

Abstract
Several lineages of influenza A viruses (IAV) currently circulate in North American pigs. Genetic diversity is further increased by transmission of IAV between swine and humans and subsequent evolution. Here, we characterized the genetic and antigenic evolution of contemporary swine H1N1 and H1N2 viruses representing clusters H1-α (1A.1), H1-β (1A.2), H1pdm (1A.3.3.2), H1-γ (1A.3.3.3), H1-δ1 (1B.2.2), and H1-δ2 (1B.2.1) currently circulating in pigs in the United States. The δ1-viruses diversified into two new genetic clades, H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2), which were also antigenically distinct from the earlier H1-δ1-viruses. Further characterization revealed that a few key amino acid changes were associated with antigenic divergence in these groups. The continued genetic and antigenic evolution of contemporary H1 viruses might lead to loss of vaccine cross-protection that could lead to significant economic impact to the swine industry, and represents a challenge to public health initiatives that attempt to minimize swine-to-human IAV transmission.
Keywords: Antigenic diversity, H1N1, H1N2, Influenza A, Swine, Virus evolution
1. Importance
The hemagglutinin (HA) protein of influenza A virus (IAV) is the primary target of protective immune responses and the major component in vaccine formulation. However, repeated introductions of non-swine IAV into swine populations and the processes of antigenic shift and drift result in virus evolution and potential mismatches between vaccines and circulating strains. Further, the increasing diversity of swine IAV represents a challenge for public health initiatives, and assessment of the risk of interspecies transmission of viruses relies on assessing antigenic diversity relative to human population immunity. In this study, we found that antigenic drift of IAV in the U.S. pig population resulted in seven distinct antigenic phenotypes of the H1 subtype currently circulating. We identified changes in the HA protein associated with the observed antigenic drift, suggesting that these amino acids may be important antigenic sites. These data demonstrate that recent evolution of H1 swine IAV resulted in novel genetic clades with distinct antigenic phenotypes that are unlikely to be protected by current vaccine formulations.
2. Introduction
Influenza A virus (IAV) is endemic in pigs globally, and three different subtypes currently co-circulate in North American swine: H1N1, H1N2, and H3N2 (Anderson et al., 2013; Lorusso et al., 2013). Swine have been indicted as a potential source of reassortant viruses that can be transmitted to humans (Brown, 2000; Vijaykrishna et al., 2011). Two major events illustrate this potential: the introduction of the swine-origin 2009 pandemic H1N1 virus (H1N1pdm09) with widespread global morbidity and mortality (Smith et al., 2009b), and the occurrence of more than 300 cases of swine-origin H3N2 IAV (H3N2v) (Jhung et al., 2013). However, the transmission of human IAV into swine populations is a frequent occurrence, and the repeated introduction and subsequent evolution of these human viral lineages in swine populations has a critical role in the genetic diversity of swine IAV (Nelson et al., 2012, 2014).
The first documented occurrence of bidirectional transmission between swine and humans dates to the 1918 H1N1 pandemic (Smith et al., 2009a), with the resulting swine strain referred to as classical H1N1 (cH1N1). The cH1N1 remained antigenically and genetically stable in the U.S. for nearly a century. In the late 1990s, a novel triple-reassortant H3N2 virus was detected in U.S. swine herds (Zhou et al., 1999) and reassorted with endemic cH1N1, leading to a period of rapid diversification of the cH1N1 hemagglutinin (HA) into three distinct HA clades: H1-α (1A.1), H1-β (1A.2), and H1-γ (1A.3.3.3) (Lorusso et al., 2011; Vincent et al., 2008b). Viruses containing HA and/or neuraminidase (NA) derived from human-seasonal IAV were introduced to U.S. pigs in the early 2000s (Karasin et al., 2006; Vincent et al., 2009), giving rise to two HA phylogenetic clades, H1-δ1 (1B.2.2) and H1-δ2 (1B.2.1) (Anderson et al., 2013; Lorusso et al., 2013). The 2009 H1N1 pandemic viruses were reintroduced from humans into U.S. pigs, resulting in reassortment with endemic swine viruses (Ducatez et al., 2011) and the establishment of a sixth swine H1 genetic clade (H1N1pdm09: 1A.3.3.2). This dynamic evolution is not restricted to the HA; N2 genes were derived from two human seasonal-lineages introduced in 1998 and 2002 or N1 genes from the classical or H1N1pdm09 lineages (Anderson et al., 2013; Lorusso et al., 2013). Moreover, the current diversity of lineages within IAV circulating in swine populations is complicated by antigenic drift of the HA (de Jong et al., 1999), and this gradual evolution may alter antigenic properties of IAV that result in vaccine failure (Both et al., 1983; Luoh et al., 1992). However, antigenic diversity is not always linearly correlated with genetic diversity, and few amino acid mutations can lead to biologically significant antigenic differences that may facilitate immune escape (Both et al., 1983; Luoh et al., 1992).
Vaccination against IAV in swine is an important tool to limit clinical disease, but it may not prevent infection and/or transmission. Products currently available in the U.S. provide good protection against homologous strains, however their efficacy against antigenically distinct viruses may diminish (Vincent et al., 2010, 2008a). Vaccine strains should be updated when there is loss in cross-reactivity against contemporary strains to improve the match between vaccine antigens and circulating antigenic diversity. Although genetic analysis of the HA gene has utility in predicting vaccine efficacy (Neher et al., 2016), antigenic cross-reactivity between field-sourced viruses and vaccine strains remains the gold standard for vaccine strain candidate selection. Thus, understanding the molecular epidemiology of IAV circulating in swine populations in association with antigenic properties are crucial to improve vaccine composition to better control IAV in pigs.
Several studies have mapped the antigenic and genetic evolution of H3N2 viruses in swine (Abente et al., 2016; de Jong et al., 2007; Lewis et al., 2014), and recently a comprehensive study characterized the high level global antigenic diversity of H1 and H3 viruses circulating in human and swine (Lewis et al., 2016). However, an extensive description of the links between genetic diversity, amino acid identity, and antigenic phenotype remains unclear for H1 viruses in the U.S. Here, we performed a comprehensive characterization of the genetic and antigenic evolution of swine H1N1 and H1N2 viruses, representing all genetic clades currently circulating in pigs in the U.S. We then conducted an in-depth antigenic characterization on contemporary strains from the H1-δ (1B.2.1 and 1B.2.2) clades due to their predominance in surveillance detections and apparent genetic expansion. Further, we examined the underlying genetic basis for significant antigenic differences among these circulating H1-δ viruses.
3. Materials and methods
3.1. Genetic evolution and amino acid sequence analysis
All available swine IAV HA and NA sequences from H1N1 and H1N2 viruses collected in the U.S. were downloaded from the Influenza Research Database (IRD) (Squires et al., 2012; Zhang et al., 2016) on December 5, 2016. To restrict our analyses to relevant field viruses, we excluded sequences with “lab” or “laboratory” host. From these data, alignments for the HA and the N1 and N2 NA genes were generated using MAFFT v7.294 (Katoh et al., 2002; Katoh and Standley, 2013). In addition, sequences with 100% identity were deleted using mothur v1.36.0 (Schloss et al., 2009), and poor quality data was removed using two criteria: a sequence was removed if > 50% of the gene was missing or if it had more than 5 nucleotide base ambiguities. This process resulted in a set of 3211 non-identical H1 HA, 1606 N1 and 1406 N2 NA swine IAV sequences that represent the full extent of the published swine H1N1 and H1N2 genetic diversity in the U.S. (Table S1). For each alignment (i.e., the HA, the NA-N1 and the NA-N2) we inferred the best-known maximum likelihood tree using RAxML (v8.2.4; (Stamatakis, 2014)) employing the rapid bootstrap algorithm, a general time-reversible (GTR) model of nucleotide substitution, and Γ-distributed rate variation among sites. The statistical support for individual branches was estimated by bootstrap analysis with the number of replicates determined automatically using an extended majority-rule consensus tree criterion (Pattengale et al., 2010). These analyses used the computational resources of the USDA-ARS computational cluster Ceres on ARS SCINet.
To determine the temporal evolution and relative genetic diversity of U.S. swine H1 HA, we implemented a time-scaled Bayesian approach on a second dataset. We downloaded complete HA H1N1 and H1N2 swine IAV genes collected from 2000 to present from the IRD on December 5, 2016. Given deep evolutionary divergence between the two major H1 HA lineages, we separated these data into the classical lineage H1 HA including H1-α (1A.1), H1-β (1A.2), H1-γ (1A.3.3.3), and H1N1pdm09 (1A.3.3.2) (n = 2208), and human-seasonal H1-δ lineage H1-δ1 (1B.2.2) and H1-δ2 (1B.2.1) (n = 1740). From each of these datasets, we randomly sampled to create smaller datasets of 750 HA genes to overcome computational limitations (i.e., we randomly sampled 707 H1-δ lineage viruses and added 43 reference antigens; similarly, we randomly sampled 713 H1-classical lineage viruses, then added the 37 reference antigens). These data were then aligned using MAFFT v7.294 (Katoh et al., 2002; Katoh and Standley, 2013), and HA genes that were duplicated due to the addition of reference antigen data were removed. The resultant data were then screened using root-to-tip regression in TempEst v.1.5 (Rambaut et al., 2016) and sequences with incongruent genetic divergence and sampling date were removed (Hicks and Duffy, 2012) resulting in a dataset of 730 and 726 HA genes for the H1-δ and H1-classical lineages respectively. The remaining data were analyzed using an uncorrelated relaxed lognormal molecular clock (Drummond et al., 2006), the SRD06 codon position model (Shapiro et al., 2006) that partitions codon positions (1 + 2 positions and 3 position) with an HKY85 + Γ substitution model applied to each partition. To reconstruct population dynamics we used the coalescent-based Gaussian Markov random field (GMRF) method with time-aware smoothing (Minin et al., 2008). The precision function in BEAUTi was used to sample uniformly within a one-year or one-month window for those viruses for which an exact date of collection was not available. All analyses were implemented in BEAST v1.8.4 (Drummond et al., 2012) with the BEAGLE library (Ayres et al., 2012) with two independent analyses of 100 million generations with sampling every 10,000 generations. Convergence of runs was checked in Tracer v1.6.0, runs were combined with LogCombiner v1.8.4, and evolutionary history was summarized and visualized using an annotated maximum clade credibility tree using TreeAnnotator v1.8.4 and FigTree v1.4.2. These analyses used the computational resources of the USDA-ARS computational cluster Ceres on ARS SCINet.
Hemagglutinin sequences used for antigenic characterization were translated to amino acids, trimmed to the HA1 domain, and aligned using MAFFT v7.294 (Katoh et al., 2002; Katoh and Standley, 2013). The HA1 amino acid alignments were used to identify amino acid substitutions likely to be clade-defining or to have resulted in antigenic differences between or within a genetic clade. We followed a criterion in which a substitution was considered pertinent as clade-defining if all (or all but one) viruses in one clade had the same amino acid at a specific position in comparison to a different amino acid in the same position of all (or all but one) viruses in another clade (Lewis et al., 2014, 2011; Smith et al., 2004). Antigenic outliers within the H1-δ (1B.2) lineage were identified as strains that were mapped at an antigenic distance greater than 2 antigenic units (AU) when compared to their respective antigenic cluster representative (centroid antigen). These outliers were then compared through pairwise comparisons of antigenic distance and amino acid similarity. Pairwise amino acid distances were inferred using a JTT model of amino acid substitution with Γ-distributed rate variation among sites; pairwise antigenic distances were extracted from the antigenic map. Those strains that had a pairwise antigenic distance > 3 AU and a pairwise amino acid distance of ≤ 4% were aligned and all pairwise amino acid mutations were determined.
3.2. Viruses
Seventy-two swine influenza H1N1 and H1N2 viruses were selected as hemagglutination inhibition (HI) test antigens and/or antigens for swine IAV-antisera production, representing 15 U.S. states (IA, MN, SD, NE, KS, MO, IL, WI, MI, OH, IN, KY, NC, OK, TX). Viruses were included to represent historical and contemporary circulating strains from each of the major H1 clades currently circulating in U.S. pigs isolated between 1930 and 2014 (Anderson et al., 2013). The HA phylogeny was generated as described above to identify genetic clades with contemporary data, then strains were selected with an emphasis on the oldest and youngest viruses in the clade, and a particular emphasis on H1-δ (1B.2.1 and 1B.2.2) clades. Viruses were also selected based on amino acid differences observed compared to other strains within clades. Selection was constrained by limitations posed by isolate availability. Viruses isolated from outbreaks of respiratory disease in pigs from routine diagnostic cases were obtained from the University of Minnesota Veterinary Diagnostic Laboratory (UMVDL, kindly provided by Dr. Marie Culhane) and from the USDA-National Animal Health Laboratory Network (NAHLN) voluntary swine IAV surveillance system repository held at the National Veterinary Service Laboratories. In addition, eight human H1 viruses were used as antigens: three H1N1pdm09 (1A.3.3.2), a human-seasonal virus related to the swine H1-δ1 and H1-δ2 clades (1B.2.2 and 1B.2.1 respectively), and four human H1 vaccine strains. Viruses were grown in Madin-Darby canine kidney (MDCK) cells or embryonated chicken’s eggs. To obtain antigens for immunization, clarified virus from cell culture supernatant or allantoic fluid was concentrated by ultracentrifugation and inactivated by ultraviolet (UV) irradiation. Inactivation of the virus was confirmed by failure to replicate in two serial passages in MDCK cells. Antigens were prepared at approximately 128 HA units per 50 μl in phosphate buffered saline (PBS) and a commercial oil-in-water adjuvant (Emulsigen D, MVP Laboratories, Inc., Ralston, NE) was added at a 1:5 ratio. Viruses used in this study are listed in Table 1.
Table 1.
Swine and human H1N1 and H1N2 influenza A virus (IAV) used as test antigens in hemagglutination inhibition assays and for the generation of antisera (in bold).
| Viruses | H1 clade | Subtype | Accession number |
|---|---|---|---|
| A/swine/Iowa/15/1930 | H1-α | H1N1 | AF091308 |
| A/swine/Iowa/1945 | H1-α | H1N1 | EU139824 |
| A/swine/Wisconsin/1/1968 | H1-α | H1N1 | EU139825 |
| A/swine/Iowa/1973 | H1-α | H1N1 | EU139826 |
| A/swine/Minnesota/02053/2008 | H1-α | H1N1 | HM461762 |
| A/swine/Minnesota/02093/2008 | H1-α | H1N1 | HM461794 |
| A/swine/North Carolina/36883/2002 | H1-β | H1N1 | EU139829 |
| A/swine/Iowa/00239/2004 | H1-β | H1N1 | EU139832 |
| A/swine/Iowa/02096/2008 | H1-β | H1N1 | HM461842 |
| A/swine/Kentucky/02086/2008 | H1-β | H1N1 | HM461786 |
| A/swine/Nebraska/02013/2008 | H1-β | H1N1 | HM461834 |
| A/swine/North Carolina/02084/2008 | H1-β | H1N1 | HM461826 |
| A/swine/Minnesota/A01076180/2009 | H1-β | H1N1 | JX042526 |
| A/swine/Minnesota/03012/2010 | H1-β | H1N1 | KY290625 |
| A/swine/Missouri/A01076190/2010 | H1-β | H1N1 | JQ809747 |
| A/swine/Minnesota/37866/1999 | H1-γ2 | H1N1 | EU139827 |
| A/swine/Minnesota/1192/2001 | H1-γ | H1N2 | CY098468 |
| A/swine/Minnesota/00194/2003 | H1-γ | H1N2 | EU139830 |
| A/swine/Kansas/00246/2004 | H1-γ | H1N2 | EU139831 |
| A/swine/Ohio/511445/2007 | H1-γ | H1N1 | EU604689 |
| A/swine/North Carolina/02023/2008 | H1-γ | H1N1 | HM461818 |
| A/swine/Ohio/02026/2008 | H1-γ | H1N1 | HM461778 |
| A/swine/Missouri/02060/2008 | H1-γ | H1N1 | HM461810 |
| A/swine/Minnesota/03011/2010 | H1-γ | H1N1 | KY290621 |
| A/swine/Indiana/3062/2010 | H1-γ | H1N1 | KY290626 |
| A/swine/Illinois/3134/2010 | H1-γ | H1N2 | KY290622 |
| A/swine/Minnesota/03003/2010 | H1-γ | H1N1 | KY290631 |
| A/California/04/2009 | pH1N1 | H1N1 | FJ966082 |
| A/Mexico/4108/2009 | pH1N1 | H1N1 | GQ149654 |
| A/New York/18/2009 | pH1N1 | H1N1 | FJ984355 |
| A/swine/Illinois/32974/2009 | pH1N1 | H1N1 | GU480922 |
| A/swine/Illinois/02991/2010 | pH1N1 | H1N1 | KY290630 |
| A/swine/Minnesota/03019/2010 | pH1N1 | H1N1 | KY290624 |
| A/swine/North Carolina/03029/2010 | pH1N1 | H1N2 | KY290623 |
| A/swine/Indiana/03053/2010 | pH1N1 | H1N2 | KY290627 |
| A/swine/Illinois/5265–1/2010 | pH1N1 | H1N1 | GU984405 |
| A/swine/Minnesota/8761/2010 | pH1N1 | H1N1 | GU984411 |
| A/swine/Illinois/07003243/2007 | H1-δ1 | H1N2 | FJ638314 |
| A/swine/Texas/01976/2008 | H1-δ1 | H1N2 | HM461850 |
| A/swine/Minnesota/02011/2008 | H1-δ1 | H1N2 | HM461802 |
| A/swine/Iowa/02039/2008 | H1-δ1a | H1N2 | HM461770 |
| A/swine/Iowa/02998/2010 | H1-δ1a | H1N2 | HM193850 |
| A/swine/Minnesota/A01134353/2011 | H1-δ1a | H1N2 | JQ906881 |
| A/swine/South Dakota/A01365260/2013 | H1-δ1a | H1N2 | KF746329 |
| A/swine/Minnesota/A01392045/2013 | H1-δ1a | H1N2 | KF715130 |
| A/swine/Nebraska/A01492366/2014 | H1-δ1a | H1N2 | KJ549771 |
| A/swine/Missouri/A01411322/2014 | H1-δ1a | H1N2 | KJ437545 |
| A/swine/Iowa/A01476931/2014 | H1-δ1a | H1N2 | KP288072 |
| A/swine/South Dakota/A01481702/2014 | H1-δ1a | H1N2 | KM013804 |
| A/swine/Illinois/A01047020/2010 | H1-δ1b | H1N2 | JQ756323 |
| A/swine/Minnesota/3128/2010 | H1-δ1b | H1N1 | CY158457 |
| A/swine/Illinois/3120/2010 | H1-δ1b | H1N2 | KY290619 |
| A/swine/Minnesota/03002/2010 | H1-δ1b | H1N2 | KY290620 |
| A/swine/Minnesota/A01301731/2012 | H1-δ1b | H1N2 | KC020465 |
| A/swine/South Dakota/A01349341/2013 | H1-δ1b | H1N2 | KC844209 |
| A/swine/Missouri/A01444664/2013 | H1-δ1b | H1N2 | KC562218 |
| A/swine/Nebraska/A01290601/2013 | H1-δ1b | H1N2 | KF791371 |
| A/swine/Iowa/A01432370/2013 | H1-δ1b | H1N2 | KC534990 |
| A/Swine/Indiana/A01260356/2013 | H1-δ1b | H1N2 | KF680052 |
| A/swine/Minnesota/A01366540/2014 | H1-δ1b | H1N2 | KJ175145 |
| A/swine/Indiana/A01260471/2014 | H1-δ1b | H1N2 | KJ650529 |
| A/swine/Illinois/A01410405/2014 | H1-δ1b | H1N2 | KJ605069 |
| A/swine/Iowa/A01410774/2014 | H1-δ1b | H1N2 | KJ859606 |
| A/swine/Illinois/00685/2005 | H1-δ2 | H1N1 | FJ638298 |
| A/swine/North Carolina/00573/2005 | H1-δ2 | H1N1 | FJ638306 |
| A/swine/Minnesota/07002083/2007 | H1-δ2 | H1N1 | FJ611898 |
| A/swine/Iowa/02955/2010 | H1-δ2 | H1N2 | KY284571 |
| A/swine/Missouri/03013/2010 | H1-δ2 | H1N2 | KY290628 |
| A/swine/Ohio/003295/2010 | H1-δ2 | H1N2 | KY284570 |
| A/swine/Minnesota/A01076626/2010 | H1-δ2 | H1N1 | JQ906871 |
| A/swine/Michigan/A01259001/2012 | H1-δ2 | H1N2 | KC508596 |
| A/swine/Ohio/A01351184/2013 | H1-δ2 | H1N2 | KF313148 |
| A/swine/North Carolina/A01290598/2013 | H1-δ2 | H1N2 | KF791368 |
| A/swine/Oklahoma/A01409770/2014 | H1-δ2 | H1N2 | KJ437589 |
| A/swine/North Carolina/A01475606/2014 | H1-δ2 | H1N2 | KM027346 |
| A/New Caledonia/20/1999 | H1-Human | H1N2 | DQ508857 |
| A/Michigan/2/2003 | H1-Human | H1N2 | CY016324 |
| A/Memphis/8/2003 | H1-Human | H1N2 | CY122316 |
| A/Solomon Islands/3/2006 | H1-Human | H1N2 | EU124177 |
| A/Brisbane/59/2007 | H1-Human | H1N2 | CY058487 |
3.3. Antisera production
Three-week-old cross-bred healthy pigs demonstrated to be free of IAV and IAV-antibodies were obtained from a high-health herd to be used for IAV antisera production. All pigs were treated with ceftiofur crystalline-free acid (Excede; Pfizer, New York, NY) and enrofloxacin (Baytril; Bayer Animal Health, Shawnee Mission, KS). Two pigs per antigen were immunized intramuscularly with adjuvanted inactivated virus. Two or three doses were given 2–3 weeks apart. When HI titers to homologous virus reached at least 1:160, pigs were humanely euthanized with pentobarbital (Fatal Plus, Vortech Pharmaceuticals, Dearborn, MI) for blood collection. Sera was obtained through centrifugation and stored at − 20 °C until use. Pigs were cared for in compliance with the Institutional Animal Care and Use Committee of the National Animal Disease Center, USDA-ARS.
3.4. Serological assay and antigenic characterization
Prior to HI testing, sera were heat inactivated at 56 °C for 30 min, then treated with a 20% suspension of kaolin (Sigma–Aldrich, St. Louis, MO) followed by adsorption with 0.5% turkey red blood cells (RBCs). HI assays were performed by testing the reference antisera panel against the 80 H1 viruses according to standard techniques (Kitikoon et al., 2014). Geometric mean titers obtained by log2 transformation of reciprocal titers were used for the comparison. Antigenic cartography was used for the quantitative analyses of the antigenic properties of swine H1 viruses as previously described (de Jong et al., 2007; Lewis et al., 2014; Lorusso et al., 2011; Smith et al., 2004).
4. Results
4.1. Genetic evolution of swine H1 influenza viruses currently circulating in the U.S
The H1 phylogeny revealed that 7 H1 genetic clades circulated during the period of study (Fig. 1A and B: Fig. S1); all of these clades were reported previously (Anderson et al., 2016). Of note, the H1-δ1 (1B.2.2) clade evolved to form two independent clades (Fig. 1A) with strong statistical support. The viruses in these clades are designated as H1-δ1a and H1-δ1b, corresponding to 1B.2.2.1 and 1B.2.2.2, respectively, following the global swine H1 HA nomenclature (Anderson et al., 2016). The H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2) clades, as well as all others, were supported by within (< 7%) and between (> 7%) clade nucleotide distance, statistical support greater than 70% at the clade defining node, and continual detection of viruses within each clade over the last 5 years (Anderson et al., 2016, 2013). Coalescent reconstruction of the H1-δ (1B.2) lineages (Fig. 1A) revealed yearly oscillation and a trend for increased relative genetic diversity following the emergence and reassortment of H1N1pdm09 (1A.3.3.2) with H1-δ viruses after 2009. In contrast, there was little evidence of yearly seasonal patterns in the relative genetic diversity in the classical swine H1 lineage (Fig. 1B). Though the overall relative genetic diversity of both lineages is similar, the relative genetic diversity of the H1-δ (1B.2) lineage appears to have consistently increased from introduction in the early 2000s to present (Fig. 1A) whereas the relative diversity within the classical lineage remained more stable (Fig. 1B).
Fig. 1.
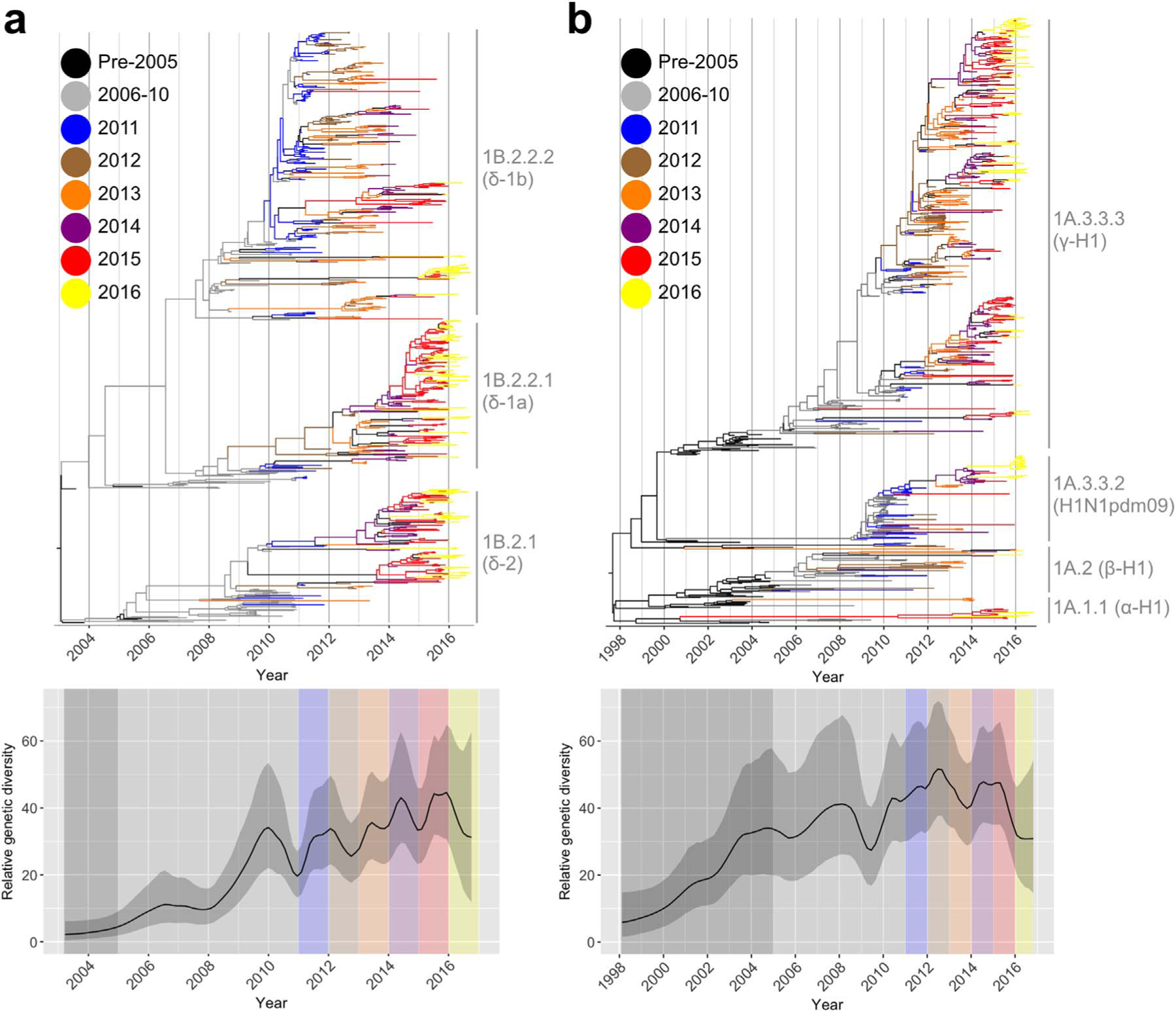
Temporal phylogenies and epidemic patterns of swine influenza A H1 viruses. Evolution of the HA genes of (a) 730 H1-δ1 and H1-δ2 lineage and, (b) 726 classical swine H1N1 viruses from a randomly sampled subset of the 3211 HA genes in this study plus the sera and antigen reference strains. Phylogenies were inferred using the uncorrelated relaxed lognormal clock model with branches colored by year of virus isolation. Relative genetic diversity was estimated using the Gaussian Markov Random Field (GMRF) model: solid black lines in the GMRF plot represent mean relative genetic diversity, with the gray shading indicating the 95% HPD intervals.
The H1 viruses circulating in pigs in the U.S. were paired with four distinct NA gene lineages: two N2 gene lineages derived from either 1998 or 2002 human-seasonal virus lineages, or an N1 of either classical swine N1 or pandemic N1 lineage. The classical N1 lineage represents the majority of the N1, whilst the pandemic N1 is becoming less frequent (Fig. S2A). Most of the H1 clades derived from the classical H1 were paired with an N1 lineage, while the majority of δ-clade viruses were paired with an N2 lineage. However, more than half of the contemporary swine H1 viruses circulating in the U.S. have an N2 gene, of which the majority is derived from the 2002 human-seasonal lineage (Fig. S2B). The δ-clades show a specific distribution pattern for either 1998 or 2002 lineage N2 genes, with H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2) paired more frequently with a 2002 N2 lineage and the H1-δ2 (1B.2.1) paired more frequently with a 1998 N2 lineage (Fig. S2B).
4.2. Antigenic diversity among contemporary swine H1 viruses
HI data (Table S2) were used to characterize the antigenic relationships of contemporary swine H1 viruses by antigenic cartography, focusing on the human-seasonal derived H1-δ viruses (Figs. 2 and S3). These data allow the calculation of antigenic distance between all pairs of antigens, with more than 2 AU between strains considered significant (1 AU is equivalent to a 2-fold difference in HI titer). Due to the antigenic diversity observed for the H1 viruses, antigenic clusters were separated based on genetic clades for improved visualization. For each antigenic cluster, the most centrally located strain with antigenic distances < 2 AU to the highest number of strains in that cluster was chosen as the antigenic reference strain. Overall, δ-clade viruses demonstrated limited cross-reactivity within each clade (H1-δ1/1B.2.2 or H1-δ2/1B.2.1), yet showed a trend to be mapped in accordance with their evolution (Figs. 2A and S3). The H1-δ1 clade (1B.2.2) was the most antigenically diverse group, and the newly emerging H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2) clades showed reduced cross-reactivity with older H1-δ1 (1B.2.2) and between each other (average within clade distance of 2.9–3 AU, and between clades of 3.2 AU). Strains of the δ-clades collected after 2012 were more antigenically diverse, with antigenic distance from the representative strain ranging from 0.4 to 4.6, and most outliers were identified among these (Fig. 2A, C–E). Classical swine-lineage H1 viruses as well as H1N1pdm09 (1A.3.3.2) were clustered antigenically according to their genetic clades, with relatively stronger cross-reactivity within a clade (average within clade distance of 1–2.4 AU). The classical swine lineage and pandemic strains formed a separate antigenic group from the H1-δ viruses (Fig. S3).
Fig. 2.
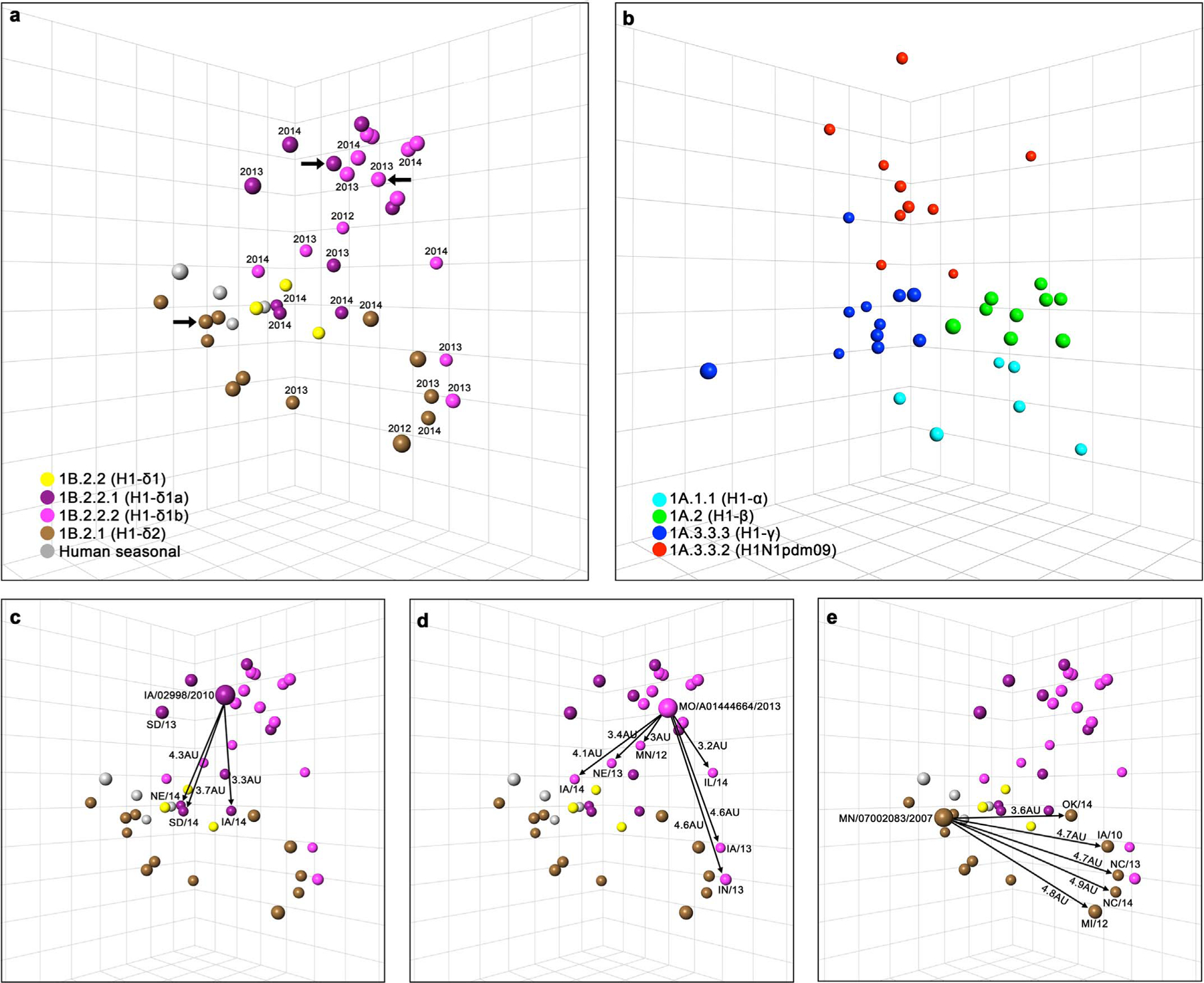
3D antigenic maps of representative historical and contemporary swine H1 influenza viruses of the 1B.2 delta (a) and 1A classical (b) lineages. Antigenic distances of outliers in comparison to antigenic cluster representative strain demonstrated for 1B.2.2.1 H1-δ1a (c), 1B2.2.2 H1-δ1b (d), and 1B.2.1 H1-δ2 (e). Isolates are shown as spheres colored by genetic clades. Each grid square represents 1 antigenic unit, corresponding to a twofold difference in HI assay titer. Year of detection is shown for contemporary delta lineage strains detected after 2012. Large arrows (a) or bigger spheres (c–e) indicate antigenic cluster representative strains A/swine/Iowa/02998/2010 (H1-δ1a), A/swine/Missouri/A01444664/2013 (H1-δ1b), A/swine/Minnesota/07002083/2007 (H1-δ2). Arrows demonstrate antigenic distance from the representative strain (c-e). Outlier strains are H1-δ1a: A/swine/Nebraska/A01492366/2014 (NE/14), A/swine/SouthDakota/A01481702/2014 (SD/14), and A/swine/Iowa/A01476931/2014 (IA/14); H1-δ1b: A/swine/Illinois/A01410405/2014 (IL/14), A/swine/Nebraska/A01290601/2013 (NE/13), A/swine/Iowa/A01410774/2014 (IA/14), A/swine/Iowa/A01432370/2013 (IA/13), A/swine/Indiana/A01260356/2013 (IN/13), and A/swine/Minnesota/A01301731/2012 (MN/12); H1-δ2: A/swine/Oklahoma/A01409770/2014 (OK/14), A/swine/Iowa/02955/2010 (IA/10), A/swine/North Carolina/A01290598/2013 (NC/13), A/swine/Michigan/A01259001/2012 (MI/12), and A/swine/North Carolina/A01475606/2014 (NC/14).
4.3. Amino acid substitutions related to antigenic diversity among H1 viruses that circulate in North American pigs
We aligned the deduced HA1 amino acid sequences of the viruses used for the antigenic cartography to investigate the genetic basis for antigenic cluster differentiation. Genetic sequences from all H1 clades were compared (Fig. S4). The H1 numbering of the mature peptide was used throughout, unless indicated otherwise (Burke and Smith, 2014). The δ-clades (1B.2) maintained the deletion of one amino acid at position 130 compared to the classical-swine (1A.1, 1A.2, 1A.3.3.3) and the H1N1pdm09 (1A.3.3.2) clades as described in the first reports of these viruses in U.S. swine (Vincent et al., 2009). The classical-swine viruses showed 14 amino acid differences from the δ-viruses that fit the clade-defining criterion (L3I, I57V, T72S, T133S, A135S, V152T, K160N, T184N, A195E, D196N, A224E, T241I, T245N, V249I; Fig. S4); a substitution was clade-defining if all, or all but one, viruses in one genetic clade had the same amino acid at a specific position in comparison to all, or all but one, viruses from a different genetic clade. The amino acids found in these 14 clade-defining positions were the same between δ-viruses and the tested human-seasonal H1 strains (Fig. S3). Six amino acid substitutions were identified as clade-defining between the H1-δ1 (1B.2.2) and the H1-δ2 (1B.2.1) viruses (A89T, I175V, T190A, W252R, D276N, I321T; Fig. S4), all of which were conserved between the swine H1-δ1 (1B.2.2) viruses and A/Michigan/2/2003, a human seasonal virus that is genetically similar to the swine δ-clade and circulated around the time the deltas emerged in pigs (Nelson et al., 2014). Differentiation between H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2) viruses was associated with four amino acid differences (E74K, S85P, D86E, G186E; Fig. S4).
We then analyzed the HA1 alignments to identify amino acid substitutions that could be involved with antigenic distance within an antigenic cluster. We focused this analysis on the H1-δ clade (1B.2.1 and 1B.2.2) because these viruses demonstrated a distinct genetic expansion and large antigenic diversity within clusters (Fig. 1A). Therefore, sequences of viruses in each H1-δ genetic clade were compared to the representative virus for each antigenic cluster separately to identify amino acid positions associated with the antigenic differences observed here (Fig. S5). Fourteen strains, most of which were strains detected after 2012, were mapped at a significant antigenic distance (> 2 AU) when compared to their respective antigenic cluster representatives (arrows in Fig. 2A), and referred to as “outliers” (Fig. 2C–E). These strains showed at least 21 unique amino acid substitutions in positions in or near the receptor binding site (RBS) and/or known antigenic sites of H1 (Caton et al., 1982; Gamblin et al., 2004) in comparison to the antigenic representative for their respective clusters (Figs. 3 and S5). Positions that were observed in more than one non-outlier virus in the cluster were not considered, only positions unique to the outliers.
Fig. 3.
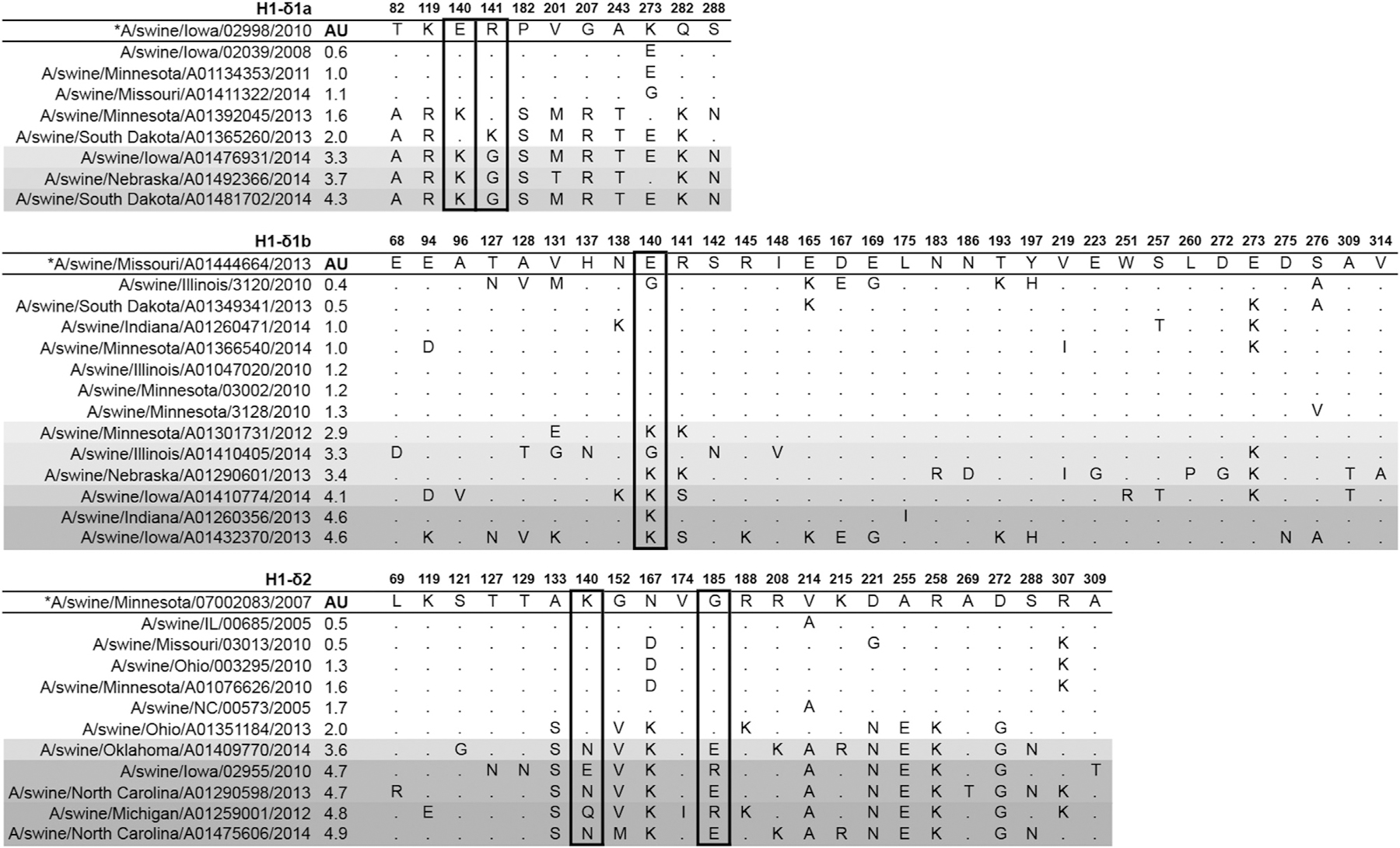
Amino acid differences identified between viruses tested in this manuscript and each respective cluster representative (*). Only amino acid positions in which outliers showed differences compared to the cluster representatives and that were located in or near the receptor binding site or antigenic sites of influenza hemagglutinin (HA) are shown, with some exceptions. Gray-scale indicates increasing antigenic distance from the representative measured as antigenic units (AU). Boxes indicate amino acid positions that were different from the cluster representatives for all outliers and that were unique to the outliers.
The three outliers for the H1-δ1a (1B.2.2.1) cluster (A/swine/Nebraska/A01492366/2014, A/swine/South Dakota/A01481702/2014 and A/swine/Iowa/A01476931/2014) were located more than 3 AU away from the H1-δ1a (1B.2.2.1) representative (Fig. 2C) and had similar amino acid changes among one another (T82A, K119R, E140K, R141G, P182S, V201M/T, G207R, A243T, Q282K, S288N), including an R141G mutation (Figs. 3 and S5). A mutation at position 141 was also present in A/swine/South Dakota/A01365260/2013 (R141K; 2 AU away from the cluster representative). An E140K mutation found in all three outliers was also found in A/swine/Minnesota/A01392045/2013 (1.6 AU away from the cluster representative), suggesting that positions 140 and 141 may have an additive effect on the antigenic differences observed for this cluster (Fig. 3).
All outliers of the H1-δ1b (1B.2.2.2) cluster (A/swine/Minnesota/A01301731/2012, A/swine/Indiana/A01260356/2013, A/swine/Iowa/A01432370/2014, A/swine/Nebraska/A01290601/2013, A/swine/Illinois/A01410405/2014, and A/swine/Iowa/A01410774/2014) were positioned 3–4.6 AU away from the cluster representative (Fig. 2D). Although these viruses showed varied numbers of amino acid differences from the representative, a common difference among them was at position 140. Positions 131 and 141 were different from the representative for at least half of these outliers (Figs. 3 and S5).
The H1-δ2 (1B.2.1) outlier strains (A/swine/Iowa/02955/2010, A/swine/Michigan/A01259001/2012, A/swine/North Carolina/A01290598/2013, A/swine/North Carolina/A01475606/2014, and A/swine/Oklahoma/A01409770/2014) were located 3.6–4.9 AU away from the cluster representative (Fig. 2E) and differed in 10 common amino acid positions. Seven of these 10 amino acid differences (except 140, 185, and 214) were also observed in A/swine/Ohio/A01351184/2013, a virus positioned at only 2 AU away from the cluster representative (Figs. 3 and S5), suggesting that the three other positions are likely the most significant. The A/swine/Indiana/A01260356/2013 H1-δ1b outlier was positioned only 0.6 AU away from two outliers of the H1-δ2 clade (NC13 and NC14); however these viruses showed less than 90% HA1 amino acid homology, and no particular amino acid similarity was observed that could affect antigenicity (Fig. S4).
To further refine antigenically-important positions, pairs of viruses with amino acid distances less than or equal to 4% and antigenic distances greater than 3 AU were identified and specific amino acid differences between these pairs were compared (Fig. 4). This criteria identified 35 pairwise comparisons between 25 viruses (Fig. 4AB). Although there were outliers fitting this criteria in all 3 δ-clades, most pairs were H1-δ1b (1B.2.2.2) clade viruses. The most frequently detected differences among the pairs of outliers were at position 140. Positions that occurred in more than ten of the pairwise comparisons included mutations at positions 131 (n = 12), 140 (n = 30), 141 (n = 17), 273 (n = 13), or 309 (n = 12) (Fig. 4C).
Fig. 4.

Scatterplot between pairwise amino acid differences and antigenic distances. (A) The relationship between pairwise amino acid distance and antigenic distance of all H1-δ (1B.2) lineage viruses (Pearson’s correlation, r = 0.45, p < 0.001: linear regression, y = 2.2 + 0.19x, r2 = 0.21). (B) Enlargement of panel A with focus on the antigenic outliers (in red circles with a pairwise antigenic distance > 3 AU and a pairwise amino acid distance of ≤ 4%. Pairwise amino acid differences were calculated using a JTT model of amino acid substitution with Γ-distributed rate variation among sites; pairwise antigenic distances were extracted from the antigenic map. Linear regression lines with 95% confidence intervals are shown. (C) Amino acid differences identified between pairs of viruses identified as outliers in the pairwise comparison. Only amino acids located in or near antigenic sites or the receptor binding site of influenza hemagglutinin (HA) are shown, with some exceptions. Positions highlighted in gray occurred in more than 10 pairwise comparisons.
5. Discussion
Following the implementation of the U.S. national swine IAV surveillance system by the U.S. Department of Agriculture in 2009, the quantity of HA and NA viral sequence data has increased substantially, revealing a diversity of viruses co-circulating in U.S. pigs. Notably, a dramatic evolution has been observed for swine IAV in the past decade (Anderson et al., 2013; Gao et al., 2017; Rajao et al., 2017). Here, we characterized the genetic and antigenic diversity of currently circulating swine H1 influenza viruses in the U.S. While the classical-swine H1 viruses maintained the relative antigenic clustering characteristics that were seen for viruses circulating prior to 2009 (Lorusso et al., 2011), the δ-viruses (1B.2.1 and 1B.2.2) became more genetically and antigenically diverse. Our analysis revealed that two new genetic sub-clades emerged from the H1-δ1 clade around 2007, H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2), sustained transmission in pigs, and had expanded antigenic diversity.
Our phylogenetic analysis demonstrated considerable genetic diversity among H1 viruses circulating in U.S. pigs, with co-circulation of both major lineages and 7 distinct genetic clades of viruses (Anderson et al., 2013; Lorusso et al., 2011). The classical-swine H1 clades circulated consistently over the past decade, with records of H1-α (1A.1), H1-β (1A.2), H1N1pdm09 (1A.3.3.2), and H1-γ (1A.3.3.3) clade viruses detected each year (Lorusso et al., 2013). The anomaly within the classical-swine lineage is the appearance and rapid decline of the genetically and antigenically distinct clade of H1-γ2 (1A.3.2) viruses (Anderson et al., 2015; Gao et al., 2017). Coalescent reconstruction of the population demographics of the classical-swine lineage (Fig. 1B) revealed an increase in relative genetic diversity from the early 2000s to 2010, followed by a period of more stable relative genetic diversity. This increase in diversity corresponds to the emergence and reassortment of the late 1990s novel triple-reassortant H3N2 virus with the endemic classical-swine H1 viruses. The δ-clade viruses (1B.2.1 and 1B.2.2), however, showed a different evolutionary trajectory with a distinct increase in genetic diversity from approximately 2008 to 2016 (Fig. 1A). Our coalescent reconstruction showed an almost linear increase in relative genetic diversity, with the more recent years demonstrating oscillating patterns that correspond to the seasonal dynamics of IAV in swine (Anderson et al., 2013). This genetic diversity resulted in two new monophyletic clades descended from the H1-δ1 (1B.2.2) clade and indicated the need for new clade designations (1B.2.2.1 and 1B.2.2.2) following criteria defined previously (Anderson et al., 2016). Antigenic characterization of representative viruses from the new clades was warranted based on the genetic distance between the two new clades. A similar pattern of increased genetic and antigenic expansion was observed for North American swine H3 viruses, in which six distinct clades evolved from the previously stable Cluster-IV (Kitikoon et al., 2013).
The molecular basis for the genetic diversity of swine H1-δ1 (1B.2.2) viruses is a result of unique human-to-swine transmission events and within-host evolution. Amino acid positions that differed between classical-swine and H1-δ remained conserved with human-seasonal reference strains. This suggests that the HA of δ-clade viruses maintains a human-seasonal genetic signature even after more than 10 years of circulation in U.S. swine herds, despite swine H1-δ viruses having higher evolutionary rates than closely related human-seasonal viruses (Nelson et al., 2014). The combination of such human-seasonal-like characteristics, particularly at or near receptor-binding sites, and waning population immunity associated with the replacement of seasonal H1N1 human strains by the H1N1pdm09 (Pica et al., 2012) may allow novel reassortant viruses and antigenically drifted strains from the 1B genetic lineage and subsequent sub-clades to spill back to human populations, underscoring the importance of monitoring the evolution of circulating swine IAV for human pandemic preparedness.
Four distinct NA lineages were identified paired with the contemporary swine H1 viruses, with some predilections between HA clade and NA clade. The classical-swine lineage virus HA’s were paired mostly with classical-swine N1 lineage, a pattern similar to that of the N1 gene of H1-α (1A.1, 1A.1.1), H1-β (1A.2), and H1-γ (1A.3.3.3) viruses circulating prior to 2008 (Lorusso et al., 2011). The H1-δ viruses were paired with N2 lineages with a high frequency, with the H1-δ2 (1B.2.1) most frequently paired with the 1998 N2 lineage, and the H1-δ1a (1B.2.2.1) and H1-δ1b (1B.2.2.2) most frequently paired with the 2002 N2 lineage. Overall, the N2–2002 lineage was detected at a much higher frequency than the N2–1998 lineage among H1N2 viruses.
Since the δ-clade viruses demonstrated a continued linear increase in relative genetic diversity, we focused on testing contemporary δ-clade viruses in HI assays and analysis by antigenic cartography to evaluate potential concurrent antigenic evolution. Contemporary viruses of the H1-δ1 (H1-δ1a/1B.2.2.1 and H1-δ1b/1B.2.2.2) and H1-δ2 (1B.2.1) clades demonstrated significant antigenic distance from previously tested viruses (Lorusso et al., 2011). Antigenic drift of IAV is frequently associated with amino acid substitutions in antigenic sites on the globular head of the HA (Koel et al., 2013). Only 6–7 amino acid changes in the HA1 domain were shown to be responsible for marked antigenic differences in human and swine H3 viruses (Koel et al., 2013; Lewis et al., 2014), however H1 viruses seem to behave differently and the patterns seen for H3 viruses do not seem to be replicated for human H1 viruses (Koel et al., 2015). Here, several amino acid differences were observed between same-clade swine H1 viruses, some of which might have an antigenic effect. Positions 131, 140, 141, 273, and 309 were identified when comparing pairs of viruses with highly similar amino acid sequences (< 4%) but substantial antigenic distance (> 3 AU). With the exception of position 309, all of the remaining positions were located at or near antigenic sites or the receptor-binding site. Mutations at position 140 were present in all viruses considered outliers. Additionally, changes at positions 131 appeared in combination with position 140 in a limited number of viruses with significant antigenic distance, while position 141 appeared in combination with 140 in more than half outliers of H1-δ1a and H1-δ1b. Position 141 is the corresponding H1 position to position 145 in swine H3N2 viruses that was associated with significant antigenic change (Abente et al., 2016; Lewis et al., 2014). Positions 152 and 185 were associated with antigenic distance among H1-δ2 viruses, which corresponds to positions 156 and 189 shown to have antigenic impact in swine H3N2 viruses. Although the swine H1-δ viruses are related to seasonal H1 viruses that circulated in humans prior to 2009 and maintain a relatively high degree of amino acid similarity, evolution in North American pigs led to the appearance of viruses that are now antigenically distinct from ancestral precursors (Fig. 2A).
The genetic evolution and consequent antigenic diversity among contemporary swine H1 viruses complicates the control of influenza infection in North American pigs through the use of vaccines. Current commercially available vaccines used against IAV in the U.S. swine industry are not frequently updated and, therefore, tend not to reflect the true genetic and antigenic diversity of contemporary circulating swine viruses (Rajao et al., 2014; Vincent et al., 2008b). Vaccine strain selection is a crucial process to obtain adequate and broad cross-protection against IAV infection in pigs, and antigenic cross-reactivity should be analyzed when doing so, since sequence similarity was not highly predictive of antigenic distance in many pairs of viruses (Fig. 4A). Thus, antigenic evaluation should be taken under consideration in conjunction with phylogenetic analysis for choosing efficacious vaccine strains. Nevertheless, faster and easier regulatory procedures are necessary to allow rapid modification of vaccines to keep up with the rapid evolution of circulating swine IAV lineages and possible emergence of reassorted strains.
In addition to swine being an important host in the general epidemiology and evolution of IAV, influenza illness also represents a significant economic burden to the swine industry. The current genetic evolution of the virus in swine populations and the resulting antigenic diversity contributes to vaccine failure, and these drifted viruses represent a potential zoonotic risk. We demonstrated that swine H1 influenza viruses underwent substantial antigenic diversification between 2010 and 2014, likely as a result of a few key amino acid changes. As genetic expansion was observed to continue from 2014 to 2016, these H1-δ viruses likely continued to drift, with obvious consequences for immune protection and vaccination. Characterizing the genetic evolution of swine IAV and understanding the implications to antigenic diversity and cross-protection is crucial to improve control measures that could lessen the consequences that this disease brings to pork production and public health.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Animal Health Laboratory Network of veterinary diagnostic laboratories that deposit swine influenza samples into publically available databases and pork producers, swine veterinarians, and laboratories for participating in the USDA Influenza A Virus in Swine Surveillance System. We wish to thank Michelle Harland and Gwen Nordholm for laboratory assistance, and Jason Huegel, Jason Crabtree, and Tyler Standley for animal care and handling assistance.
This study was supported by USDA-ARS, USDA-APHIS, and by an NIH-National Institute of Allergy and Infectious Diseases (NIAID) interagency agreement associated with CRIP (Center of Research in Influenza Pathogenesis), an NIAID funded Center of Excellence in Influenza Research and Surveillance (CEIRS, HHSN272201400008C) to ALV. TKA was supported by an appointment to the USDA-ARS Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and USDA under Contract no. DE-AC05–06OR23100. NSL was funded by USDA-ARS SCA Agreement no. 58–3625-2–103F and CRIP (CEIRS, HHSN272201400008C). This research used resources provided by the SCINet project of the USDA Agricultural Research Service, ARS Project no. 0500–00093-001–00-D. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA, DOE, or ORISE. USDA is an equal opportunity provider and employer.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2018.02.006.
References
- Abente EJ, Santos J, Lewis NS, Gauger PC, Stratton J, Skepner E, Anderson TK, Rajao DS, Perez DR, Vincent AL, 2016. The molecular determinants of antibody recognition and antigenic drift in the H3 hemagglutinin of swine influenza A virus. J. Virol 90, 8266–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TK, Campbell BA, Nelson MI, Lewis NS, Janas-Martindale A, Killian ML, Vincent AL, 2015. Characterization of co-circulating swine influenza A viruses in North America and the identification of a novel H1 genetic clade with antigenic significance. Virus Res 201, 24–31. [DOI] [PubMed] [Google Scholar]
- Anderson TK, Macken CA, Lewis NS, Scheuermann RH, Van Reeth K, Brown IH, Swenson SL, Simon G, Saito T, Berhane Y, Ciacci-Zanella J, Pereda A, Davis CT, Donis RO, Webby RJ, Vincent AL, 2016. A phylogeny-based global nomenclature system and automated annotation tool for H1 hemagglutinin henes from swine influenza A viruses. mSphere 1, e00275–00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TK, Nelson MI, Kitikoon P, Swenson SL, Korslund JA, Vincent AL, 2013. Population dynamics of cocirculating swine influenza A viruses in the United States from 2009 to 2012. Influenza Other Respir. Virus 7, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP, Rambaut A, Suchard MA, 2012. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol 61, 170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both GW, Shi CH, Kilbourne ED, 1983. Hemagglutinin of swine influenza virus: a single amino acid change pleiotropically affects viral antigenicity and replication. Proc. Natl. Acad. Sci. USA 80, 6996–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown IH, 2000. The epidemiology and evolution of influenza viruses in pigs. Vet. Microbiol 74, 29–46. [DOI] [PubMed] [Google Scholar]
- Burke DF, Smith DJ, 2014. A recommended numbering scheme for influenza A HA subtypes. PLoS One 9, e112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton AJ, Brownlee GG, Yewdell JW, Gerhard W, 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31, 417–427. [DOI] [PubMed] [Google Scholar]
- de Jong JC, Smith DJ, Lapedes AS, Donatelli I, Campitelli L, Barigazzi G, Van Reeth K, Jones TC, Rimmelzwaan GF, Osterhaus AD, Fouchier RA, 2007. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J. Virol 81, 4315–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JC, van Nieuwstadt AP, Kimman TG, Loeffen WL, Bestebroer TM, Bijlsma K, Verweij C, Osterhaus AD, Class EC, 1999. Antigenic drift in swine influenza H3 haemagglutinins with implications for vaccination policy. Vaccine 17, 1321–1328. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ho SY, Phillips MJ, Rambaut A, 2006. Relaxed phylogenetics and dating with confidence. PLoS Biol 4, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A, 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol 29, 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducatez MF, Hause B, Stigger-Rosser E, Darnell D, Corzo C, Juleen K, Simonson R, Brockwell-Staats C, Rubrum A, Wang D, Webb A, Crumpton JC, Lowe J, Gramer M, Webby RJ, 2011. Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg. Infect. Dis 17, 1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ, 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303, 1838–1842. [DOI] [PubMed] [Google Scholar]
- Gao S, Anderson TK, Walia RR, Dorman KS, Janas-Martindale A, Vincent AL, 2017. The genomic evolution of H1 influenza A viruses from swine detected in the United States between 2009 and 2016. J. Gen. Virol 98, 2001–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks AL, Duffy S, 2012. One misdated sequence of rabbit hemorrhagic disease virus prevents accurate estimation of its nucleotide substitution rate. BMC Evol. Biol 12, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhung MA, Epperson S, Biggerstaff M, Allen D, Balish A, Barnes N, Beaudoin A, Berman L, Bidol S, Blanton L, Blythe D, Brammer L, D’Mello T, Danila R, Davis W, de Fijter S, Diorio M, Durand LO, Emery S, Fowler B, Garten R, Grant Y, Greenbaum A, Gubareva L, Havers F, Haupt T, House J, Ibrahim S, Jiang V, Jain S, Jernigan D, Kazmierczak J, Klimov A, Lindstrom S, Longenberger A, Lucas P, Lynfield R, McMorrow M, Moll M, Morin C, Ostroff S, Page SL, Park SY, Peters S, Quinn C, Reed C, Richards S, Scheftel J, Simwale O, Shu B, Soyemi K, Stauffer J, Steffens C, Su S, Torso L, Uyeki TM, Vetter S, Villanueva J, Wong KK, Shaw M, Bresee JS, Cox N, Finelli L, 2013. Outbreak of variant influenza A (H3N2) virus in the United States. Clin. Infect. Dis 57, 1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasin AI, Carman S, Olsen CW, 2006. Identification of human H1N2 and human-swine reassortant H1N2 and H1N1 influenza A viruses among pigs in Ontario, Canada (2003–2005). J. Clin. Microbiol 44, 1123–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T, 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM, 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitikoon P, Gauger PC, Vincent AL, 2014. Hemagglutinin inhibition assay with swine sera. In: Spackman E (Ed.), Animal Influenza Virus Springer, New York, NY, pp. 295–301. [DOI] [PubMed] [Google Scholar]
- Kitikoon P, Nelson MI, Killian ML, Anderson TK, Koster L, Culhane MR, Vincent AL, 2013. Genotype patterns of contemporary reassorted H3N2 virus in US swine. J. Gen. Virol 94, 1236–1241. [DOI] [PubMed] [Google Scholar]
- Koel BF, Burke DF, Bestebroer TM, van der Vliet S, Zondag GC, Vervaet G, Skepner E, Lewis NS, Spronken MI, Russell CA, Eropkin MY, Hurt AC, Barr IG, de Jong JC, Rimmelzwaan GF, Osterhaus AD, Fouchier RA, Smith DJ, 2013. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science 342, 976–979. [DOI] [PubMed] [Google Scholar]
- Koel BF, Mogling R, Chutinimitkul S, Fraaij PL, Burke DF, van der Vliet S, de Wit E, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Smith DJ, Fouchier RA, de Graaf M, 2015. Identification of amino acid substitutions supporting antigenic change of influenza A(H1N1)pdm09 viruses. J. Virol 89, 3763–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NS, Anderson TK, Kitikoon P, Skepner E, Burke DF, Vincent AL, 2014. Substitutions near the hemagglutinin receptor-binding site determine the antigenic evolution of influenza A H3N2 viruses in U.S. swine. J. Virol 88, 4752–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NS, Daly JM, Russell CA, Horton DL, Skepner E, Bryant NA, Burke DF, Rash AS, Wood JL, Chambers TM, Fouchier RA, Mumford JA, Elton DM, Smith DJ, 2011. Antigenic and genetic evolution of equine influenza A (H3N8) virus from 1968 to 2007. J. Virol 85, 12742–12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NS, Russell CA, Langat P, Anderson TK, Berger K, Bielejec F, Burke DF, Dudas G, Fonville JM, Fouchier RA, Kellam P, Koel BF, Lemey P, Nguyen T, Nuansrichy B, Peiris JM, Saito T, Simon G, Skepner E, Takemae N, Webby RJ, Van Reeth K, Brookes SM, Larsen L, Watson SJ, Brown IH, Vincent AL, 2016. The global antigenic diversity of swine influenza A viruses. eLife 5, e12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso A, Vincent AL, Gramer ME, Lager KM, Ciacci-Zanella JR, 2013. Contemporary epidemiology of North American lineage triple reassortant influenza A viruses in pigs. In: Richt JA, Webby RJ (Eds.), Swine Influenza Springer, Berlin, Heidelberg, DE, pp. 113–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso A, Vincent AL, Harland ML, Alt D, Bayles DO, Swenson SL, Gramer MR, Russell CA, Smith DJ, Lager KM, Lewis NS, 2011. Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J. Gen. Virol 92, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoh SM, McGregor MW, Hinshaw VS, 1992. Hemagglutinin mutations related to antigenic variation in H1 swine influenza viruses. J. Virol 66, 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin VN, Bloomquist EW, Suchard MA, 2008. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol 25, 1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher RA, Bedford T, Daniels RS, Russell CA, Shraiman BI, 2016. Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses. Proc. Natl. Acad. Sci. USA 113, E1701–E1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MI, Gramer MR, Vincent AL, Holmes EC, 2012. Global transmission of influenza viruses from humans to swine. J. Gen. Virol 93, 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MI, Wentworth DE, Culhane MR, Vincent AL, Viboud C, LaPointe MP, Lin X, Holmes EC, Detmer SE, 2014. Introductions and evolution of human-origin seasonal influenza a viruses in multinational Swine populations. J. Virol 88, 10110–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A, 2010. How many bootstrap replicates are necessary? J. Comput. Biol 17, 337–354. [DOI] [PubMed] [Google Scholar]
- Pica N, Hai R, Krammer F, Wang TT, Maamary J, Eggink D, Tan GS, Krause JC, Moran T, Stein CR, Banach D, Wrammert J, Belshe RB, Garcia-Sastre A, Palese P, 2012. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. USA 109, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajao DS, Anderson TK, Gauger PC, Vincent AL, 2014. Pathogenesis and vaccination of influenza A virus in swine. In: Compans RW, Oldstone MBA (Eds.), Influenza Pathogenesis and Control Springer, New York, NY, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Rajao DS, Walia RR, Campbell B, Gauger PC, Janas-Martindale A, Killian ML, Vincent AL, 2017. Reassortment between swine H3N2 and 2009 pandemic H1N1 in the United States resulted in influenza A viruses with diverse genetic constellations with variable virulence in pigs. J. Virol 91, e01763–e017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG, 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2, vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF, 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B, Rambaut A, Drummond AJ, 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol 23, 7–9. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA, 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376. [DOI] [PubMed] [Google Scholar]
- Smith GJ, Bahl J, Vijaykrishna D, Zhang J, Poon LL, Chen H, Webster RG, Peiris JS, Guan Y, 2009a. Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. USA 106, 11709–11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A, 2009b. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125. [DOI] [PubMed] [Google Scholar]
- Squires RB, Noronha J, Hunt V, Garcia-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN, Ramsey A, Zhou L, Zaremba S, Kumar S, Deitrich J, Klem E, Scheuermann RH, 2012. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir. Virus 6, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna D, Smith GJ, Pybus OG, Zhu H, Bhatt S, Poon LL, Riley S, Bahl J, Ma SK, Cheung CL, Perera RA, Chen H, Shortridge KF, Webby RJ, Webster RG, Guan Y, Peiris JS, 2011. Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473, 519–522. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Ciacci-Zanella JR, Lorusso A, Gauger PC, Zanella EL, Kehrli ME Jr., Janke BH, Lager KM, 2010. Efficacy of inactivated swine influenza virus vaccines against the 2009 A/H1N1 influenza virus in pigs. Vaccine 28, 2782–2787. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Lager KM, Janke BH, Gramer MR, Richt JA, 2008a. Failure of protection and enhanced pneumonia with a US H1N2 swine influenza virus in pigs vaccinated with an inactivated classical swine H1N1 vaccine. Vet. Microbiol 126, 310–323. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Ma W, Lager KM, Gramer MR, Richt JA, Janke BH, 2009. Characterization of a newly emerged genetic cluster of H1N1 and H1N2 swine influenza virus in the United States. Virus Genes 39, 176–185. [DOI] [PubMed] [Google Scholar]
- Vincent AL, Ma W, Lager KM, Janke BH, Richt JA, 2008b. Swine influenza viruses: a North American perspective. In: Maramorosch K, Shatkin AJ, Murphy FA (Eds.), Adv. Virus Res Academic Press, Burlington, MA, pp. 127–154. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Aevermann BD, Anderson TK, Burke DF, Dauphin G, Gu Z, He S, Kumar S, Larsen CN, Lee AJ, Li X, Macken C, Mahaffey C, Pickett BE, Reardon B, Smith T, Stewart L, Suloway C, Sun G, Tong L, Vincent AL, Walters B, Zaremba S, Zhao H, Zhou L, Zmasek C, Klem EB, Scheuermann RH, 2016. Influenza research database: an integrated bioinformatics resource for influenza virus research. Nucleic Acids Res 45, D466–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou NN, Senne DA, Landgraf JS, Swenson SL, Erickson G, Rossow K, Liu L, Yoon K, Krauss S, Webster RG, 1999. Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J. Virol 73, 8851–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.