

Abstract
REV7 is a small multifunctional protein that participates in multiple DNA repair pathways, most notably translesion DNA synthesis and double strand break (DSB)repair. While the role of REV7 in translesion synthesis has been known for several decades, its function in DSB repair is a recent discovery. Investigations into the DSB repair function of REV7 have led to the discovery of a new DNA repair complex known as Shieldin. Recent studies have also highlighted the importance of REV7’s HORMA domain, an ancient structural motif, in REV7 function and have identified the HORMA regulators, TRIP13 and p31, as novel DNA repair factors. In this review, we discuss these recent findings and their implications for repair pathway choice, both at DSBs and replication forks. We suggest that REV7, in particular the activation state of its HORMA domain, can act as a critical determinant of mutagenic vs. error-free repair in multiple contexts.
Keywords: REV7, Shieldin, TRIP13, DNA Repair, Fanconi Anemia, Double Strand Break Repair, Translesion Synthesis
REV7: A multifunctional coordinator of DNA repair
REV7 (Also known as MAD2L2, MAD2B or FANCV) is a highly conserved protein, first identified in Saccharomyces cerevisiae as a gene promoting ultraviolet (UV) induced mutagenesis [1], but was soon found to be a highly conserved component of the DNA Polymerase ζ (see Glossary) translesion synthesis (TLS) complex [2, 3]. Recently, surprising new findings have dramatically expanded the scope of REV7 research [4, 5]. In a role independent of its known TLS binding partners, REV7 controls double strand break (DSB) repair pathway choice. REV7 has also been identified as a Fanconi Anemia protein (FANCV) [6]. These and other recent findings have led to a reevaluation of REV7 function and suggest previously unappreciated connections between replication-associated TLS and DSB repair. In this review we discuss recent findings regarding REV7 function and regulation and discuss these in the context of the unique structural biology of REV7 as a HORMA domain protein. Furthermore, we elaborate on the multifunctional nature of REV7 and propose cohesive models for understanding REV7 function.The HORMA family of proteins
REV7 is a founding member of the HORMA family of proteins; named for its three founding members: HOp1, a meiotic chromosome axis factor, REV7 and MAd2. Although first identified on the basis of primary sequence similarity in yeast [7], their similarity in tertiary structure is far more striking [8–10] and widely conserved across eukaryotes. More recently, studies have identified the spindle assembly checkpoint (SAC) inhibitor p31comet [11] and the autophagy proteins, Atg13/Atg101 [12, 13] as additional HORMA family members. In addition, a recent study unveiled HORMA proteins as peptide recognition modules mediating bacteriophage immunity in bacteria [14], suggesting an even deeper evolutionary history of HORMA proteins than previously thought.
HORMA proteins as regulatory modules
While HORMA proteins have widely disparate cellular functions, they have highly conserved protein-protein interaction modules and regulatory mechanisms. A key characteristic of HORMA proteins is a C-terminal region known as the seatbelt, due to its unique ability to latch onto binding partners [15] (Figure 1A). Although not all HORMA proteins have active seatbelts, such as p31comet, Atg13, and Atg101 whose seatbelts are locked in either a static “open” or “closed” state [11, 13], a dynamic seatbelt that can latch and unlatch is a common feature among many HORMA proteins [14, 16–18].
Figure 1: REV7 is a HORMA protein and is regulated through stable structural rearrangement.
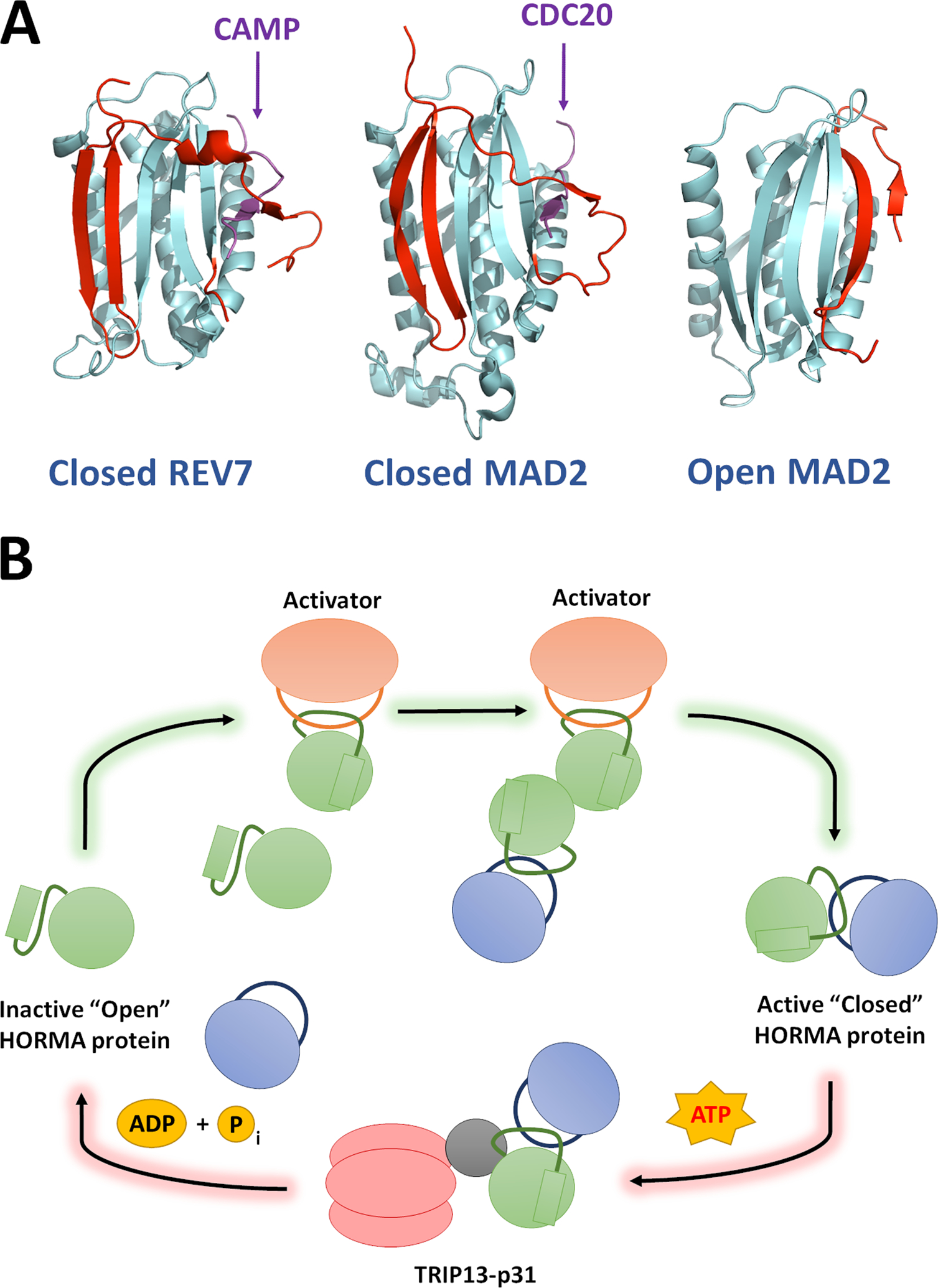
(A) Structures of REV7 and MAD2 in their closed, active forms (PDB ID: 5XPT and 4AEZ, respectively) and MAD2 in the open conformation. The seatbelt region of each protein is highlighted in red. REV7 and MAD2 show high structural similarity and bind to their cognate seatbelt binding partners via an identical mechanism. Small seatbelt-binding fragments of CAMP and CDC20 (Purple) are depicted in these representative structures. All known HORMA seatbelt binding partners associate though the same mechanism. (B) The “templating” model for HORMA protein activation: HORMA protein activation proceeds through a chain reaction mechanism, beginning when the first HORMA molecules are closed through their interaction with a dedicated activation partner. HORMA proteins can also be closed through dimerization with an already closed protein, hence the signal can be rapidly propagated by a small number of activators. The reverse reaction is well-understood and proceeds through ATP-dependent remodeling of HORMA proteins from closed to open via the TRIP13 ATPase and the p31 adaptor subunit.
There is a large energetic barrier between the open and closed states of HORMA proteins due to the requirement for significant structural rearrangement, which renders the conformations relatively stable in cells. This stability allows HORMA protein state to constitute a persistent signal in the cell, but also requires dedicated mechanisms to promote conformational transition. The AAA+ ATPase, TRIP13 along with its substrate adapter p31comet, promotes the conversion from closed to open of at least three distinct HORMA proteins: Mad2 [19], REV7 [18, 20] and Hop1 orthologues [21]. Recent structural work has additionally raised the possibility of a p31-indendent action of TRIP13 on REV7, dependent on the Shieldin complex [22].TRIP13-like ATPases are also present in some bacteria and appear to similarly regulate HORMA proteins [14].
The most well-characterized seatbelt-dependent interaction is that between Mad2 and the anaphase-promoting complex (APC) factor, Cdc20 [16], which inhibits the APC by sequestering Cdc20, hence preventing anaphase progression until the mitotic spindle has assembled properly. The Hop1 HORMA domain is utilized in a different manner, by binding to other Hop1 molecules in order to self-assemble into large oligomeric structures as part of the meiotic chromosome axis [10, 17].
REV7 is an abundant cellular protein and is unique among HORMA proteins, both in its large number of unique seatbelt binding partners and in its involvement in multiple distinct pathways. The REV7 seatbelt has been definitively shown to bind to the Polζ component, REV3 [9, 23], the Shieldin component, SHLD3 [18, 24–26], and the poorly understood mitotic factor, CAMP [27, 28]. Some studies have also suggested a seatbelt-dependent interaction between REV7 and the Cdc20-related APC factor, Cdh1 [29, 30], although recent results indicate that this interaction is seatbelt-independent [31].
Activation of HORMA proteins
The action of p31-TRIP13 in opening the seatbelt of HORMA proteins dissociates seatbelt-dependent complexes. While the fundamental features of this process are well-understood, the inverse process of closing HORMA proteins in order to form these complexes is more enigmatic. Although open Mad2 (O-Mad2) slowly converts to closed (C-Mad2) over time in vitro [16], its activity in vivo is dependent on activation by Mad1 [32]. REV7, on the other hand, does not spontaneously close in vitro [18], suggesting a requirement for other protein factors for its closing in vivo. The leading model for Mad2 activation by Mad1 is the templating model. In this case, a subpopulation of Mad2 is activated and forms a closed complex with Mad1 at unattached kinetochores, forming a C-Mad2 template. These templates can then activate further Mad2 modules through homodimerization (Figure 1B) [33, 34].
REV7 also homodimerizes in a manner analogous with Mad2 [26, 35], suggesting that REV7 activation could also proceed through a templating mechanism (Figure 1B); however, there is no concrete evidence to support this. The mechanism of REV7 activation through closing is one of the most pressing questions in the field. If the templating model is applicable, the key question then becomes: what seeds this activation, analogous to Mad1’s activation of Mad2? In our in vitro studies, we were unable to observe any transition of O-REV7 to C-REV7 in the presence of full length SHLD3 or high concentrations of the seatbelt-binding peptide derived from SHLD3 [18], demonstrating that SHLD3 alone cannot function as a seed to promote REV7 activation. It will be useful going forward to assay other known REV7 binding partners for their ability to promote REV7 closing in a Mad1-like manner.
Alternatively, REV7 may be activated by an entirely distinct mechanism, for example, posttranslational modification. There is some precedent for this as Mad2 conformation has been shown to be influenced by phosphorylation in the vicinity of the seatbelt region [36, 37]. While these modifications are thought to exert a minor influence relative to Mad1 activation, similar alterations in REV7 could conceivably play a more major role. Indeed, a cluster of potential phosphorylation sites is present in the vicinity REV7’s seatbelt, including a consensus site for the DNA damage response kinases ATM, ATR and DNA-PK at threonine 103. The functional significance of these sites, however, remains entirely unexplored.
REV7 in translesion DNA synthesis
The more well-understood function of REV7 is its role as the small non-catalytic subunit of the DNA Polymerase ζ (Polζ) complex. Pol ζ is one of several human polymerases specialized for synthesizing DNA across lesions in the template strand, a process known as translesion synthesis (TLS). TLS is more mutagenic than normal replication [38, 39]. In particular, Polymerase ζ, in conjunction with its partner, REV1, are responsible for the majority of spontaneous and damage-induced mutations during DNA replication [40, 41]. Mechanistically, REV1 is thought to act as a recruitment module and insertor polymerase, introducing a single nucleotide across from a lesion. REV1 insertion is highly mutagenic as it generally adds a cytosine regardless of the template base [42]. Polζ is an extension TLS polymerase, carrying out longer stretches of templated DNA synthesis using a damaged primer terminus [43].
REV7 interacts with REV3, the large catalytic subunit of Polζ, via two distinct seatbelt-binding motifs (SBMs, Figure 2A) [23, 35, 44], an interaction that is dependent on REV7 adopting the closed conformation [18]. The primary function of REV7 is to mediate the interaction between REV3 and REV1, although recent structural work has demonstrated potential interactions between REV7 and Pol32 (POLD3 in mammals), a regulatory subunit that is shared between Polζ and the replicative Polymerase δ (Figure 2A) [44, 45]. By linking REV3 to REV1, REV7 acts as a lynchpin between the REV1 recruitment module and REV3’s catalytic activity.
Figure 2: REV7 Controls Pathway Choice at Stalled Replication Forks.
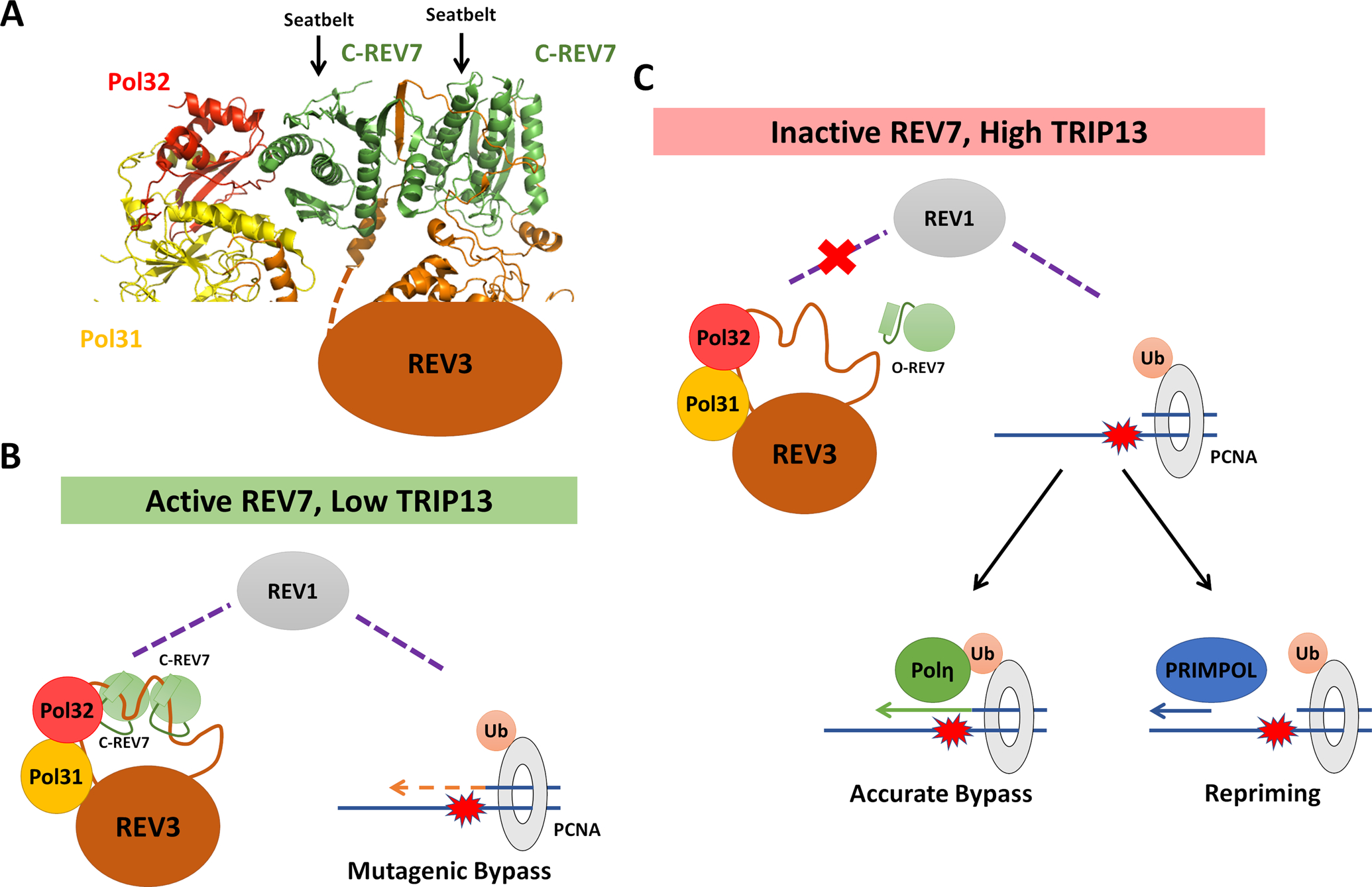
(A) Cryo-electron microscopy structure of the S. cerevisiae DNA Polymerase ζ holoenzyme, consisting of REV3, Pol31, Pol32 and two REV7 molecules (PDB ID: 6V93). REV7 homodimerizes in a head-to-tail manner, forming closed seatbelt-dependent interactions with two seatbelt binding motifs in REV3. One molecule of REV7 makes apparent contract with the Pol32 subunit (POLD3 in mammals). (B) When REV7 is active, whether due to low TRIP13 or other factors, active Polζ is formed with C-REV7 mediating its recruitment to stalled replication forks via REV1. Polζ, together with REV1, catalyzes mutagenic bypass of DNA lesions. (C) When levels of closed REV7 are low, Polζ recruitment is expected to be decreased due to its inability to interact with REV1. In this case, alternative pathways including accurate translesion synthesis and re-priming are expected to predominate in lesion bypass.
Regulation of REV7-mediated translesion DNA synthesis
Given the mutagenic nature of TLS, its regulation is of the utmost importance in maintaining genomic stability; however, this process remains poorly understood. A critical step in promoting TLS, both by Polζ and others, is the monoubiquitination of the replication clamp, PCNA, by Rad6-Rad18 [46, 47] (Figures 2B and C). REV1 is thought to interact directly with PCNA and ubiquitin via its BRCT motifs and ubiquitin binding motifs (UBMs) [48, 49], thereby recruiting Polζ in a PCNA-Ub-dependent manner through REV7. The requirement for PCNA, and especially its ubiquitination, in recruiting Polζ is still debated, as several studies have suggested that TLS can occur in the absence of PCNA-Ub [50–53]. Indeed, REV1 BRCT mutants that cannot interact with PCNA still localize to sites of DNA damage and partially restore tolerance to DNA damage [48], consistent with some additional abilities to recruit Polζ and carry out TLS.
One alternative pathway of Polζ recruitment, at least in mammals, is through the Fanconi Anemia (FA) core complex [54, 55], via a direct interaction between REV1 and the FANCA-binding partner FAAP20 [56]. Whether this allows for PCNA- or PCNA-Ub-independent TLS activation is an unexplored question. Recent experiments using in vitro reconstitution of the budding yeast replisome have also shed new light on this process [57]. In this simplified system, TLS Polymerase η can bypass a specific lesion regardless of the ubiquitination state of PCNA. The main inhibitor of TLS in this system was found to be Polδ, regardless of whether the lesion is on the leading or lagging strand. The ubiquitination of PCNA may thus push the balance away from Polδ and towards TLS polymerases. The regulation of Polζ is more complex, as it requires REV1 to mediate its recruitment. Polζ also competes with Polδ, not only for PCNA binding but for interaction with shared accessory subunits as well.
Implications of TLS regulation through REV7 and alternative pathways
We have shown that altering REV7 conformational dynamics affects both damage-induced mutagenesis and DNA damage tolerance [18, 20]. Despite the obvious regulatory implications of REV7’s HORMA structure, little is known about how this regulation works under normal conditions and what utility it provides to the organism. Although PCNA ubiquitination and REV7 activation promote TLS, they do so in subtly different ways, likely resulting in different outcomes. In the absence of PCNA-Ub, Polδ likely remains a potent inhibitor of all forms of TLS. In contrast, the absence of closed REV7 is not expected to affect the recruitment of REV1. REV1 interacts with multiple other TLS polymerases through its C-terminus, including Pols η, ι and κ [58] and therefore is expected to promote TLS even in the absence of REV7. While Polζ is not the only TLS polymerase, it is responsible for the majority of TLS-induced mutations [40, 41]. A case study in contrast to Polζ is Polymerase η which has been shown to carry out highly accurate TLS across a specific subset of lesions [59, 60]. One can therefore envision that, by inactivating REV7, the cell may favor the usage of other TLS polymerases, which in mammals also includes polymerases ι and κ, thus reducing mutagenesis at the expense of versatility (Figures 2B and C).
While some lesions can be bypassed in the absence of Polζ, its activity is not dispensable as its loss leads to severe developmental defects and DNA damage sensitivity [6, 61]. An alternative pathway to resume replication in the face of an impassable lesion is to simply skip over it and leave a short region of the chromosome unreplicated. On the lagging strand, it is trivial to resume replication ahead of the lesion due to the inherently discontinuous nature of synthesis. On the leading strand, the enzymatic activity required to re-prime and restart synthesis past a lesion is provided by the primase polymerase PRIMPOL [62–64] (Figure 2C). Of course, this is not a true solution to the problem, merely a delay tactic. However, this tactic allows the cell to potentially activate Polζ at a later time, after the bulk of replication has been completed and the potential for mutagenesis is lower.
REV7 in double strand break repair
The involvement of REV7 in DSB repair was uncovered through the surprising finding that its loss conferred strong PARP inhibitor resistance to BRCA1-deficient cancer cells [5] and suppressed telomere fusions in shelterin-deficient cells [4]. Further work demonstrated that REV7 participates in a previously unknown complex known as Shieldin, which operates downstream of the core DSB response factor, 53BP1 [24, 25, 65–69]. Several groups noted that SHLD3, the most upstream Shieldin component, contains motifs in its N-terminus reminiscent of the REV7 SBMs in REV3 and CAMP, suggesting that the REV7-SHLD3 interaction is similarly dependent on REV7 closure [24, 25]. Subsequent biochemical [18] and structural [26, 70] studies confirmed this hypothesis (Figure 3A).
Figure 3: REV7 controls pathway choice at DSBs.
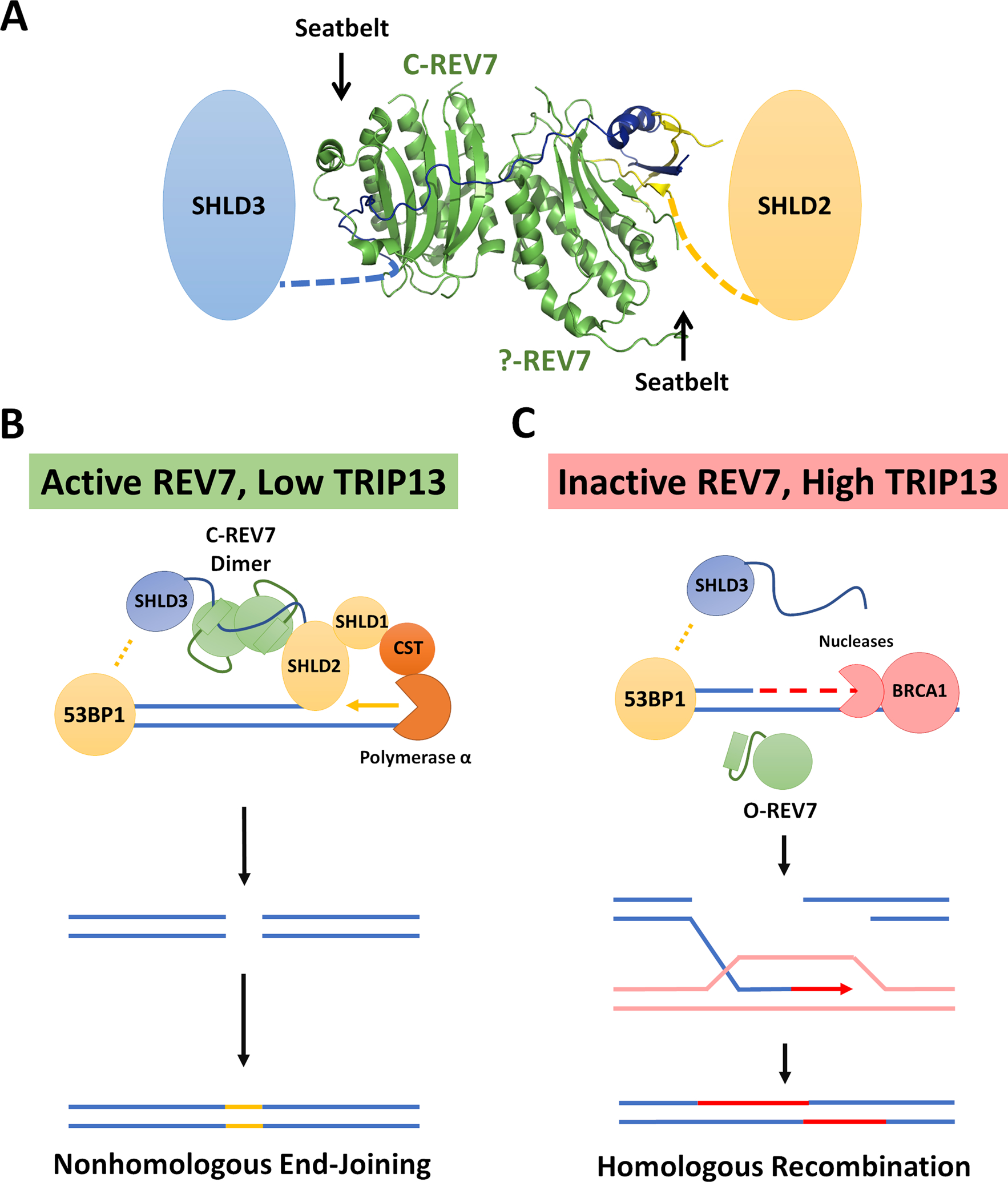
(A) Crystal structure of REV7 in complex with SHLD2 and SHLD3 fragments (PDB ID: 6KTO). REV7 forms a head-to-head homodimer in the context of the Shieldin complex. Only one molecule of REV7 forms a canonical closed-seatbelt interaction with the seatbelt binding motif in the N-terminus of SHLD3. “?-REV7” indicates a novel form of REV7 identified by Liang et al. The functional significance of which is not fully understood. (B) When REV7 is active, the full Shieldin complex can be assembled. REV7 mediates the recruitment of SHLD2-SHLD1 via SHLD3, inhibiting resection and subsequently recruiting CST-Polα for fill-in synthesis. (C) Inactive REV7 fails to bind to SHLD3, leading to an inability to recruit downstream Shieldin factors. In the absence of Shieldin, resection can occur at high levels and HR is expected to predominate.
Mechanistically, the Shieldin complex acts at DSBs by inhibiting 5’−3’ DNA end resection. Such resection generates the ssDNA overhangs required for repair through homologous recombination (HR) [71]. By inhibiting end resection and HR, Shieldin promotes repair through non-homologous end joining (NHEJ), a process that prefers blunt or minimally processed ends. Normally 53BP1, and hence the Shieldin complex, are removed through the action of BRCA1 at DSBs [72]. Shieldin has been proposed to inhibit resection directly through physical binding to short stretches of ssDNA and by recruiting the CST complex which promotes the “fill-in” of resected regions by Primase-Polα [73]. Many details of Shieldin function, however, remain to be sorted out.
One thing that is clear regarding Shieldin is that the anti-resection activity resides in the SHLD2/SHLD1 business end of the complex. REV7, once again, acts as the lynchpin, connecting the SHLD2/SHLD1 effector module to the recruitment module, SHLD3. Recent structural work has clarified the mechanism through which REV7 mediates this interaction, which is distinct from other HORMA protein interactions [26]. REV7 interacts directly with the N-terminal SBMs of SHLD3 and the N-terminus of SHLD2. In order to bring them together, REV7 forms a head-to-head homodimer with one moiety bound to the SHLD3 SBM in a conventional manner and the other adopting a novel seatbelt conformation distinct from that seen in other HORMA proteins. Furthermore, the SHLD3 N-terminus straddles both REV7 units, obstructing the interface that mediates the interaction between REV7 and REV1 (Figure 3A).
Despite this unique organization in the Shieldin complex, at least one unit of closed REV7 appears to be required for its integrity. This observation aligns well with evidence that perturbing REV7 conformation through TRIP13/p31 affects both TLS and DSB repair similarly [18, 20]. Therefore, REV7 is positioned to play a key role in regulating HR usage by controlling end resection. Some aspects of the regulation of end resection are well-understood, while others are not. For example, resection is restricted during G1 phase, thereby preventing the cell from attempting HR when a sister chromatid is not present [74, 75]. In contrast, the regulation of DSB repair pathway choice during S phase is much less clear. It is certainly not necessary to use HR for every break in S phase, and indeed NHEJ remains active throughout the cell cycle [76].
A role for REV7 in controlling S phase DSB repair
One means by which HR is controlled during S phase is by the selective binding of 53BP1 to histone H4 methylated on lysine 20. This results in resection being permitted only in replicated regions where this mark has been diluted by new histone deposition [77]. The control of REV7 conformation offers another avenue to control HR vs. NHEJ usage during S phase. Unlike previously described regulation of 53BP1, the control of REV7 is more fine-tuned, as it inhibits only the Shieldin complex (Figures 3B and C). 53BP1 has other functions such as promoting DSB mobility within the nucleus [78], and it can also inhibit resection through a Shieldin-independent pathway involving PTIP [79, 80]. These additional functions of 53BP1 are poorly understood, making it difficult to speculate as to what advantage a cell gains by regulating the Shieldin complex specifically through REV7 activity. Further research into these roles will enhance our understanding, not only of REV7 function, but also of DSB repair pathway choice as a whole.
REV7 in other contexts
Several other proteins have been recurrently identified through REV7 interactome studies, but the functional importance of these proteins remains unknown. Notable among this group are the HP1 heterochromatin binding proteins [18, 65], which associate with the REV7 binding proteins CAMP and POGZ [18, 65, 66, 81, 82]. The strong association of REV7 with at least five distinct heterochromatin-associated proteins suggests a functional connection, yet this area is entirely unexplored.
CAMP and POGZ have been linked to mitotic progression. CAMP-deficient cells exhibit impaired kinetochore-microtubule attachments, persistent SAC activation, and impaired mitosis [27]. Importantly, however, this function does not appear to require the REV7-interacting domain of CAMP, suggesting a REV7-independent function [27]. POGZ knockdown, on the other hand, causes SAC failure in addition to chromosome misalignment [82]. Whether this reflects a truly distinct function of POGZ as opposed to CAMP, or merely off-target siRNA effects will require further research.
POGZ has also been independently associated with two other recurrent REV7 interactors: LEDGF and HDGFRP2 [18, 83, 84]. All three of these proteins have been reported to promote end resection and HR repair at DSBs [83, 84]. HDGFRP2 and LEDGF are both members of the HDGF family of Tudor domain-containing proteins, which recognize methylated histones [85]. One can imagine that active REV7, perhaps in complex with CAMP, inhibits the pro-resection activity of POGZ, LEDGF, and HDGFRP in specific chromatin environments. Alternatively, REV7 binding to these proteins may act as a sink, passively preventing REV7 from forming productive complexes with Shieldin or Polζ. Lastly, these factors may operate in conjunction with TRIP13-p31, thereby promoting the active inhibition of REV7 by opening the REV7 seatbelt.
It warrants mentioning that these factors score more weakly in DNA damage-related genetic screens than REV7 itself, or than Shieldin/Polζ components. There are many reasons why certain factors may be missed in screens, such as poor or absent guide RNAs, and these issues should be addressed in future targeted studies. In our interactome studies and others, CAMP and POGZ are among the strongest REV7 interacting partners, surpassing the Shieldin and Polζ components. While this may reflect the overall abundance of these proteins, it also suggests that these interactions are likely functionally significant. Furthermore, recent crystallographic work has unambiguously demonstrated that CAMP is a bona fide REV7 seatbelt-binding partner [28]. Unraveling the functions of these enigmatic REV7 partners will likely prove a key piece in solving the overall puzzle of REV7.
An evolutionary history of REV7
The HORMA domain is an ancient protein structural motif. Evolutionary analyses suggest that REV7 and all other human HORMA proteins evolved from a common ancestral HORMA protein, related to a class of bacterial HORMA proteins [86]. While the function of these proteins diverged widely, from bacteriophage immunity to their varied roles in mammalian cells, their basic mechanism of function is conserved [14]. Bacterial HORMA proteins also adopt open and closed conformations, bind tightly to specific peptide motifs in their closed conformation, and activate a binding partner only in the closed conformation [14]. While eukaryotes no longer use HORMA proteins in anti-viral immunity, they apparently co-opted useful features of the HORMA domain for various functions.
REV7 specifically is conserved across Eukaryota, having first been discovered in the fungus S. cerevisiae and subsequently found in mammals, insects, plants and others [1, 2, 87, 88]. Among the known REV7 binding partners, only the TLS-related factors, namely REV3 and REV1, seem to be widely conserved. The Shieldin complex appears to be a much more recent evolutionary innovation, found only in vertebrates. The Shieldin complex appears to have co-evolved with the process of immunoglobulin class-switch recombination (CSR), a process which depends on its anti-resection activity [25].
What is intriguing about this evolutionary timeline is that it suggests that the ancestral function of REV7 is in the TLS pathway and that some property of REV7 made it useful to be co-opted by the Shieldin complex. Notably, REV7 is the glue that holds the Shieldin complex together, suggesting that Shieldin evolved around REV7 rather than merely adjusting to augment an existing function. Taken together, this evolutionary evidence is strongly suggestive that the shared usage of REV7 in Shieldin, Polζ and other complexes is no coincidence. The co-opting of this highly conserved REV7 regulatory module in evolutionarily recent complexes is provocative and ripe for further exploration.
REV7 as a key determinant of mutagenic repair
Although TLS and DSB repair have historically been viewed as independent processes, their coordinate regulation by REV7 conformation calls for a reevaluation of their relationship. Indeed, there are other important connections between these two processes both functional and spatiotemporal: (1) In both contexts, the active REV7 complex promotes the activity of rapid, but potentially mutagenic, pathways even when a higher fidelity option is available. (2) Both processes play an important role during S phase, when the bulk of DNA replication is carried out and when HR is a viable DSB repair option. (3) Resection and HR-like pathways are utilized at stalled replication forks as well as DSBs, raising the possibility that Shieldin and Polζ components could cooperate at the same substrate.
Coordinate regulation model for REV7-dependent complexes
One possible utility of a shared REV7 subunit is for coordinate regulation through REV7 signaling, similar to the manner in which MAD2 is used to signal the activation of the SAC. This model raises two key questions: What is the stimulus that activates REV7 signaling? And what advantage is there to regulating DSB repair and TLS using the same signal? One possibility is that the degree of REV7 activation can be used as a dial to control how much mutagenesis a cell is willing to tolerate in order to complete cell division as quickly as possible (Figure 4A). There are many reasons that a cell may want to adjust this dial. First, certain cell types may prioritize fidelity (i.e. self-renewing stem cells), while other cell types favor rapid expansion (i.e. immune cells in response to an infection). Second, cells may dial down REV7 activity early in S phase but increase it in late S/G2 in order to rapidly complete DNA replication. An important corollary to this model is that cancer cells would have a strong interest in hijacking this dial to promote mutagenesis and rapid growth in the case of high REV7 activity, while retaining the ability to turn the dial in the other direction to resist DNA-damaging chemotherapy.
Figure 4: Two models for coordination between TLS and DSB Repair by REV7.
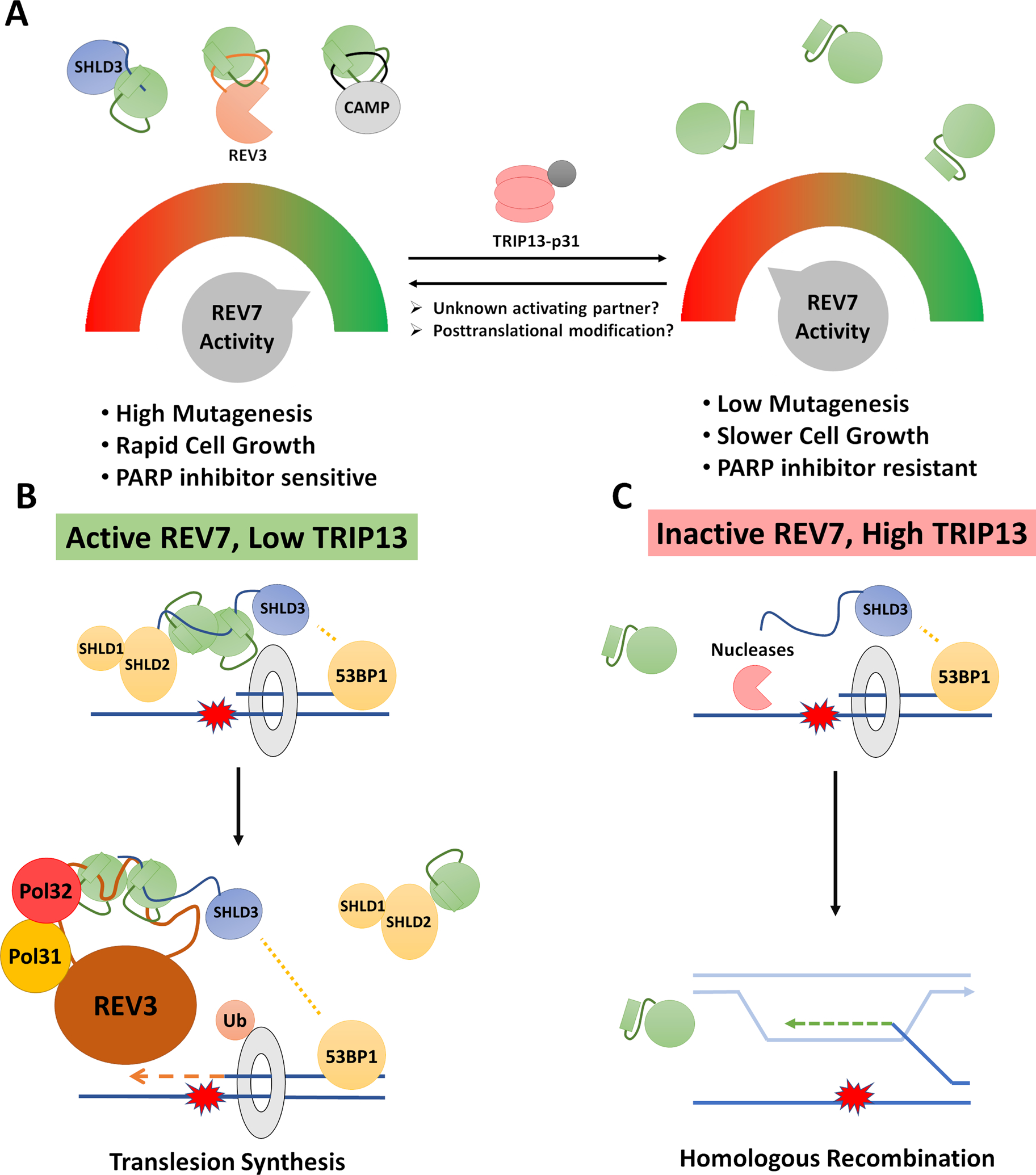
(A) Coordinate regulation: By modulating the activity of REV7, a cell can simultaneously coordinate two mutagenic DSB repair processes. This would provide the cell with a dial to exert significant control over mutagenic processes with a single signal. Such a dial could be adjusted based on cell type, cell cycle phase or external stimuli. It is also expected to be hijacked in cancer cells to either promote rapid growth and mutagenesis or resist chemotherapy. (B and C) Cooperation at stalled replication forks: translesion synthesis and double strand break repair factors are both known to act at certain types of stalled replication forks, hence direct interaction and cooperation between Shieldin and Polζ may be useful in this circumstance to inhibit homologous recombination and promote translesion synthesis. The ability of REV7 to interact with multiple partners simultaneously and form different types of homodimers allows for many possible higher order structures including hybrid Shieldin-Polζ complexes. While these have not been experimentally observed, they have not been ruled out. One such hypothetical hybrid complex is shown in (B).
Cooperation model for REV7-dependent complexes
Another intriguing possibility is that REV7 could act to coordinate direct physical interactions between the TLS machinery and the Shieldin complex at stalled replication forks. HR occurs at stalled replication forks, and although the mechanism is likely somewhat different than that at DSBs, many of the same factors are involved [89, 90]. Resection factors are also recruited to stalled replication forks, and aberrant resection-like activity at stalled forks has been linked to genomic instability [91, 92]. Although the role of Shieldin has not been fully evaluated in inhibiting resection at stalled forks, some studies have implicated the upstream component, 53BP1, in this context [93, 94].
NHEJ is not a viable alternative to HR at stalled forks since a “second end” of the DSB is generally not available for ligation. Instead, HR can be used to switch templates to the opposing nascent strand in order to pass a damaged region, after which normal replication can resume (Figure 4C). Hence, at a stalled replication fork, HR competes directly with TLS to bypass a polymerase-blocking lesion (Figures 4B and C). This is an ideal scenario for the cooperation of Shieldin with Polζ. In cases of high REV7 activity, Shieldin would first be recruited via 53BP1 to protect the stalled replication fork from nuclease processing. REV7 could then mediate a hand-off by disengaging from SHLD2-SHLD1 and engaging with REV3, allowing for Polζ-mediated TLS (Figure 4B). Such a mechanism would activate REV7 closure, inhibit stalled fork processing by HR, and promote speedy bypass through TLS.
Concluding Remarks
While great progress has been made in recent years in understanding REV7 biology, many open questions remain. Recent large-scale genetic screens [95] and interactome studies [96] have the potential to shine new light on many aspects of biology including REV7 and DNA repair pathway choice. A REV7 activator, if it exists, is expected to correlate strongly with REV7 in DNA damage related genetic screens. Furthermore, if the templating model for REV7 activation is correct, then this activator must also be a member of the REV7 physical interactome. High-throughput proteomics studies will also prove useful in identifying regulatory posttranslational modifications of REV7, TRIP13 and p31.
An essential tool for deciphering the regulatory dynamics of REV7, that is currently lacking, is an assay to identify open and closed pools of REV7 in cells. While biochemical fractionation through ion exchange chromatography is useful to study REV7 in isolation, it is not adequate to study the variety of REV7 complexes that form in a physiological setting. One promising approach may be to develop conformation specific monoclonal antibodies - an approach that has been used for MAD2 [97]. This invaluable tool would not only aid in unraveling the regulatory network controlling REV7 conformation but may also be developed into a clinical assay to predict the DSB repair and TLS capabilities in tumors.
Here, we have reviewed the current knowledge of REV7 and its roles in multiple repair processes. We have described how REV7, as a HORMA protein, can exist in a distinct open or closed conformation. Like at least two other HORMA proteins, MAD2 and Hop1, REV7 is a substrate of the ATPase TRIP13 which inactivates REV7, releasing seatbelt-dependent binding partners. We have shone a particular spotlight on the open questions surrounding REV7’s conformational dynamics - including its activation, regulation and connections between its multiple functions. We hope that this discussion and the approaches outlined above will serve to guide the next iteration of REV7 research.
Highlights.
REV7 is a critical component of multiple complexes including well-characterized DNA repair complexes: DNA polymerase ζ and Shieldin, and other poorly understood protein complexes.
REV7 is a HORMA family protein, characterized by a distinctive tertiary structure and an associated regulatory mechanism – where activity is controlled by stable conformational changes.
The TRIP13 ATPase and p31 regulatory subunit remodel REV7 to the inactive HORMA conformation, inhibiting both Polymerase ζ-mediated DNA synthesis and the activity of the Shieldin complex in promoting non-homologous end joining repair.
REV7 plays a similar role in both Shieldin and Polymerase ζ by connecting the upstream recruitment modules and downstream effector modules when REV7 is in its closed, active conformation.
In both Shieldin and Polymerase ζ, REV7 promotes the choice of a potentially mutagenic DNA repair pathway over higher fidelity alternatives. We propose that REV7 activity may be a powerful mechanism for a cell to fine-tune the amount of mutagenesis it is willing to tolerate.
Outstanding Questions.
How is REV7 activated? Does it utilize a templating mechanism analogous to its HORMA cousin MAD2? Or does posttranslational modification play a larger role in REV7 activation?
What is/are the endogenous stimuli that determine REV7 activation and inactivation?
What are the other functions of REV7 aside from Polζ and the Shieldin complex? What is the function of the REV7-CAMP-POGZ complex?
Is REV7 activity used to coordinately control TLS and DSB repair in a physiological context?
Does REV7 interact with the Shieldin complex and Polζ simultaneously in order to coordinate repair at stalled replication forks?
Acknowledgments
We apologize to those whose work we were unable to acknowledge due to space constraints. We thank Prabha Sarangi for critically reading the manuscript and for many helpful discussions. This work was supported by grants from the U.S. National Institutes of Health (R01HL052725, P01HL048546), the U.S. Department of Defense (BC151331P1), the Breast Cancer Research Foundation, the Ludwig Center at Harvard, and the Smith Family Foundation to A.D.D.
Glossary
- HORMA
A protein family named for founding members HOP1, REV7 and MAD2. HORMA proteins have a common tertiary structure and regulatory mechanism. HORMA proteins adopt two stable conformations known as closed and open which are active and inactive, respectively.
- TRIP13-p31m
TRIP13 is a AAA+ ATPase that acts specifically on HORMA family proteins. TRIP13 unwinds target proteins to promote their conformational transition from closed to open, thereby inactivating them. p31 is an adaptor protein that links TRIP13 with its substrates.
- Translesion Synthesis (TLS)
A process used during DNA replication where cells bypass a damaged segment of DNA by using specialized polymerases. TLS is often more mutagenic than normal DNA replication and is therefore highly restricted under normal circumstances. TLS is one of several pathways that can be used to handle DNA damage during replication, along with replication fork reversal and re-priming. Pathway choice at damaged replication forks is not fully understood, but there is a clear role for ubiquitination of the replicative clamp, PCNA, in determining pathway usage.
- DNA Polymerase ζ
A translesion polymerase consisting of the large catalytic subunit REV3 and the small subunit REV7. REV7 can only form the Polymerase ζ complex in its closed, active conformation. REV7 promotes the activity of Polymerase ζ by linking the catalytic subunit, REV3, to the upstream recruitment module, REV1.
- DSB Repair
The process through which cells repair double strand breaks in DNA. Our cells possess several mechanisms to repair DSBs including homologous recombination and non-homologous end joining. The choice between DSB repair pathways is highly complex and essential for the maintenance of genomic stability.
- Homologous Recombination (HR)
A double strand break repair pathway that uses the sister chromatid as a template to ensure high-fidelity repair.
- DNA End Resection
The first step in homologous recombination repair. Nucleases act on the ends of the double strand break to generate long, single-stranded 3’-overhangs. These overhangs are then coated by the recombinase, RAD51, which promotes homology search and annealing to the complementary strand in the sister chromatid. End resection is considered a decisive point is DSB repair pathway choice as it commits repair to homologous recombination.
- Non-homologous End Joining (NHEJ)
A double strand break repair pathway that ligates blunt or minimally processed DNA ends. Resected DNA ends are not compatible with NHEJ.
- Shieldin
A complex consisting of SHLD3, REV7, SHLD2 and SHLD1 which acts downstream of 53BP1 to inhibit DNA end resection. Closed, active REV7 links the upstream SHLD3 recruitment module to the effector module, SHLD2/SHLD1. The Shieldin complex cannot form without closed REV7.
Footnotes
Declaration of Interests
A.D.D. is a consultant/advisory board member for Lilly Oncology, Merck-EMD Serono, Cyteir Therapeutics, Ideaya Inc., and Cedilla Therapeutics Inc., and is a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir.
References
- 1.Lawrence CW et al. (1985) REV7, a new gene concerned with UV mutagenesis in yeast. Mol Gen Genet 200 (1), 80–5. [DOI] [PubMed] [Google Scholar]
- 2.Murakumo Y et al. (2000) A human REV7 homolog that interacts with the polymerase zeta catalytic subunit hREV3 and the spindle assembly checkpoint protein hMAD2. J Biol Chem 275 (6), 4391–7. [DOI] [PubMed] [Google Scholar]
- 3.Nelson JR et al. (1996) Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science 272 (5268), 1646–9. [DOI] [PubMed] [Google Scholar]
- 4.Boersma V et al. (2015) MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 521 (7553), 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu G et al. (2015) REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521 (7553), 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bluteau D et al. (2016) Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 126 (9), 3580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravind L and Koonin EV (1998) The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23 (8), 284–6. [DOI] [PubMed] [Google Scholar]
- 8.Luo X et al. (2000) Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol 7 (3), 224–9. [DOI] [PubMed] [Google Scholar]
- 9.Hara K et al. (2010) Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem 285 (16), 12299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim Y et al. (2014) The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Dev Cell 31 (4), 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang M et al. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131 (4), 744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki SW et al. (2015) Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A 112 (11), 3350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki H et al. (2015) Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struct Mol Biol 22 (7), 572–80. [DOI] [PubMed] [Google Scholar]
- 14.Ye Q et al. (2020) HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol Cell 77 (4), 709–722 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg SC and Corbett KD (2015) The multifaceted roles of the HORMA domain in cellular signaling. J Cell Biol 211 (4), 745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo X et al. (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11 (4), 338–45. [DOI] [PubMed] [Google Scholar]
- 17.West AMV et al. (2018) Conformational dynamics of the Hop1 HORMA domain reveal a common mechanism with the spindle checkpoint protein Mad2. Nucleic Acids Res 46 (1), 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clairmont CS et al. (2020) TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol 22 (1), 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eytan E et al. (2014) Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet). Proc Natl Acad Sci U S A 111 (33), 12019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarangi P et al. (2020) p31(comet) promotes homologous recombination by inactivating REV7 through the TRIP13 ATPase. Proc Natl Acad Sci U S A 117 (43), 26795–26803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balboni M et al. (2020) COMET Functions as a PCH2 Cofactor in Regulating the HORMA Domain Protein ASY1. Curr Biol 30 (21), 4113–4127 e6. [DOI] [PubMed] [Google Scholar]
- 22.Xie W et al. (2021) Molecular mechanisms of assembly and TRIP13-mediated remodeling of the human Shieldin complex. Proc Natl Acad Sci U S A 118 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomida J et al. (2015) REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res 43 (2), 1000–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghezraoui H et al. (2018) 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560 (7716), 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta R et al. (2018) DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173 (4), 972–988 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang L et al. (2020) Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat Commun 11 (1), 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh G et al. (2011) CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J 30 (1), 130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hara K et al. (2017) Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment-maintaining phosphoprotein (CAMP). J Biol Chem 292 (43), 17658–17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J and Fang G (2001) MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev 15 (14), 1765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Listovsky T and Sale JE (2013) Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J Cell Biol 203 (1), 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pernicone N et al. (2020) CDH1 binds MAD2L2 in a Rev1-like pattern. Biochem Biophys Res Commun 531 (4), 566–572. [DOI] [PubMed] [Google Scholar]
- 32.Sironi L et al. (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J 20 (22), 6371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H (2006) Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J Cell Biol 173 (2), 153–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Antoni A et al. (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15 (3), 214–25. [DOI] [PubMed] [Google Scholar]
- 35.Rizzo AA et al. (2018) Rev7 dimerization is important for assembly and function of the Rev1/Polzeta translesion synthesis complex. Proc Natl Acad Sci U S A 115 (35), E8191–E8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wassmann K et al. (2003) Mad2 phosphorylation regulates its association with Mad1 and the APC/C. EMBO J 22 (4), 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim S et al. (2010) Phosphorylation of the spindle checkpoint protein Mad2 regulates its conformational transition. Proc Natl Acad Sci U S A 107 (46), 19772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tissier A et al. (2000) poliota, a remarkably error-prone human DNA polymerase. Genes Dev 14 (13), 1642–50. [PMC free article] [PubMed] [Google Scholar]
- 39.Ohashi E et al. (2000) Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J Biol Chem 275 (50), 39678–84. [DOI] [PubMed] [Google Scholar]
- 40.Diaz M et al. (2003) Decreased frequency and highly aberrant spectrum of ultraviolet-induced mutations in the hprt gene of mouse fibroblasts expressing antisense RNA to DNA polymerase zeta. Mol Cancer Res 1 (11), 836–47. [PubMed] [Google Scholar]
- 41.Gibbs PE et al. (2000) The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc Natl Acad Sci U S A 97 (8), 4186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair DT et al. (2005) Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309 (5744), 2219–22. [DOI] [PubMed] [Google Scholar]
- 43.Martin SK and Wood RD (2019) DNA polymerase zeta in DNA replication and repair. Nucleic Acids Res 47 (16), 8348–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malik R et al. (2020) Structure and mechanism of B-family DNA polymerase zeta specialized for translesion DNA synthesis. Nat Struct Mol Biol 27 (10), 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pustovalova Y et al. (2016) Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polzeta Suggests a Mechanism of Polymerase Exchange upon Rev1/Polzeta-Dependent Translesion Synthesis. Biochemistry 55 (13), 2043–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoege C et al. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419 (6903), 135–41. [DOI] [PubMed] [Google Scholar]
- 47.Stelter P and Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425 (6954), 188–91. [DOI] [PubMed] [Google Scholar]
- 48.Guo C et al. (2006) REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell 23 (2), 265–71. [DOI] [PubMed] [Google Scholar]
- 49.Guo C et al. (2006) Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol 26 (23), 8892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ross AL et al. (2005) Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res 33 (4), 1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmutz V et al. (2010) Role of the ubiquitin-binding domain of Poleta in Rad18-independent translesion DNA synthesis in human cell extracts. Nucleic Acids Res 38 (19), 6456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendel A et al. (2011) PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet 7 (9), e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edmunds CE et al. (2008) PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30 (4), 519–29. [DOI] [PubMed] [Google Scholar]
- 54.Mirchandani KD et al. (2008) The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair (Amst) 7 (6), 902–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Budzowska M et al. (2015) Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J 34 (14), 1971–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H et al. (2012) Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol 19 (2), 164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guilliam TA and Yeeles JTP (2020) Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat Struct Mol Biol 27 (5), 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo C et al. (2003) Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J 22 (24), 6621–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Washington MT et al. (2000) Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. Proc Natl Acad Sci U S A 97 (7), 3094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haracska L et al. (2000) Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase eta. Nat Genet 25 (4), 458–61. [DOI] [PubMed] [Google Scholar]
- 61.Wittschieben J et al. (2000) Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol 10 (19), 1217–20. [DOI] [PubMed] [Google Scholar]
- 62.Bianchi J et al. (2013) PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52 (4), 566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Gomez S et al. (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52 (4), 541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mouron S et al. (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20 (12), 1383–9. [DOI] [PubMed] [Google Scholar]
- 65.Findlay S et al. (2018) SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 37 (18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noordermeer SM et al. (2018) The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560 (7716), 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomida J et al. (2018) FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J 37 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao S et al. (2018) An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat Commun 9 (1), 3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dev H et al. (2018) Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20 (8), 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai Y et al. (2020) Structural basis for shieldin complex subunit 3-mediated recruitment of the checkpoint protein REV7 during DNA double-strand break repair. J Biol Chem 295 (1), 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ceccaldi R et al. (2016) Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol 26 (1), 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chapman JR et al. (2012) BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 125 (Pt 15), 3529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mirman Z et al. (2018) 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560 (7716), 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aylon Y et al. (2004) The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23 (24), 4868–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ira G et al. (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 (7011), 1011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karanam K et al. (2012) Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47 (2), 320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simonetta M et al. (2018) H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle 17 (1), 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mirman Z and de Lange T (2020) 53BP1: a DSB escort. Genes Dev 34 (1–2), 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Callen E et al. (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153 (6), 1266–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Callen E et al. (2020) 53BP1 Enforces Distinct Pre- and Post-resection Blocks on Homologous Recombination. Mol Cell 77 (1), 26–38 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vermeulen M et al. (2010) Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142 (6), 967–80. [DOI] [PubMed] [Google Scholar]
- 82.Nozawa RS et al. (2010) Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol 12 (7), 719–27. [DOI] [PubMed] [Google Scholar]
- 83.Daugaard M et al. (2012) LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol 19 (8), 803–10. [DOI] [PubMed] [Google Scholar]
- 84.Baude A et al. (2016) Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res 44 (5), 2214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maurer-Stroh S et al. (2003) The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28 (2), 69–74. [DOI] [PubMed] [Google Scholar]
- 86.Tromer EC et al. (2019) Mosaic origin of the eukaryotic kinetochore. Proc Natl Acad Sci U S A 116 (26), 12873–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takeuchi R et al. (2004) Purification of Drosophila DNA polymerase zeta by REV1 protein-affinity chromatography. Biochem J 382 (Pt 2), 535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi S et al. (2005) Roles of Arabidopsis AtREV1 and AtREV7 in translesion synthesis. Plant Physiol 138 (2), 870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Willis NA et al. (2014) BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510 (7506), 556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Willis NA et al. (2018) Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS Genet 14 (7), e1007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schlacher K et al. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145 (4), 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ray Chaudhuri A et al. (2016) Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535 (7612), 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmid JA et al. (2018) Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol Cell 71 (6), 897–910 e8. [DOI] [PubMed] [Google Scholar]
- 94.Her J et al. (2018) 53BP1 Mediates ATR-Chk1 Signaling and Protects Replication Forks under Conditions of Replication Stress. Mol Cell Biol 38 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olivieri M et al. (2020) A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182 (2), 481–496 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huttlin EL et al. (2021) Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sedgwick GG et al. (2016) Conformation-specific anti-Mad2 monoclonal antibodies for the dissection of checkpoint signaling. MAbs 8 (4), 689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]