic acid levels was variable and did not result in a statistically significant increase in average concentration (Fig. 5B); however, folic acid supplementation was sufficient to support growth of MTR knockout tumors (Fig. 5D), arguing that MTR is essential for tumor growth in vivo because it is required to generate THF.
Figure 5. Physiological folates prevent growth of MTR knockout tumors and cause methotrexate resistance.
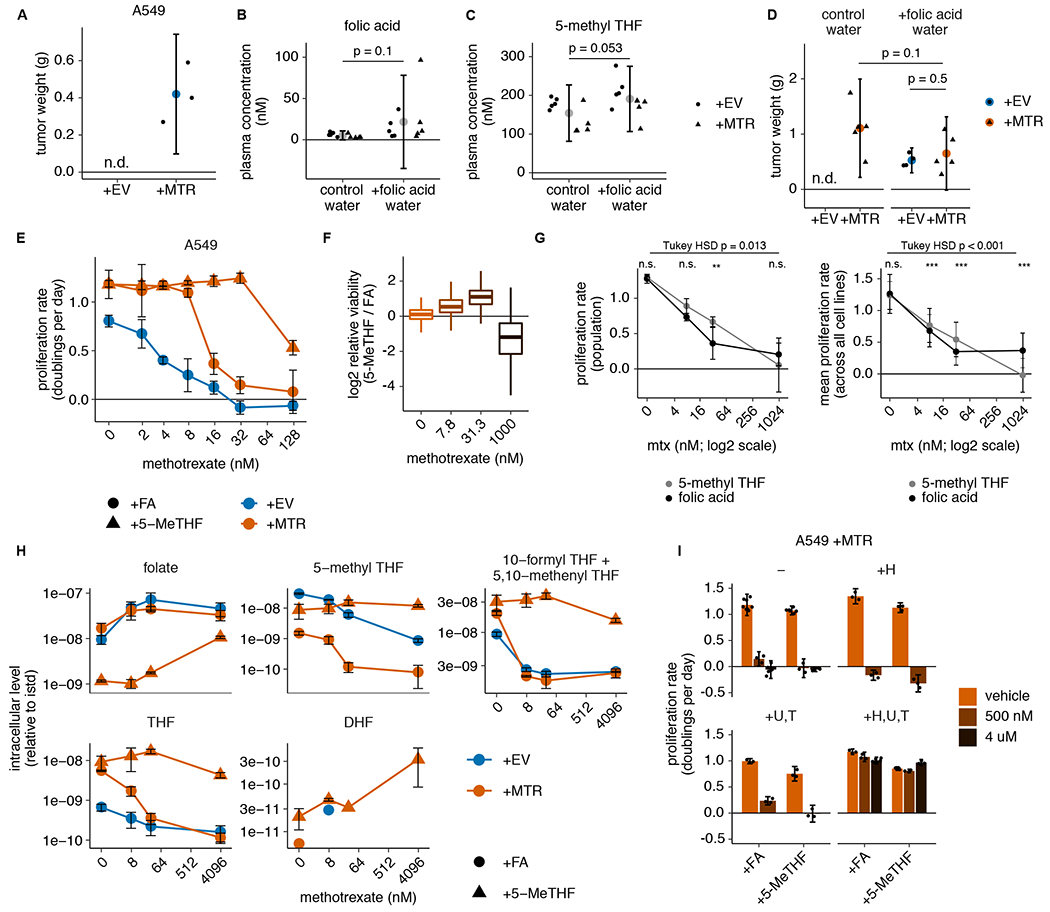
(A) Tumor weight of subcutaneous xenografts generated by injection of 100,000 A549 MTR knockout cells without (+EV) or with (+MTR) MTR expression into the flanks of NSG mice. Tumors were harvested after 4 months (n=3 mice per condition; n.d.: not detected). (B-C) Plasma concentration of folic acid (B) and 5-methyl THF (C) in NSG mice provided with unlimited water (control water) or water containing 0.1 g/L folic acid (+folic acid water) for 3 weeks (n=10 mice per condition; folic acid p = 0.1; 5-methyl THF p = 0.053). (D) Tumor weight of subcutaneous xenografts formed by injecting 1,000,000 A549 MTR knockout cells +EV or +MTR into the flanks of NSG mice provided with control or folic acid water as in B-C since the day of injection. Tumors were harvested after 3 months (n=5 mice per condition; +MTR control vs. folic acid water p = 0.1, +EV vs +MTR folic acid water p = 0.5; n.d.: not detected). (E) Proliferation rates of A549 MTR knockout cells +EV or +MTR cultured in folic acid (+FA) or 5-methyl THF (5-MeTHF) for 4 days across a range of methotrexate doses (n = 3 independent samples). (F) Overall population log2 fold change in relative viability of 489 cell lines cultured in 5-methyl THF versus folic acid as in E at the indicated methotrexate doses. Median (box center line), interquartile range (IQR) (box), and 1.5*IQR (whiskers) are plotted (n = 489 cell lines). (G) Overall pooled 489 cell line population bulk proliferation rate in doublings per day (left panel, n = 4 measurements of 1 pooled sample; overall folic acid vs. 5-methyl THF dose-response p-value = 0.013, pairwise comparison adjusted p-values: 0 nM p = 1, 7.8 nM p = 0.233, 31.3 nM p = 0.001, 1000 nM p = 0.28) and averaged proliferation rates across all cell lines (right panel, n = 489 cell lines; overall folic acid vs. 5-methyl THF dose-response p-value = 3.5*10−5, pairwise comparison adjusted p-values: 0 nM p = 0.85, 7.8 nM p = 5.2*10−5, 31.3 nM p ~ 0, 1000 nM p ~ 0) cultured in 5-methyl THF or folic acid as in E at the indicated methotrexate doses. Tukey HSD and pairwise comparison p-values at each dose were calculated by correcting a two-way ANOVA test for multiple comparisons between the dose response curves in the two folate sources. (H) LC/MS measurement of intracellular folate, 5-methyl THF, combined 5,10-methenyl THF/10-formyl THF, THF, and DHF levels in A549 MTR knockout cells +EV or +MTR cultured in the indicated folate as in E across a range of methotrexate doses (n = 3 independent samples). Data are normalized to protein concentration and an internal standard. (I) Proliferation rates of A549 MTR knockout cells +MTR cultured in the indicated folate as in E with or without the addition of 100 μM each of the indicated nucleotide precursors hypoxanthine (H), uridine (U), and thymidine (T), as well as methotrexate or vehicle (DMSO) (n = 3 independent samples except n=6 for samples +FA − vehicle, − 4 uM, and +H,U,T +4 uM and +5-MeTHF − vehicle). For the +H and +U,T conditions, only the lower dose (500 nM) of methotrexate was assessed. (A-I) Mean +/− SD error bars are displayed. p-values are derived from a two-tailed, unpaired Welch’s t test except as otherwise indicated in G (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
Physiological folates render cells resistant to methotrexate
MTR is not required for proliferation in standard culture conditions because THF can be produced from folic acid by the enzyme DHFR (Fig. 1A). We hypothesized that excess environmental folic acid might create a non-physiological dependence on DHFR activity, perhaps accounting for the observation that cancer cells are more sensitive to the DHFR inhibitor methotrexate in culture than in human patients46. Indeed, both wild-type and complemented MTR knockout cells were more sensitive to methotrexate in folic acid than 5-methyl THF medium (Fig. 5E; Extended Data Fig. 5A,B). Furthermore, A549 MTR knockout cells were more sensitive to methotrexate than A549 +MTR cells in folic acid medium (Fig. 5E), likely due to the baseline reduction in folate and nucleotide levels we observed in MTR knockout cells even in folic acid medium (Fig. 2C, Extended Data Fig. 2B, Fig. 3A–D). However, differential sensitivity to methotrexate with and without MTR was not observed in T.T cells (Extended Data Fig. 5B), suggesting that MTR expression and environmental folate source are independent influences on methotrexate sensitivity.
To assess whether cancer cells are generally more resistant to methotrexate in physiological folate medium, we performed a PRISM multiplexed barcoded proliferation assay with 489 cell lines cultured in folic acid or 5-methyl THF and treated with several methotrexate doses chosen based on the dose response curves for wild-type A549 and T.T cells (Extended Data Fig. 5A). At a population level (Fig. 5F,G; Extended Data Fig. 5D), and across individual cancer types (Extended Data Fig. 5C), cells were more resistant to low doses of methotrexate when grown in 5-methyl THF, despite similar viabilities in folic acid and 5-methyl THF without methotrexate (Fig. 1G; replotted in first panel Extended Data Fig. 5C). At a very high methotrexate dose, cells did not proliferate in either medium (Fig. 5G, Extended Data Fig. 5D), and the resulting low barcode counts showed increased variance; however, cells grown in folic acid may be less sensitive than those grown in 5-methyl THF at that methotrexate dose (Fig 5F,G; Extended Data Fig. 5C,D). Together, these results suggest that the folate source broadly affects methotrexate sensitivity in cells derived from diverse tumor types.
To determine the metabolic basis for the folate source-specific response to methotrexate, we performed LC/MS to measure intracellular folates and polar metabolites across a range of methotrexate doses in folic acid and 5-methyl THF media. In folic acid medium, intracellular folates were depleted in concert with proliferation arrest in response to methotrexate (Fig. 5E,H), consistent with the drug acting as a DHFR inhibitor to prevent environmental folate processing into THF and downstream folate species. However, intracellular folates were only slightly reduced by methotrexate treatment in 5-methyl THF, even at doses of methotrexate that completely arrested proliferation; in this context, DHF accumulated as proliferation rate declined (Fig. 5E,H). These data are consistent with methotrexate inhibiting folic acid and DHF reduction to THF without impacting utilization of 5-methyl THF.
Purine and pyrimidine synthesis, as assessed by purine and thymidylate levels and by accumulation of substrates directly upstream of folate-utilizing steps, appeared diminished at all methotrexate doses that inhibited proliferation (Extended Data Fig. 5E). Supplementation of both purine and pyrimidine precursors also rescued proliferation (Fig. 5I), indicating that methotrexate ultimately arrests proliferation due to nucleotide deficiency in either folate source. Intracellular amino acids accumulated as proliferation rate declined, suggesting that neither methionine levels nor access to serine for one-carbon units were specifically compromised by methotrexate treatment (Extended Data Fig. 5F). Methotrexate accumulated in cells in a dose-dependent manner, suggesting that its import was not affected by folate source (Extended Data Fig. 5G). Given that at high levels, DHF may act as a competitive inhibitor of folate-utilizing enzymes47, DHF accumulation (Figure 5H) rather than folate depletion may account for the toxicity of higher methotrexate doses in conditions where 5-methyl THF is the predominant folate source.
Discussion
Culturing cancer cells in a physiological folate environment modifies cell metabolism and the response to antifolate drugs, and provides insight into the coupling between two distinct metabolic pathways that can both be critical for cancer cell metabolism: the methionine and folate cycles. Of note, independent work from Wang et al. co-published with this study38 reached similar conclusions about the role of MTR in methionine and folate metabolism, providing direct evidence that MTR is important for THF regeneration rather than methionine production in vivo and highlighting the critical function of MTR to support the folate cycle for nucleotide synthesis. This orthogonal study demonstrated a key role for MTR in tumor formation and folate metabolism using a set of cell lines derived from different cancer tissues of origin than those reported in our study, suggesting that the role of MTR is likely generalizable across many cancers.
Methionine is an essential amino acid for mammals, but can be re-synthesized from precursors involved in one-carbon and polyamine metabolism48. Although MTR inhibition has been reported to acutely reduce methionine and SAM levels in mice21, other pathways can compensate for methionine synthesis in the absence of MTR11,21, and we observe that MTR is not essential for maintaining intracellular methionine levels in cancer cell lines. This finding is consistent with the co-published study of Wang et al.38, which demonstrated using stable isotope tracing in vitro and in vivo that the majority of methionine in tumor cells is not derived from re-methylation of homocysteine by MTR. However, SAM, which is synthesized from methionine and ATP, is nevertheless depleted by loss of MTR. This decoupling between methionine and SAM pools is likely explained by MTR loss stalling the folate cycle and reducing intracellular ATP levels, underscoring a critical link between intracellular methylation donors and purine nucleotide synthesis40. Although changes to cytoplasmic methylation state and SAM levels alone could not explain the proliferation defect of MTR knockout cells in physiological folates, it remains possible that MTR activity influences the epigenomic methylation state indirectly through this ATP-mediated effect on SAM levels.
Although MTR appears to play a minor role in net methionine production, we find that MTR is critical for regenerating the THF backbone from 5-methyl THF to support nucleotide synthesis. Interestingly, dietary methionine limitation that inhibits tumor growth can therapeutically synergize with nucleotide synthesis inhibition in some cancers35,49. Of note, dietary methionine limitation has been found to inhibit de novo methionine synthesis35, likely indicating lower MTR activity. Our results suggest a possible mechanism through which methionine restriction could impact tumor growth in synergy with throttled nucleotide synthesis, as limited THF regeneration due to reduced MTR activity could further impair nucleotide synthesis.
We found that MTR expression is essential for cancer cells to form tumors in mice, and can affect sensitivity to antifolate drugs. As suggested decades ago9, MTR inhibition might be useful as a cancer therapy and warrants renewed consideration for drug development. Effective targeting of MTR would require that tumor cells have a higher demand for MTR activity than other tissues. The success of other therapies that influence folate metabolism50–53 suggests that this may be the case, and tumor types with a high demand for nucleotide synthesis might be expected to be most sensitive to MTR inhibition. Notably, MTR inhibition may not have the same effect as other anti-folates. For instance, the anti-folate pemetrexed is an effective therapy for non-squamous non-small cell lung cancer (NSCLC)54, and increased thymidylate synthase expression in squamous NSCLC55,56 correlates with reduced sensitivity to pemetrexed57. However, squamous NSCLC cells do not exhibit MTR overexpression58, possibly due to the complexities of MTR synthesis and cofactor regulation59,60; MTR function and stability are supported by multiple enzymes that process and transport cobalamin61, as well as a protein that specifically chaperones62 and regenerates MTR63. Indeed, though we observed higher MTR protein levels in MTR-complemented cells compared to wild-type cells (Fig. 2A), these pairs of cell lines were similarly sensitive to methotrexate (Fig. 5E compared to Extended Data Fig. 5A, left panel; Extended Data Fig. 5B compared to Extended Data Fig. 5A, right panel) and grew at the same rate (Fig. 1C and Extended Data Fig.1C compared to Fig. 2B). Indeed, we observed no significant correlation between MTR expression level and proliferation rate in physiological folates across 489 cancer cell lines (Extended Data Fig. 1K,L). As varying the environmental folate source rewires intracellular folate metabolism, altering dependence on MTR and response to antifolate drugs, the use of culture models with physiological folates will be critical to improve efforts to target folate metabolism with new and existing drugs.
METHODS
Cell culture
Cells were passaged in RPMI-1640 (Corning Life Sciences, Tewksbury, MA) with 10% fetal bovine serum (FBS) that had been heat inactivated for 30 min. at 56°C (VWR Seradigm, Lot 120B14). All cells were cultured in a Heracell (Thermofisher) humidified incubators at 37°C and 5% CO2. A549 (RRID:CVCL_0023) cells were obtained from ATCC (Manassas, VA). A549 cells are derived from a lung adenocarcinoma in a male patient. T.T cells (RRID:CVCL_3174) were derived from an oral metastasis of an esophageal squamous cell carcinoma in a male patient. All cell lines were regularly tested for mycoplasma contamination using the Mycoprobe mycoplasma detection kit (R and D Systems, Minneapolis, MN). All cell lines were validated using Short Tandem Repeat (STR) testing most recently in December 2016. For experiments, cells were grown in RPMI-1640 without phenol red with 10% FBS that has been dialyzed to remove small molecules (Gibco, 26400044; batch 1: Lot #1841165, batch 2: Lot #2093857). We noted that different lots of dialyzed serum yielded subtle differences in sensitivity to folate starvation, so comparisons were only made between experiments carried out using the same lot of dialyzed serum. RPMI-1640 lacking folic acid was made using the method outlined previously64. Briefly, enough of all of the components of RPMI-1640 media except for folic acid were weighed out to make 25 L of media, then the resulting powder was homogenized using an electric blade coffee grinder (Hamilton Beach, Glen Allen, VA, 80365) that had been washed with methanol then water. The resulting powder was resuspended in water to make RPMI-1640 media lacking folic acid and the pH was adjusted to ~7.4 with 1M HCl to improve solubilization. Folic acid (Sigma-Aldrich, St. Louis, MO, F8758) or (6S)-5-Methyl-5,6,7,8-tetrahydrofolic acid (5-methyl THF) (Schircks Laboratories, Bauma, Switzerland, 16.236) was dissolved in water to make 44-fold concentrated 100 uM stock solutions, then added back to RPMI-1640 media lacking folic acid to yield a final concentration of 2.26 μM, which is the standard concentration of folic acid in RPMI-1640. Methotrexate (MedChemExpress, HY-14519) was dissolved in DMSO.
Proliferation rates
Cellular proliferation rate in different media conditions was determined as previously described65, and outlined below. Cell lines expressing MTR were pre-starved of all folates for 3 days, and MTR knockout cell lines were prestarved of all folates for 1 day; these time periods were empirically determined to be the longest folic acid starvation period that still allowed for full recovery of maximal proliferation rate when the folate source was restored. Cells were plated into RPMI-1640 lacking folic acid at a density of 300,000 cells per 10 cm plate. 4 days after plating, cells were trypsinized, counted and plated into six well dishes (Corning Life Sciences) in 2.5 mL of RPMI-1640 medium lacking folic acid and allowed to adhere to the plate overnight. Initial seeding density was 30,000-50,000 cells/well. The next day, a six well plate of cells was trypsinized and counted to provide a number of cells at the start of the experiment. Cells were then washed once with 2 mL of phosphate buffered saline (PBS), and 2 mL of the indicated medium was added.
For rescue experiments, 1 mM methionine, 1 mM serine, or 1 mM formate from 100 or 1000X stocks in water were added to the experimental media. SAM (Sigma-Aldrich, A7007) was dissolved directly in experimental media to 1 mM, which was then filter sterilized. This concentration of SAM was chosen based on evidence that it can rescue mTORC1 signaling through the specific sensor SAMTOR in methionine starved cells43. 100 μM each uridine and thymidine from 1000X stocks in water, and hypoxanthine from a 200X stock in 0.1 N NaOH, were added to the experimental media. Uridine, thymidine, and hypoxanthine were chosen for nucleotide precursor rescue experiments because they are readily taken up and utilized by cells, and can replace all de novo nucleotide synthesis pathways that require folates66,67. Cells were then trypsinized and counted 4 days after adding the indicated medium. Proliferation rate was determined using the following formula: doublings per day = log2(final cell count / initial cell count) / 4 days. Cells were counted using a Cellometer Auto T4 Plus Cell Counter (Nexcelom Bioscience, Lawrence, MA). All proliferation experiments were carried out in biological triplicate unless otherwise indicated on figure legends.
For the serial passaging proliferation assay in nucleotide precursor-supplemented medium shown in Extended Data Fig. 3F, cells were trypsinized, counted, and re-plated directly into fresh treatment media every 4 days for 16 days in total.
Generation of MTR knockout cells
MTR knockout using CRISPR-Cas9 was accomplished using the pLenti-CRISPR v2 plasmid (Addgene Plasmid 49535)68. sgRNAs were designed based on previously described algorithms69, and MTR knockout was carried out using the following sgRNA sequence: TGGCATTGATCTCATCCCGC. Cell lines were diluted in 96 well plates to obtain single cell clones, and loss of MTR expression in individual clones was confirmed by western blot. sgRNA resistant cDNA of MTR was ordered from VectorBuilder (Shenandoah, TX) with silent mutations in the region of the gene targeted by the sgRNA.
Western blot analysis
Cells from a 6 well plate were once washed in PBS and scraped into 300 μL RIPA buffer [25 mM Tris-Cl, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, cOmplete protease inhibitor (Roche, Basel, Switzerland), phosSTOP (Sigma-Aldrich)]. The resulting lysate was clarified by centrifugation at 21000 x g for 10 min. Protein concentration of the lysate was determined by BCA assay (Thermofisher). ~10 ug lysate in 1X NuPAGE LDS sample loading buffer (Novex) and 5 mM dithiothreitol was denatured at 70°C for 10 min and resolved by SDS-PAGE using NuPAGE 4-12% Bis Tris gels (Novex) and 1X MES Buffer (Novex) running at 150-200V until the dye front left the gel. After SDS-PAGE resolution, protein extracts were transferred to 0.45 um nitrocellulose at 18V for 1 hour using a Trans-Blot® SD Semi-Dry Electrophoretic Transfer Cell (Bio-Rad). Membranes were blocked in 5% non-fat dry milk or 5% BSA for phospho-epitope antibodies, incubated in primary antibodies to MTR (Abcam, Cambridge, UK, ab66039, 1:1000), β-actin (Cell Signaling Technology, Danvers, MA, 8457 clone D6A8, 1:2500), phospho-T389 S6K (Cell Signaling Technology, Danvers, MA, 9234 clone 108D2, 1:1000) S6K (Cell Signaling Technology, Danvers, MA, 9202, 1:1000), phospho-S235/6 RPS6 (Cell Signaling Technology, Danvers, MA, 4858 clone D57.2.2E, 1:1000), RPS6 (Cell Signaling Technology, Danvers, MA, 2217 clone 5G10, 1:1000), or mono/dimethyl lysine residues (Abcam, Cambridge UK, ab23366, 1:1000) overnight and detected using HRP-conjugated anti-rabbit IgG secondary antibodies (Cell Signaling Technology, Danvers, MA, 7074, 1:5000) and chemiluminescence with the Western Lightning Plus-ECL Reagent (Perkin Elmer).
For quantification, ImageJ was used for densitometry. To calculate a phosphorylation index the phospho-epitope signals for S6K and RPS6 were divided by the total protein signal, which was obtained after stripping the phospho-epitope antibody for 15 minutes Restore Western Blot Stripping Reagent (Thermo Scientific) and 30 minutes with 30% hydrogen peroxide at 37°C, and then re-blocking and re-probing the same blot with a total protein antibody. Phosphorylation indices from triplicate blots from independent wells of cells lysed on the same day but processed for Western blotting on different days were averaged and used to calculate a mean and standard deviation.
Metabolite extraction
For analysis of mouse plasma folates, 10 μL of plasma was mixed with 90 μL extraction buffer (80:20 methanol:water with 2.5 mM sodium ascorbate, 25 mM ammonium acetate, 100 nM aminopterin). Samples were vortexed for 10 minutes at 4 °C, then centrifuged at 21000 x g at 4°C for 10 minutes. Supernatant was removed and dried under nitrogen.
For extraction of polar metabolites, 5 μL of a standard composed of a 13C-labeled amino acid mix (Cambridge Isotopes, Tewksbury, MA, MSK-A2-1.2) diluted to a concentration of 200 μM per amino acid was added to 600 μL 80% HPLC grade methanol (Sigma-Aldrich, 646377-4X4L) and added to cells after draining media and washing with 2-3 mL ice cold saline. Cells were then scraped, transferred to an Eppendorf tube, vortexed for 10 minutes at 4 °C, then centrifuged at 21000 x g at 4°C for 10 minutes. 400 μL of sample was removed and dried under nitrogen.
Extraction of intracellular folates was performed according to previously described methods70. 1 mL 80% HPLC grade methanol with 2.5 mM sodium ascorbate and 25 mM ammonium acetate, with a 100 nM aminopterin standard and 500 nM each of a 13C-labeled amino acid mix, was added to cells in a 6 cm dish on ice after draining media and washing twice with ice cold saline. Cells were then scraped, transferred to an Eppendorf tube, vortexed for 60 seconds, and centrifuged at 21000 x g at 4°C for 10 minutes. 600-800 μL of sample was removed for analysis of folates and 100 uL was removed for analysis of polar metabolites; both were dried under nitrogen. Folate samples were resuspended in 500 uL rat serum (Sigma-Aldrich, R9759) for 2 hours at 37°C that had been stripped twice with 0.5 g activated charcoal (Sigma-Aldrich, C9157) per 10 mL serum, centrifuged at 3200 x g at 4°C for 10 minutes to sediment charcoal and filtered each time through a 0.45 uM PES filter. 5 uL 1:1 formic acid : water was added to folate samples to adjust pH, and samples were cleaned up on SPE columns (Agilent Technologies, 14102062) according to the manufacturer’s instructions and dried under nitrogen.
LC/MS analysis of non-folate metabolites
Dried samples were resuspended in 100 μL HPLC grade water. LC-MS analysis was performed using a QExactive orbitrap mass spectrometer using an Ion Max source and heated electro-spray ionization (HESI) probe coupled to a Dionex Ultimate 3000 UPLC system (Thermofisher). External mass calibration was performed every 7 days. Polar metabolite samples were separated by chromatography by injecting 10 μL of sample on a SeQuant ZIC-pHILIC 2.1 mm x 150 mm (5 μm particle size) column. Flow rate was set to 150 mL/min. and temperatures were set to 25°C for the column compartment and 4°C for the autosampler tray. Mobile phase A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide. Mobile phase B was 100% acetonitrile. The chromatographic gradient was: 0–20 min.: linear gradient from 80% to 20% mobile phase B; 20–20.5 min.: linear gradient from 20% to 80% mobile phase B; 20.5 to 28 min.: hold at 80% mobile phase B. The mass spectrometer was operated in full scan, polarity-switching mode and the spray voltage was set to 3.0 kV, the heated capillary held at 275°C, and the HESI probe was held at 350°C. The sheath gas flow rate was 40 units, the auxiliary gas flow was 15 units and the sweep gas flow was one unit. The MS data acquisition was performed in a range of 70–1000 m/z, with the resolution set set at 70,000, the AGC target at 1x106, and the maximum injection time at 20 msec. Relative quantitation of metabolites was performed with XCalibur QuanBrowser 2.2 (Thermo Fisher Scientific) using a 5-7 ppm mass tolerance and referencing an in-house library of chemical standards. Peak areas were normalized to cell number and 13C-amino acid standard peak areas. All LC/MS measurements of non-folate metabolites were carried out in biological triplicate, unless otherwise indicated in the figure legend.
LC/MS analysis of folate species
Detection of folate species was performed on the same instrumentation described above, as outlined in previous work70. LC-MS measurements were initiated on the same day as extraction. In general, instrument settings remained the same unless specified. Samples were resuspended in 25-50 μL water and 15 μL was injected onto an Ascentis® Express C18 HPLC column (2.7 μm × 15 cm × 2.1 mm; Sigma Aldrich). The column oven and autosampler tray were held at 30°C and 4°C, respectively. The following conditions were used to achieve chromatographic separation: Buffer A was 0.1% formic acid; buffer B was acetonitrile with 0.1% formic acid. The chromatographic gradient was run at a flow rate of 0.250 mL/min as follows: 0-5min.: gradient was held at 5% B; 5-10 min.: linear gradient of 5% to 36% B; 10.1-14.0 min.: linear gradient from 36%-95% B; 14.1-18.0 min.: gradient was returned to 5% B. The mass spectrometer was operated in full-scan, positive ionization mode. MS data acquisition was performed using three narrow-range scans: 438-450 m/z; 452-462 m/z; and 470-478 m/z, with the resolution set at 70,000, the AGC target at 10e6, and the maximum injection time of 150 msec. Relative quantitation of folate species was performed with XCalibur QuanBrowser 2.2 (Thermo Fisher Scientific) using a 5 ppm mass tolerance. Folate species were identified using chemical standards, and 5,10-methenyl THF and 10-formyl THF were pooled during analysis due to the possibility of interconversion of the species in each pool after lysis30. LC/MS measurements of folates were carried out in biological triplicate, unless otherwise indicated in the figure legend.
Mouse tumor studies
Cells were trypsinized and either 100,000 or 1,000,000 cells were resuspended in 100 μL PBS as indicated on figure legends. Cells were injected into the flanks of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Mice were euthanized according to institutional guidelines prior to tumors reaching 1 cm3 in size, and tumor weight was recorded. 0.1 g/L folic acid water was prepared by resuspending folic acid in tap water, then filtering the mixture through a 0.22 μm filter to sterilize the water. All animal studies were carried out in male mice aged 12 weeks from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in an environment with controlled temperature, humidity, and light cycle (18-23°C, 40-60% humidity). All experiments were performed according to MIT Committee on Animal Care guidelines.
Blood collection from mice
Blood was collected from fed, anesthetized mice by retro-orbital bleeding at 11 AM. Blood was placed directly into EDTA coated collection tubes (Sarstedt, Nümbrecht, Germany, 41.1395.105) and centrifuged 10 minutes at 845 x g; the supernatant of plasma was transferred to another tube.
PRISM multiplexed cell proliferation assay
The PRISM multiplexed barcoded cell line assay was carried out as previously described28 with the following modifications. A pool of 1.5 million cells consisting of 489 cell lines at equal representation was thawed directly into a 15 cm plate. The next day, the medium was replaced with RPMI-1640 lacking folic acid to prestarve cells. 3 days later, cells were trypsinized with 0.25% trypsin-EDTA, counted and 150,000 cells were plated into 10 cm plates (Corning Life Sciences) in 25 mL of RPMI-1640 medium lacking folic acid and allowed to adhere to the plate overnight. The next day, triplicate plates of cells were lysed in a genomic DNA prep lysis buffer [20 mM Tris HCl pH 8.4, 50 mM KCl, 0.45% NP-40, 0.45% Tween-20, 10% proteinase K] and one plate of cells was counted to provide a number of cells and barcode representation at the start of the experiment. The medium was then replaced with RPMI-1640 with folic acid, 5-methyl THF, and methotrexate (MedChemExpress, HY-14519) or DMSO vehicle added at the indicated doses. After 4 days of growth, parallel triplicate plates were lysed in the genomic DNA prep lysis buffer, heated at 60°C for 1 hour, and lysate was flash frozen. An accompanying plate of cells was counted to provide a number of cells at the end of the experiment, and cells were lysed at 100K cells / mL lysis buffer. Lysates were denatured at 95°C and DNA barcodes were amplified from lysates directly by PCR using a 2X KAPA polymerase master mix and primers containing Illumina flow-cell binding and adapter sequences. Resulting PCR products were confirmed for single-band amplification using gel electrophoresis (E-Gel™ 48 Agarose, 2%, Thermo Fisher Scientific), pooled, purified using the Zymo Select-a-Size DNA Clean & Concentration kit, quantitated using 1 μl on a Qubit 3 Fluorometer (Qubit dsDNA HS Assay Kit, Invitrogen Q32851), and then sequenced on MiSeq (50 cycles, single read) as previously described (final library concentration 10pM with a 25% PhiX spike-in) 28,71. Fastq files were de-multiplexed using the Illumina software bcl2fastq (v2.20). Note that only 1 PRISM assay was performed; the untreated control data is presented in Figure 1 and again in Figure 5 as a comparator for methotrexate-treated cells.
PRISM analysis
The multiplexed barcoded cell line assay analysis was carried out as previously described28,71 with the following modifications. Raw barcode read counts were first normalized to a set of 10 invariant control barcodes, which are spiked into samples at known amounts before PCR to control for non-uniform amplification during PCR.
To adjust for differences in the total cell numbers between plates after 4 days of growth at varying doses of methotrexate, barcode counts for each sample were secondarily corrected by computing a sample fraction for each cell line and multiplying it by the population-level corrected cell count for that sample. The population-level cell count for each sample was corrected for counting errors by counting cells on a parallel plate before sample harvesting, lysing 100K cells per sample, and adjusting the raw cell counts based on the measured mean-normalized gDNA concentration from the sample, assuming that all lysed samples should have a mean-normalized gDNA concentration of 1 if counting was accurate (cell count / mean-normalized gDNA concentration).
Relative cell line viability in 5-methyl THF vs. folic acid was calculated for each condition as follows:
| (1) |
where BC5m and BCFA are the mean normalized corrected barcode read counts for a given cell line in 5-methyl THF and folic acid, respectively. Proliferation rate of each cell line, reported as doublings per day, was calculated for each condition as follows:
| (2) |
where BC4 is the mean normalized corrected barcode read count of a given cell line at day 4, and BC0 is the mean normalized corrected barcode read count of that cell line at day 0 before beginning the experiment (but after the folate pre-starvation period).
Statistics & Reproducibility
Sample size for animal experiments was determined by power analysis. No data were excluded from the analyses. Samples were randomized for LC/MS experiments. Investigators were blinded to identity of mice in all animal studies. Investigators were not blinded to allocation during all other experiments and outcome assessment.
Data analysis
Raw data was collated into csv files using Excel v16.53 and then subject to simple manipulations or statistical tests described in detail in the accompanying methods section/figure legends and plotted using scripts written in jupyter notebook (ipython v5.8.0; jupyter-notebook v5.7.9) in R (v3.6.1) using the package collection tidyverse (v1.3.0) including ggplot (v3.3.2).
Extended Data
Extended Data Fig. 1. Characterization of cells cultured in 5-methyl THF.
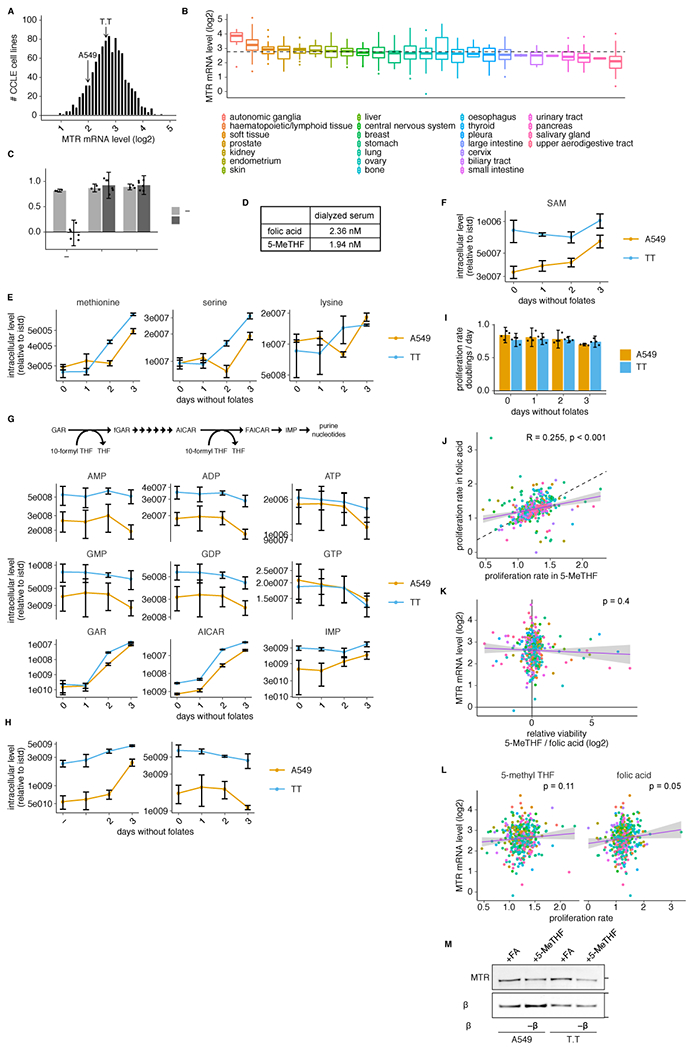
(A-B) Histogram (A) or box plot (B) of log2 MTR mRNA levels for cells in the Cancer Cell Line Encyclopedia (CCLE)24 (n = 1019/1457 cell lines with detectable MTR). Median (box center line), interquartile range (IQR) (box), 1.5*IQR (whiskers), and outliers (points) are plotted; dashed line represents the overall median. (C) Proliferation rate of T.T cells in media containing no folate source (−), folic acid (+FA), or 5-methyl THF (+5-MeTHF) before (0 days, n = 3 independent samples) or after (3 days, n = 6 independent samples) of culture with no folic acid (“prestarved”). (D) LC/MS measurement of folic acid and 5-methyl THF concentrations in dialyzed fetal bovine serum. (E-H) LC/MS measurement of intracellular methionine, serine, lysine (E), SAM (F), AMP, ADP, ATP, GMP, GDP, GTP, GAR, AICAR, IMP (G), dUMP and dTMP (H) in A549 and T.T cells cultured for up to 3 days in medium lacking folates (n = 3 independent samples). Data are normalized to protein concentration and an internal standard. (I) Proliferation rate of A549 or T.T cells cultured in medium lacking folates for up to 3 days (n = 4 independent samples). (J-L) Correlation between the proliferation rate in 5-methyl THF and folic acid medium (J; dashed line: y=x), log2 MTR mRNA level and log2 relative viability in 5-methyl THF versus folic acid medium (K), or log2 MTR mRNA level and proliferation rate in 5-methyl THF / folic acid (L) for 489 barcoded cell lines (Pearson’s product moment correlation coefficient / p-value for J: R = 0.255, p = 2e-07; K: R = −0.041 p = 0.4; L: 5-methyl THF R = 0.08, p = 0.11 and folic acid R = 0.1, p = 0.05). Purple line is a linear regression fit, shading represents a 95% confidence interval around the mean. (M) Western blot analysis of MTR expression in A549 or T.T cells cultured in medium with the indicated folate source (representative of 1 experiment). (A-M) Mean +/− SD error bars are displayed.
Extended Data Fig. 2. MTR knockout affects levels of intracellular folate species and proliferation in different folate sources.
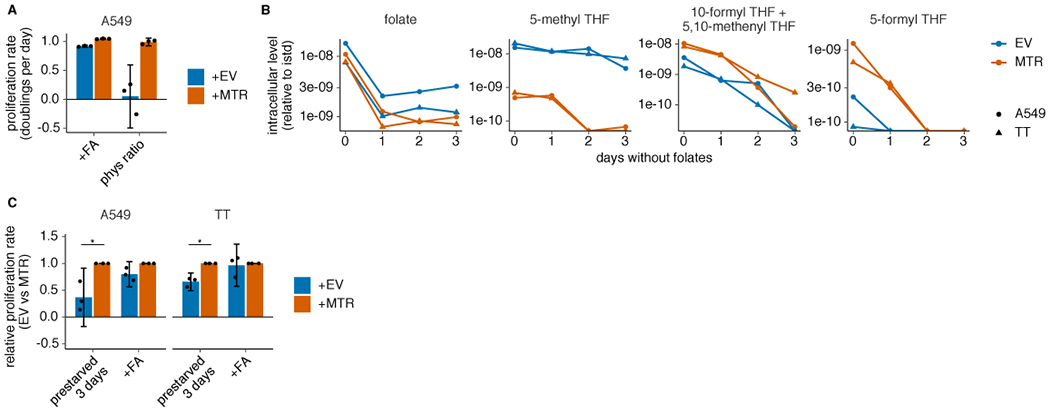
(A) Proliferation rates of A549 MTR knockout cells without (+EV) or with MTR expression (+MTR) cultured in folic acid (+FA) or 96% 5-methyl THF, 4% folic acid (phys ratio)4 for 4 days after a folate pre-starvation period (n = 3 independent samples). (B) LC/MS measurement of intracellular folate, 5-methyl THF, combined 5,10-methenyl THF/10-formyl THF, and 5-formyl THF levels in A549 and T.T MTR knockout cells +EV or +MTR cultured up to 3 days in medium lacking folates (n = 1 independent sample for each time point). Data are normalized to protein concentration and an internal standard. (C) Proliferation rate of A549 or T.T MTR knockout cells +EV or +MTR cultured in medium lacking folates (prestarved) or with folic acid for 3 days (n = 3 independent samples; A549: p = 0.011; T.T: p = 0.046). p values indicated are derived from a two-tailed, unpaired Welch’s t test (* = p < 0.05). (A,C) Mean +/− SD error bars are displayed.
Extended Data Fig. 3. MTR knockout results in folate insufficiency and impaired nucleotide synthesis in 5-methyl THF.
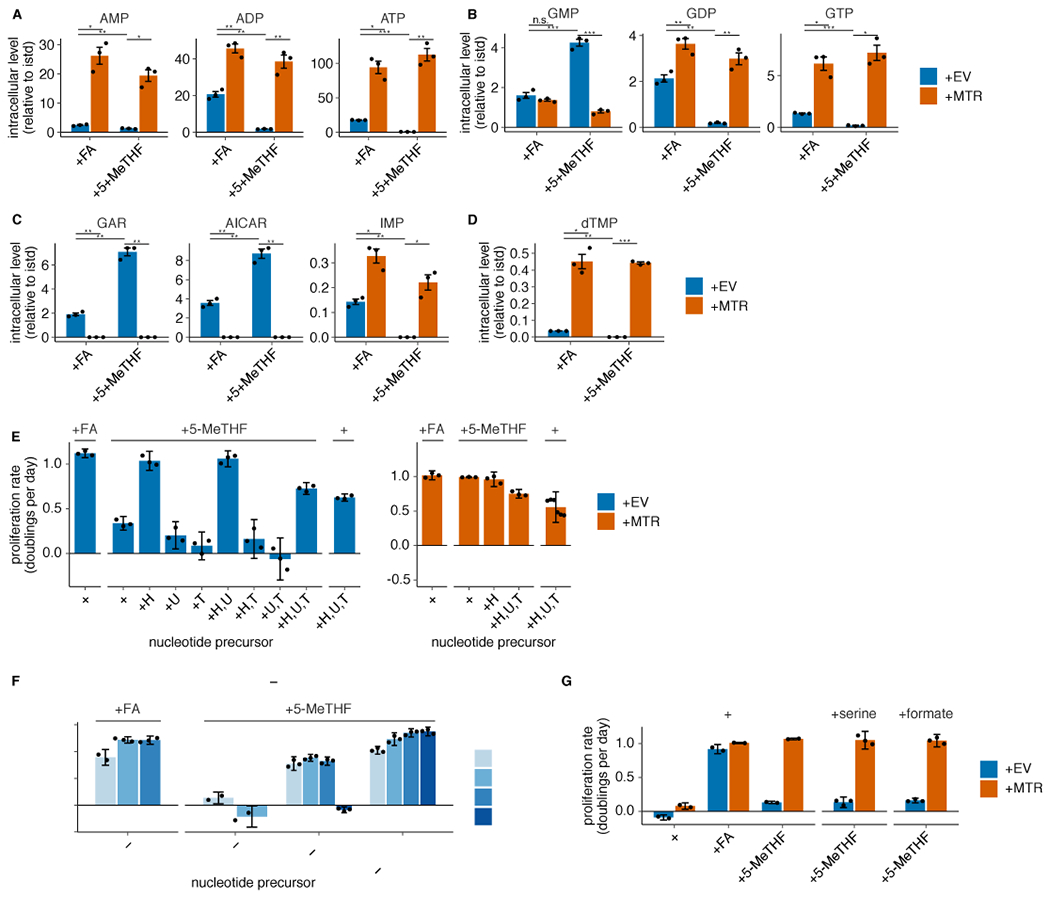
(A-D) LC/MS measurement of intracellular AMP, ADP, ATP (A), GMP, GDP, GTP (B), GAR, AICAR, IMP (C), and dTMP (D) levels in T.T MTR knockout cells without (+EV) or with MTR expression (+MTR) cultured for 4 days in folic acid (+FA) or 5-methyl THF (+5-MeTHF) after a prestarvation period (n = 3 independent samples; +EV +FA vs +MTR +FA p-values: AMP: 0.014, ADP: 0.002, ATP: 0.014, GMP: 0.26, GDP: 0.008, GTP: 0.017, GAR: 0.004, AICAR: 0.005, IMP: 0.014, dTMP: 0.01; +EV +5-methyl THF vs +MTR +5-methyl THF p-values: AMP: 0.012, ADP: 0.0097, ATP: 0.007, GMP: 0.0006, GDP: 0.008, GTP: 0.012, GAR: 0.002, AICAR: 0.003, IMP: 0.022, dTMP: 2.51*10−5; +EV +FA vs +EV +5-methyl THF p-values: AMP: 0.007, ADP: 0.006, ATP: 5.26*10−5, GMP: 0.0004, GDP: 0.006, GTP: 2.94*10−5, GAR: 0.002, AICAR: 0.003, IMP: 0.008, dTMP: 0.005). Data are normalized to protein concentration and to an internal standard. Mean +/− SEM error bars are displayed. (E) Proliferation rates of T.T MTR knockout cells +EV or +MTR cultured as in A-D in the indicated folate with or without the addition of 100 μM each of the indicated nucleotide precursors hypoxanthine (H), uridine (U), and thymidine (T) (n = 3 independent samples; except +MTR +H,U,T n = 6). (F) Proliferation rates of A549 MTR knockout cells +EV or +MTR passaged in the indicated folate with the addition of nucleotide precursors for up to 16 days (n = 3 independent samples). (G) Proliferation rates of T.T MTR knockout cells +EV or +MTR cultured in the indicated folate as in A-D, with or without 1 mM serine or formate (n = 3 independent samples). (E-G) Mean +/− SD error bars are displayed. p-values are derived from a two-tailed, unpaired Welch’s t test (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
Extended Data Fig. 4. Neither SAM nor methionine levels are limiting for proliferation in MTR knockout cells in 5-methyl THF.
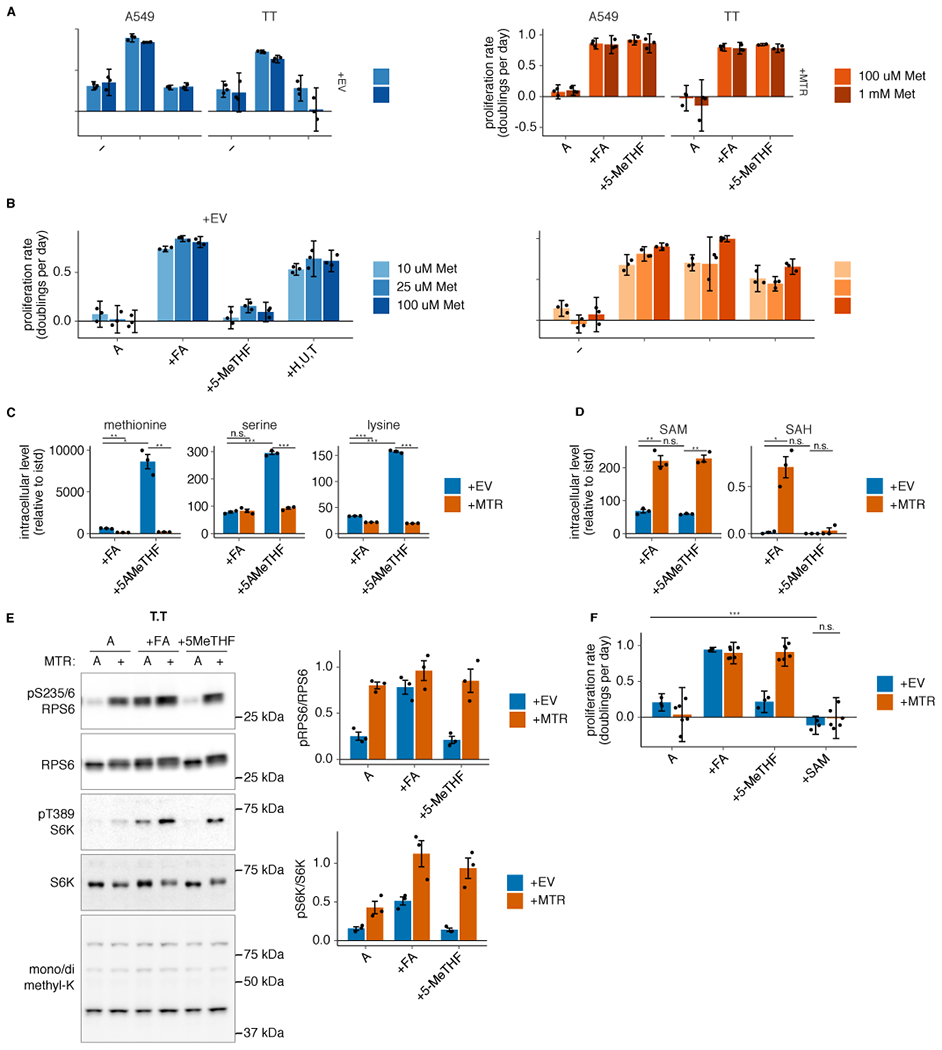
(A-B) Proliferation rates of A549 and T.T MTR knockout cells without (+EV) or with MTR expression (+MTR) cultured in folic acid (+FA), 5-methyl THF (5-MeTHF), or no folate (−) for 4 days after a prestarvation period, with or without 900 uM added methionine (A) or T.T MTR knockout cells +EV or +MTR cultured in the indicated folate or 100 uM each nucleotide precursor (hypoxanthine, uridine, and thymidine) at varied extracellular methionine concentrations (B). (C-D) LC/MS measurement of intracellular methionine, serine, lysine (C), SAM, and SAH levels (D) in T.T MTR knockout cells +EV or +MTR cultured in the indicated folate source as in A (n = 3 independent samples; +EV +FA vs +MTR +FA p-values: methionine: 0.002, serine: 0.57, lysine: 9.94*10−6, SAM: 0.007, SAH: 0.024; +EV +5-methyl THF vs +MTR +5-methyl THF p-values: methionine: 0.01, serine: 2.71*10−5, lysine: 0.0001, SAM: 0.003, SAH: 0.47; +EV +FA vs +EV +5-methyl THF p-values: methionine: 0.011, serine: 5.57*10−5, lysine: 0.0001, SAM: 0.24, SAH: 0.27). Data are normalized to protein concentration and an internal standard. (E) Western blots to assess phosphorylation of mTORC1 targets and levels of mono- or dimethyl lysine-containing proteins in T.T MTR knockout cells +EV or +MTR cultured in the indicated folate as in A. The ratio of phospho-protein to total protein signal from 3 independent replicates is shown. (F) Proliferation rates of T.T MTR knockout cells +EV or +MTR cultured in the indicated folate for 16 hours, with or without the addition of 1 mM SAM (n = 3 independent samples for +EV, n = 6 for +MTR; +EV −folate vs +EV +SAM p = 1.6*10−6, +EV +SAM vs +MTR +SAM p = 0.11). (A,B,F) Mean +/− SD error bars are displayed. (C-E) Mean +/− SEM error bars are displayed. p-values are derived from a two-tailed, unpaired Welch’s t test (* = p < 0.05, ** = p < 0.01, *** = p < 0.001).
Extended Data Fig. 5. 5-methyl THF medium blunts methotrexate efficacy but not import.
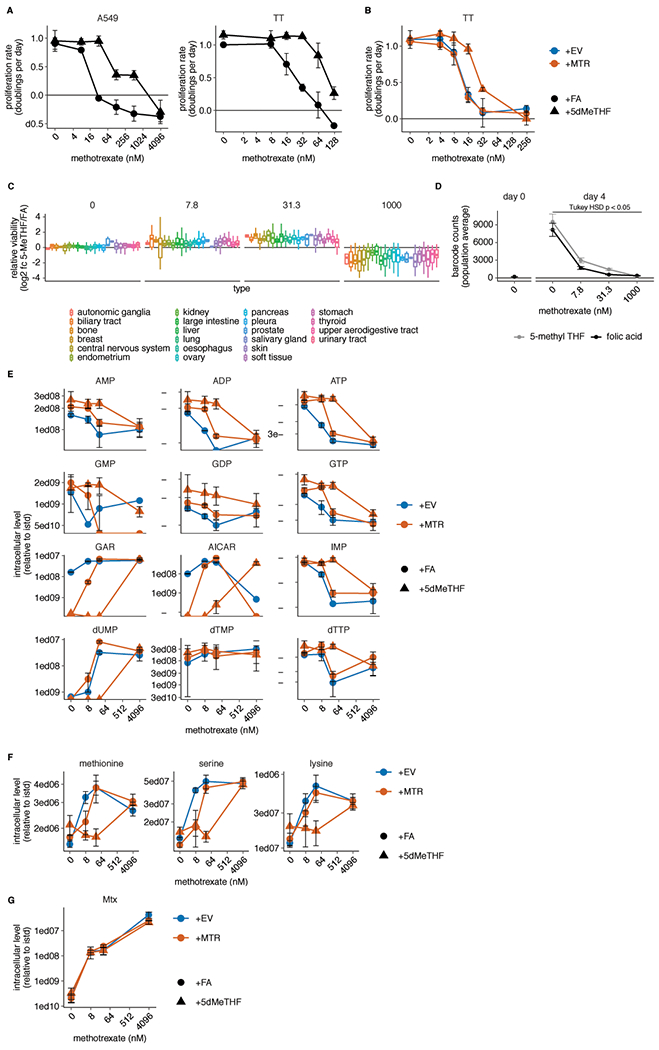
(A-B) Proliferation rates of A549 and T.T cells (A) or T.T MTR knockout cells without (+EV) or with MTR expression (+MTR) (B) cultured in folic acid (+FA) or 5-methyl THF (+5-MeTHF) for 4 days across a range of methotrexate doses (n = 3 independent samples). (C) Log2 fold change in relative viability of 489 cell lines cultured in 5-methyl THF versus folic acid as in A-B (log2 fc 5-MeTHF/FA) at the indicated methotrexate doses, grouped by tissue of origin (note that the 0 dose is the same data plotted in Fig. 1G). Median (box center line), interquartile range (IQR) (box), and 1.5*IQR (whiskers) are plotted. (D) Mean barcode read counts for the total pool of cancer cell lines, before (day 0) or after (day 4) culture in the indicate folate/methotrexate doses as in A-B. Tukey HSD p-value was calculated by correcting a two-way ANOVA test for multiple comparisons between the dose response curves in the two folates (n = 489 cell lines; folic acid vs. 5-methyl THF dose response p = 0.049). (E-G) LC/MS measurement of intracellular AMP, ADP, ATP, GMP, GDP, GTP, GAR, AICAR, IMP, dUMP, dTMP, dTTP (E), methionine, serine, lysine (F), and intracellular methotrexate (Mtx) (G) levels in A549 MTR knockout cells +EV or +MTR cultured in the indicated folate across a range of doses of methotrexate as in A-B (n = 3 independent samples). Data are normalized to cell number and an internal standard. (A-G) Mean +/− SD error bars are displayed, except in D where mean +/− SEM is displayed.
Supplementary Material
Supplementary Table 1 Raw PCR amplicon sequencing barcode counts for each cancer cell line our PRISM barcoded pool or control spike in, in folic acid or 5-methyl THF medium across a range of methotrexate doses. Individual biological and technical replicates, population level cell counts, and mean normalized gDNA yields for correcting cell counts (as described in Methods) are included. No sequenced barcodes have been removed (therefore not all are exact matches to the barcodes associated with each cell line/condition/replicate group due to sequencing errors, etc.
ACKNOWLEDGEMENTS
We thank Naama Kanarek for her comments and technical advice on folate measurements. We acknowledge all of the members of the Vander Heiden lab for input and advice on the manuscript. M.R.S. was supported by T32-GM007287 and by an MIT Koch Institute Graduate Fellowship. A.M.D. was supported by a Jane Coffin Childs Postdoctoral Fellowship. M.G.V.H. acknowledges support from R35CA242379, R01CA201276, P30CA014051, the Ludwig Center at MIT, the MIT Center for Precision Cancer Medicine, SU2C, the Emerald Foundation, the Lustgarten Foundation, and a Faculty Scholars Grant from HHMI.
Footnotes
CODE AVAILABILITY
Any code used to analyze or plot data in this manuscript is available from the corresponding author upon request.
COMPETING INTERESTS STATEMENT
M.G.V.H. is on the scientific advisory board of Agios Pharmaceuticals, Aeglea Biotherapeutics, iTeos Therapeutics, Faeth Therapeutics, Sage Therapeutics, DRIOA Ventures, and Auron Therapeutics. The other authors declare no competing interests.
DATA AVAILABILITY
Unprocessed western blot images, integrated peak areas for metabolite level quantification, and processed data from comparative analysis of barcode counts of 489 human cancer cell lines in each condition in the PRISM multiplexed growth assay are provided as source data for Figures 1–5 and Extended Data Figures 1–5. PCR amplicon sequencing results including raw barcode counts for each cancer cell line in the PRISM 489 cell line pool in each condition/replicate are provided in Supplementary Table 1.
REFERENCES
- 1.Chiang PK et al. S-Adenosylmethionine and methylation. FASEB J 10, 471–480 (1996). [PubMed] [Google Scholar]
- 2.Lane AN & Fan TW Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485, doi: 10.1093/nar/gkv047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ducker GS & Rabinowitz JD One-Carbon Metabolism in Health and Disease. Cell Metab 25, 27–42, doi: 10.1016/j.cmet.2016.08.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfeiffer CM et al. Folate status and concentrations of serum folate forms in the US population: National Health and Nutrition Examination Survey 2011–2. Br J Nutr 113, 1965–1977, doi: 10.1017/S0007114515001142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman ID The characteristics of the membrane transport of amethopterin and the naturally occurring folates. Ann N Y Acad Sci 186, 400–422, doi: 10.1111/j.1749-6632.1971.tb46996.x (1971). [DOI] [PubMed] [Google Scholar]
- 6.Ritchie C, Cordova AF, Hess GT, Bassik MC & Li L SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell 75, 372–381 e375, doi: 10.1016/j.molcel.2019.05.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muir A, Danai LV & Vander Heiden MG Microenvironmental regulation of cancer cell metabolism: implications for experimental design and translational studies. Dis Model Mech 11, doi: 10.1242/dmm.035758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rancati G, Moffat J, Typas A & Pavelka N Emerging and evolving concepts in gene essentiality. Nat Rev Genet 19, 34–49, doi: 10.1038/nrg.2017.74 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Banerjee RV & Matthews RG Cobalamin-dependent methionine synthase. FASEB J 4, 1450–1459 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Stover PJ Vitamin B12 and older adults. Curr Opin Clin Nutr Metab Care 13, 24–27, doi: 10.1097/MCO.0b013e328333d157 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chanarin I, Deacon R, Lumb M, Muir M & Perry J Cobalamin-folate interrelations: a critical review. Blood 66, 479–489 (1985). [PubMed] [Google Scholar]
- 12.Fujii K, Nagasaki T & Huennekens FM Accumulation of 5-methyltetrahydrofolate in cobalamin-deficient L1210 mouse leukemia cells. J Biol Chem 257, 2144–2146 (1982). [PubMed] [Google Scholar]
- 13.Walker PR et al. Induction of apoptosis in neoplastic cells by depletion of vitamin B12. Cell Death Differ 4, 233–241, doi: 10.1038/sj.cdd.4400225 (1997). [DOI] [PubMed] [Google Scholar]
- 14.McLean GR et al. Cobalamin analogues modulate the growth of leukemia cells in vitro. Cancer Res 57, 4015–4022 (1997). [PubMed] [Google Scholar]
- 15.Corcino JJ, Zalusky R, Greenberg M & Herbert V Coexistence of Pernicious Anaemia and Chronic Myeloid Leukaemia: An Experiment of Nature Involving Vitamin BI2 Metabolism. Br J Haematol 20, 511 (1971). [DOI] [PubMed] [Google Scholar]
- 16.Eastwood DW, Green CD, Lambdin MA & Gardner R Effect of Nitrous Oxide on the White-Cell Count in Leukemia. New England Journal of Medicine 268, 297–299, doi: 10.1056/nejm196302072680607 (1963). [DOI] [Google Scholar]
- 17.Ikeda K et al. Antileukemic effect of nitrous oxide in a patient with chronic myelogenous leukemia. Am J Hematol 30, 114 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Matthews JH Cyanocobalamin [c-lactam] Inhibits Vitamin B12 and Causes Cytotoxicity in HL60 Cells: Methionine Protects Cells Completely. Blood 89, 4600–4607 (1997). [PubMed] [Google Scholar]
- 19.Liteplo RG, Hipwell SE, Rosenblatt DS, Sillaots S & Lue-Shing H Changes in cobalamin metabolism are associated with the altered methionine auxotrophy of highly growth autonomous human melanoma cells. J Cell Physiol 149, 332–338, doi: 10.1002/jcp.1041490222 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Boss GR Cobalamin inactivation decreases purine and methionine synthesis in cultured lymphoblasts. J Clin Invest 76, 213–218, doi: 10.1172/JCI111948 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lumb M et al. Effects of nitrous oxide-induced inactivation of cobalamin on methionine and S-adenosylmethionine metabolism in the rat. Biochim Biophys Acta 756, 354–359 (1983). [DOI] [PubMed] [Google Scholar]
- 22.van der Westhuyzen J, Fernandes-Costa F & Metz J Cobalamin inactivation by nitrous oxide produces severe neurological impairment in fruit bats : protection by methionine and aggravation by folates. Life Sci 31, 2001–2010 (1982). [DOI] [PubMed] [Google Scholar]
- 23.Scott JM, Dinn JJ, Wilson P & Weir DG Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet 2, 334–337 (1981). [DOI] [PubMed] [Google Scholar]
- 24.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607, doi: 10.1038/nature11003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen MF, Jensen SO, Fuchtbauer EM & Martensen PM High folic acid diet enhances tumour growth in PyMT-induced breast cancer. Br J Cancer 116, 752–761, doi: 10.1038/bjc.2017.11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farber S et al. The Action of Pteroylglutamic Conjugates on Man. Science 106, 619–621, doi: 10.1126/science.106.2764.619 (1947). [DOI] [PubMed] [Google Scholar]
- 27.Farber S, Diamond LK, Mercer RD, Sylvester RF & Wolff JA Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteryl-glutamic acid (aminopterin). New England Journal of Medicine 238, 787–793 (1948). [DOI] [PubMed] [Google Scholar]
- 28.Yu C et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 34, 419–423, doi: 10.1038/nbt.3460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer AM, Kamynina E, Field MS & Stover PJ Folate rescues vitamin B12 depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc Natl Acad Sci U S A 114, E4095–E4102, doi: 10.1073/pnas.1619582114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Ducker GS, Lu W, Teng X & Rabinowitz JD An LC-MS chemical derivatization method for the measurement of five different one-carbon states of cellular tetrahydrofolate. Anal Bioanal Chem, doi: 10.1007/s00216-017-0514-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stover P & Schirch V Evidence for the accumulation of a stable intermediate in the nonenzymatic hydrolysis of 5,10-methenyltetrahydropteroylglutamate to 5-formyltetrahydropteroylglutamate. Biochemistry 31, 2148–2155, doi: 10.1021/bi00122a036 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH & Maddocks ODK Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7, 1248–1258, doi: 10.1016/j.celrep.2014.04.045 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Misselbeck K et al. A hybrid stochastic model of folate-mediated one-carbon metabolism: Effect of the common C677T MTHFR variant on de novo thymidylate biosynthesis. Sci Rep 7, 797, doi: 10.1038/s41598-017-00854-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bjursell G & Reichard P Effects of Thymidine on Deoxyribonucleoside Triphosphate Pools and Deoxyribonucleic Acid Synthesis in Chinese Hamster Ovary Cells. Journal of Biological Chemistry 248, 3904–3909, doi: 10.1016/s0021-9258(19)43819-2 (1973). [DOI] [PubMed] [Google Scholar]
- 35.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401, doi: 10.1038/s41586-019-1437-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan MR et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab 29, 1410–1421 e1414, doi: 10.1016/j.cmet.2019.02.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cynober LA Plasma amino acid levels with a note on membrane transport: characteristics, regulation, and metabolic significance. Nutrition 18, 761–766, doi: 10.1016/s0899-9007(02)00780-3 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Wang JZ, Ghergurovich JM, Yang L & Rabinowitz JD Methionine synthase supports tumor tetrahydrofolate pools. Nature Metab (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mato JM, Alvarez L, Ortiz P & Pajares MA S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther 73, 265–280 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Maddocks OD, Labuschagne CF, Adams PD & Vousden KH Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell 61, 210–221, doi: 10.1016/j.molcel.2015.12.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su X, Wellen KE & Rabinowitz JD Metabolic control of methylation and acetylation. Curr Opin Chem Biol 30, 52–60, doi: 10.1016/j.cbpa.2015.10.030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho HS et al. Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nat Commun 3, 1072, doi: 10.1038/ncomms2074 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu X et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818, doi: 10.1126/science.aao3265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reeves PG Components of the AIN-93 Diets as Improvements in the AIN-76A Diet. J Nutr 127, 838–841 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Bailey RL et al. Total folate and folic acid intake from foods and dietary supplements in the United States: 2003–2006. Am J Clin Nutr 91, 231–237, doi: 10.3945/ajcn.2009.28427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaukroger J et al. Paradoxical response of malignant melanoma to methotrexate in vivo and in vitro. Br J Cancer 47, 671–679, doi: 10.1038/bjc.1983.105 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu E, Drake JC, Boarman D, Baram J & Allegra CJ Mechanism of thymidylate synthase inhibition by methotrexate in human neoplastic cell lines and normal human myeloid progenitor cells. Journal of Biological Chemistry 265, 8470–8478, doi: 10.1016/s0021-9258(19)38912-4 (1990). [DOI] [PubMed] [Google Scholar]
- 48.Sanderson SM, Gao X, Dai Z & Locasale JW Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat Rev Cancer 19, 625–637, doi: 10.1038/s41568-019-0187-8 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Chaturvedi S, Hoffman RM & Bertino JR Exploiting methionine restriction for cancer treatment. Biochem Pharmacol 154, 170–173, doi: 10.1016/j.bcp.2018.05.003 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Sakura T et al. High-dose methotrexate therapy significantly improved survival of adult acute lymphoblastic leukemia: a phase III study by JALSG. Leukemia 32, 626–632, doi: 10.1038/leu.2017.283 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Baldwin CM & Perry CM Pemetrexed: A Review in its Use in the Management of Advanced Non-Squamous Non-Small Cell Lung Cancer. Drugs 69, 2279–2302 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Gonen N & Assaraf YG Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat 15, 183–210, doi: 10.1016/j.drup.2012.07.002 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Visentin M, Zhao R & Goldman ID The antifolates. Hematol Oncol Clin North Am 26, 629–648, ix, doi: 10.1016/j.hoc.2012.02.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Wei S & Chen J Critical appraisal of pemetrexed in the treatment of NSCLC and metastatic pulmonary nodules. Onco Targets Ther 7, 937–945, doi: 10.2147/OTT.S45148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otake Y et al. Expression of thymidylate synthase in human non-small cell lung cancer. Jpn J Cancer Res 90, 1248–1253 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ceppi P et al. Squamous cell carcinoma of the lung compared with other histotypes shows higher messenger RNA and protein levels for thymidylate synthase. Cancer 107, 1589–1596, doi: 10.1002/cncr.22208 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Takezawa K et al. Thymidylate synthase as a determinant of pemetrexed sensitivity in non-small cell lung cancer. Br J Cancer 104, 1594–1601, doi: 10.1038/bjc.2011.129 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y et al. Transcriptomic and functional network features of lung squamous cell carcinoma through integrative analysis of GEO and TCGA data. Sci Rep 8, 15834, doi: 10.1038/s41598-018-34160-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gherasim C, Lofgren M & Banerjee R Navigating the B(12) road: assimilation, delivery, and disorders of cobalamin. J Biol Chem 288, 13186–13193, doi: 10.1074/jbc.R113.458810 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oltean S & Banerjee R Nutritional modulation of gene expression and homocysteine utilization by vitamin B12. J Biol Chem 278, 20778–20784, doi: 10.1074/jbc.M300845200 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Jarrett JT, Goulding CW, Fluhr K, Huang S & Matthews RG Purification and assay of cobalamin-dependent methionine synthase from Escherichia coli. Methods Enzymol 281, 196–213, doi: 10.1016/s0076-6879(97)81026-9 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Yamada K, Gravel RA, Toraya T & Matthews RG Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc Natl Acad Sci U S A 103, 9476–9481 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olteanu H & Banerjee R Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J Biol Chem 276, 35558–35563, doi: 10.1074/jbc.M103707200 (2001). [DOI] [PubMed] [Google Scholar]
- 64.Muir A et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, doi: 10.7554/eLife.27713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sullivan LB et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563, doi: 10.1016/j.cell.2015.07.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao H, French JB, Fang Y & Benkovic SJ The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chem Commun (Camb) 49, 4444–4452, doi: 10.1039/c3cc41437j (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okesli A, Khosla C & Bassik MC Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr Opin Biotechnol 48, 127–134, doi: 10.1016/j.copbio.2017.03.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784, doi: 10.1038/nmeth.3047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hart T et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526, doi: 10.1016/j.cell.2015.11.015 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Kanarek N et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559, 632–636, doi: 10.1038/s41586-018-0316-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin X et al. A metastasis map of human cancer cell lines. Nature 588, 331–336, doi: 10.1038/s41586-020-2969-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Raw PCR amplicon sequencing barcode counts for each cancer cell line our PRISM barcoded pool or control spike in, in folic acid or 5-methyl THF medium across a range of methotrexate doses. Individual biological and technical replicates, population level cell counts, and mean normalized gDNA yields for correcting cell counts (as described in Methods) are included. No sequenced barcodes have been removed (therefore not all are exact matches to the barcodes associated with each cell line/condition/replicate group due to sequencing errors, etc.
Data Availability Statement
Unprocessed western blot images, integrated peak areas for metabolite level quantification, and processed data from comparative analysis of barcode counts of 489 human cancer cell lines in each condition in the PRISM multiplexed growth assay are provided as source data for Figures 1–5 and Extended Data Figures 1–5. PCR amplicon sequencing results including raw barcode counts for each cancer cell line in the PRISM 489 cell line pool in each condition/replicate are provided in Supplementary Table 1.