

Abstract
Intricate relationships between endocytosis and cellular signaling, first recognized nearly 40 years ago through the study of tyrosine kinase growth factor receptors, are now known to exist for multiple receptor classes and to affect myriad physiological and developmental processes. This review summarizes our present understanding of how endocytosis orchestrates cellular signaling networks, with an emphasis on mechanistic underpinnings and focusing on two receptor classes—tyrosine kinase and G protein–coupled receptors—that have been investigated in particular detail. Together, we believe that these examples provide a useful survey of the current consensus, uncertainties, and controversies in this rapidly advancing area of cell biology.
Keywords: receptor tyrosine kinases, G protein–coupled receptors, endocytosis, signaling, endosomes, downregulation
INTRODUCTION
Signaling receptors transit secretory and endocytic pathways similarly to many other integral membrane proteins. Accordingly, they depend on generic membrane trafficking mechanisms. Evidence for a more elaborate and specific relationship emerged from studies of two receptor tyrosine kinases (RTKs), the EGF receptor (EGFR) and insulin receptor (IR), whose endocytic rates were found to be strongly increased in response to ligand-induced activation (1). These receptors internalize in complex with their activating ligand and then are packaged into vesicles within the endosome lumen and delivered to lysosomes for degradation. In the interim, ligand-bound receptors with their activating tyrosine residues phosphorylated reside in the endosome-limiting membrane and remain capable of binding cytoplasmic signaling adaptors. Together, these seminal observations established the concept that endocytosis can affect cellular signaling by receptors not only by attenuating cellular ligand responsiveness through receptor downregulation but also by mediating temporally and spatially distinct responses from the surface of endosomes.
Intricate relationships between endocytosis and cellular signaling have now been observed for many receptor classes, as reviewed previously elsewhere (2–4). Among these are G protein–coupled receptors (GPCRs), the largest receptor family overall and an important group of drug targets. Ligand-induced internalization and downregulation of GPCRs were discovered only slightly later than the corresponding processes for RTKs, with both classes of receptors sharing key trafficking machineries and pathways (5). However, GPCRs use additional mechanisms for control of receptor signaling and transit through the endocytic network, and their elucidation evolved from different starting assumptions about whether signaling does (RTKs) or does not (GPCRs) occur from endosomes.
This review considers how endocytic membrane trafficking affects cellular signaling by RTKs and GPCRs in mammalian cells. We start by summarizing the present mechanistic framework of receptor endocytic trafficking and then discuss what is currently known about the effects of RTK and GPCR endocytosis on signaling to downstream effectors, considering both negative and positive effects and highlighting knowledge gaps and areas of controversy. We believe that this scope, while admittedly limited, captures key aspects of the evolving view of trafficking–signaling relationships more broadly.
ENDOCYTOSIS OF RECEPTOR TYROSINE KINASES
Analysis of the ligand-induced endocytosis of growth factor receptors began in the early 1980s, immediately after receptors for EGF and, subsequently, for other growth factors and insulin were discovered and their intrinsic tyrosine kinase activity was demonstrated. This research was built on, and benefited from, pioneering methods for analysis of labeled EGF and insulin uptake in cells (6, 7). These studies have shown that the constitutive endocytosis of most ligand-free RTKs is slow, and therefore, they accumulate at the cell surface and are accessible to extracellular ligands. Ligand binding robustly accelerates the endocytosis of many RTKs. First EGFR and then other RTKs were shown to use the clathrin-mediated endocytosis (CME) pathway for their ligand-induced internalization (8; reviewed in 1). Ligand-induced, clathrin-independent endocytosis (CIE) of several RTK subfamilies has also been demonstrated (see below).
Despite thousands of studies, the molecular mechanisms of RTK endocytosis are not well understood. The literature reports such large differences in rates and mechanisms of endocytosis that a unified mechanistic model has yet to emerge. For example, although ligand binding dramatically accelerates EGFR endocytosis, the closest relative of EGFR, ErbB2, is not efficiently internalized after its activation by overexpression or heteromerization in many types of cancer cells (9, 10). At the same time, efficient ErbB2 endocytosis has been reported in other cell types (11, 12).
EGFR has been the most studied RTK for historical reasons and because of the commercial availability of labeled EGF and other experimental tools. Nonetheless, a unified model for EGFR endocytosis has yet to emerge. Ligand-induced EGFR endocytosis combines high-affinity uptake that is easily saturated with lower-affinity uptake that has higher capacity (13). Mechanistically, the high-affinity pathway is mediated by CME, and this pathway is predominantly used under conditions when a relatively small number of EGFRs, typically 10,000–20,000 per cell, are ligand occupied at the cell surface. Low-affinity uptake is mediated by slower (but still ligand-induced) CIE pathways, and these significantly contribute to the overall uptake of EGFR when large amounts of ligand-bound EGFR are present (14). The nature of the limited capacity of EGFR CME is unknown. Recruitment of active EGFR into clathrin-coated pits (CCPs) is considered to be a rate-limiting step of CME. Whether receptors are recruited into preexisting CCPs or promote assembly of new CCPs is under debate. Two mechanisms, (a) direct interaction with the clathrin adaptor complex AP-2 via receptor tyrosine-based (YxxΦ) and/or dileucine motifs and (b) ubiquitination, have been proposed to mediate EGFR recruitment into CCPs in a redundant fashion (15) (Figure 1). EGFR is ubiquitinated by two highly homologous E3 ubiquitin ligases, c-Cbl and Cbl-b, which bind to phosphorylated tyrosines of the activated receptor directly or indirectly through the adaptor protein Grb2 (16–18). Ubiquitin moieties on EGFR are thought to interact with ubiquitin-binding domains of accessory proteins, such as epsins and Eps15R, that bind to AP-2 or directly to clathrin. However, mutations in multiple ubiquitin conjugation sites and AP-2 binding motifs did not fully inhibit the CME of EGFR, suggesting the existence of yet another redundant mechanism for EGFR recruitment into CCPs (15). The contribution of these redundant mechanisms varies in different cell types.
Figure 1.
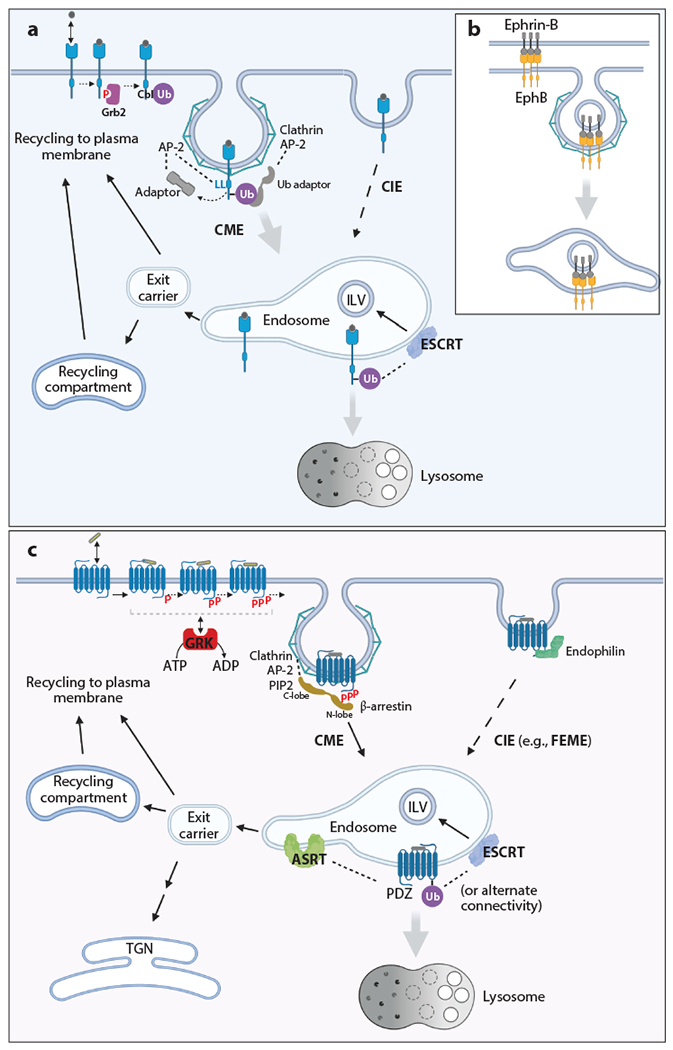
Pathways of endocytosis and postendocytic sorting of RTKs and GPCRs. (a) Simplified scheme for regulated endocytosis and postendocytic sorting of RTKs. Ligand binding results in activation of the receptor kinase; tyrosine phosphorylation of the C-terminal tail (red P); binding of Cbl, either directly to the receptor pTyr or through Grb2; and RTK ubiquitination. RTK is recruited into CCPs either via an interaction between ubiquitin moieties and adaptors in CCPs (which bind to AP-2 or clathrin), direct binding of internalization motifs (LL motif is shown) to AP-2, or an indirect interaction between AP-2 and additional adaptor proteins. RTKs internalized through CIE and CME accumulate in the same early/sorting endosomes containing EEA1 and Rab5. Ubiquitinated RTK is sorted to ILVs in MVEs via interactions with ESCRTs, whereas nonubiquitinated RTK is capable of recycling back to the plasma membrane via a fast route involving Rab4 or a slower route involving additional recycling compartments containing Rab11 or Rab25. (b) Trans-endocytosis of the Ephrin-B:EphB complex. Binding of Ephrin-B expressed in a donor cell to EphB expressed in the acceptor cell results in CME of the ligand together with the surrounding plasma membrane of the donor cell into the acceptor cell. Postendocytic sorting pathways of EphRs are similar to those shown in panel a. (c) Simplified scheme for regulated endocytosis and postendocytic sorting of GPCRs. Endocytosis is regulated through agonist-dependent Ser/Thr phosphorylation of the GPCR tail (red P), which promotes recruitment of β-arrestin from the cytoplasm and a conformational change in β-arrestin that exposes latent endocytic motifs. Agonists can selectively regulate endocytosis through an allosteric selection process mediated by distinct GRK recruitment modes that encode different phosphorylation patterns into a modular Ser/Thr substrate sequence (or barcode) in the receptor tail. Internalized receptors traffic to lysosomes by engaging ESCRTs, either through receptor ubiquitination or alternative protein connectivity not requiring receptor ubiquitination, as described in the text. A number of GPCRs are sorted by sequence-directed recycling; a PDZ motif–directed mechanism that is engaged by β2AR is depicted. There is considerable diversity among GPCRs in their overall postendocytic itinerary and kinetics of transit. Abbreviations: β2AR, β-2 adrenergic receptor; ASRT, actin–SNX27–retromer tubule; CCP, clathrin-coated pit; CIE, clathrin-independent endocytosis; CME, clathrin-mediated endocytosis; ESCRT, endosomal sorting complex required for transport; FEME, fast endophilin-mediated endocytosis; GPCR, G protein–coupled receptor; GRK, GPCR kinase; ILV, intraluminal vesicle; MVE, multivesicular endosome; P, phosphate; PIP2, phosphatidylinositol 4,5-phosphate; RTK, receptor tyrosine kinase; TGN, trans-Golgi network; Ub, ubiquitin. Figure adapted from images created with BioRender.com.
Ubiquitination and AP-2 interactions have been implicated in the endocytosis of several other RTKs, such as platelet-derived growth factor (PDGF), c-Met, fibroblast growth factor, vascular endothelial growth factor, Trk, Axl, IR, and the Eph receptor (EphR) subfamily, although the molecular mechanisms by which the ubiquitination and internalization motifs of these other RTKs facilitate CME have not been, to our knowledge, well established (reviewed in 1). An interesting alternative mechanism of IR endocytosis has been recently proposed (19, 20). These studies showed that a novel MIM motif in IR binds to a complex of mitotic checkpoint regulators (MAD2–CDC20–BUBR1) that interacts with AP-2. In addition, the NPxY motif in IR, when phosphorylated on tyrosine, binds to IRS1/2 adaptors, which in turn recruit AP-2 through their YxxΦ motifs. Both of these adaptor-based mechanisms can lead to CME of IR. Thus, a general mechanism of RTK recruitment into CCPs is either direct binding to AP-2 or, more commonly, an indirect interaction with AP-2 through RTK ubiquitination and/or additional adaptor proteins (Figure 1a).
Accumulating evidence supports the existence of multiple CIE mechanisms of RTK internalization. Massive endocytosis (pinocytosis) of labeled EGF without any apparent involvement of CCPs was first noticed in early studies using A-431 cells that express extraordinarily high levels of EGFR (21). Several mechanisms of EGFR CIE have been proposed that involve membrane ruffling (22), fast endophilin-mediated endocytosis (FEME) (23), reticulon-3 (24), and other proteins and processes. Likewise, CIE has been demonstrated for PDGF (25), TrkA (26), and EphR RTKs (27). However, the molecular mechanisms by which RTKs are specifically recruited into forming endocytic intermediates are unknown. Moreover, the relative contribution of CIE to RTK endocytosis in physiologically relevant experimental models has not been rigorously determined.
The endocytic mechanisms of the EphR subfamily of RTKs are unique. This family consists of EphA and EphB, which are activated by the membrane-associated ligands glycophosphatidylinositol (GPI)-anchored ephrins (A type) or transmembrane type B ephrins, respectively (reviewed in 28, 29). Binding of ephrin-A, which is expressed on the surface of the donor cell, to EphA in the acceptor cell leads to the proteolysis of the GPI-anchored ligand by metalloproteases, such as ADAM10, and subsequent endocytosis of the cleaved portion of the extracellular domain of ephrin-A bound to the receptor in the acceptor cell. Endocytosis of EphB receptors involves so-called trans-endocytosis, during which the ligand, together with the surrounding plasma membrane of the donor cell, is internalized into the EphB-expressing acceptor cell (Figure 1b). Similar trans-endocytosis of the Ephrin-A1:EphA2 complex has also been demonstrated (30). CME mechanisms have been predominantly implicated in EphR internalization and trans-endocytosis (30–32). An important property of EphR CME is its strong dependence on actin polymerization and the activity of the small GTPase Rac1 (30, 31). A fascinating feature of the ephrin-EphR system is that signaling processes and endocytosis are bidirectional, i.e., they can be triggered in the donor cell by the ligand and in the acceptor cell by the receptor (reviewed in 28, 29).
Summarizing what is known about RTK internalization reveals striking redundancy among the putative mechanisms, which is indicative of the robustness of the process. This redundancy is one of the major reasons why the molecular mechanisms governing internalization of RTKs, even the extensively studied EGFR, have been difficult to define and experimentally manipulate.
ENDOCYTOSIS OF G PROTEIN–COUPLED RECEPTORS
Many GPCRs undergo agonist-stimulated endocytosis. CME is widely used and is the best understood mechanism, with the β-2 adrenergic receptor (β2AR) considered a representative example. Within several minutes after activation, this GPCR is internalized via CCPs, with the rate controlled primarily by the degree of receptor clustering into existing CCPs (33, 34), and this, in turn, is regulated by β-arrestin, which is recruited from the cytosol after agonist-induced activation of the receptor (35, 36). β-arrestin acts as an endocytic adaptor by binding also to clathrin heavy chain, β2-adaptin, and phosphatidylinositol 4,5-phosphate (PIP2) (35, 37, 38) (Figure 1c). Binding of β-arrestin is promoted by both an activating conformational change of the GPCR and regulated phosphorylation of the receptor’s cytoplasmic tail, typically by GPCR kinases (GRKs). GRKs are Ser/Thr kinases related to rhodopsin kinase (GRK1) that recognize GPCR substrates according to their conformation (reviewed in 39).
β-arrestins are paralogs of visual arrestin, a protein expressed in photoreceptor cells of the eye that is named for its ability to arrest G protein signaling by binding to the light-activated GPCR rhodopsin (40). Arrestins share a bilobed structure with a flexible interdomain loop that occupies a cavity opened in the receptor’s transmembrane bundle after activation (41, 42). This core interaction is mutually exclusive with the G protein interaction required for signaling, explaining rapid signal termination. The phosphorylated cytoplasmic tail of the GPCR binds to a charged groove in the arrestin N-terminal lobe. This tail interaction displaces the arrestin C terminus, which is bound intramolecularly in the inactive state (43). In β-arrestins, but not visual arrestin, the C-terminal sequence that is displaced harbors clathrin- and AP-2-binding motifs, and it is flanked by the PIP2-binding patch. Accordingly, β-arrestins function as regulated endocytic adaptors through a concerted conformational change that is allosterically coupled to GPCR activation and phosphorylation, resulting in the exposure of latent endocytic motifs in β-arrestin.
Many GPCRs undergo regulated CME similarly to β2AR, but not all GPCRs comport with this model. For example, mammals express three paralogous β-adrenergic receptor subtypes, each activated by the same physiological agonist and sharing the ability to signal via the G protein Gs. β2AR typically internalizes rapidly via CME after activation, but the β-3 subtype (β3AR) remains at the plasma membrane (44). The β-1 subtype (β1AR) exhibits different and variable behavior depending on the system—the β1AR is relatively resistant to agonist-induced endocytosis in some cells (45), is rapidly internalized by CME in others (46), and is rapidly internalized by CIE (FEME) in still others (23).
Multiple mechanisms contribute to such receptor-specific differences. One mechanism involves differential phosphorylation that affects β-arrestin binding. β3AR is poorly phosphorylated after activation compared to β2AR and engages β-arrestin weakly. Mutations in the β3AR cytoplasmic tail that increase receptor phosphorylation also increase β-arrestin binding and internalization (44). Another mechanism involves differential binding to proteins that inhibit endocytosis. The β1AR C-terminal tail has a PDZ motif that binds actin-associated scaffold proteins including PSD-95. This is thought to stabilize receptors at the plasma membrane because mutations that disrupt the PDZ motif increase receptor internalization (47). A third mechanism involves receptor binding to alternate adaptors. The β1AR can bind endophilin via a pro line-rich motif present in the receptor’s third cytoplasmic loop. This interaction is thought to promote CIE of the β1AR using endophilin as a specific endocytic adaptor protein for FEME (23).
Another level of specificity in the regulated endocytosis of GPCRs involves differential control by agonists. For example, opioid receptors (ORs) are activated both by endogenous peptide agonists and various chemically distinct, nonpeptide agonists used as drugs. Drugs can differ widely in their ability to stimulate OR internalization (48). This is determined fundamentally by agonist-specific receptor states (49) that produce differential phosphorylation in the cytoplasmic tail by GRKs (50, 51). GRK2 was shown recently to act as an allosteric detector of agonist-selective OR states via discrete regulated binding modes (52). A first mode involves GRK2 recruitment to the plasma membrane by G protein βγ subunits; in this indirect mode, binding is induced by all agonists and depends only on the strength of G protein activation by the OR. A second mode involves an association between GRK2 and the OR; binding in this direct mode does not require G protein activation and is induced only by strongly internalizing agonists. Both binding modes appear to be needed to produce the multisite phosphorylation code in the OR tail that drives strong β-arrestin engagement and CME.
POSTENDOCYTIC TRAFFICKING OF RECEPTOR TYROSINE KINASES AND G PROTEIN–COUPLED RECEPTORS
After ligand-dependent internalization, RTKs and GPCRs are routed through the endosomal compartment system. Sorting to the lysosomal degradation pathway is a well-known fate of internalized RTKs, although RTKs are also capable of recycling back to the plasma membrane after their passage through early endosomes (reviewed in 1). The mechanisms underlying efficient sorting of RTKs to the degradative pathway involve the accumulation of activated RTKs in intraluminal vesicles (ILVs) within multivesicular endosomes or bodies (MVEs), which terminates their ability to recycle back to the cell surface. MVEs eventually target receptor-bearing ILVs for lysosomal degradation (Figure 1). Members of virtually all RTK subfamilies studied thus far have been found to be ubiquitinated (53–59). Ubiquitin moieties are recognized by ubiquitin-binding domains of the endosomal sorting complex required for transport 0 (ESCRT-0) on the cytoplasmic side of the endosomal membrane. ESCRT-I, -II, and -III then facilitate inward endosomal membrane invagination, capturing RTKs in the forming ILV (reviewed in 1). The internal content of the MVE is eventually degraded by lysosomal enzymes delivered to an MVE/late endosome. This classical model of the ubiquitination-dependent sorting of RTKs in MVEs was first proposed and then established in great detail at morphological, biochemical, and molecular mechanistic levels for the EGFR (17, 21, 60–62). In addition, degradation of active c-Met through a mechanism whereby ATG8 family member LC3C promotes trafficking of the c-Met to an autophagic compartment has been recently demonstrated (63).
Recycling of RTKs can occur through two pathways: (a) from early endosomes with relatively fast kinetics (involving Rab4) and (b) via perinuclear-located endosomes and/or the recycling compartment with slower kinetics (involving Rab11 and other related Rabs, such as Rab25) (64, 65) (Figure 1a). Recycling is thought to be simply a default RTK sorting pathway i.e., receptors that are not ubiquitinated, and thus remain in the limiting membrane of MVEs, are capable of recycling back to the cell surface. However, the participation of specific adaptors, such as GGA3 in c-Met recycling (66) and sortilin-related receptor 1 (SORLA) in ErbB2 recycling (12), has also been reported.
The RTK sorting model just described has, however, a major limitation. Even for the most studied system, sorting of EGFR, it is not clear whether lysosome degradation has a major role in postendocytic trafficking of this receptor in the presence of low, physiological concentrations of EGFR ligands, which are present in most mammalian tissues and body fluids accessible to EGFR. Several studies reported weak ubiquitination of EGFR and ineffective ligand-induced degradation of EGF:EGFR complexes under such conditions in cultured cells and tumors in vivo (67–69), even when c-Cbl is overexpressed (68).
Some GPCRs, much like RTKs, are delivered to lysosomes via the ESCRT-dependent MVE pathway, while others selectively recycle and can do so repeatedly during prolonged activation. Pronounced differences in their postendocytic itinerary are evident even between closely related GPCR paralogs, such as the δ-OR, which selectively traffics to lysosomes, and μ-OR, which selectively recycles. Even alternatively spliced μ-OR variants, which are otherwise indistinguishable in functional properties, differ significantly in their sorting between the lysosomal and recycling pathways after agonist-induced CME (reviewed in 70).
For a number of GPCRs, lysyl-ubiquitination of receptors and ubiquitin-directed transfer into ILVs is the dominant mechanism determining their postendocytic fate. For example, the CXCR4 chemokine receptor requires ubiquitination of its cytoplasmic tail for accumulation in ILVs and efficient delivery to lysosomes (71). Other GPCRs, such as the δ-OR and PAR1 protease-activated receptors, accumulate in ILVs and are delivered to lysosomes at a similar rate to wild-type receptors, even after blocking receptor ubiquitination by mutating lysines. Such ubiquitination-independent trafficking is believed to occur through additional cellular proteins that mediate alternative connectivity to ESCRT, for example, GRASP1 (also called GASP1) in the case of δ-OR (72, 73) and ALIX in the case of PAR1 (74, 75).
GPCRs can also recycle via multiple pathways (Figure 1c) including Rab4- and Rab11-dependent routes, as well as a third route that is available after retrograde traffic to the trans-Golgi network (TGN). In addition, some GPCRs are sorted for rapid recycling from discrete endosomes that are marked by APPL1 but not EEA1 (76).
Mechanistic aspects of GPCR recycling are also diverse. For some GPCRs, such as the CXCR4 chemokine receptor, ubiquitin-directed sorting to ILVs primarily determines the postendocytic itinerary of the receptors, similarly to RTKs, with receptor recycling as a bypass route when ubiquitination is blocked (71). Other GPCRs, e.g., β2AR and μ-OR, are specifically sorted into the recycling pathway (77, 78).
Presently, the best understood mechanism for sequence-directed recycling is that engaged by a PDZ binding motif in the β2AR tail (72), which directs actin-dependent recycling within tubular transport carriers. The PDZ domain in sorting nexin 27 (SNX27) recognizes this motif in the β2AR tail (79) and assembles along with a WASH–Arp2/3 actin nucleation complex and retromer to form the actin–SNX27–retromer tubule (ASRT) machinery, a protein coat that mediates the selective accumulation of cargo into tubular domains of the endosome-limiting membrane associated with a dynamic, branched actin network (80, 81). Receptor-containing transport carriers are generated through membrane scission events that occur along the tubule at sites of dynamic actin polymerization (80, 81). ASRT-generated carriers can support direct (endosome to plasma membrane) and retrograde (endosome to TGN) recycling pathways depending on the receptor cargo, with the latter requiring an additional downstream sorting step (82). The existence of a direct route is particularly clear in neurons, where it underlies a system for rapid and local insertion of selected receptors into and adjacent to synapses (83, 84).
The specificity of sequence-directed GPCR recycling is further emphasized in its precise regulation. Phosphorylation of a serine in the distal β2AR tail by GRK5 blocks SNX27 binding and receptor recycling, effectively rerouting internalized receptors to lysosomes (77). Phosphorylation of a distinct PKA site in the proximal β2AR tail, while not needed for SNX27 binding or to determine the overall β2AR itinerary, specifically modulates the kinetics of Rab4-dependent recycling (85). A third site, in a midportion of the β2AR tail, increases SNX27 binding and enhances the efficiency of receptor recycling (86).
ATTENUATION OF SIGNALING BY ENDOCYTOSIS: RECEPTOR DOWNREGULATION BY LYSOSOMAL DEGRADATION
The concept of a negative effect of endocytosis on RTK signaling developed as a consequence of the demonstration that activation of EGFR by EGF in cultured cells rapidly reduces cell-surface and total cellular levels of the EGFR protein (6, 87). A role for endocytosis as part of the negative feedback regulation of signaling was further supported by studies of a noninternalizable EGFR mutant that exhibited elevated signaling (88). This EGFR mutant, however, lacked the entire C-terminal domain, and its signaling mechanisms were aberrant, e.g., signaling was most likely mediated through heterodimerization of the mutant EGFR with ErbB2. Early studies in genetically tractable organisms provided evidence for the downregulating function of endocytosis in RTK signaling in vivo. For example, screening for gain-of-function mutants regulating vulvar development in Caenorhabditis elegans, which requires signaling by EGFR (let-23), revealed genes encoding proteins involved in the endocytic downregulation of let-23 (89, 90). Removal or mutation of HRS (ESCRT-0 protein) in Drosophila embryos inhibited RTK degradation and increased RTK signaling (91, 92).
At present, the prevailing view among RTK researchers is that endocytosis plays a role in RTK signaling as a negative feedback regulatory mechanism. Mechanistically, acceleration of receptor degradation upon ligand binding decreases receptor concentration in the cell, thereby reducing the number of active receptors and attenuating signaling. While this general principle is well established, numerous mechanisms exist that can modulate the extent and rate of ligand-induced RTK downregulation. In the EGFR system, the receptor degradation rate is regulated physiologically, depending on which of seven EGFR ligands activates the receptor. Whereas EGF remains bound to the receptor in endosomes, EGFR ligands with low affinity and/or pH sensitivity of binding dissociate from the receptor in endosomes at acidic pH, which results in dephosphorylation, deubiquitination, and, thus, rerouting of internalized EGFR to the recycling pathway at the expense of degradation (93). Moreover, similar to the internalization step, the process of EGFR degradation is saturable: The apparent degradation rate decreases as the concentration of internalized EGFR in sorting endosomes increases (94, 95). The extent of EGFR ubiquitination appears to be a limiting factor, because overexpression of c-Cbl elevates EGFR degradation (17). The limited capacity of the degradation pathway translates into an inability of ligand-induced endocytosis to significantly downregulate EGFR and attenuate its signaling in cancer cells that highly overexpress this receptor. EGFR downregulation is attenuated by overexpression of its heterodimerization partners that are internalization impaired, such as ErbB2 (96, 97).
The internalization and sorting decision in endosomes, and therefore, the overall efficiency of RTK downregulation and signal transduction intensity, are also regulated by various modulatory proteins and signaling processes, which exhibit physiological and pathophysiological variabilities and alterations, rather than by the core trafficking machinery, which is rarely mutated or amplified (98). At the cell surface, numerous mechanisms of RTK retention from endocytosis have been described, including physical association with plasma membrane resident proteins and the direct inhibition of internalization (99–102). Regulators of E3 ubiquitin ligases, such as Sprouty proteins (103), and deubiquitination enzymes (DUBs) that may act directly on RTKs (104, 105) or on components of the endocytic machinery (106) control RTK ubiquitination and their rate of degradation, and therefore, the intensity of signaling (Figure 2). In the endosomal compartment, in addition to ubiquitination, regulation of core sorting proteins, such as Rabs and ESCRTs, by phosphorylation alters the degradation and signaling output of RTKs. For example, tyrosine phosphorylation of Rab7 in EGF-stimulated cells has been proposed to be necessary for efficient degradation of EGFR (107). FCHSD2 (FCH and double SH3 domain-containing protein) also promotes recycling of RTKs by inhibiting Rab7 activity, which results in increased ERK1/2 activity in lung cancer cells (108). The Rab11 effector Rab-coupling protein (RCP or Rab11-FIP1) binds to EGFR and increases its recycling, leading to elevated Akt activity, which contributes to increased invasion and metastasis (109). Overexpression of other recycling Rabs, such as Rab25, has also been associated with upregulation of EGFR oncogenic signaling (110). In Drosophila, the sorting nexin SH3PX1, in cooperation with several components of the endocytic and autophagic machinery, promotes lysosomal targeting of EGFR, which restrains intestinal stem cell division and maintains gut epithelial homeostasis. Inhibition of the SH3PX1 pathway reroutes EGFR to the Rab11 recycling pathway, leading to hyperactivated ERK and calcium signaling, overstimulation of intestinal stem cell proliferation, and dysregulation of gut epithelia (111).
Figure 2.
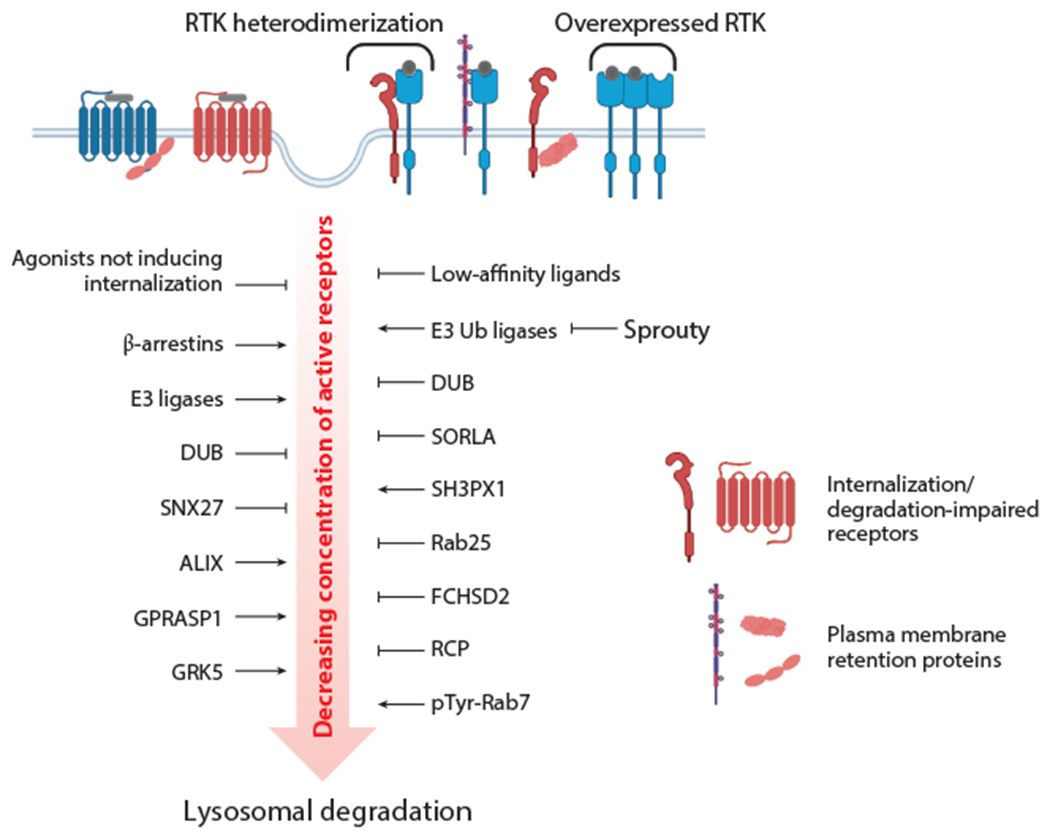
Downregulation of RTK and GPCR signaling by endocytic trafficking to lysosomes and examples of regulatory mechanisms controlling signal attenuation. Receptor ligands, proteins, and posttranslational modifications are shown that augment or impair the endocytosis and postendocytic targeting for lysosomal degradation of GPCRs (left) and RTKs (right) by retaining receptors at the cell surface and modulating receptors or the core trafficking machinery. Downregulation of signaling by an RTK can also be minimized by its overexpression and through heterodimerization with an internalization- or degradation-impaired RTK. Abbreviations: DUB, deubiquitination enzyme; FCHSD2, FCH and double SH3 domain-containing protein; GPCR, G protein–coupled receptor; GRK, GPCR kinase; pTyr, phosphorylated tyrosine; RCP, Rab-coupling protein; RTK, receptor tyrosine kinase; SORLA, sortilin-related receptor 1; Ub, ubiquitin. Figure adapted from images created with BioRender.com.
It is important to recognize that the prevailing view of ligand-induced receptor degradation as the negative regulator of signal transduction, while widely accepted, is not without a caveat. The contribution of RTK degradation to signaling attenuation mechanisms in mammals in vivo, and specifically in the context of tumors, is largely unknown. As discussed in the section titled Postendocytic Trafficking of Receptor Tyrosine Kinases and G Protein–Coupled Receptors, activation of EGFR with physiological ligand concentrations has little, if any, effect on the total EGFR expression level. Therefore, continuous receptor activation and endocytic degradation may not negatively affect signaling intensity in normal tissues and tumors. Nevertheless, the importance of ubiquitin-mediated degradation of RTKs in mammals in vivo is supported by the observation of the dramatic downregulation of several RTKs and severe cell growth defects in the mouse knockout of the deubiquitination enzyme USP8 (112). However, these effects could be the consequence of downregulation of the ESCRT-0 proteins HRS and STAM2, which are USP8 substrates and necessary for MVE sorting. Clearly, more mechanistic studies of the endocytosis and signaling of RTKs using experimental models that recapitulate the tissue microenvironment in vivo are needed.
While many GPCRs recycle efficiently after endocytosis, there is also clear evidence for proteolytic downregulation through endocytic delivery to lysosomes. GPCR downregulation typically occurs more slowly than EGFR downregulation. Downregulation of the δ-OR, for example, requires more than an hour in the continuous presence of agonist (113). Yet, δ-OR transit to lysosomes occurs via typical MVEs requiring ESCRT, with receptors accumulating in ILVs over tens of minutes before arriving in lysosomes (114). Also, as discussed in the section titled Postendocytic Trafficking of Receptor Tyrosine Kinases and G Protein–Coupled Receptors, GPCRs are increasingly recognized to engage various sorting interactions in endosomes that stabilize receptors in the limiting membrane during endocytic transit (Figure 2). Accordingly, GPCRs use the conserved MVE/ESCRT network to downregulate, similarly to RTKs. However, GPCRs transit with variable and generally slower kinetics, with some having a considerably prolonged residence time in the limiting membrane.
INTERNALIZATION DISRUPTS COLOCALIZATION AND SPATIAL PROXIMITY OP RECEPTORS WITH PLASMA MEMBRANE SIGNALING COMPONENTS
The second principal mechanism of RTK signal attenuation by internalization is the spatial separation of signaling and enzymatic complexes from their downstream effectors and substrates (Figure 3a). In the case of RTKs, a classical example of such separation is translocation of the phosphatidylinositol 3-kinases (PI3Ks) and phospholipase Cγ1 (PLCγ1) bound to RTKs through their Src homology 2 (SH2) domains to endosomes (115,116). RTKs activate class I PI3K through direct binding or through tyrosine phosphorylation of scaffolding adaptors, such as GAB1 and IRS1, which then bind and activate PI3K. PI3K phosphorylates PIP2 to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). PLCγ1 also binds to RTKs and cleaves PIP2 to produce the second messengers diacylglycerol and IP3. PIP2 is present predominantly in the plasma membrane but not in early or late endosomes. Therefore, these two enzymes transduce their signals to activate downstream pathways at the plasma membrane, and this signaling is terminated as a consequence of internalization of RTK:PI3K and RTK:PLCγ1 complexes. This physical separation of the enzyme and the substrate may play a key role in the kinetics of signaling through Ras, another major signaling pathway triggered by RTKs (117) (see discussion in the section titled Receptor Tyrosine Kinase Signaling from Endosomes). It is likely that some other signaling processes initiated from the plasma membrane are attenuated due to RTK endocytosis. For example, signaling by integrins is triggered by their binding to the extracellular matrix, and this signaling is augmented by RTKs, which may physically interact with integrins or cooperate in activation of downstream signaling (reviewed in 118). Rapid ligand-induced endocytosis of RTKs terminates this concomitant and collective signaling involving integrins at the cell surface. Another signaling event that occurs at the plasma membrane within the first few seconds of cell stimulation with EGF is the activation of plasma membrane Ca2+ channels (119). The time course of EGF-stimulated Ca2+ influx tightly correlates with the time course of EGFR passing through the plasma membrane during ligand-induced endocytosis, suggesting that EGFR endocytosis leads to termination of this signaling process as well.
Figure 3.
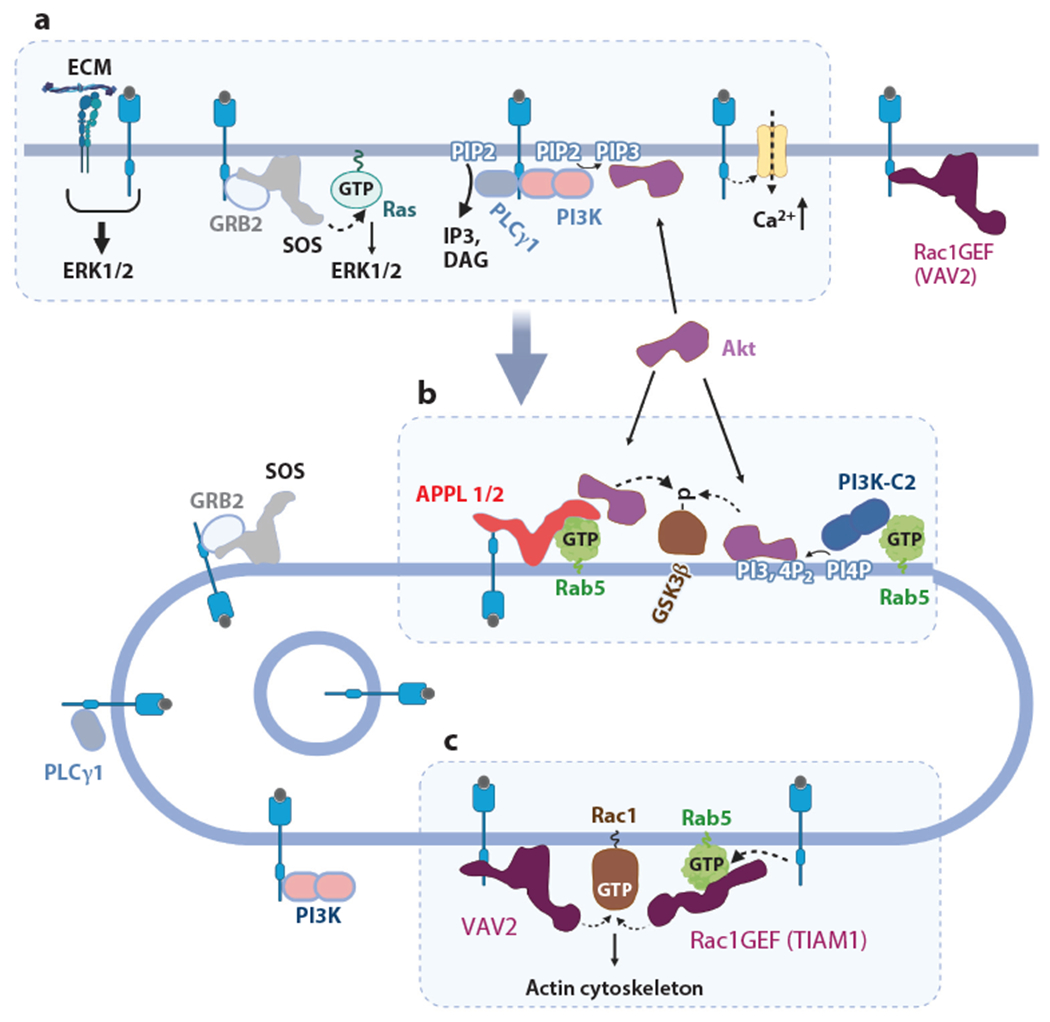
Endocytosis disrupts or facilitates the proximity of RTKs and their downstream signaling effectors. (a) Examples of the spatial separation of RTKs from their effectors during endocytosis are shown, such as separation of the plasma membrane Ca2+ channel and RTK–enzymatic complexes (with PI3K and PLCγ1 or Grb2–SOS) from their substrates (PIP2 or Ras) and disruption of cooperative RTK–integrin signaling (b) Example of endosomal signaling 1. Akt is activated in endosomes through binding to APPL1/2 and/or PI3,4P22. Liver-specific PI3K–C2γ binds to activated (GTP-loaded) Rab5 and converts PI4P to PI3,4P2. Therefore, activation of Akt results in the inhibitory phosphorylation of GSK3β in APPL- and Rab5-containing endosomes. (c) Example of endosomal signaling 2. Rac1 is located in endosomes and activated by its GEF, VAV2, translocated to endosomes in a complex with an activated RTK or TIAM1, and recruited to GTP–Rab5, whose activity is increased by RTK signaling. Abbreviations: ECM, extracellular matrix; GEF, guanine exchange factor; GTP, guanosine triphosphate; PI3K, phosphatidylinositol 3-kinases; PI3,4P2, phosphatidylinositol 3,4-bisphosphate; PIP2, phosphatidylinositol 4,5-phosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PLC, phospholipase C; RTK, receptor tyrosine kinase. Figure adapted from images created with BioRender.com.
GPCR signaling from the plasma membrane is similarly attenuated by internalization because these reactions classically require the receptor, G protein, and effector to be associated with the same membrane and in close proximity (reviewed in 120). Thus, G protein–dependent signaling reactions that involve effectors restricted to the plasma membrane—such as signaling via some ion channels, adenylyl cyclases, and PLC isoforms that use PIP2 as substrate—are thought to be terminated by endocytosis. GPCR endocytosis is also tightly linked to desensitization of G protein activation by its shared dependence on receptor phosphorylation and binding to β-arrestin (35, 36, 40), although desensitization does not require (and can precede) internalization (121).
RECEPTOR TYROSINE KINASE SIGNALING FROM ENDOSOMES
The first evidence for the signaling endosome hypothesis—sustained activity of RTKs in endosomes—was reported in studies of IR and EGFR in liver cells in vitro and ex vivo at approximately the same time as the concept of the downregulating function of endocytosis in RTK signaling was developing (122, 123). That many RTKs remain fully active and capable of signaling in endosomes has been undisputedly demonstrated (reviewed in 124). Moreover, RTKs are found associated with adaptors containing SH2 or phosphotyrosine binding (PTB) domains and their interacting proteins, such as Grb2, She, Crk, GAB1, IRS, FRS, and others, in endosomes (reviewed in 124) (Figure 3). These endosomal complexes have been implicated in mediating signaling through Ras/ERK1/2, PI3K/Akt, STATs, the Rho small GTPase family that controls the actin cytoskeleton, and other signaling pathways. The idea of an endosome as a signaling compartment makes sense, considering the typical RTK trafficking itinerary and ligand–receptor binding pharmacology. First, even though some RTK ligands dissociate from their receptors in the acidic environment of endosomes, many ligand–RTK complexes remain intact. Second, given that ligand concentration is several magnitudes higher in endosomes than in extracellular fluids, equilibrium is shifted toward ligand binding to receptors in endosomes. Third, the residence time of an activated RTK is significantly longer in endosomes than in the plasma membrane because internalization rates are much higher than rates of receptor sorting for lysosomal degradation. Therefore, continuous signaling from endosomes could be a means to extend a signaling process in time and space. A classic example of such an extension is the survival of signaling in large neurons that requires endocytosis of activated TrkA in the axonal terminus and retrograde transport of TrkA-containing endosomes to the neuronal soma, where TrkA activates ERK5, leading to phosphorylation of the transcription factor CREB (125, 126). The concept of neuronal retrograde transport, which developed in the late 1990s, emphasizes another useful property of endosomes—the ability to move their cargo in a regulated and directed fashion with high speed by using microtubule motor–mediated transport.
Studies of TrkB regulation by the Rap GTPase-activating protein (RapGAP) SIPA1L2, which interacts with dynein and the autophagosomal protein LC3, illustrate the fine-tuning of the local versus spatially distant (retrograde) activities of TrkB signaling endosomes (127). Ligand-activated TrkB is internalized at the synapse of hippocampal neurons and routed to the autophagic pathway accumulating in autophagosomes fused with late endosomes (also called amphisomes). SIPA1L2 appears to bind TrkB and connect TrkB amphisomes to a dynein motor, allowing their retrograde transport and signaling. LC3 regulates the RapGAP activity of SIPA1L2, which in turn regulates SIPA1L2’s interaction with dynein. Induction of presynaptic plasticity causes the dissociation of amphisomes from dynein in the synaptic areas, enabling local signaling and promoting neurotransmitter release. Thus, TrkB-signaling endosomes (amphisomes) are capable of both retrograde transport and local signaling in the context of presynaptic plasticity (127). Notably, involvement of the autophagic machinery in the trafficking of internalized Trk receptors and a signaling role for amphisomes is a novel mechanism of RTK regulation (128).
It is important to recognize that the presence of active RTK signaling complexes in endosomes does not automatically indicate that these complexes trigger downstream signaling. In fact, MVEs are the compartments where signaling is terminated as soon as an RTK is sequestered in the ILV and becomes inaccessible to cytosolic effectors. Because EGFRs remain ligand bound and, therefore, tyrosine phosphorylated in ILVs (60, 61), they are identified as active receptors by biochemical methods and confocal microscopy, even though these receptors are incapable of signaling. The rates of RTK incorporation in ILVs have not been defined. Recent live-cell microscopy studies of the dynamics of endogenously labeled ESCRT proteins suggested that the formation of ILVs starts early after the appearance of a cargo like EGFR in endosomes, and the kinetics of ILV formation are relatively fast (129). Whether a significant number of active RTKs remain on the limiting membrane for an extended time in cells expressing physiological levels of receptors and under conditions of stimulation with low ligand concentrations is unknown. It is generally assumed that the entrapment of RTKs in ILVs is irreversible, although the possibility of back-fusion of ILVs to the limiting membrane of MVEs was proposed in studies of p38/MAPK-dependent endocytic trafficking of EGFR (130).
The activity of RTKs at the limiting membrane of MVEs also gradually decreases because of increased dissociation of the ligand during continuing acidification of maturing endosomes and partial proteolysis of ligands and also due to the activity of protein tyrosine phosphatases (PTPs). Interestingly, it has been shown that EGFR is dephosphorylated in MVEs at their contact sites with the endoplasmic reticulum (ER) by the ER-associated phosphatase PTP1B (131). However, the importance of PTP1B for the dephosphorylation of endosomal EGFR has not been demonstrated in other studies (132, 133). More recently, screening for EGFR-specific PTPs revealed that EGFR activity in endosomes is mainly controlled by another ER-localized PTP, PTPN2 (134).
While the localization of active RTKs and their associated adaptors in endosomes is well documented, it is less clear whether the downstream effectors of those receptor complexes are also present in the same endosome. In the extensively studied RTK/Ras/Raf/MEK/ERK1/2 signaling axis, Ras is the most downstream membrane-associated component. Therefore, localization of GTP-loaded Ras determines where in the cell signaling to ERK1/2 is propagated. Ras was found in endosomal fractions that also contained EGFR in the liver (135) and was also cofractionated with activated TrkA receptor in clathrin-containing vesicles in PC12 cells (136). Overexpressed H-Ras and K-Ras were detected in EGFR-containing endosomes by fluorescence microscopy (137). These and other similar findings provide strong support for the endosomal signaling hypothesis. However, more recent live-cell microscopy studies of endogenous H-Ras tagged with fluorescent protein by gene editing showed that H-Ras is predominantly located in the plasma membrane. H-Ras was also present in small quantities in tubular recycling endosomes but was not found in any significant amounts in early/sorting endosomes containing EGFR associated with the complex of Grb2 and SOS [the guanine exchange factor (GEF) for Ras] (117). Moreover, endogenously labeled c-Raf1, an effector of GTP-Ras, translocated upon EGF stimulation to the plasma membrane but not to endosomes (138). Such spatial separation of endosomal RTK signaling complexes from Ras suggested that signaling through the Ras/ERK pathway is predominantly initiated from the plasma membrane, and the contribution from endosomal signaling is minimal, if any, at least in the HeLa cells used in studies with gene-edited proteins. Countless studies propose the existence of signaling to ERK1/2 in endosomes. However, we are not aware of a demonstration to date of a significant colocalization of RTK:SOS complexes and endogenous Ras in the same endosome. Whether endosomal RTKs can sustain the activity of the plasma-membrane Ras through soluble intermediates or bypass Ras to sustain ERK1/2 activity remains to be investigated. B-Raf activation by the small GTPase Rap1 is one possible mechanism for Ras-independent signaling to ERK1/2 (139).
By contrast with the Ras/ERK pathway, the PI3K/Akt signaling pathway has traditionally been viewed as being associated exclusively with the plasma membrane. RTKs activate class I PI3K, which phosphorylates PIP2 to PIP3 in the plasma membrane. Akt and PDK1 bind to PIP3 at the plasma membrane, and PDK1 phosphorylates the activation loop of Akt (reviewed in 140). However, accumulating evidence suggests that Akt can be activated in endosomes. Akt binds the endosomal scaffold proteins APPL1 and APPL2, which are Rab5 effectors (141). APPLs presumably through their PTB domains, are capable of binding to RTKs, such as EGFR and TrkA. Transient recruitment of Akt to APPL1/2- and RTK-containing endosomes has been proposed to promote Akt activity. APPLs have been shown to mediate antiapoptotic and cell-survival signaling in zebrafish development, and this signaling is facilitated by the proximity of activated Akt to its substrate GSK3β, and possibly other substrates, in APPL-containing endosomes (142, 143) (Figure 3b).
In another example of Akt activation in endosomes, insulin signaling triggers the association of the liver-specific class II PI3K isoform γ (PI3K-C2γ) with Rab5-GTP and its recruitment to Rab5- and APPL-positive early endosomes (144). In these endosomes, PI3K-C2γ produces phosphatidylinositol 3,4-bisphosphate, and it has been suggested that binding to this lipid in endosomes mediates delayed and sustained Akt2 activation. Loss of PI3K-C2γ selectively reduces Akt2 activation and its inhibitory phosphorylation of GSK3β but not that of other substrates (Figure 3b). As a consequence, PI3K-C2γ-deficient mice display severely reduced liver accumulation of glycogen and develop hyperlipidemia, adiposity, and insulin resistance. Therefore, PI3K-C2γ supports a specific branch of IR signaling to an endosomal pool of Akt2 that is required for glucose homeostasis (144).
Recent studies revealed that CME and recycling processes can be dysregulated in cancer cells via expression of dynamin-1 and in cells with an excess of clathrin light chain (CLC) b over CLCa (145). In such cells, the rate of rapid recycling of EGFR, but not that of transferrin receptors, was found to be accelerated (146). There was also an increase in the abundance of APPL1-positive endosomes and enhanced Akt signaling, specifically, the phosphorylation of GSK3β (147). Thus, APPL1/2-containing endosomes are emerging as the key site of intracellular Akt activity (Figure 3b). However, given the highly transient (within the first 1–2 min of cell stimulation with growth factors) recruitment of small pools of Akt and GSK3γ to APPL-positive endosomes, the contribution of endosomal activation to total Akt activity is likely to be relatively small (143). It has been proposed that Akt can be activated in CCPs, which would involve the EGF- and hepatocyte growth factor (HGF, a c-Met ligand)-stimulated phosphorylation of the GAB1 adaptor (148, 149). It would be revealing to directly monitor the dynamics of the translocation to CCP and endosomes of endogenous Akt, GSK3γ, and other Akt substrates to determine the extent of the regulation of these dynamics by dynamin-1 and CLCb.
RTK signaling to control actin cytoskeleton remodeling is another branch of signal transduction that can be initiated from endosomes. Several studies implicate endosomes as the site of Rac1 activation by EGFR and c-MET (Figure 3c). CME of EGFR is proposed to be required for Rac1 activation in early endosomes, where Tiam1, a Rac1 GEF, is recruited through binding to Rab5 (150). Subsequent translocation of GTP-loaded Rac1 to the plasma membrane ensures localized formation of actin-based membrane protrusions in cultured cells and in primordial germinal cells during zebrafish development. In cells stimulated with HGF, recruitment to perinuclear endosomes of another Rac1 GEF, VAV2, was proposed to be necessary to mediate optimal Rac1 signaling to the actin cytoskeleton and promote cell migration (151). Trans-endocytosis of Ephrin-EphR complexes also results in signaling to Rac1 in endosomes involving Tiam1 and VAV2 (reviewed in 28, 29). Conversely, Rac1 activity appears to be necessary for efficient endocytosis of Ephrin-EphR complexes (30–32). Nerve growth factor (NGF)-induced internalization of TrkA is known to be required for local TrkA signaling to promote neurite elongation and branching events and distant TrkA survival signaling in neurons (reviewed in 152). NGF–TrkA-containing endosomes carry activated Rac1 GTPase and cofilin, an actin filament–severing protein; both are necessary for promoting retrograde trafficking as well as trophic signaling of TrkA receptors in distal axons of sympathetic neurons (153). Thus, Rac1 appears to be the nexus of endosome-specific RTK signaling to the actin cytoskeleton (Figure 3c).
G PROTEIN–COUPLED RECEPTOR SIGNALING FROM ENDOSOMES
Thinking in the field about potential GPCR signaling from endosomes essentially followed the opposite historical path relative to RTKs. Since 1980, the light-activated GPCR rhodopsin had been known to mediate G protein (transducin) activation on internal membranes (154), and these membranes are formed by a specialized type of CIE (155). Moreover, endosomes were long recognized to be sites at which ligand-activated GPCRs such as β2AR are functionally resensitized by dephosphorylation (156, 157). Nevertheless, for many years there was virtually no evidence for internal membrane signaling by ligand-activated GPCRs and a number of arguments against it (reviewed in 158). This began to change approximately 20 years ago, with evidence emerging for endosomal signaling by some GPCRs via β-arrestin. However, at that time, endosomal signaling was thought to apply only to β-arrestin pathways independent of G proteins. Ligand-dependent GPCR signaling via heterotrimeric G proteins—arguably the major signaling mechanism—was thought to be entirely restricted to the plasma membrane (159, 160).
Early evidence for ligand-dependent G protein signaling from endosomes emerged from a study in yeast of the mating response initiated by activation of the Ste2p polypeptide pheromone receptor (161). A screen of yeast knockout strains identified genes encoding endocytosis-related proteins as necessary for a late component of the Ste2p response. It was concluded that Ste2p can activate the G protein α subunit Gpa1 from endosomes through a distinct heterotrimeric complex in which the βγ subcomplex that acts at the plasma membrane is replaced by PI3K (Vps34p) in complex with its activating protein kinase (Vps15p).
Support for G protein signaling after endocytosis in mammalian cells emerged from investigation of the prolonged time course of anti-inflammatory signaling mediated by the sphingosine-1 phosphate lipid receptor (S1PR). Here, a sustained component of the Gi-dependent S1PR response was correlated with internalized receptor localization in a Golgi-associated compartment (162). A similar correlation was noted between Gs-dependent cAMP signaling and internal localization of two polypeptide hormone receptors, the parathyroid hormone receptor (PTHR) 1 in endosomes (163) and the thyroid-stimulating hormone receptor (TSHR) in Golgi-associated membranes (164). In addition, endocytic inhibitors were shown to change the normally persistent cAMP elicited by receptor activation to a transient response, with cell fractionation experiments providing support for an internal source of TSH-induced cAMP generation.
Evidence for the broader relevance of endosomal G protein activation by GPCRs emerged from a study of cAMP signaling initiated by the D1 dopamine receptor (DRD1). In contrast to sustained polypeptide hormone signaling, this Gs-dependent response is transient and reversible. Endocytic inhibitors were found to only slightly reduce global cytoplasmic cAMP accumulation, but a downstream cAMP-dependent response controlling the electrical excitability of neurons was blocked (165). Gs-dependent cAMP accumulation initiated by β2AR, another GPCR that signals transiently, is also only slightly reduced by endocytic inhibition. However, with the development of conformational biosensors of localized G protein activation, both β2AR and DRD1 were found to mediate agonist-dependent activation of Gs in endosomes as well as the plasma membrane (166).
It is now clear that GPCR–G protein activation is not restricted to the plasma membrane for many GPCRs (Figure 4). It is also evident that activation can occur at additional internal sites, with current data supporting considerable diversity and specificity in the subcellular organization of GPCR–G protein activation. For example, β2AR mediates internal activation of Gs primarily from the limiting membrane of EEA1-marked endosomes (82, 166, 167), but the TSHR does so primarily from a distinct membrane compartment that is associated with, or en route to, the Golgi apparatus (168). Further, the β1AR mediates internal activation of Gs primarily from the Golgi apparatus rather than endosomes, and the source of these receptors is the biosynthetic rather than endocytic pathway (169). Moreover, activation of the Golgi-associated β1AR pool requires a facilitated transmembrane transport process to enable ligand access to this compartment (169). Gi-coupled ORs have also been shown to undergo activation in endosomes and Golgi membranes with different spatiotemporal profiles depending on the agonist (170), and subcellular control of GPCR activation is gaining interest as a means to improve the selectivity of therapeutic drugs (169–173).
Figure 4.
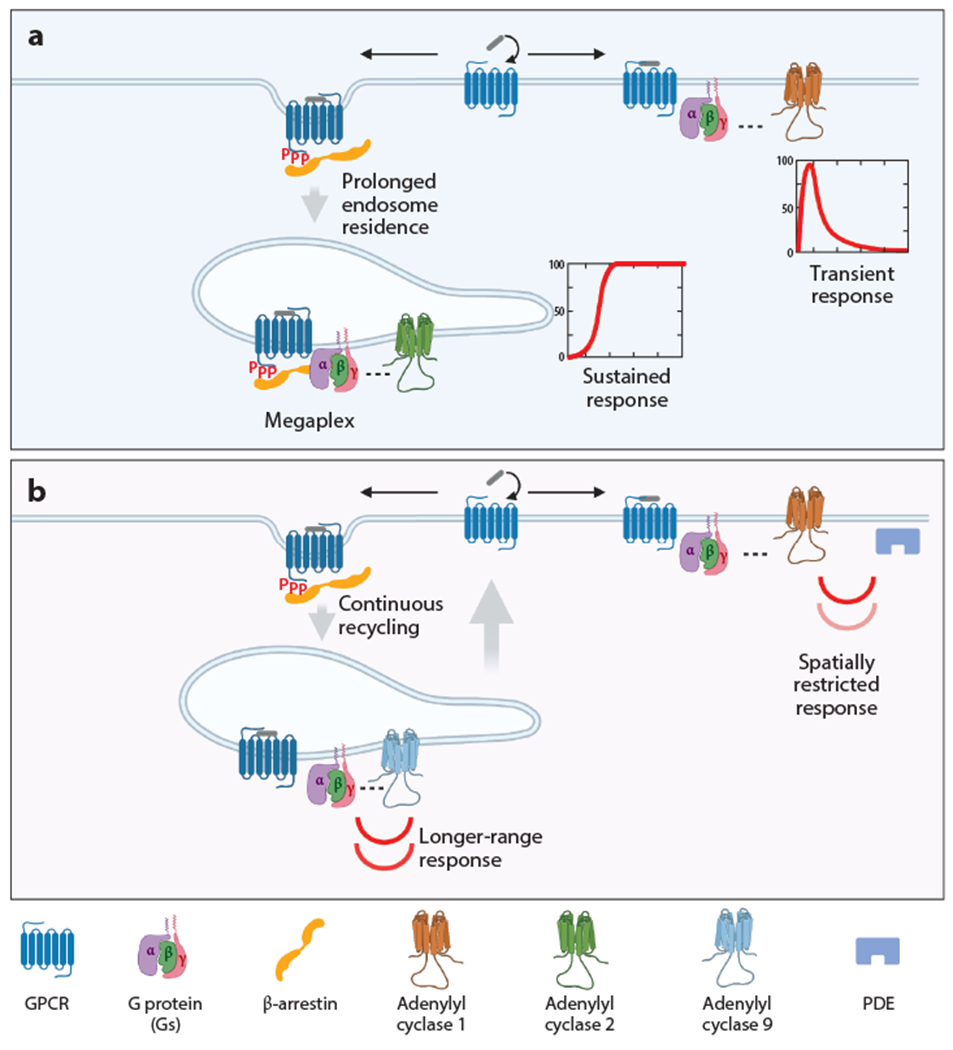
Current models for how endocytosis affects GPCR–G protein signaling via cAMP. (a) Example of temporal control using endocytosis to produce a sustained cAMP response. GPCRs remain phosphorylated and bound to β-arrestin after endocytosis. The GPCR-β-arrestin complex assembles with Gs in an alternate signaling complex (megaplex) on the endosome-limiting membrane. This prolongs GPCR residence in endosomes and increases the efficiency of Gs activation, producing a sustained cAMP response that persists even after agonist removal from the extracellular milieu. In contrast, cAMP production from the plasma membrane is transient due to desensitization mediated by receptor phosphorylation, binding to β-arrestin, and endocytic removal from the plasma membrane. (b) Example of spatial control using endocytosis to change proximity to adenylyl cyclase isoforms and phosphodiesterases. GPCRs are dephosphorylated and dissociated from β-arrestin during or shortly after arrival in the endosome-limiting membrane. This enables a second phase of Gs activation and cAMP production in endosomes that is transient due to receptor recycling. Iterative cycling that occurs with prolonged agonist exposure drives repeated bursts of cAMP production from the plasma membrane and endosomes. cAMP produced from endosomes can signal over a longer range due to reduced local proximity to PDEs, and cAMP production from endosomes differs in its control by other pathways due to isoform-specific localization and trafficking of adenylyl cyclase. Abbreviations: cAMP, cyclic adenosine monophosphate; GPCR, G protein–coupled receptor; P, phosphate; PDE, phosphodiesterase. Figure adapted from images created with BioRender.com.
FUNCTIONAL IMPACT OF G PROTEIN–COUPLED RECEPTOR–G PROTEIN SIGNALING AFTER ENDOCYTOSIS
As in RTK signaling, a key question raised by the recognition of GPCR–G protein activation in endomembranes is its functional significance relative to signaling from the plasma membrane. The study of pheromone signaling in yeast again provided early insight. Activation of the Gpa1 G protein on the plasma membrane stimulates protein kinase activity, but activation on endosomes stimulates a PI3K (Vps34p) that is localized to this compartment (161). Moreover, because phosphatidylinositol 3-phosphate (PI3P) is an inherently membrane-delimited mediator, the PI3P produced by Vps34p stimulation is also restricted to the endosomal membrane. Biochemical segregation of signaling mediators thus suggests one basic difference between G protein signaling from endosomes relative to the plasma membrane. In this system, the endosome-restricted PI3P response then produces a selectively delayed and prolonged activation of downstream MAP kinase signaling relative to protein kinase activation at the plasma membrane.
A persistent or sustained cellular response after endocytosis is also characteristic of cAMP signaling stimulated by Gs through mammalian polypeptide hormone receptors (163, 164, 174). A similar effect is observed for inhibitory cAMP signaling via OR activation of Gi in endosomes (170). However, unlike PI3P, which is compartmentalized, cAMP is diffusible. Sustained cytoplasmic cAMP changes generated by signaling from endosomes have been shown to drive long-range cell-cycle control in tissues by cAMP diffusion through gap junctions that enables the response to spread between cells (175) and to promote sustained transcriptional responses within individual cells through sustained global cAMP elevation (176).
The mechanistic basis by which some GPCRs produce a persistent response from endosomes, while others signal transiently is incompletely understood. It has been noted that some GPCRs that produce sustained cAMP elevation, such as the vasopressin-2 receptor (V2R) and PTHR, stably associate with β-arrestin in the limiting membrane. This contrasts with DRD1 and β2AR, which signal transiently and associate with β-arrestin primarily at the plasma membrane. There is now evidence that β-arrestin, despite its undisputed role in rapid signal termination at the cell surface, has an opposite function in endosomes by increasing G protein activation. This signal-sustaining function of β-arrestin is proposed to be mediated through the formation of a multiprotein complex on the endosome-limiting membrane that includes the GPCR and Gs and enables Gs to be activated more rapidly or repeatedly from endosomes (177). This hypothesis is presently under intensive investigation, with a structural basis for a β-arrestin-scaffolded endosomal signaling megaplex (Figure 4a) having been proposed recently (178).
Functional differences between sustained and transient global activation are widely observed in cellular signaling pathways, and a mechanism linking sustained cytoplasmic cAMP elevation to signaling via PKA in the nucleus has been described (179). Given the diffusible nature of cAMP, an interesting question concerns how GPCRs that elevate cAMP transiently achieve selective responses from endosomes. For example, cAMP-dependent transcriptional control initiated by β2AR activation is endocytosis dependent, despite this GPCR producing only transient global cAMP elevations. Studies using optogenetic control of local cAMP production indicate that this selectivity is achieved by changing the location of cAMP production rather than its overall amount (Figure 4b) and is due, in part, to more rapid cAMP hydrolysis at the plasma membrane (180, 181). Spatial separation of effectors may also contribute to signaling selectivity. For example, adenylyl cyclase type 9 (ADCY9) localizes to the limiting membrane of endosomes that contain activated β2ARs and Gs (182); ADCY9 is activated by Gs but is insensitive to inhibitory control by Gi/o-subclass G proteins, PKC, or calcium (183).
CONCLUSIONS, EXPERIMENTAL HURDLES, AND FUTURE DIRECTIONS
While endosomes have been recognized as potential sites of RTK signaling for many years, evidence for ligand-dependent GPCR signaling via heterotrimeric G proteins after endocytosis is relatively recent. Nevertheless, the available data already suggest several themes regarding how signaling initiated by internalized receptors may confer functional advantages that cross receptor classes.
One such common theme is the potential for signaling from endosomes to be extended in both time and space. For RTKs, endocytosis can sustain some signals initiated at the plasma membrane or move activated receptor complexes through the cell, such as from distal axons to the cell body, which enables long-range transcriptional control. For GPCRs, examples include selectively sustaining cAMP signaling produced by internalized polypeptide hormone receptors.
A second emerging theme is that endocytosis can reorganize signaling cascades through enforced proximity. For RTK signaling, endocytosis can terminate some signaling reactions and initiate others, depending on differences in receptor proximity to other essential signal mediators and substrates. For GPCR signaling via cAMP, the rate of cAMP hydrolysis appears to differ at internal membrane locations relative to the plasma membrane, and emerging evidence indicates that GPCR–G protein activation in endosomes is coordinated with endocytic membrane trafficking of specific ADCY isoforms that differ in regulation.
A third emerging theme is that endocytosis can mediate or support higher-order signal processing and integration functions. For RTKs, these include using activation in endosomes to control the signal dynamics differently, depending on the ligand, and possibly also contributing to the generation of switch-like responses to graded inputs of some ligands (133). For GPCRs, these include using endocytosis not only to control signaling dynamics and discriminate among distinct agonists, but also as a selectivity filter to confer uniformity on downstream responses (184).
A main barrier for further understanding the cross talk between endocytosis and signaling is that experimental evidence for the negative or positive impact of endocytosis on signaling is difficult to obtain, resulting in vast inconsistencies among different model cell systems. Controversies in the interpretation of experiments where the effects of RTK and GPCR endocytosis on Ras/ERK1/2 signaling are examined by inhibiting major components of the endocytic machinery, such as clathrin and dynamin, are discussed in previous reviews (e.g., 2, 124). In the RTK world, a myriad of proteins have been implicated in the regulation of their endocytosis. However, none has been shown to specifically and uniquely regulate the endocytosis of a single RTK or an RTK subfamily, whereas all of them can regulate other receptors or have functions unrelated to trafficking. Hence, experimental manipulations with such proteins often lead to pleiotropic effects on membrane trafficking and signaling networks. For example, although Cbls are often considered to be specific regulators of EGFR endocytosis, they ubiquitinate multiple RTKs and a variety of other receptor types and integral membrane proteins, as well as nonreceptor Src family kinases. In another example, important evidence for the role of late endosomes in ERK1/2 activation by EGFR was obtained in experiments with the p14/MP1 scaffold complex in which knockdown of these proteins was shown to inhibit sustained activation of ERK1/2 (185). However, further investigations revealed that p14/LAMTOR2 and MP1/LAMTOR3 are components of the Ragulator complex, which mediates binding of mTORC1 to the late endosome and lysosome membranes (186). Therefore, the effects of the knockdown and knockout of these proteins on ERK activity could be indirect, resulting from the perturbation of late endosome and lysosome function. The lack of mutant versions of EGFR and other RTKs that are not internalized but maintain all of the signaling properties of a native receptor is the major obstacle for defining the role of endocytosis in RTK signaling. Similar limitations apply to GPCRs. Dissecting the signaling consequences of GPCR endocytosis by manipulating β-arrestin, for example, is complex because β-arrestin has functions in addition to its role as a regulated endocytic adaptor for CME. These include the originally identified role of β-arrestin in attenuating G protein activation from the plasma membrane (desensitization) and its more recently proposed functions in mediating G protein-independent signaling pathways and positively regulating G protein activation in endosomes. These limitations warrant further efforts to advance our mechanistic understanding of the endocytic trafficking of this class of receptors as well as RTKs.
Many discrepancies, inconsistencies, and controversies with regard to the mechanisms and functional role of endocytosis that have been emphasized in this review can be overcome by expanding the use of new or emerging methodological approaches. Microscopic analysis of the subcellular localization of signaling receptors and their effectors needs to be performed with endogenous proteins, preferably in living cells to avoid artifacts of overexpression and to track the dynamics of localization. The number of studies of endogenous components fused to fluorescent proteins or other tags in gene-edited cells is increasing, although these efforts so far cover only a few signaling and endocytosis proteins and a few signaling pathways. Detection of endogenous proteins by labeled nanobodies is an alternative approach that has the additional advantage of sensing protein conformations. This approach has been extensively used in GPCR research, and its expansion to studies of RTK signaling and endocytosis will be instrumental. Furthermore, knowing and accounting for the cellular abundances of receptors and their effectors may help to identify rate-limiting steps of signaling pathways and elucidate mechanisms by which endocytosis controls these pathway bottlenecks. Copy numbers of the entire HeLa cell proteome have been quantitated using mass spectrometry (187), and a targeted analysis was performed to quantitate copy numbers of major signaling proteins in multiple human mammary cell lines (188). Studying the signaling and endocytosis of GPCRs and RTKs in mammalian organisms in vivo is the next inevitable, yet feasible, step in this research area. The first attempts to examine the behavior of labeled endogenous EGFR in mice demonstrated the feasibility of the mechanistic analysis and unveiled conceptually different properties of the EGFR system in vivo (67, 189). We anticipate that a combination of all of these approaches with high-end systems biology, including genomics, proteomics, and mathematical modeling analyses, will enable mechanisms by which endocytosis controls physiological signaling by receptors to be defined precisely and the integrated consequences for cellular physiology to be delineated. This will enable the identification of disease-related aberrations in endocytosis signaling networks that can then be therapeutically targeted. Such aberrations may define a rational basis for improving the therapeutic specificity and efficacy of drugs through the specific targeting of select signaling reactions based on subcellular organization.
ACKNOWLEDGMENTS
We thank all of the scientists who have contributed to this important research area over the years, in our laboratories and many others, and we regret being able to cite only a subset of relevant publications in this review due to space limitations. This work was supported by grants from the National Institute on Drug Abuse (NIDA), the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke (M.v.Z.) and from the National Cancer Institute, NIDA the National Institute of General Medical Sciences, and the National Science Foundation (A.S.).
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Goh LK, Sorkin A. 2013. Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 5(5):a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorkin A, von Zastrow M. 2009. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10(9):609–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. 2012. Endocytosis and signaling: Cell logistics shape the eukaryotic cell plan. Physiol. Rev. 92(1):273–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergeron JJM, Di Guglielmo GM, Dahan S, Dominguez M, Posner BI. 2016. Spatial and temporal regulation of receptor tyrosine kinase activation and intracellular signal transduction. Annu. Rev. Biochem. 85:573–97 [DOI] [PubMed] [Google Scholar]
- 5.Irannejad R, Tsvetanova NG, Lobingier BT, von Zastrow M. 2015. Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol. 35:137–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter G, Cohen S. 1976.125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J. Cell Biol. 71(1):159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlessinger J, Shechter Y, Willingham MC, Pastan I.1978. Direct visualization of binding, aggregation, and internalization of insulin and epidermal growth factor on living fibroblastic cells. PNAS 75(6):2659–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gorden P, Carpentier JL, Cohen S, Orci L. 1978. Epidermal growth factor: morphological demonstration of binding, internalization, and lysosomal association in human fibroblasts. PNAS 75(10):5025–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorkin A, Di Fiore PP, Carpenter G. 1993. The carboxyl terminus of epidermal growth factor receptor/erbB-2 chimerae is internalization impaired. Oncogene 8(11):3021–28 [PubMed] [Google Scholar]
- 10.Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. 1996. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 271(9):5251–57 [DOI] [PubMed] [Google Scholar]
- 11.Worthylake R, Opresko LK, Wiley HS. 1999. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 274(13):8865–74 [DOI] [PubMed] [Google Scholar]
- 12.Pietilä M, Sahgal P, Peuhu E, Jäntti NZ, Paatero I, et al. 2019. SORLA regulates endosomal trafficking and oncogenic fitness of HER2. Nat. Commun. 10(1):2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lund KA, Opresko LK, Starbuck C, Walsh BJ, Wiley HS. 1990. Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J. Biol. Chem. 265(26):15713–23 [PubMed] [Google Scholar]
- 14.Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, et al. 2005. Clathrin-independent endocytosis of ubiquitinated cargos. PNAS 102(8):2760–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortian A, Dionne LK, Hong SH, Kim W, Gygi SP, et al. 2015. Endocytosis of ubiquitylation-deficient EGFR mutants via clathrin-coated pits is mediated by ubiquitylation. Traffic 16(11):1137–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lill NL, Douillard P, Awwad RA, Ota S, Lupher ML Jr., et al. 2000. The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 275(1):367–77 [DOI] [PubMed] [Google Scholar]
- 17.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, et al. 1999. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4(6):1029–40 [DOI] [PubMed] [Google Scholar]
- 18.Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, et al. 2002. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 21(3):303–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi E, Zhang X, Xing C, Yu H. 2016. Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell 166(3):567–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi E, Kikuchi S, Gao H, Brodzik K, Nassour I, et al. 2019. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat. Commun. 10(1):1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haigler HT, McKanna JA, Cohen S. 1979. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J. Cell Biol. 83(1):82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orth JD, Krueger EW, Weller SG, McNiven MA. 2006. A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res. 66(7):3603–10 [DOI] [PubMed] [Google Scholar]
- 23.Boucrot E, Ferreira APA, Almeida-Souza L, Debard S, Vallis Y, et al. 2015. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 517(7535)460–65 [DOI] [PubMed] [Google Scholar]
- 24.Caldieri G, Barbieri E, Nappo G, Raimondi A, Bonora M, et al. 2017. Reticulon 3–dependent ER-PM contact sites control EGFR nonclathrin endocytosis. Science 356(6338):617–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jastrzębski K, Zdżalik-Bielecka D, Mamińska A, Kalaidzidis Y, Hellberg C, Miaczynska M. 2017. Multiple routes of endocytic internalization of PDGFRβ contribute to PDGF-induced STAT3 signaling. J. Cell Sci. 130(3):577–89 [DOI] [PubMed] [Google Scholar]
- 26.Valdez G, Akmentin W, Philippidou P, Kuruvilla R, Ginty DD, Halegoua S. 2005. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J. Neurosci. 25(21):5236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vihanto MM. 2006. Caveolin-1 is required for signaling and membrane targeting of EphB1 receptor tyrosine kinase. J. Cell Sci. 119(11):2299–309 [DOI] [PubMed] [Google Scholar]
- 28.Pitulescu ME, Adams RH. 2010. Eph/ephrin molecules—a hub for signaling and endocytosis. Genes Dev. 24(22):2480–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kania A, Klein R. 2016. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 17(4):240–56 [DOI] [PubMed] [Google Scholar]
- 30.Valenzuela JI, Perez F. 2020. Localized intercellular transfer of ephrin-As by trans-endocytosis enables long-term signaling. Dev. Cell 52(1):104–17.e5 [DOI] [PubMed] [Google Scholar]
- 31.Marston DJ, Dickinson S, Nobes CD. 2003. Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nat. Cell Biol. 5(10):879–88 [DOI] [PubMed] [Google Scholar]
- 32.Evergren E, Cobbe N, McMahon HT. 2018. Eps15R and clathrin regulate EphB2-mediated cell repulsion. Traffic 19(l):44–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Zastrow M, Kobilka BK. 1994. Antagonist-dependent and -independent steps in the mechanism of adrenergic receptor internalization. J. Biol. Chem. 269(28):18448–52 [PubMed] [Google Scholar]
- 34.von Zastrow M, Kobilka BK. 1992. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J. Biol. Chem. 267(5):3530–38 [PubMed] [Google Scholar]
- 35.Goodman OB Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, et al. 1996. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383(6599):447–50 [DOI] [PubMed] [Google Scholar]
- 36.Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Ménard L, Caron MG. 1996. Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271(5247):363–66 [DOI] [PubMed] [Google Scholar]
- 37.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. 1999. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 18(4):871–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SSG, et al. 1999. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. PNAS 96(7):3712–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komolov KE, Benovic JL. 2018. G protein-coupled receptor kinases: past, present and future. Cell Signal. 41:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. 1990. β-Arrestin: a protein that regulates β-adrenergic receptor function. Science 248(4962):1547–50 [DOI] [PubMed] [Google Scholar]
- 41.Kang Y, Zhou XE, Gao X, He Y, Liu W, et al. 2015. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523(7562):561–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang W,Masureel M, Qianhui Q, Janetzko J, Inoue A, et al. 2020. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579(7798):303–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, et al. 2013. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497(7447):137–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liggett SB, Freedman NJ, Schwinn DA, Lefkowitz RJ. 1993. Structural basis for receptor subtype-specific regulation revealed by a chimeric β3/β2-adrenergic receptor. PNAS 90(8):3665–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eichel K, Jullié D, von Zastrow M. 2016. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 18(3):303–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang W, Curran PK, Hoang Q, Moreland RT, Fishman PH. 2004. Differences in endosomal targeting of human β1- and β2-adrenergic receptors following clathrin-mediated endocytosis. J. Cell Sci. 117(5):723–34 [DOI] [PubMed] [Google Scholar]
- 47.Hu LA, Tang Y, Miller WE, Cong M, Lau AG, et al. 2000. β1-adrenergic receptor association with PSD-95. Inhibition of receptor internalization and facilitation of β1-adrenergic receptor interaction with N-methyl-d-aspartate receptors. J. Biol. Chem. 275(49):38659–66 [DOI] [PubMed] [Google Scholar]
- 48.Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, et al. 1998. μ-Opioid receptor internalization: Opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol. Pharmacol. 53(3):377–84 [PubMed] [Google Scholar]
- 49.Von Zastrow M, Keith DE Jr., Evans CJ. 1993. Agonist-induced state of the δ-opioid receptor that discriminates between opioid peptides and opiate alkaloids. Mol. Pharmacol. 44(1):166–72 [PubMed] [Google Scholar]
- 50.Just S, Illing S, Trester-Zedlitz M, Lau EK, Kotowski SJ, et al. 2013. Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol. Pharmacol. 83(3):633–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau EK, Trester-Zedlitz M, Trinidad JC, Kotowski SJ, Krutchinsky AN, et al. 2011. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci. Signal. 4(185):ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoeber M, Jullié D, Li J, Chakraborty S, Majumdar S, et al. 2020. Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife 9:e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galcheva-Gargova Z, Theroux SJ, Davis RJ. 1995. The epidermal growth factor receptor is covalently linked to ubiquitin. Oncogene 11(12):2649–55 [PubMed] [Google Scholar]
- 54.Mori S, Heldin CH, Claesson-Welsh L. 1993. Ligand-induced ubiquitination of the platelet-derived growth factor β-receptor plays a negative regulatory role in its mitogenic signaling. J. Biol. Chem. 268(l):577–83 [PubMed] [Google Scholar]
- 55.Mori S, Heldin CH, Claesson-Welsh L. 1992. Ligand-induced ubiquitination of the platelet-derived growth factor β-receptor. J. Biol. Chem. 267(9):6429–34 [PubMed] [Google Scholar]
- 56.Vecchione A, Marchese A, Henry P, Rotin D, Morrione A. 2003. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol. Cell. Biol. 23(9):3363–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haugsten EM,Malecki J, Bjørklund SMS, Olsnes S, Wesche J. 2008. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell. 19(8):3390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valverde P 2005. Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem. Biophys. Res. Commun. 333(1):180–85 [DOI] [PubMed] [Google Scholar]
- 59.Geetha T, Jiang J, Wooten MW. 2005. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 20(2):301–12 [DOI] [PubMed] [Google Scholar]
- 60.McKanna JA, Haigler HT, Cohen S. 1979. Hormone receptor topology and dynamics: morphological analysis using ferritin-labeled epidermal growth factor. PNAS 76(11):5689–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller K, Beardmore J, Kanety H, Schlessinger J, Hopkins CR. 1986. Localization of the epidermal growth factor (EGF) receptor within the endosome of EGF-stimulated epidermoid carcinoma (A431) cells. J. Cell Biol. 102(2):500–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, et al. 2013. Lysine 63-linked polyubiquitination is required for EGF receptor degradation. PNAS 110(39):15722–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell ES, Coelho PP, Ratcliffe CDH, Rajadurai CV, Peschard P, et al. 2019. LC3C-mediated autophagy selectively regulates the Met RTK and HGF-stimulated migration and invasion. Cell Rep. 29(12):4053–68.e6 [DOI] [PubMed] [Google Scholar]
- 64.Sorkin A, Krolenko S, Kudrjavtceva N, Lazebnik J, Teslenko L, et al. 1991. Recycling of epidermal growth factor-receptor complexes in A431 cells: identification of dual pathways. J. Cell Biol. 112(1):55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grant BD, Donaldson JG. 2009. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10(9):597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parachoniak CA, Luo Y, Abella JV, Keen JH, Park M. 2011. GGA3 functions as a switch to promote Met receptor recycling, essential for sustained ERK and cell migration. Dev. Cell 20(6):751–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinilla-Macua I, Grassart A, Duvvuri U, Watkins SC, Sorkin A. 2017. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. eLife 6:e31993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sigismund S, Argenzio E, Tosoni D, Cavallaro E, Polo S, Di Fiore PP. 2008. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15(2):209–19 [DOI] [PubMed] [Google Scholar]
- 69.Sigismund S, Algisi V, Nappo G, Conte A, Pascolutti R, et al. 2013. Threshold-controlled ubiquitination of the EGFR directs receptor fate. EMBO J. 32(15):2140–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hislop JN, von Zastrow M. 2011. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic 12(2):137–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. 2003. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5(5):709–22 [DOI] [PubMed] [Google Scholar]
- 72.Whistler JL, Enquist J, Marley A, Fong J, Gladher F, et al. 2002. Modulation of postendocytic sorting of G protein-coupled receptors. Science 297(5581):615–20 [DOI] [PubMed] [Google Scholar]
- 73.He C, Wei Y, Sun K, Li B, Dong X, et al. 2013. Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell 154(5):1085–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dores MR, Lin H, Grimsey NJ, Mendez F, Trejo J. 2015. The α-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 26(25):4660–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dores MR, Chen B, Lin H, Soh UJK, Paing MM, et al. 2012. ALIX binds a YPX33L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 197(3):407–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sposini S, Jean-Alphonse FG, Ayoub MA, Oqua A, West C, et al. 2017. Integration of GPCR signaling and sorting from very early endosomes via opposing APPL1 mechanisms. Cell Rep. 21(10):2855–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. 1999. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature 401(6750):286–90 [DOI] [PubMed] [Google Scholar]
- 78.Tanowitz M, von Zastrow M. 2003. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J. Biol. Chem. 278(46):45978–86 [DOI] [PubMed] [Google Scholar]
- 79.Lauffer BEL,Melero C, Temkin P, Lei C, Hong W, et al. 2010. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J. Cell Biol. 190(4):565–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Puthenveedu MA, Lauffer B, Temkin P, Vistein R, Carlton P, et al. 2010. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell 143(5):761–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Temkin P, Lauffer B, Jäger S, Cimermancic P, Krogan NJ, von Zastrow M. 2011. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 13(6):715–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Varandas KC, Irannejad R, von Zastrow M. 2016. Retromer endosome exit domains serve multiple trafficking destinations and regulate local G protein activation by GPCRs. Curr. Biol. 26(23):3129–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choy RW-Y, Park M, Temkin P, Herring BE, Marley A, et al. 2014. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 82(1):55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jullié D, Stoeber M, Sibarita J-B, Zieger HL, Bartol TM, et al. 2019. A discrete presynaptic vesicle cycle for neuromodulator receptors. Neuron 105(4):663–77.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yudowski GA, Puthenveedu MA, Henry AG, von Zastrow M. 2009. Cargo-mediated regulation of a rapid Rab4-dependent recycling pathway. Mol. Biol. Cell 20(1l):2774–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clairfeuille T, Mas C, Chan ASM, Yang Z, Tello-Lafoz M, et al. 2016. A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat. Struct. Mol. Biol. 23(10):921–32 [DOI] [PubMed] [Google Scholar]
- 87.Stoscheck CM, Carpenter G. 1984. Down regulation of epidermal growth factor receptors: direct demonstration of receptor degradation in human fibroblasts. J. Cell Biol. 98(3):1048–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wells A, Welsh J, Lazar C, Wiley H, Gill G, Rosenfeld M. 1990. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science 247(4945):962–64 [DOI] [PubMed] [Google Scholar]
- 89.Yoon CH, Lee J, Jongeward GD, Sternberg PW. 1995. Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science 269(5227):1102–5 [DOI] [PubMed] [Google Scholar]
- 90.Hopper NA, Lee J, Sternberg PW. 2000. ARK-1 inhibits EGER signaling in C. elegans. Mol. Cell 6(1):65–75 [PubMed] [Google Scholar]
- 91.Jékely G, Rørth P. 2003. Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Rep. 4(12):1163–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ. 2002. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell 108(2):261–69 [DOI] [PubMed] [Google Scholar]
- 93.Roepstorff K, Grandal MV, Henriksen L, Knudsen SLJ, Lerdrup M, et al. 2009. Differential effects of EGER ligands on endocytic sorting of the receptor. Traffic 10(8):1115–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reddy CC, French AR, Wiley HS, Wells AS, Lauffenburger DA. 1996. Growth factor/receptor trafficking effects on cell proliferation responses: the EGF system as a paradigm for insight. Cardiovasc. Pathol. 5(5):293 [Google Scholar]
- 95.French AR, Sudlow GP, Wiley HS, Lauffenburger DA. 1994. Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J. Biol. Chem. 269(22):15749–55 [PubMed] [Google Scholar]
- 96.Lenferink AEG, Pinkas-Kramarski R, van de Poll ML, van Vugt MJ, Klapper LN, et al. 1998. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 17(12):3385–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, Zhang L, Yeung TK, Chen X. 1999. Endocytosis deficiency of epidermal growth factor (EGL) receptor-ErbB2 heterodimers in response to EGL stimulation. Mol. Biol. Cell 10(5):1621–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lanzetti L, Di Fiore PP. 2017. Behind the scenes: endo/exocytosis in the acquisition of metastatic traits. Cancer Res. 77(8):1813–17 [DOI] [PubMed] [Google Scholar]
- 99.Asp N, Kvalvaag A, Sandvig K, Pust S. 2016. Regulation of ErbB2 localization and function in breast cancer cells by ERM proteins. Oncotarget 7(18):25443–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pust S, Klokk TI, Musa N, Jenstad M, Risberg B, et al. 2013. Flotillins as regulators of ErbB2 levels in breast cancer. Oncogene 32(29):3443–51 [DOI] [PubMed] [Google Scholar]
- 101.Gur G, Rubin C, Katz M, Amit I, Citri A, et al. 2004. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 23(16):3270–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Muratoglu SC, Mikhailenko I, Newton C, Migliorini M, Strickland DK. 2010. Low density lipoprotein receptor-related protein 1 (LRP1) forms a signaling complex with platelet-derived growth factor receptor-β in endosomes and regulates activation of the MAPK pathway. J. Biol. Chem. 285(19):14308–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong ESM, Fong CW, Lim J, Yusoff P, Low BC, et al. 2002. Sprouty2 attenuates epidermal growth factor receptor ubiquitylation and endocytosis, and consequently enhances Ras/ERK signalling. EMBO J. 21(18):4796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Komada M 2008. Controlling receptor downregulation by ubiquitination and deubiquitination. Curr. Drug Discov. Technol. 5(1):78–84 [DOI] [PubMed] [Google Scholar]
- 105.Pareja F, Ferraro DA, Rubin C, Cohen-Dvashi H, Zhang F, et al. 2012. Deubiquitination of EGFR by Cezanne-1 contributes to cancer progression. Oncogene 31(43):4599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Savio MG, Wollscheid N, Cavallaro E, Algisi V, Di Fiore PP, et al. 2016. USP9X controls EGFR fate by deubiquitinating the endocytic adaptor Eps15. Curr. Biol. 26(2):173–83 [DOI] [PubMed] [Google Scholar]
- 107.Francavilla C, Papetti M, Rigbolt KTG, Pedersen A-K, Sigurdsson JO, et al. 2016. Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nat. Struct. Mol. Biol. 23(6):608–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao G-Y, Schmid SL. 2020. FCHSD2 controls oncogenic ERK1/2 signaling outcome by regulating endocytic trafficking. PLOS Biol. 18(7):e3000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caswell PT, Chan M, Lindsay AJ, McCaffrey MW, Boettiger D, Norman JC. 2008. Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 183(1):143–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jo U, Park KH, Whang YM, Sung JS, Won NH, et al. 2014. EGFR endocytosis is a novel therapeutic target in lung cancer with wild-type EGFR. Oncotarget 5(5):1265–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang P, Holowatyj AN, Roy T, Pronovost SM, Marchetti M, et al. 2019. An SH3PX1-dependent endocytosis-autophagy network restrains intestinal stem cell proliferation by counteracting EGFR-ERK signaling. Dev. Cell 49(4):574–89.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Niendorf S, Oksche A, Kisser A, Löhler J, Prinz M, et al. 2007. Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol. Cell. Biol. 27(13):5029–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Law PY, Hom DS, Loh HH. 1984. Down-regulation of opiate receptor in neuroblastoma × glioma NG108–15 hybrid cells. Chloroquine promotes accumulation of tritiated enkephalin in the lysosomes. J. Biol. Chem. 259(7):4096–104 [PubMed] [Google Scholar]
- 114.Henry AG, White IJ, Marsh M, von Zastrow M, Hislop JN. 2011. The role of ubiquitination in lysosomal trafficking of δ-opioid receptors. Traffic 12(2):170–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haugh JM, Schooler K, Wells A, Wiley HS, Lauffenburger DA. 1999. Effect of epidermal growth factor receptor internalization on regulation of the phospholipase C-γ1 signaling pathway. J. Biol. Chem. 274(13):8958–65 [DOI] [PubMed] [Google Scholar]
- 116.Haugh JM, Meyer T. 2002. Active EGF receptors have limited access to PtdIns(4,5)P2 in endosomes: implications for phospholipase C and PI 3-kinase signaling. J. Cell Sci. 115(2):303–10 [DOI] [PubMed] [Google Scholar]
- 117.Pinilla-Macua I, Watkins SC, Sorkin A. 2016. Endocytosis separates EGF receptors from endogenous fluorescently labeled HRas and diminishes receptor signaling to MAP kinases in endosomes. PNAS 113(8):2122–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ivaska J, Heino J. 2011. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 27:291–320 [DOI] [PubMed] [Google Scholar]
- 119.Marquèze-Pouey B, Mailfert S, Rouger V, Goaillard J-M, Marguet D. 2014. Physiological epidermal growth factor concentrations activate high affinity receptors to elicit calcium oscillations. PLOS ONE 9(9):e106803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Calebiro D, Sungkawom T. 2018. Single-molecule imaging of GPCR interactions. Trends Pharmacol. Sci. 39(2):109–22 [DOI] [PubMed] [Google Scholar]
- 121.Harden TK, Cotton CU, Waldo GL, Lutton JK, Perkins JP. 1980. Catecholamine-induced alteration in sedimentation behavior of membrane bound beta-adrenergic receptors. Science 210(4468):441–43 [DOI] [PubMed] [Google Scholar]
- 122.Khan MN, Savoie S, Bergeron JJ, Posner BI. 1986. Characterization of rat liver endosomal fractions. In vivo activation of insulin-stimulable receptor kinase in these structures. J. Biol. Chem. 261(18):8462–72 [PubMed] [Google Scholar]
- 123.Lai WH, Cameron PH, Doherty JJ, Posner BI, Bergeron JJ. 1989. Ligand-mediated autophosphorylation activity of the epidermal growth factor receptor during internalization. J. Cell Biol. 109(6):2751–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sorkin A, Von Zastrow M. 2002. Signal transduction and endocytosis: close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 3(8):600–14 [DOI] [PubMed] [Google Scholar]
- 125.Grimes ML, Beattie E, Mobley WC. 1997. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. PNAS 94(18):9909–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. 1997. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 277(5329):1097–100 [DOI] [PubMed] [Google Scholar]
- 127.Andres-Alonso M, Ammar MR, Butnaru I, Gomes GM, Acuña Sanhueza G, et al. 2019. SIPA1L2 controls trafficking and local signaling of TrkB-containing amphisomes at presynaptic terminals. Nat. Commun. 10(1):5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kononenko NL, Claßen GA, Kuijpers M, Puchkov D, Maritzen T, et al. 2017. Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat. Commun. 8(1):14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Quinney KB, Frankel EB, Shankar R, Kasberg W, Luong P, Audhya A. 2019. Growth factor stimulation promotes multivesicular endosome biogenesis by prolonging recruitment of the late-acting ESCRT machinery. PNAS 116(14):6858–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tomas A, Vaughan SO, Burgoyne T, Sorkin A, Hartley JA, et al. 2015. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 6:7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eden ER, White IJ, Tsapara A, Futter CE. 2010. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 12(3):267–72 [DOI] [PubMed] [Google Scholar]
- 132.Li C, Baquiran G, Gu F, Tremblay ML, Fazel A, et al. 2006. Insulin receptor kinase-associated phosphotyrosine phosphatases in hepatic endosomes: assessing the role of phosphotyrosine phosphatase-1B. Endocrinology 147(2):912–18 [DOI] [PubMed] [Google Scholar]
- 133.Villaseñor R, Nonaka H, Del Conte-Zerial P, Kalaidzidis Y, Zerial M. 2015. Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. eLife 4:e06156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stanoev A, Mhamane A, Schuermann KC, Grecco HE, Stallaert W, et al. 2018. Interdependence between EGFR and phosphatases spatially established by vesicular dynamics generates a growth factor sensing and responding network. Cell Syst. 7(3):295–309.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pol A, Calvo M, Enrich C. 1998. Isolated endosomes from quiescent rat liver contain the signal transduction machinery. Differential distribution of activated Raf-1 and Mek in the endocytic compartment. FEBS Lett. 441(1):34–38 [DOI] [PubMed] [Google Scholar]
- 136.Howe CL, Valletta JS, Rusnak AS, Mobley WC. 2001. NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 32(5):801–14 [DOI] [PubMed] [Google Scholar]
- 137.Jiang X, Sorkin A. 2002. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol. Biol. Cell 13(5):1522–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Surve SV, Myers PJ, Clayton SA, Watkins SC, Lazzara MJ, Sorkin A. 2019. Localization dynamics of endogenous fluorescently labeled RAF1 in EGF-stimulated cells. Mol. Biol. Cell 30(4):506–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Marshall CJ. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80(2):179–85 [DOI] [PubMed] [Google Scholar]
- 140.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129(7):1261–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Saito T, Jones CC, Huang S, Czech MP, Pilch PF. 2007. The interaction of Akt with APPL1 is required for insulin-stimulated Glut4 translocation. J. Biol. Chem. 282(44):32280–87 [DOI] [PubMed] [Google Scholar]
- 142.Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, et al. 2004. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 116(3):445–56 [DOI] [PubMed] [Google Scholar]
- 143.Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, et al. 2008. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133(3):486–97 [DOI] [PubMed] [Google Scholar]
- 144.Braccini L, Ciraolo E, Campa CC, Perino A, Longo DL, et al. 2015. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 6:7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schmid SL. 2017. Reciprocal regulation of signaling and endocytosis: implications for the evolving cancer cell. J. Cell Biol. 216(9):2623–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen P-H, Bendris N, Hsiao Y-J, Reis CR, Mettlen M, et al. 2017. Crosstalk between CLCb/Dyn1-mediated adaptive clathrin-mediated endocytosis and epidermal growth factor receptor signaling increases metastasis. Dev. Cell 40(3):278–88.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Reis CR, Chen P-H, Srinivasan S, Aguet F,Mettlen M, Schmid SL. 2015. Crosstalk between Akt/GSK3β signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J. 34(16):2132–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Garay C, Judge G, Luearelli S, Bautista S, Pandey R, et al. 2015. Epidermal growth factor-stimulated Akt phosphorylation requires clathrin or ErbB2 but not receptor endocytosis. Mol. Biol. Cell 26(19):3504–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lucarelli S, Pandey R, Judge G, Antonescu CN. 2016. Similar requirement for clathrin in EGF- and HGF- stimulated Akt phosphorylation. Commun. Integr. Biol. 9(3):e1175696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Palamidessi A, Frittoli E, Garré M, Faretta M, Mione M, et al. 2008. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 134(1):135–47 [DOI] [PubMed] [Google Scholar]
- 151.Ménard L, Parker PJ, Kermorgant S. 2014. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 5:3907. [DOI] [PubMed] [Google Scholar]
- 152.Cosker KE, Segal RA. 2014. Neuronal signaling through endocytosis. Cold Spring Harb. Perspect. Biol. 6(2):a020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harrington AW, St Hillaire C, Zweifel LS, Glebova NO, Philippidou P, et al. 2011. Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell 146(3):421–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kwok-Keung Fung B, Stryer L. 1980. Photolyzed rhodopsin catalyzes the exchange of GTP for bound GDP in retinal rod outer segments. PNAS 77(5):2500–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Salinas RY, Pearring JN, Ding J-D, Spencer WJ, Hao Y, Arshavsky VY. 2017. Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J. Cell Biol. 216(5):1489–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pippig S, Andexinger S, Lohse MJ. 1995. Sequestration and recycling of beta 2-adrenergic receptors permit receptor resensitization. Mol. Pharmacol. 47(4):666–76 [PubMed] [Google Scholar]
- 157.Krueger KM, Daaka Y, Pitcher JA, Lefkowitz RJ. 1997. The role of sequestration in G protein-coupled receptor resensitization. Regulation of β2-adrenergic receptor dephosphorylation by vesicular acidification. J. Biol. Chem. 272(1):5–8 [DOI] [PubMed] [Google Scholar]
- 158.Tsvetanova NG, Irannejad R, von Zastrow M. 2015. G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J. Biol. Chem. 290(11):6689–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, et al. 1998. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J. Biol. Chem. 273(2):685–88 [DOI] [PubMed] [Google Scholar]
- 160.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. 2007. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 69:483–510 [DOI] [PubMed] [Google Scholar]
- 161.Slessareva JE, Routt SM, Temple B, Bankaitis VA, Dohlman HG. 2006. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein α subunit at the endosome. Cell 126(1):191–203 [DOI] [PubMed] [Google Scholar]
- 162.Mullershausen F, Zeeri F, Cetin C, Billich A, Guerini D, Seuwen K. 2009. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat. Chem. Biol. 5(6):428–34 [DOI] [PubMed] [Google Scholar]
- 163.Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, et al. 2009. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5(10):734–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, et al. 2009. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLOS Biol. 7(8):e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. 2011. Endocytosis promotes rapid dopaminergic signaling. Neuron 71(2):278–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, et al. 2013. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495(7442):534–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bowman SL, Shiwarski DJ, Puthenveedu MA. 2016. Distinct G protein-coupled receptor recycling pathways allow spatial control of downstream G protein signaling.J. Cell Biol. 214(7):797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Godbole A, Lyga S, Lohse MJ, Calebiro D. 2017. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat. Commun. 8(1):443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, et al. 2017. Functional selectivity of GPCR-directed drug action through location bias. Nat. Chem. Biol. 13(7):799–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Stoeber M,Jullié D,Lobingier BT, Laeremans T, Steyaert J,et al. 2018. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 98(5):963–76.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nash CA, Wei W, Irannejad R, Smrcka AV. 2019. Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. eLife 8:e48167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ramírez-García PD, Retamal JS, Shenoy P, Imlach W, Sykes M, et al. 2019. A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain. Nat. Nanotechnol. 14(12):1150–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, et al. 2020. Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. PNAS 117(26):15281–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, et al. 2016. GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166(4):907–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lyga S, Volpe S, Werthmann RC, Götz K, Sungkaworn T, et al. 2016. Persistent cAMP signaling by internalized LH receptors in ovarian follicles. Endocrinology 157(4):1613–21 [DOI] [PubMed] [Google Scholar]
- 176.Ho PWM, Chan AS, Pavlos NJ, Sims NA, Martin TJ. 2019. Brief exposure to full length parathyroid hormone-related protein (PTHrP) causes persistent generation of cyclic AMP through an endocytosis-dependent mechanism. Biochem. Pharmacol. 169:113627. [DOI] [PubMed] [Google Scholar]
- 177.Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga J-P. 2013. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. PNAS 110(4):1530–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang L-Y, et al. 2019. Structure of an endosomal signaling GPCR-G protein-β-arrestin megacomplex. Nat. Struct. Mol. Biol. 26(12):1123–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Clister T, Greenwald EC, Baillie GS, Zhang J. 2019. AKAP95 organizes a nuclear microdomain to control local cAMP for regulating nuclear PKA. Cell Chem. Biol. 26(6):885–91.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tsvetanova NG, von Zastrow M. 2014. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 10(12):1061–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.O’Banion CP, Vickerman BM, Haar L, Lawrence DS. 2019. Compartmentalized cAMP generation by engineered photoactivated adenylyl cyclases. Cell Chem. Biol. 26(10):1393–406.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lazar AM, Irannejad R, Baldwin TA, Sundaram AB, Gutkind JS, et al. 2020. G protein-regulated endocytic trafficking of adenylyl cyclase type 9. eLife 9:e58039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sunahara RK, Taussig R. 2002. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol. Interv. 2(3):168–84 [DOI] [PubMed] [Google Scholar]
- 184.Tsvetanova NG, Trester-Zedlitz M, Newton BW, Riordan DP, Sundaram AB, et al. 2017. G protein-coupled receptor endocytosis confers uniformity in responses to chemically distinct ligands. Mol. Pharmacol. 91(2):145–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Teis D, Wunderlich W, Huber LA. 2002. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 3(6):803–14 [DOI] [PubMed] [Google Scholar]
- 186.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141(2):290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. 2014. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11(3):319–24 [DOI] [PubMed] [Google Scholar]
- 188.Shi T, Niepel M, McDermott JE, Gao Y, Nicora CD, et al. 2016. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci. Signal 9(436):rs6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yang Y-P, Ma H, Starchenko A, Huh WJ, Li W, et al. 2017. A chimeric Egfr protein reporter mouse reveals Egfr localization and trafficking in vivo. Cell Rep. 19(6):1257–67 [DOI] [PMC free article] [PubMed] [Google Scholar]