

Abstract
Neuronal ceroid Lipofuscinosis (NCL), inherited disorders of lysosomal storage disorders, constitute the most common progressive encephalopathies with an incidence of 1.3 to 7 in 100,000 live births. We reported clinical, electrophysiological, radiological, ultrastructural, and molecular genetic features of NCL. This is a retrospective review, in a tertiary care center from January 2016 to December 2019. All children with clinical features of NCL and confirmed by pathogenic mutation and/or enzyme assay were included. A total of 60 children (male:female = 3:1) were studied. The commonest type was CLN 2 (41.7%). Neuroregression, seizures, and ataxia were present in all cases. Retinal arterial attenuation was seen in 38.33% cases. Magnetic resonance imaging (MRI) brain was abnormal in all patients, thalamic and caudate nucleus atrophy common in CLN1 (62%). Electroencephalography was abnormal in all children, but photoparoxysmal response at low intermittent photic stimulation frequencies was seen in four children of CLN2. Electron microscopy done in 43 children revealed abnormal inclusions in 20 (46.52%) children. Enzyme study showed low levels in 36 (78%) out of 46 cases. Of these, 21 had low tripeptidyl peptidase and 15 had low palmitoyl protein thioesterase levels. Molecular testing done in 26 cases showed pathogenic variant in 23 (88%) cases. Infantile onset with thalamic atrophy on MRI is common in CLN1 and refractory epilepsy, visual impairment and specific EEG changes are common in CLN2. These features are helpful in selecting enzyme assay for CLN1 versus CLN2. Electron microscopy helped in the diagnosis and genetic testing in subtyping. Thus, a multimode approach played a role in the diagnosis of NCL.
Keywords: cerebellar atrophy, CLN genetic loci, involuntary movements, myoclonic epilepsy, neuronal ceroid lipofuscinoses, neuroregression
Introduction
Neuronal ceroid lipofuscinosis (NCL) is a group of autosomal recessive inherited lysosomal neurodegenerative diseases characterized by the intracellular accumulation of fluorescent lipopigments, ceroid, and lipofuscin that causes progressive neurologic degeneration. 1 The clinical course includes progressive dementia, seizures, movement abnormalities, and progressive visual failure. Incidence ranges from 1.3 to 7 in 100,000 live births and varies among geographic ethnic regions. 1 The term “NCL” was coined by Zeman et al to distinguish the familial cerebromacular degeneration clinically and pathologically from gangliosidosis. 2 Based on age at onset, six basic subtypes, consisting of congenital (CLN10), infantile (CLN1), late infantile (CLN2), variant late infantile (CLN5, CLN6, CLN7, and CLN8), juvenile (CLN3), and adult (CLN4 or Kufs disease) has been reported. 3 However, there is variation in age of onset and disease progression among the various groups posing difficulty in categorizing to a particular subtype. In the recent past, with the advent of molecular diagnosis and identification of causative genes, a novel NCL nomenclature has been proposed taking into consideration the mutation with key clinical and pathological features. Thus, in the novel nomenclature, the gene mutated denotes the specific subtype, followed by the clinical presentation based on symptomatology, age of onset, and EM findings, (e.g., CLN2 disease and late infantile type). 4 Currently, 14 NCL gene loci are identified.
The NCL is often diagnosed late due to lack of awareness and clinical features are not specific. Over the past decade, there have been improvements in the understanding of the underlying pathophysiology of NCLs, development of reliable animal models, and therapeutic success in these animal models. In April 2017, Food and Drug administration approved cerliponase alfa recombinant human tripeptidyl peptidase (TPP)1 for the treatment of NCL2, which is an innovative treatment available for this rare disease. 5 Hence, early diagnosis is important. The NCLs share common clinical features such as epilepsy, neuro-regression, visual impairment, and premature death. It is imperative to gain adequate information of various subtypes, clinical, and laboratory parameters as treatments become available. Hence, this study was planned.
Methods
This is a retrospective review of all cases diagnosed with NCL in a tertiary care center, Bengaluru, India from January 2016 to December 2019. All children with clinical features of NCL and confirmed gene mutation and/or enzyme assay were included. Detailed clinical history, examination, and investigations which include MRI of brain, skin biopsy, ophthalmological examination, enzyme assay, and molecular analysis were taken from case records in all patients.
For enzyme assay, the whole blood samples (4 mL) were collected in sodium heparin and subjected to hemolysis by the treatment with a hypotonic solution to obtain a leukocyte pellet. The leukocyte pellet is sonicated under the ice to make a protein homogenate. The protein content of this homogenate was measured by bicinchoninic acid assay where in the protein-based reduction of Cu +2 to Cu +1 in the alkaline solution is measured. For TPP assay, the protein homogenate at 6× dilution was incubated with 100 µL 0.25 mM Ala-Ala-Phe 7-amido-4-methylcoumarin substrate in 75 mM sodium acetate buffer, pH 4.0 for one hour at 37°C. The reaction is quenched by addition of 150 µL of 0.2 M sodium phosphate/0.1 M citrate buffer, pH 4.5. Tritonx-100 was present in both the buffers at a concentration of 0.025% w/v to serve as surfactant. Using the standard 7-amido-4-methylcoumarin fluorescence as a reference, the fluorescence was measured at 440 nm and accordingly enzyme activity is expressed and activity less than 10% was taken as abnormal. For palmitoyl protein thioesterase assay, the protein homogenate (10 µL) was incubated with 20 µL substrate (0.64 mM methylumbelliferyl-6-thio-palmitoyl-β-D-glucoside, including β glucosidase, triton X 100 + DTT/BSA in 0.2 M sodium phosphate/0.1 M citrate buffer, pH 4.0) for one hour at 37°C. The reaction is quenched by the addition of 200 µL carbonate buffer (0.5 M sodium carbonate buffer, pH 10.7 with 0.025% triton x-100). Enzyme activity was calculated based on a calibration curve of methylumbelliferone standard and activity less than 10% was taken as abnormal.
Genetic testing was done by targeted gene sequencing and confirmed by Sanger sequencing. DNA extracted from blood was used to perform targeted gene capture using a custom capture kit. The libraries were sequenced to mean >80 to 100× coverage on illumina sequencing platform as per the manufacturer protocol. Sequences obtained were aligned to the human reference genome (GRCh37/hg19), QC, data mapping, variant calling, and annotation of variants with external and internal data sources were achieved with a customized genome Analysis Toolkit framework. Gene/variant annotation was achieved using the variant effect predictor program 6 against the ensemble release 91-human gene model. 7 Each transcript listed, the analyzed region includes coding exons and ± 10 base pair of flanking intronic region on both sides of each exon. Clinically, relevant mutations were annotated using published variants in literature and a set of diseases databases— ClinVar, OMIM (updated on November 21, 2018), GWAS, HGMD (v2018.3), and SwissVar. 8 9 10 11 12 Common variants are filtered based on allele frequency in 1000Genome Phase 3, ExAC (v1.0), gnomAD (v2.1), EVS, dbSNP (v151), 1000 Japanese Genome, and Indian population database. 13 14 15 16 Nonsynonymous variants effect was calculated using multiple algorithms such as PolyPhen-2, SIFT, MutationTaster2, and LRT. Only nonsynonymous and splice site variants found in the targeted gene capture were used for clinical interpretation and synonymous, and deep intronic variants without being reported elsewhere were not considered for the analysis. QIAGEN Ingenuity Variant Analysis was used to identify variants which are relevant to the clinical indication and were classified as per the American College of Medical Genetics and Genomics (ACMG) guidelines. 17 The variants were classified based on the standard guidelines of ACMG/Association for Molecular Pathology, and are scored based on the evidence and strength of each criteria as described in the ACMG guidelines. The detailed description of ACMG criteria for classifying pathogenic variants are mentioned in the supplementary document.
Based on the clinical, enzyme, and molecular analysis, the patients were categorized into various subtypes. The study was approved by the institutional ethical committee.
Results
A total of 60 patients with diagnosis of NCL were analyzed. There was male preponderance of 68.33%. Out of 60 patients, enzyme assay was done in 46 patients, mutation analysis was done in 26 patients, and electron microscopy in 43. The various subtypes and age of presentation is depicted in Table 1 . Detailed clinical features and laboratory findings are mentioned in Table 2 . Consanguineous marriage was noted in 80% with a positive family history of siblings affected or death due to similar complaints in seven. History of seizures and neuroregression, ataxia was present in all patients. Fig. 1 shows retinal arterial attenuation in a child having CLN2.
Table 1. Various subtypes and age of presentation of neuronal ceroid lipofuscinoses.
| CLN types | Number of children n = 60 (%) | Mean age of presentation | Range of presentation |
|---|---|---|---|
| CLN 1 | 21 (35) | 9.9 months | 2 months to 3.4 years |
| CLN 2 | 25 (42) | 2 years 10 months | 0 to 7 years |
| CLN 3 | 01 (02) | 7 years | 7 years |
| CLN 5 | 01 (02) | 7 years | 7 years |
| CLN 6 | 05 (08) | 4 years 2 months | 3 to 5 years |
| CLN 7 | 03 (05) | 2 years 3 months | 2 to 7 years |
| CLN 8 | 03 (05) | 3 years 2 months | 18 months to 4 years |
| CLN 11 | 01 (02) | 13 years | 13 years |
Table 2. Various clinical and laboratory features of neuronal ceroid lipofuscinoses.
| Parameter | CLN1 n = 21 (35%) |
CLN2 n = 21 (41.66%) |
CLN 6 n = 5 (8%) |
CLN7 n = 3 (5%) |
CLN8 n = 3 (5%) |
|---|---|---|---|---|---|
| Mean age of presentations | 9.9 months | 2 years 10 months | 4 years 2 months | 2 years 3 months | 3 years 2 months |
| Developmental delay | 12 (57) | 16 (64) | 3 (60) | 2 (66) | 2 (66) |
| Regression | 21 (100) | 25 (100) | 5 (100) | 3 (100) | 3 (100) |
| Abnormal eye findings | 15 (71) | 23 (92) | 2 (40) | 3 (100) | 2 (66) |
| Epilepsy | 21 (100) | 25 (100) | 5 (100) | 3 (100) | 3 (100) |
| Myoclonic epilepsy | 18 (86) | 25 (100) | 5 (100) | 3 (100) | 3 (100) |
| Involuntary movements | 21 (100) | 25 (100) | 5 (100) | 3 (100) | 3 (100) |
| Electroencephalography | |||||
| PPR at low IPS frequencies of 1 to 3 Hz | 0 | 04 (16) | 0 | 0 | 0 |
| Periodic discharges | 0 | 1 | 0 | 0 | 1 |
| Multifocal epilepsy | 13 (21) | 20(33) | 4(6) | 2(3) | 2(3) |
| Magnetic resonance imaging of brain | |||||
| Thalamus | 10 (47) | 2 (8) | 0 | 0 | 01 (33) |
| Caudate | 13 (62) | 2 (8) | 0 | 0 | 0 |
| Cerebellar and cerebral atrophy | 21 (100) | 25 (100) | 5 (100) | 3 (100) | 3 (100) |
| Electron microscopy done in | 16 | 22 | 4 | 0 | 1 |
| Abnormal | 6 (37) | 10 (45) | 2 (50) | 1 (100) | |
| Curvilinear inclusions | 4 | 8 | 0 | 1 | |
| GRODS | 1 | 2 | 2 | ||
| Curvilinear + GRODS | 1 | 0 | 0 | ||
| Enzyme assay done | 20 | 26 | 5 | 1 | 3 |
| Low levels of enzyme | 15 | 21 | Normal | Normal | Normal |
| Pathogenic variant by genetic testing | 4 | 5 | 4 | 3 | 3 |
Abbreviations: GRODS, granular round osmiophilic deposits; IPS, intermittent photic stimulation; PPR, photoparoxysmal response.
Fig. 1.
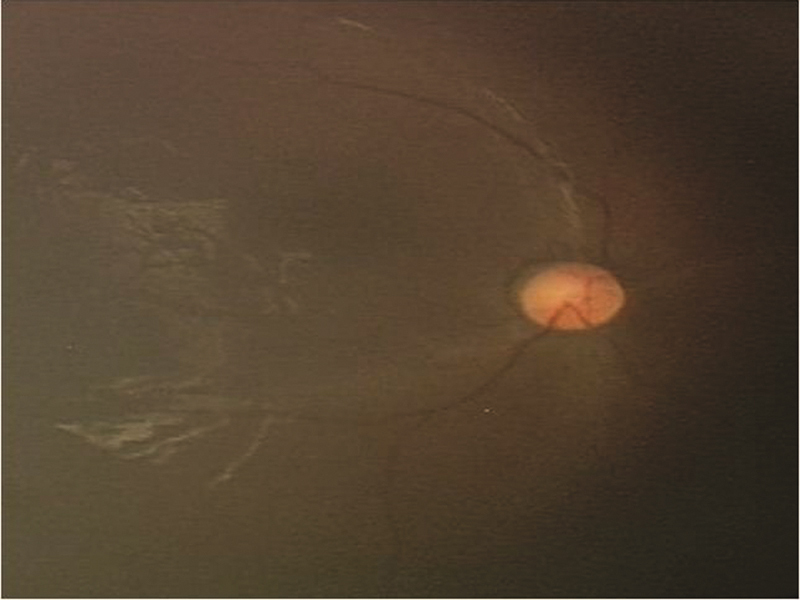
Showing of fundus of CLN2 child with retinal arterial attenuation.
Neuroimaging (MRI brain) revealed cerebral and cerebellar atrophy in all patients. It is noteworthy that two children with family history of similar complaints and early death whose initial brain MRI was normal, later developed cerebellar atrophy. Fig. 2 shows MRI of the brain, with thalamic, cerebral, and cerebellar atrophy with periventricular white matter signal changes in a child with CLN1. EEG was abnormal in all children. EEG showed early photosensitivity in the form of photoparoxysmal response (PPR) at low intermittent photic stimulation (IPS) frequencies of 1 to 3 Hz in four children of CLN2 subtype.
Fig. 2.
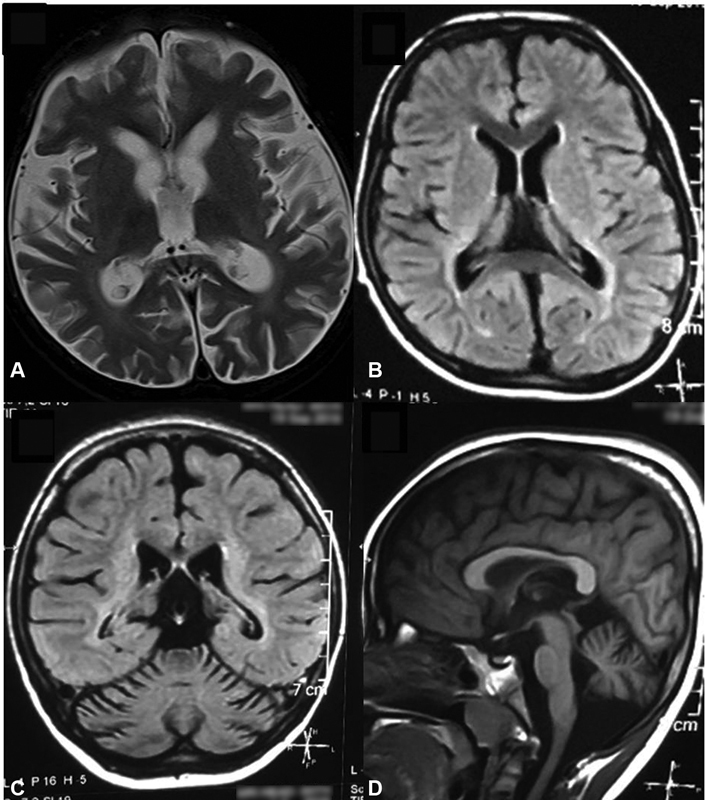
Magnetic resonance imaging of brain showing. ( A ) T1W and ( B ) FLAIR thalamic and cerebral atrophy with signal changes in periventricular white matter and posterior limb of internal capsule. ( C, D ) Cerebellar atrophy in coronal and sagittal sections, respectively.
Axillary skin biopsy done in 43 patients, revealed curvilinear inclusions in 13 (65%), five (18.51%) had granular round osmiophilic deposits (GRODS), one (5%) had both curvilinear and GRODS, and one (5%) had curvilinear plus fingerprint inclusions. The inclusions in different subtypes of NCL are GRODS seen in five cases of CLN1, curvilinear in 13 cases of CLN2, curvilinear and GROD in one case of CLN2, and curvilinear and fingerprint in one case of CLN3. Fig. 3 shows Electron micrograph picture of curvilinear inclusions, membrane-bound fingerprint, and curvilinear profiles and GRODS.
Fig. 3.
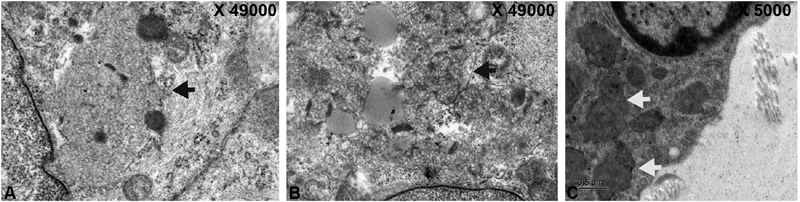
Electronmicrograph showing. ( A ) Curvilinear Inclusions within the myoepithelium of the gland (black arrow). ( B ) Membrane bound fingerprint and curvilinear profiles (black arrow). ( C ) Granular round osmiophilic deposits (white arrows).
One case each in CLN3, CLN5, and CLN11 were noted. All of them had seizures, regression, and ataxia. CLN3, a 5-year-old male child, had visual impairment and multifocal epilepsy on EEG. CLN5, 7-year-old male child with retinal artery attenuation and retinal pigmentary changes, generalized epileptiform discharges on EEG. CLN11, 14-year-old female child myoclonic jerks since the age of 13 year, and multifocal epileptiform discharges on EEG.
Enzyme study done in 46 patients had low enzyme levels in 36 (78%). While low TPP level was noted in 21, and low palmitoyl protein thioesterase (PPT) level was seen in 15. Genomic DNA from twenty-six patients analyzed by NGS showed pathogenic variant in 23 ( Table 3 ). The pathogenic variants identified include five each in TPP1 and CLN6, four in PPT1, three in each MFSD8 and CLN 8, one each in CLN3, CLN5 and GRN gene. Six novel pathogenic variants noted, are mentioned in Table 3 . Two children with CLN2 mutation succumbed due to refractory epilepsy and aspiration pneumonia.
Table 3. Various pathogenic variants in different subtypes of neuronal ceroid lipofuscinosis.
| Patients (P) | Gene | Location | Variant | Zygosity | NCL Subtype | Classification (ACMG) |
|---|---|---|---|---|---|---|
| P1 | CLN3 | Exon 6 | NM_001042432.2:c.388G > A (V130I p.Val130Ile) | HOM | NCL3 | VUS |
| P2 | CLN5 | Exon3 | ENST00000377453.3:c.634G > A (p.Ala163Thr) | HOM | NCL5 | VUS |
| P3 | CLN6 | Exon 4 | NM_017882.3:c.476C > T (p.Pro159Leu) | HOM | NCL6 | VUS |
| P4 | Intron 6 | NM_017882.3:c.665 + 1G > T | HOM | NCL6 | P | |
| P5–P6 | Exon 7 | NM_017882.3:c.679G > A (p.Glu227Lys) | HOM | NCL6 | LP | |
| P7 | Exon 7 | NM_017882.3:c.775G > C (p.Gly259Arg) | HOM | NCL6 | LP | |
| P8–P9 | CLN8 | Exon 2 | NM_018941.4:c.522C > G (p.Cys174Trp) | HOM | NCL8 | LP |
| P10 | Exon 3 | NM_018941.4:c.592delA (p.Ile198PhefsTer10) | CH | NCL8 | P | |
| P10 | Exon 3 | NM_018941.4:c.610C > T (p.Arg204Cys) | CH | NCL8 | P | |
| P11 | GRN | Exon 9 | NM_002087.3:c.912G > A (p.Trp304Ter) | HOM | NCL11 | P |
| P12 | MFSD8 | Intron 2 | NM_152778.3:c.63-1G > A | HOM | NCL7 | P |
| P13 | Exon 5 | NM_152778.3:c.268G > C (p.Ala90Pro) | HOM | NCL7 | VUS | |
| P14 | Exon 10 | NM_152778.3:c.894T > G (p.Tyr298Ter) | HOM | NCL7 | P | |
| P15–P18 | PPT1 | Exon 7 | NM_000310.4:c.713C > T (p.Pro238Leu) | HOM | NCL1 | LP |
| P19 | TPP1 | Exon 1 | NM_000391.4:c.2T > C (p.Met1Thr) | HOM | NCL2 | P |
| P20 | Exon 8 | NM_000391.4:c.1015C > T (p.Arg339Trp) | HOM | NCL2 | P | |
| P21–P22 | Exon 9 | NM_000391.4:c.1080C > A (p.Asp360Glu) | HOM | NCL2 | LP | |
| P23 | Exon 12 | NM_000391.4:c.1544T > G (p.Leu515Arg) | HOM | NCL2 | LP |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; CH, compound heterozygous; HOM, homozygous; LP, likely pathogenic; NCL, neuronal ceroid lipofuscinosis; P, pathogenic; PPT, palmitoyl protein thioesterase; TPP, tripeptidyl peptidase; VUS, variant of unknown significance.
Three children of CLN2 with Epilepsia partialis continua (EPC) required intensive care treatment for three-week durations. One child with CLN2 was initially treated as autistic spectrum disorder. One child who presented initially with ataxia, subsequently with hyperammonemic encephalopathy with sodium valproate and treated as metabolic disorder was confirmed as CLN2. Encephalopathy improved with discontinuation of valproate. All NCL children required more than two anti-epileptic drugs to control seizures, except three cases in CLN1 group who were controlled well with sodium valproate alone. Antenatal diagnosis was offered for six families of which 4 families had affected fetus hence medical termination of pregnancy was advised.
Discussion
NCL is a rare autosomal recessive lysosomal storage disorder characterized by refractory seizures, progressive loss of vision and developmental delay or regression. 18 NCL is more common in males. In this study male constitutes 41 (68.33%) of cases, similar to that reported by other studies. 19 20
CLN1 usually presents between 6-months to 2-years of age, however it also has late onset variants, such as late infantile, juvenile, and adult onset variants. The plausible reason for late onset presentation is compound heterozygotes for a typically “severe” mutation and a “mild” mutation. All children ( n = 21) with infantile onset classified as CLN1 type had age at onset less than 2 years, except in two children who had onset after 2-years of age. However, we did not notice any compound heterozygous mutations in CLN1 gene.
The common presentation was developmental delay, ataxia, and regression of milestones, which was found in all patients in this study. The ophthalmological examination revealed attenuation of retinal vessels in 38.33%, retinal pigmentary changes in 18.33%, optic disc pallor in 18.33%, macular dystrophy in 15%, optic atrophy in 10%, and bull's eye maculopathy in 1.66%.
An important feature on MRI brain was cerebral and cerebellar atrophy seen in all 60 children. In addition, small thalamus, and cerebellar dentate nucleus hyperintensities were noted as reported in other studies. 21 22 Thalamic and caudate nucleus abnormalities are more common in CLN1 compared with CLN2. Autti et al emphasized that a decrease in T2 signal intensity in the thalami might be a sign of lysosomal storage disease. 23 It is noteworthy that two children with family history of similar complaints and early death whose initial brain MRI was normal later developed cerebellar atrophy.
EEG was abnormal in all. The most common abnormality seen in 72% was multifocal epilepsy with disorganised background. Characteristic finding of early photosensitivity in the form of PPR at low intermittent photic stimulation (IPS) frequencies of 1–3 Hz was seen in 4/25 (16%) children of CLN2 subtype. In a study using a standardized IPS method, Specchio et al, 24 reported PPR to be the hallmark feature of early CLN2 disease in most patients (93%). This finding contradicts with previous studies reporting that PPR is not a feature in all CLN2 patients 25 26 including the present study. The probable reason could be that these studies failed to use standardized EEG/IPS procedures and did not collect a comprehensive data, and likely that the phenomenon was present but not captured. When this occurs in a child presenting with any type of seizure, and particularly accompanied by delayed speech and/or ataxia, or MRI abnormalities (posterior white matter signal alteration or cerebellar atrophy) are present, a diagnosis of CLN2 should be considered. Ana Christina et al 20 showed that disorganized background activity was the earliest change in EEG in NCL in their study.
Skin biopsy done in 43 patients as part of diagnostic procedure subjected to ultrastructural study was abnormal in 20 patients (46.52%). GRODS was noted in 25%, curvilinear deposits in 65%, combined curvilinear and fingerprint inclusions in one (5%) and curvilinear combined with GRODS in one patient (5%). In a study by Ana Christina et al, 20 it was seen that skin biopsies showed osmiophilic granular deposits with fingerprint profiles. Curvilinear inclusions are more common in CLN2. However, biopsy when positive suggests NCL but does not always differentiate between types of NCL and negative biopsy does not rule out disease. 27
Epilepsy is less common in CLN1 as compared with CLN2. MRI shows thalamic atrophy in CLN1 in comparison to only cerebellar and cerebral atrophy in other subgroups. None of CLN1 cases showed PPR in LF PS in EEG. EM in this group showed GROD in six children. All children in CLN2 presented with refractory epilepsy. Three children had Epilepsia partialis continua. One child earlier was treated as a case of autistic spectrum disorders. Involuntary movements were more common in CLN2. EM showed curvilinear bodies (CL), in 8 children in CLN2. Above findings help in selecting cases to perform an enzyme assay before genetic testing. CLN3 usually presents with progressive visual loss followed by neurological involvement. EM shows presence of FP or FP/ CL/GROD.
Of the 46 patients subjected for enzymatic assay, 21 patients had low levels of the enzyme tripeptidyl peptidase (TPP), while 15 had low levels of PPT. Genetic analysis showed pathogenic variants in five each in TPP1 and CLN6 , four in PPT1 , three each in MFSD8 and CLN 8 , and one each in CLN3 , CLN5 , and GRN gene. In this series, we reported six novel pathogenic variants. Kousi et al, 4 in their update, reported 365 NCL causing mutations in 14 genes. In an analysis of 34 patients from India, Sheth et al 28 reported 12 CLN1 and 22 CLN2 cases. There is complex correlation between phenotypes and genotype. Mutations in CLN5 , CLN6 , and CLN8 can mimic CLN2 phenotype. CLN8 genotype can cause severe CLN2 phenotype or mild progressive epilepsy with mental retardation.
We detected six novel pathogenic variants in this study. Pathogenic variant in TPP1 gene at Ex12:c.1544T > G/p.L515R, presented with encephalopathy due to hyperammonamia following valproate administration. Second pathogenic mutation in TPP1 gene at Ex3:c.1080C > A/p.Asp360Glu presented with autistic regression. Pathogenic variant in CLN5 at Ex3:c.634G > A/p.Ala212Thr, had classical phenotype, is the first case of CLN5 reporting from India. Pathogenic variant in MFSD8, In2:c.63–1G > A/3′ splice site had small thalamus and periodic complexes in the EEG not been described previously. Pathogenic variant in CLN6, at In6:c.665 + 1G > G/5′split site and pathogenic variant in CLN8 at Ex2:c.522C > G/p.Cys174Trp had classical phenotype.
In the present study, five children initially suspected to be having CLN2 were later turned out be CLN1 positives, following enzyme and genetic testing. Similarly, two children with initial clinical diagnosis of CLN1 turned out to be CLN2 after enzyme and genetic confirmation. One child in CLN2 in targeted exome sequencing did not reveal any pathogenic variant. Electron microscopy done in 43 showed evidence of inclusions in twenty (46.52%). Targeted exome sequencing was done in 26. Pathogenic variant was noted in 23 (88.46%). Enzyme level was low in all children those confirmed CLN1 and CLN2 subtypes by genetic testing. Three children who could not be clinically subtyped nor enzyme assay performed was sent for NGS. The results are CLN2 in two and CLN1 in one child. Two children clinically suspected with CLN2 tested for TPP levels showed normal level; however, molecular testing revealed CLN8 subtype. Thus, a multimodal investigative approach played a role in the diagnosis of NCL.
EM though helpful in confirmation of diagnosis cannot be used as a tool for subtyping CLN. It is useful when diagnosis is not clear before considering enzyme analysis and genetic testing. In the present study enzyme analysis, in younger age group proved to be useful noninvasive technique for CLN1 and CLN2 genetic testing is better for prognostication and antenatal diagnosis. In older children, progressive course of myoclonic epilepsy is unclear; hence, EM can be considered for confirmation followed by targeted genetic testing for subtyping. We provided algorithm in Fig. 4 for diagnosis of NCL in resource poor setting.
Fig. 4.
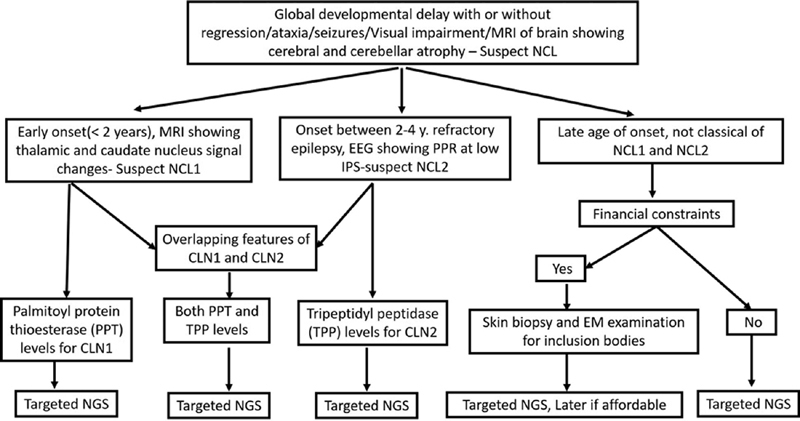
Diagnostic algorithm for diagnosis of neuronal ceroid lipofuscinoses in resource poor setting.
NCL is known to present with cognitive regression, refractory epilepsy, ataxia, visual impairment, cerebral and cerebellar atrophy, and photosensitivity to low-frequency photic stimulation in EEG. Our study added the following findings to the existing literature. NCL can present as developmental delay without regression. It can also present as autistic regression, EPC, and chorea-athetosis. MRI of brain can be normal in the initial stages of the disease. Multifocal epileptiform discharges are most common EEG abnormalities compared with photosensitivity to low frequency photic stimulation reported in the literature. We also noted periodic complexes in CLN8 subtype of NCL. CLN5 subtype is the first case reporting from India. We have also found six novel pathogenic variants of NCL.
To conclude, this was the largest cohort subtyping NCL in pediatric age group using multimodal investigative approach in a resource limited center. Our study identified six novel pathogenic variants. Diagnosis of NCL should be considered in children with global developmental delay with or without regression of milestones, involuntary movements, and refractory seizures. Thalamic and caudate nucleus changes with cerebellar atrophy on MRI brain in correlation with clinical features help in suspecting CLN1 compared with other subtypes. Refractory epilepsy and EEG showing PPR at low IPS frequencies of 1 to 3 Hz are suggestive of CLN2. Enzyme assay should be considered in clinically suspected CLN1 and CLN2 particularly in younger age group. Genetic studies are useful to subtype, for prognostication, and genetic counselling, especially for non CLN1 and CLN2 as there no biochemical tests available currently. Genetic testing is the key toward diagnosis of all cases of NCL, enzyme testing and EM are useful in the resource limited settings.
Conflict of Interest None declared.
Authors' Contributions
V.K.G. dedicated in supervision, guidance, and reviewing the manuscript. H.V. and K.S. were involved in the management of the child and the preparation of the manuscript. V.M.S. supported in the collection of data and the preparation of the manuscript. G.N. and R.S. were involved in diagnosis and final manuscript preparation. M.B., S.B., and M.R.N. provided valuable inputs in the diagnosis and final manuscript preparation.
References
- 1.Glykys J, Sims K B. Edinburgh: Elsevier saunders; 2018. The neuronal ceroid lipofuscinosis disorders; pp. 390–404. [Google Scholar]
- 2.Zeman W, Donanne S, Dyken P, Green J. Amsterdam: NorthHolland Publishers; 1970. The NCL (Batten-Vogt syndrome) pp. 588–679. [Google Scholar]
- 3.Mole S E, Williams R E, Goebel H H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6(03):107–126. doi: 10.1007/s10048-005-0218-3. [DOI] [PubMed] [Google Scholar]
- 4.Kousi M, Lehesjoki A-E, Mole S E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33(01):42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- 5.Lewis G, Morrill A M, Conway-Allen S L, Kim B. Review of cerliponase alfa: recombinant human enzyme replacement therapy for late-infantile neuronal ceroid lipofuscinosis type 2. J Child Neurol. 2020;35(05):348–353. doi: 10.1177/0883073819895694. [DOI] [PubMed] [Google Scholar]
- 6.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zerbino D R, Achuthan P, Akanni W.Ensembl 2018 Nucleic Acids Res 201846(D1):D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landrum M J, Lee J M, Benson M.ClinVar: public archive of interpretations of clinically relevant variants Nucleic Acids Res 201644(D1):D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKusick V A. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007;80(04):588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welter D, MacArthur J, Morales J.The NHGRI GWAS Catalog, a curated resource of SNP-trait associations Nucleic Acids Res 201442(Database issue):D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stenson P D, Mort M, Ball E V. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(06):665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mottaz A, David F P, Veuthey A L, Yip Y L. Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics. 2010;26(06):851–852. doi: 10.1093/bioinformatics/btq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.1000 Genomes Project Consortium Auton A, Brooks L D, Durbin R M.A global reference for human genetic variation Nature 2015526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Exome Aggregation Consortium Lek M, Karczewski K J, Minikel E V.Analysis of protein-coding genetic variation in 60,706 humans Nature 2016536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ToMMo Japanese Reference Panel Project . Nagasaki M, Yasuda J, Katsuoka F. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018. doi: 10.1038/ncomms9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherry S T, Ward M H, Kholodov M. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(01):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamate M, Prashanth G P, Hattiholi V. Clinico-investigative profile of infantile and late-infantile neuronal ceroid lipofuscinoses. Neurol India. 2012;60(03):316–320. doi: 10.4103/0028-3886.98524. [DOI] [PubMed] [Google Scholar]
- 19.Sinha S, Satishchandra P, Santosh V, Gayatri N, Shankar S K. Neuronal ceroid lipofuscinosis: a clinicopathological study. Seizure. 2004;13(04):235–240. doi: 10.1016/S1059-1311(03)00163-8. [DOI] [PubMed] [Google Scholar]
- 20.Puga A CS, Jardim L B, Chimelli L, De Souza C F, Clivati M.Neuronal ceroid lipofuscinoses: a clinical and morphological study of 17 patients from southern Brazil Arq Neuropsiquiatr 200058(3A):597–606. [DOI] [PubMed] [Google Scholar]
- 21.Kamate M, Hattiholi V. Novel neuroimaging finding in PPT1-Related neuronal ceroid lipofuscinosis. Pediatr Neurol. 2012;46:325–328. doi: 10.1016/j.pediatrneurol.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 22.Jadav R H, Sinha S, Yasha T C. Magnetic resonance imaging in neuronal ceroid lipofuscinosis and its subtypes. Neuroradiol J. 2012;25(06):755–761. doi: 10.1177/197140091202500616. [DOI] [PubMed] [Google Scholar]
- 23.Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology. 2007;49(07):571–578. doi: 10.1007/s00234-007-0220-6. [DOI] [PubMed] [Google Scholar]
- 24.Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. 2017;58(08):1380–1388. doi: 10.1111/epi.13820. [DOI] [PubMed] [Google Scholar]
- 25.Pérez-Poyato M S, Marfa M P, Abizanda I F. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28(04):470–478. doi: 10.1177/0883073812448459. [DOI] [PubMed] [Google Scholar]
- 26.Jadav R H, Sinha S, Yasha T C. Clinical, electrophysiological, imaging, and ultrastructural description in 68 patients with neuronal ceroid lipofuscinoses and its subtypes. Pediatr Neurol. 2014;50(01):85–95. doi: 10.1016/j.pediatrneurol.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Acharya J N, Satishchandra P, Shankar S K. PME and related disorders, a clinical and pathological appraisal. Neurol India. 1992;40:145–153. [Google Scholar]
- 28.Sheth J, Mistri M, Bhavsar R. Batten disease: biochemical and molecular characterization revealing novel PPT1 and TPP1 gene mutations in Indian patients. BMC Neurol. 2018;18(01):203. doi: 10.1186/s12883-018-1206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]