

Abstract
Seminoma is the most common subtype of testicular germ cell tumor, with an increasing incidence worldwide. Clusterin (CLU) expression was found to be downregulated in testicular seminoma in our previous study. We now expanded the sample size, and further indicated that CLU expression correlates with tumor stage. Tcam-2 cell line was used to investigate the CLU function in testicular seminoma, and CLU was found to inhibit the proliferation and metastasis abilities. Besides, extracellular matrix protein COL15a1 was demonstrated as the downstream of CLU to affect the epithelial-mesenchymal transition (EMT) process via competitively binding to DDR1 with COL1A1 and inhibiting the phosphorylation of PYK2. MEF2A was found to interact with CLU and bind to the promoter of COL15a1 and so upregulate its expression. This is the first study using testicular xenografts in situ to simulate testicular seminoma metastatic and proliferative capacities. In conclusion, CLU acts as a tumor suppressor to inhibit the metastasis of testicular seminoma by interacting with MEF2A to upregulate COL15a1 and blocking the EMT process.
Keywords: clusterin, seminoma, EMT, COL15a1, MEF2A, testicular xenografts in situ
Graphical abstract
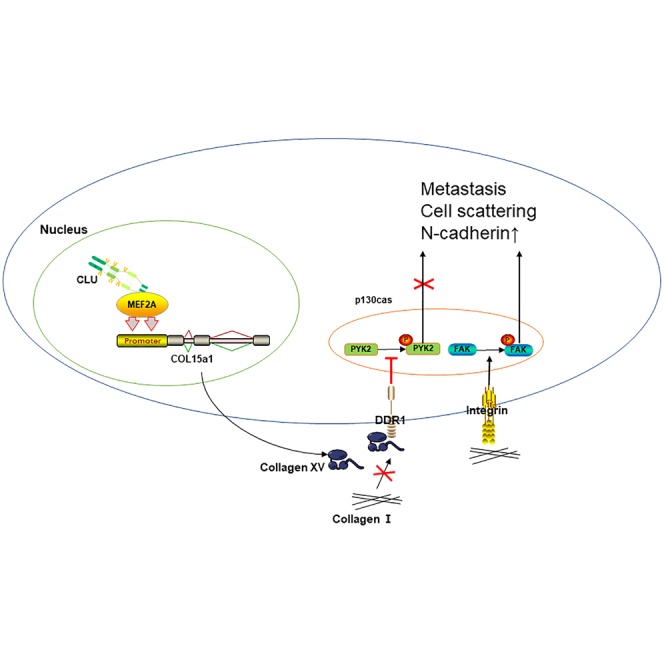
They used testicular xenograft in situ for the first time to prove the tumor suppressor role of clusterin (CLU) in testicular seminoma. Briefly, CLU could bind to the translation factor MEF2A and regulate its ability to promote COL15a1's expression. Eventually, CLU achieves the inhibition effect of tumor metastases.
Introduction
Testicular germ cell tumor (TGCT), which is mostly emerged in developed countries, has a low but increasing incidence.1 However, it is also the commonest solid tumor in men aged 15–44 years, predominantly manifesting as seminoma in 60% of patients.2 In the United States, 9,610 new cases are estimated in 2020, sharply increased from the 8,430 cases in 2015.3 Despite its increase in incidence, TGCT has seen a mortality greatly decreased by modern surgery combined with radiotherapy or chemotherapy.4,5 The cure rate of stage I disease approaches 100% in developed countries.6 About 30% of patients are diagnosed with metastatic disease.7 Even as TGCT metastasizes, the cure rates for poor-, intermediate-, and good-risk disease are approximately 48%,8 70%,9 and 80%,7,10 respectively. However, chemotherapy resistance or TGCT relapse makes some patients highly untreatable. The largest study showed an overall relapse rate of 16.8% in unselected patients.11 Hence, it is of great urgency to discover specific therapeutic targets.
Clusterin (CLU), widely present in cerebrospinal fluid, milk, blood, urine, and semen,10,12 plays an important role in many pathophysiological processes, such as carcinogenesis, atherosclerosis, neurodegeneration, and tissue damage.12 Besides, recent studies have revealed that CLU is upregulated in most stressed conditions and related to numerous tumor-associated signaling networks, such as epithelial-mesenchymal transition (EMT), metastasis, tumor progression, therapy resistance, and programmed cell death.13 On the contrary, there were also some studies showing that the expression of CLU in tumors of different genetic origins is lower than in corresponding normal tissues.14,15 In non-small cell lung cancer, CLU was found to be less expressed in tumor tissues than in normal tissues. Regarding the mechanism, CLU was reported to block the formation of TRAF6/TAB2/TAK1 complex via inhibiting TGFBR1 and thus inhibiting activation of the TAK1-NF-κB axis.16 These findings lead us to speculate that CLU may function in seminoma progression.
In our previous study, CLU was demonstrated to be downregulated in testicular seminoma.17,18 In this study, we report that CLU is a potent and clinically relevant tumor-suppressive gene in testicular seminoma. CLU inhibits seminoma's metastasis and proliferation in vivo and in vitro. Mechanistically, CLU upregulates the expression of collagen type XV alpha 1 (COL15a1) via interacting with myocyte enhancer factor 2A (MEF2A), which can bind to the promoter of COL15a1, and thus COL15a1 competitively binds to the discoidin domain receptor tyrosine kinase 1(DDR1) with collagen I (COL1A1) to block the phosphorylation of proline-rich tyrosine kinase 2 (PYK2) and inhibit the EMT process. Our finding is expected to present a novel potential therapeutic target for seminoma.
Results
CLU is lowly expressed in TGCT
The data downloaded from Oncomine were grouped and are shown in Figure 1A. The expression of CLU was lower in TGCT than in normal testis, and the lowest in seminoma compared with other subtypes of TGCT especially normal testis (p < 0.0001). To further verify the expression difference, we collected seminoma tissues and normal testes to perform polymerase chain reaction analysis, and seminoma tissues and adjacent non-tumor tissues to perform western blot and immunohistochemistry (IHC) analysis. As shown in Figures 1B, 1C, and 1D, CLU expression was lower in seminoma tissues than in normal testicular or adjacent non-tumor tissues. For the analysis of correlation between CLU expression and clinicopathological features, including age, tumor-node-metastasis (TNM) stage, and tumor size, we graded the CLU expression in 50 pieces of seminoma tissue according to the IHC staining. As shown in Table 1, lower expression of CLU was significantly associated with TNM stage (p = 0.036), but not with age (p = 0.770) and tumor size (p = 0.139).
Figure 1.

CLU was significantly downregulated in seminoma according to database and clinical tissues
(A) Data downloaded from Oncomine were redrawn by GraphPad software. (B) qRT-PCR analyses were performed with 38 seminoma tissues and 12 normal testes to explore the mRNA expression of CLU. (C) Western blot analyses were performed with 10 pairs of seminoma tumor tissues and adjacent normal tissues to explore the protein expression of CLU. (D) IHC analyses were further used to identify the expression difference. Scale bar: 100 μm. (E) Representative stains of seminoma tissues by IHC, 1 (negative), 2 (weak brown), 3 (moderate brown), 4 (strong brown). Data are represented as mean ± SD. Scale bar: 100 μm, ∗∗∗∗p < 0.0001.
Table 1.
Correlations between CLU expression and clinicopathological features in 50 seminoma patients
| Characteristics | Case | CLU expression |
p value |
|
|---|---|---|---|---|
| Low | High | |||
| All cases | 50 | 37 | 13 | |
| Age (years) | 0.770 | |||
| <30 | 19 | 15 | 4 | |
| ≥30 | 31 | 22 | 9 | |
| TNM | 0.036a | |||
| T1 | 33 | 28 | 5 | |
| T2–4 | 17 | 9 | 8 | |
| Tumor size (cm) | 0.139 | |||
| ≤4 | 28 | 23 | 5 | |
| >4 | 22 | 14 | 8 | |
p < 0.05.
CLU weakens the proliferation ability in vitro and in vivo
Because CLU expression was decreased in seminoma samples, we first tested whether CLU had an effect on the proliferation of seminoma cells. Knockdown and overexpression of CLU in Tcam-2 cells were respectively confirmed by PCR and western blot assays (Figure 2A). According to the cell counting kit-8 (CCK-8) assay, we found that CLU-overexpression cells (oeCLU) retarded proliferation compared with the negative control (oe-NC) group. Meanwhile, decreased expression of CLU (shCLU-1 and -2) accelerated cell proliferation (Figure 2B). Additionally, colony numbers of CLU-overexpressing cells were significantly lower than those of the oe-NC group (p < 0.05; Figure 2C). Knocking down CLU significantly enhanced colony formation compared with that in the sh-NC group (p < 0.001, Figure 2C).
Figure 2.

CLU curbed Tcam-2 cells proliferation in vitro and in vivo through arresting cell cycle in G1 phase
(A) Stable CLU-knockdown and -overexpression cell lines were established successfully. (B) The optical density (OD) values detected in the CCK-8 proliferation assay were performed after 1–4 days seeding, and higher OD values indicate higher cell viability. (C) Colony formation was photographed 2 weeks after seeding and quantified by ImageJ software. (D) Xenografts in mice in vivo and their ki-67 staining (E), volumes (F), and weights (G). (H) Flow cytometric cell cycle analysis of shCLU, oeCLU, and NC cells showed the percentage of cells in G1 or G2+S phase. Data are represented as mean ± SD. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scale bar: 100 μm.
To explore the effects of CLU on tumorigenesis and tumor progression in vivo, xenograft models were successfully established with nude mice (Figures 2D, 2F, and 2G). Mice that received subcutaneous injection of shCLU cells developed tumors with significantly larger volume and weight than those injected with negative control (NC) cells after 5 weeks. On the contrary, the tumors were markedly smaller and lighter in oeCLU group compared with those in the NC group. The IHC staining of these tumors showed that more positive expression of Ki-67 could be observed in the shCLU group (Figure 2E).
Given the essential role of CLU in cell proliferation, we performed flow cytometry assays to evaluate the cell cycle distribution and cell apoptosis in cells with CLU overexpression and knockdown. As shown in Figure 2H, the knockdown of CLU increased the percentage of cells in G2+S phase, thereby inducing cell cycle arrest at G2+S phase and enhancing cell division. However, for CLU-overexpression cells, the cell cycle was arrested at G1 phase.
These findings suggest that CLU weakened the proliferation in vitro and in vivo via arresting cell cycle at G1 phase.
CLU inhibits invasive, migrative, and wound healing abilities of Tcam-2 cells
The results of global seminoma patients' follow-up showed that the 5-year overall survival (OS) of non-pulmonary visceral metastases or mediastinal primary metastases was 72%, but, for non-metastatic TGCT, the OS rose to 93%.19 Therefore, to explore the relationship between CLU expression and Tcam-2 cells' metastatic ability, we performed wound healing and transwell assays. The CLU-knockdown cells showed stronger healing ability than NC cells (Figure 3A; p < 0.05). On the contrary, elevated CLU expression retarded the wound healing (p < 0.001). For invasive and migrative abilities, transwell experiments were performed with or without Matrigel, respectively. As shown in Figure 3B, both shCLU-1 and shCLU-2 cells exhibited obvious enhanced migrative and invasive capabilities (p < 0.01). However, for CLU overexpression, these abilities reduced significantly (p < 0.01).
Figure 3.

CLU knockdown enhanced the invasion, migration, and wound healing of Tcam-2 cells
(A) Photographs at 0 and 12 h after scratching. (B) Transwell assays of CLU-knockdown and -overexpression cells. The migration assays were performed without Matrigel and photographed 12 h after seeding. The invasion assays were performed with Matrigel and photographed 48 h after seeding. Data are represented as mean ± SD. Scale bar: 20 μm. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
These results revealed that CLU could inhibit the invasion and migration of Tcam-2 cells.
CLU affects extracellular matrix organization
Given that CLU exhibited a lower expression in seminoma tissue and inhibited the proliferation, invasion, and migration abilities of Tcam-2 cells, we subsequently performed RNA sequencing (RNA-seq) with CLU overexpression and NC cells. Gene Ontology (GO) analysis showed that extracellular matrix (ECM) organization was significantly enriched (Figure 4A). To further verify the RNA-seq results, we conducted gene set enrichment analysis (GSEA) analysis based on data from The Cancer Genome Atlas (TCGA) (Figure 4B). The enrichment results were basically consistent with RNA-seq results, and the ECM was also the top enriched pathway. Venn diagram (Figure 4C) showed three significantly upregulated genes (VCAN, COL15a1, COL7a1, p < 0.05, fold-change [FC] ≥1.5) screened out by overlapping ECM gene lists of GO (21 genes) and GSEA analyses (115 genes). Furthermore, Spearman's correlation analysis was performed in the database: Gene Expression Profiling Interactive Analysis (GEPIA) to calculate the correlation between ECM genes (VCAN, COL7A1, and COL15a1) and CLU (Figure 4D) based on TGCT samples. qRT-PCR assays were performed to verify the database results with our 50 seminoma tissues and cell lines, respectively (Figures 4E and 4F; VCAN, R = 0.6691; COL15a1, R = 0.7125; COL7A1, R = 0.6297). These ECM genes' mRNA expression was also increased in oeCLU cells.
Figure 4.

The expression of ECM-associated COL15A1 and COL7A1 was significantly changed with CLU knockdown or overexpression
(A) The most enriched GO terms based on the RNA-seq data of oeCLU and oe-NC groups. (B) Data downloaded from TCGA were submitted to GSEA and extracellular matrix organization was also significantly enriched in samples with higher CLU expression. (C) 21 and 115 upregulated ECM-associated genes from GO and GSEA terms were overlapped. COL15a1, COL7A1, and VCAN were screened out. (D) The co-expression of CLU with COL15a1, COL7A1, and VCAN based on the database: GEPIA. (E) The verification of co-expression relationship with 50 seminoma tissues. (F and G) mRNA and protein expression of COL15a1, COL7A1, and VCAN in pretreated Tcam-2 cells. Data are represented as mean ± SD. ∗p < 0.05.
We then explored the protein level expression of these ECM genes with CLU expression changed. For COL15a1 and COL7a1, lower protein expression can be found in CLU-knockdown cells and higher expression in CLU-overexpression cells (p < 0.05). However, the same results cannot be observed in the expression of VCAN. We speculated that the post-translational modification of VCAN had made the difference in its protein and transcription levels.
EMT is significantly activated in CLU-knockdown cells
After reviewing literature, we know that ECM genes participate extensively in the EMT process. High levels of COLXV could suppress endogenous levels of N-Cadherin (N-Cad) and inhibit EMT progression in pancreatic adenocarcinoma cells.20 Loss of Col VII was found to increase EMT in squamous cell carcinoma.21 Hence, we hypothesized that EMT induced by COL15a1 or COL7A1 absence could damage CLU-knockdown cells and lead to metastasis. The signals of EMT-related markers (including E-cad, N-cad, Vimentin, β-catenin, and MMP3) were detected with western blot assay. As shown in Figure 5A, E-cad was lowly expressed in CLU-knockdown cells but highly expressed in CLU-overexpression cells. Conversely, N-cad, Vimentin, β-catenin, and MMP3 showed higher expression in CLU-knockdown and lower expression in CLU-overexpression cells.
Figure 5.

EMT pathway was significantly inhibited in CLU-overexpression cells
(A) Western analysis of EMT markers (E-cad, N-cad, Vimentin, β-catenin, and MMP3) in CLU-knockdown and -overexpression cells. (B) 3D spheroid invasion of oeCLU, shCLU, and NC cells 24 h and 72 h after Matrigel addition. The aggressive cells are marked with red arrow. Data are represented as mean ± SD. Scale bar: 100 μm. ∗∗∗∗p < 0.0001.
Given that the ECM genes played an essential role in CLU-regulated EMT, we further performed a three-dimensional (3D) spheroid invasion experiment to simulate cancer cell invasion in vivo (Figure 5B). Cultured in the same manner, NC cells were more invasive at 72 h compared with those at 24 h after the addition of Matrigel. CLU-overexpression cells showed no obvious invasion having been cultured for 3 days. However, for CLU-knockdown cells, the invasion capacity was significantly augmented.
COL15a1 but not COL7a1 acts downstream of CLU to affect the invasion ability
To find out which ECM gene is the key regulator of the signal axis by which CLU affects the EMT process, COL15a1- and COL7a1-knockdown cells were established, respectively. Transwell and western blot assays were performed next. As shown in Figure 6A, the knockdown of COL15a1 (shCOL15a1) could significantly rescue the inhibition of cells' invasive ability (p < 0.001). Meanwhile, E-cad expression was decreased. Vimentin and N-cad expressions were increased in oeCLU + shCOL15a1 cells compared with cells overexpressed CLU only (p < 0.0001). However, the knockdown of COL7a1(shCOL7a1) cannot recover the invasive ability, and the invasion-related proteins (N-cad and Vimentin) had no tendency to increase (Figures 6C and 6D).
Figure 6.

COL15a1 competitively binds to the DDR1 with collagen I to block the phosphorylation of PYK2 and so inhibit the EMT process
(A) Tcam-2 cells (EV) and Tcam-2 cells with high level of CLU (oeCLU) were transfected with NC and shCOL15a1 lentivirus. The invasion abilities of pretreated cells were determined by the transwell assay. (B) The expression of indicated proteins was detected by western blot. (C) COL7a1 was also downregulated as above methods, and the transwell assays were followed subsequently. (D) Western blot assays were performed to detect the indicated proteins level. (E) Pretreated cells were coated or non-coated with collagen I. The cell aggregation indicated their scattering ability. (F) The downstream markers of collagen I (pPYK2, pFAK) and collagen XV (pPYK2) were detected by western blot. (G) In vivo imaging of testicular xenografts in situ injected with four groups of pretreated cells. Metastasis occurred in the lung, medial iliac, mediastinal, and inguinal lymph nodes. The number of metastatic foci and the tumor volumes were recorded. (H) Photographs and H&E staining of testicular xenografts in situ. (I) The protein markers of EMT (E-cad, N-cad, and Vimentin) in group 2 primary tumor and group 4 pulmonary metastasis were detected by IHC. Data are represented as mean ± SD. Scale bar: 20 μm.
COL15a1 was reported to compete with COL1a1 to interact directly with DDR1 and so inhibit the phosphorylation of PYK2 (pPYK2).20 Along with the phosphorylation of FAK (pFAK), pPYK2 can promote cells' scattering and metastasis, which happen to the advantage of expression of COL1a1.22 Therefore, we used collagen Ⅰ (COL Ⅰ) to coat pretreated cells. For non-coat and COL Ⅰ coat groups, the knockdown of COL15a1 could accelerate the scattering of oeCLU cells significantly (Figure 6E). For the oeCLU + shCOL15a1 group, the cells appeared to be more dispersed when coated with COL Ⅰ. Besides, for protein expression, in non-coat and COL Ⅰ coat groups, pPYK2 expression in shCOL15a1 cells was both higher than that in just CLU-overexpression cells (Figure 6F; p < 0.0001). In the same transfected cells, COL Ⅰ coat could increase the expression of both pPYK2 and pFAK. Moreover, COL Ⅰ coat could also increase the N-cad expression.
To verify the above results in vivo, we conducted testicular xenografts in situ (Figure 6H). Compared with NC group (group 1), oeCLU group mice (group 2) presented fewer metastatic foci (Figure 6G; p < 0.01) and smaller tumor volumes (p < 0.0001). In addition, the knockdown of COL15a1 after overexpressing CLU (group 4) could increase the metastatic focus compared with mice injected into cells just overexpressing CLU (group 2, p < 0.01). However, shCOL15a1 could not affect the tumor proliferation ability. Hematoxylin-eosin (H&E) staining was further performed to validate the cells’ growth status in testes. Different from the group 1 testes, the Tcam-2 cells were sparser in group 2 testes. Moreover, IHC staining of group 4 (oeCLU + shCOL15a1) pulmonary metastasis tissues showed higher staining of Vimentin and N-cad than E-cad (Figure 6I). On the contrary, higher E-cad staining was observed in testis of group 2 mice. The in vivo study revealed that the overexpression of CLU could inhibit the invasion and proliferation ability of Tcam-2 cells in testis. Meanwhile, the knockdown of COL15a1 could rescue the inhibition of invasion, driving cells to metastasis to lymph nodes and pulmonary tissues, increasing N-cad and Vimentin expressions.
CLU upregulates COL15a1 via transcription factor MEF2A
We first used the database: Jaspar to predict the potential transcription factors (TFs) that can bind to the promoter of COL15a1, and the MEF family members MEF2A and MEF2D were found. We then performed chromatin immunoprecipitation (ChIP) assay in pretreated Tcam-2 cells (Figure 7A). The results revealed that MEF2A could bind to the COL15a1 promoter located on the −124 to −110 nt (Jaspar: MA0052.4). Besides, cells that overexpressed CLU showed an increased fold enrichment of targeted regions (p < 0.001). Luciferase reporter assays indicated that MEF2A knockdown significantly diminished the luciferase activity of MEF2A. Consistently, COL15a1 mRNA level was also relatively altered (Figure 7B), downregulated in shMEF2A groups and upregulated in oeCLU group. However, knockdown of MEF2A after overexpression of CLU would counteract the effect of CLU overexpression on COL15a1 expression, leaving no significant difference between them and the NC group.
Figure 7.

CLU interacted with MEF2A to upregulate COL15a1
(A) Jaspar database was used to estimate the potential TFs that could bind to the promoter of COL15a1, and ChIP assay was performed to prove this in oeCLU and NC groups. (B) Luciferase reporter assay was performed in shMEF2A cells to detect the luciferase activity. The mRNA expression of COL15a1 was then explored with qRT-PCR in shMEF2A, oeCLU, and oeCLU + shMEF2A groups. (C) The mutated promoter of COL15a1 was constructed. The luciferase activity of MEF2A was then detected in WT and mutant groups with cells transfected by shCLU, shMEF2A, oeCLU + shMEF2A, and NC. (D) Immunoprecipitation assay was performed with total, nucleus, and cytoplasm protein respectively to study the directly interaction between CLU and MEF2A. (E) Co-localized IF staining displayed the distribution of CLU and MEF2A. (F) The analysis of COL15a1 expression in seminoma based on UALCAN database. (G) The mechanism of CLU in inhibiting testicular seminoma metastasis.
We then constructed the mutated promoter region of COL15a1 to perform luciferase reporter assays (Figure 7C). In the wild-type (WT) group, shCLU and shMEF2A can both decrease luciferase activity. In oeCLU + shMEF2A cell line, the luciferase activity showed no significant difference with the NC group. However, the mutation of COL15a1's promoter displayed no statistical luciferase activity difference among NC, shCLU, shMEF2A, and oeCLU + shMEF2A groups. This means that the transcriptional regulation of MEF2A on COL15a1 required the participation of CLU.
Taken together, abnormal CLU in seminoma could modulate MEF2A's translational activity and promote its enrichments at the promoter sites of COL15a1, thus increasing COL15a1's expression.
To further verify the interaction between CLU and MEF2A, we performed immunoprecipitation assay with total protein, nucleus protein, and cytoplasm protein respectively. We found that the protein blot of MEF2A can be detected in the protein complexes pulled down by CLU antibody, which were performed with total protein and nucleus protein. However, this phenomenon did not occur in cytoplasm protein samples. Hence, CLU could directly interact with MEF2A in the nucleus of Tcam-2 cells (Figure 7D). Furthermore, we identified that CLU was primarily distributed in the whole-cell chamber, while MEF2A conservatively localized in cell nucleus (Figure 7E).
If COL15a1 expression is regulated by CLU, given the lowly expression of CLU in TGCT, we then analyzed COL15a1 expression in TGCT patients with the database: UALCAN23 (http://ualcan.path.uab.edu/). As expected, COL15a1 is also lowly expressed in seminoma significantly (Figure 7F). Collectively, we speculated that CLU could bind to the effector domain of MEF2A to regulate its activity and so promote COL15a1 expression (Figure 7G).
Discussion
As described above, it is easy to manage early-stage testicular seminoma with an almost 100% cure rate under surgery combined or not with radiotherapy and chemotherapy.6 However, there are also 30% of patients with metastatic disease, including relapsed and refractory patients who will not benefit from the first-line cisplatin-based chemotherapy.19,24 Due to the lack of novel targeted therapies, no alternative treatment options are available for testicular seminoma patients not being cured by current treatment strategies, highlighting the need for investigating novel molecular targeted therapies that could block the metastasis process of tumor cells.
The important role of ECM in regulating cell migration, proliferation, and apoptosis has been highlighted in recent years.25 It is now known that the ECM not only carries on continuous active remodeling but also induces biophysical and biochemical signals to affect cell migration and adhesion.26 As the most significant ECM component, collagen determines the main functional properties of matrix. Changes in the degradation or deposition of collagen can result in the loss of ECM homeostasis and so affect cancer progression.27,28 As a non-fibrillar collagen, collagen XV is found in the basement membrane (BM) zone of testicular tissue cells.29 After it was first proposed in 2003 that collagen XV might be a tumor suppressor,30 many studies have been performed to prove this point. Collagen XV functioned as a dose-dependent suppressor of tumorigenicity in an in vivo model system, using cervical carcinoma cells.31 Besides, loss of collagen XV precedes invasion and metastasis of aggressive breast tumors, melanomas, and colon carcinomas suggest this protein may have an essential role in stabilizing the ECM and so preventing distal metastasis.32, 33, 34 In our present study, COL15a1 was proved as the downstream gene of CLU to affect the EMT process. The knockdown of COL15a1 after overexpressing CLU could rescue the inhibition of Tcam-2 cells' invasion ability and lead to the increasing expression of N-cad and Vimentin. The subsequent in vivo study confirmed this point. The group 4 mice (oeCLU + shCOL15a1) showed a higher probability of metastasis to lymph node or lung than mice in group 2 (oeCLU + con). Besides, COL15a1 was reported to inhibit EMT via interacting directly with DDR1 and blocking the phosphorylation of PYK2 in pancreatic adenocarcinoma cells. This interaction could be seized by COL Ⅰ when its expression takes advantage and so induces the phosphorylation of PYK2.20 Hence, we designed the collagen Ⅰ coat experiment to investigate whether this phenomenon exists in testicular tumor cells. Interestingly, the overexpressing of CLU (similarly to overexpressing COL15a1) could inhibit the cell scattering, and the phosphorylation level of PYK2 could be elevated with the knockdown of COL15a1. On the contrary, the COL Ⅰ coat (similarly to overexpressed COL1A1) could promote Tcam-2 cells' scattering significantly. In the group of cells with knocked down COL151 and coated with COL Ⅰ, the cells were most dispersed. These results disclose the mechanism that the function of upregulating of COL15a1 in inhibiting the EMT process, which is induced by CLU.
Most eukaryotic TFs are thought to act by recruiting cofactors, including coactivators and corepressors, which have been identified as mediators of TF effector activity.35 They commonly contain domains involved in nucleosome remodeling, chromatin binding, and/or covalent modification of histones or other proteins.36 Depending on the interaction with coactivators or corepressors partners, MEF2 TFs were reported to support both pro-oncogenic and tumor-suppressive activities.37 In our study, we found that MEF2A had the potential to bind to the promoter of COL15a1. Further ChIP assay revealed that MEF2A could significantly enriched in the −124 to −110 nt of COL15a1's promoter. Besides, with the overexpression of CLU, more enriched signal could be detected. The luciferase reporter assay further confirmed that MEF2A acted as an upstream TF of COL15a1 and played a regulatory role in the expression of COL15A1. This regulation could be influenced by CLU directly. MEF2A was next found to interact with CLU by immunoprecipitation and immunofluorescence (IF) assays. These results suggested that CLU may bind to the effector domain of MEF2A to mediate its activity and so regulate the expression of COL15a1.
Testicular xenograft in situ has not been reported before. This is the first study using this xenograft technique to evaluate the metastatic and proliferative activity of pretreated tumor cells in testis. Comparing with xenograft in a subcutaneous, orthotopic tumor model can better simulate the conditions of human tumor growth, especially metastasis. In humans, testicular tumors are mainly lymph node metastases, which are common in internal iliac, common iliac, paraaortic, and mediastinal lymph nodes. Distant metastasis is common in lung.7 In our orthotopic tumor model, metastasis occurred in the lung, medial iliac, mediastinal, and inguinal lymph nodes. It is highly consistent with human testicular tumor metastasis status and has great significance for the study of testicular tumors.
Conclusions
This is the first study to discover the functional role of CLU-MEF2A-COL15a1-EMT axis in the invasion and migration of seminoma cells. Briefly, CLU interacts with the effector domain of MEF2A to regulate its activity. MEF2A can bind to the promoter of COL15a1 to mediate its expression. COL15a1 is demonstrated to combine with DDR1 and so inhibit the phosphorylation of PYK2, which was reported to be the inducing factors of EMT phenotype. Besides, the testicular xenograft model in situ was first performed to study the testicular seminoma's metastasis status in nude mice. We provide a more solid and effective model for the study of testicular tumor. Collectively, CLU may serve as a potential therapeutic target in seminoma treatment.
Materials and methods
Online analysis
The CLU expression data in different subtypes of TGCT were downloaded from the database: Oncomine (https://www.oncomine.org/). Seven subtypes were submitted to Korkola's human male germ cell tumors study (GSE3218): embryonal carcinoma (n = 15), teratoma (n = 16), choriocarcinoma (n = 2), mixed germ cell tumor (n = 44), seminoma (n = 13), yolk sac tumor (n = 10), and normal testis (n = 6).
Fragments per kilobase of transcript per million fragments mapped (FPKM)-standardized RNA-seq data from the TGCT cohort were downloaded from the database: TCGA. Subsequently, GSEA was performed. The C5 (c5.all.v7.1.symbols.gmt) from the Molecular Signatures Database (MSigDB) was used as the reference sequences. The patients were classified into high-CLU and low-CLU group. The gene set permutation was repeated 1,000 times. p value < 0.05 was set to screen the significant signaling.
The databases: Jaspar (http://jaspar.genereg.net/) and UCSC (http://genome.ucsc.edu/) were used to estimate the potential TFs. The relative profile score threshold was set as 95%.
Sample collection
TGCT samples were collected from 50 patients (average age 34 ± 4 years, range 21~43 years) from 2011 to 2020 in The First Affiliated Hospital of Nanjing Medical University. They received orchiectomy and were diagnosed with testicular seminoma by postoperative pathology. Normal testes were collected from 12 patients (average age 70.5 ± 1.9 years, range 65–81 years) receiving surgical castration to control prostate cancer progression. Pathology confirmed that the cancer did not invade the testis. Samples for IHC analysis were fixed with formalin. Samples for protein and RNA extraction were freshly frozen in liquid nitrogen and stored at −80°C. The design and protocol of this study were approved by the ethics committee of The First Affiliated Hospital of Nanjing Medical University. Informed consent was obtained from all participants.
RNA extraction, qRT-PCR, and ChIP assay
Total RNA was extracted from testis tissues and cell lines using TRIzol reagent (Invitrogen, Carlsbad, CA). The total RNA was reverse transcribed into cDNA using HiScript II (Vazyme, Shanghai, China). qRT-PCR was performed using SYBR Green I (Vazyme, Shanghai, China) on an ABI 7900 system (Applied Biosystems, Carlsbad, CA) and the primers were as listed: CLU (forward, CCAATCAGGGAAGTAAGTACGTC; reverse, CTTGCGCTCTTCGTTTGTTTT); COL15a1 (forward, GGATCATCCTCTACTACACGGAG; reverse, CCTGCATTGCCCATGAAGATT); COL7A1 (forward, ACCCAGTACCGCATCATTGTG; reverse, TCAGGCTGGAACTTCAGTGTG); VCAN (forward, GTAACCCATGCGCTACATAAAGT; reverse, GGCAAAGTAGGCATCGTTGAAA); β-actin (forward, GAAGATCAAGATCATTGCTCCT; reverse, TACTCCTGCTTGCTGATCCA).
The ChIP Assay Kit (Beyotime biotechnology, Shanghai, China) was used to perform ChIP assays according to the manufacturer's protocols. Briefly, cells were collected and purified for subsequent antibody immunoprecipitation, antibodies against MEF2A and NC immunoglobulin G (IgG) were used in the ChIP assays. ChIP DNA products were amplified with specific primer of the COL15a1 promoter (5′-TCTAATAATAGA-3′ and 5′-TACTAAAAATACAAA-3′).
Western blotting
Cell lines and testis tissues were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Biotechnology, Shanghai, China). For immunoprecipitation assay, the proteins were extracted with nuclear and cytoplasmic protein extraction kit (Beyotime Biotechnology, Shanghai, China). Proteins were harvested and quantified using the bicinchoninic acid (BCA) kit (Beyotime Biotechnology, Shanghai, China), separated on a 10% gel using sodium dodecyl sulfate (SDS)-PAGE, and transferred onto polyvinylidene fluoride (PVDF) membranes (Sigma-Aldrich, St Louis, MO). The membranes were blocked in Tris-buffered saline (TBS) containing 5% non-fat milk for 2 h. After incubation with primary antibody overnight at 4°C, the membranes were washed three times with TBS containing 0.1% Tween 20 (TBS-T). Subsequently, the membranes were incubated in a secondary antibody solution at room temperature for 2 h. After washing, the signals were detected using the chemiluminescence system and analyzed with Image Lab software. The primary antibodies used in this study: CLU (sc-6419, Santa Cruz); COL15a1(ab150463, Abcam); Ki-67 (27309-1-AP, Proteintech); COL7a1 (ab223639, Abcam); VCAN (ab19345, Abcam); β-tubulin (2146, CST); GAPDH antibody (ab9485, Abcam): Vimentin (5741, CST); N-Cadherin (13,116, CST); E-Cadherin (14,472, CST); β-Catenin (8480, CST); MMP-3 (14,351, CST); FAK (71,433, CST); Phospho-FAK (8556, CST); Pyk2 (3480, CST); Phospho-PYK2 (3291, CST); and MEF2A (sc-17785, Santa Cruz).
Cell culture and lentiviral infection
Testicular seminoma-derived Tcam-2 cell line was obtained from Department of Urology of Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, and cultured in DMEM medium (Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY) at 37°C with 5% CO2. Two different short hairpin RNA (shRNA) sequences targeting different regions of CLU (shCLU-1, shCLU-2), MEF2A (shMEF2A-1, shMEF2A-2), shCOL15a1, shCOL7a1, and oeCLU were obtained from GenePharma (Shanghai, China) along with the scramble control. Stable cell lines were generated by lentiviral infection of Tcam-2 cells overnight in media (DMEM and 10% FBS and 4 μg/mL polybrene). After infection, the cells were incubated in growth medium (DMEM and 10% FBS) and selected with 4 mg/mL puromycin. The knockdown or overexpression efficiency of each lentivirus was evaluated by western blot and qRT-PCR analysis, respectively. The cell lines with lentiviral transfection efficiency over 90% were frozen in liquid nitrogen and fresh cells were thawed for subsequent experiments.
Collagen I scatter assays were performed as described previously22 using 50 mg/mL collagen I solution from rat tails (Corning).
RNA-seq and gene expression analysis
Total RNA was extracted from three oeCLU stable cell lines groups and three corresponding control cell lines groups using TRIzol reagent (Invitrogen, United States). Primary RNA-seq was carried out by Personal (Shanghai, China). Raw sequence data were mapped to hg19 and expression values were determined as reads per kilo bases per million reads (RPKM) using RSeQC (http://rseqc.sourceforge.net/). GO analysis was performed for the RNA-seq data and the results were plotted in R 4.0.0.
Immunoprecipitation
CLU-overexpression cells were harvested in 500 μL of binding buffer (PBS containing 1 mM NaVO3, 50 mM NaF, 10 mM Na+ -pyrophosphate, 1% Triton X-100, and pH7.4). Lysates were centrifuged at 30,000 × g for 20 min and subsequently incubated with 1 μg of anti-CLU antibody overnight at 4°C. Then, 50 μL of protein G Sepharose 4B beads (Thermo Fisher Scientific, United States) was added to the mixture and incubated for an additional 4 h to capture the immune complexes. Beads were washed three times with binding buffer. Proteins were released from the beads with 50 μL of SDS loading buffer and subjected to immunoblotting.
IHC and IF
IHC staining was performed as previously described.38 Immunoreactive score of the Remmele and Stegner (IRS) system was performed by two experienced pathologists to determine the protein expression level. A final score >1 was considered as high CLU expression; otherwise, it was considered as low CLU expression.
Tcam-2 cells were plated in chamber dishes 24 h prior to the experiment. Cells on coverslips were washed three times in PBS, then fixed with 4% paraformaldehyde for 15 min. After being permeabilized with 0.5% Triton X-100 at room temperature for 20 min, cells were washed three times in PBS and blocked with 2% BSA for 30 min. Then, cells were incubated with the indicated primary and secondary antibodies at 4°C overnight or at room temperature for 2 h, respectively. The nuclei were stained with DAPI and slides were observed using a fluorescence microscope (Nikon).
Wound healing assay
The cells were seeded into six-well plates (3 × 105 cells/well) and cultured until confluence rose to 90%–100%. After being scratched by a 10-μL pipette tip, the wells were washed twice with PBS to remove cell debris and residual serum. Subsequently, the cells were cultured in DMEM without serum and incubated for an additional 12 h. The wound edges were photographed using DP-BSW software (10× objective) 12 h after injury and measured using ImageJ software.
Transwell and 3D invasion assay
Cells (1 × 105/200 μL) were plated on the upper chamber (8-μm pore size) and coated with or without 50 μL of Matrigel (BD Biosciences) in serum-free medium. DMEM with 20% FBS was added to the lower chambers as an attractant. After incubation at 37°C for 12 h for migration and 48 h for invasion, the chambers were transformed in 4% paraformaldehyde for 30 min to fix the cells that had migrated to the bottom of the membrane, and then stained with 2% crystal violet for 1 h. Non-migrating or non-invading cells were gently removed and cells having migrated toward the outer chamber were counted in five representative (200×) fields.
For 3D invasion assay, the transfected Tcam-2 cells were collected and seeded into ultra-low adhesion (ULA) 96-hole round base plate at a density of 4 × 103/200 μL and cultured for 4 days. After removing 100 μL of medium from each well using a cold pipette tip, 100 μL of Matrigel matrix (Corning) diluted to 3 mg/mL was added to the hole in the bottom of the well. The ULA 96-hole round base plate was transferred to the incubator at 37°C to solidify the Matrigel. One hour later, we added 100 μL of medium to each well. Cell masses were imaged after 24 h and 72 h to compare invasive abilities. The invasion percentage was calculated using the formula: invasion percentage = (72 h area − 24 h area)/24 h area.
Cell proliferation assay and colony formation assay
To evaluate the proliferative capacities of cells, the transfected Tcam-2 cells were seeded into 96-well plates (2 × 103 cells per well). We added CCK-8 system (Dojindo, Japan) into each well after 0, 1, 2, 3, and 4 days of culture according to the manufacturer's instructions. We read the absorbance of each well at 450 nm using a microplate reader (Tecan, Switzerland) to determine the cells' viability.
For colony formation assay, approximately 1 × 103 transfected cells were seeded into six-well dishes and cultured in DMEM medium containing 10% FBS. Two weeks later, the colonies were fixed with paraformaldehyde, stained with 0.1% crystal violet, and imaged and counted.
Cell cycle assays
For cell cycle analysis, the transfected Tcam-2 cells were collected and washed three times with cold PBS. After being fixed with 75% ethanol overnight, the cells were treated with RNAse (KeyGEN BioTECH), stained with 50 μg/mL propidium iodide, and incubated in the dark for 30 min. Cell cycle was analyzed using flow cytometry (Beckman, United States).
Luciferase reporter assays
The promoter of COL15a1 was constructed into pGL3-reporter plasmid for luciferase reporter assay. Tcam-2 cells with NC or shMEF2A were transiently transfected with COL15a1 luciferase reporter using X-tremeGENE transfection reagent (Sigma, United States). Luciferase activity was measured using a ONE-Glo luciferase assay system (Promega, United States) according to the manufacturer's instructions.
Xenografts in mice
The Tcam-2 cells were stably transfected, collected, and mixed with Matrigel 1:1. The cells were injected subcutaneously or into the testis of nude mice (male, 4–6 weeks old, five mice per group). Tumor length (L) and width (W) were monitored every week with calipers, and the tumor volume (V) was calculated using the formula: V = (L × W2)/2. Five weeks after injection, the nude mice were sacrificed and the tumor masses were isolated for weight measuring within 4 h. The metastatic focus of testicular xenografts in situ were assessed by In Vivo Imaging System. The suspicious metastatic foci were confirmed by H&E staining. The samples were fixed and processed as paraffin tissue sections. The animal studies complied with the institutional ethics guidelines and were approved by the animal management committee of Nanjing Medical University.
Statistical analysis
All the analyses were performed with GraphPad Prism 6 or SPSS 16.0 software and p value <0.05 was considered to be statistically significant. The Student's t test or chi-square test was used to analyze the difference between two groups. The correlations between genes were analyzed using the Spearman's correlation.
Acknowledgments
We thank all the participants who were involved in this study. Particularly, we are grateful for the generous offer of the Tcam-2 cell line from Professor Gonghui Li of Sir Run Run Shaw Hospital, Zhejiang University School of Medicine.
The study was supported by the Six Talent Peak Project of High-level Talents in Jiangsu Province (WSW-017); 333 High-level Talents Training Project in Jiangsu Province, Professional from Six-Pronged Top-Talent Program (LGY2018053); Qing Lan Project of Jiangsu University (JX2161015100); The Fifth Batch of Outstanding Young and Middle-aged Teachers Support Program of Nanjing Medical University; a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (JX10231802); Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_1159); and International Exchange and Cooperation Program for Postgraduates of Nanjing Medical University.
Author contributions
Y.C., C.M., and Z.W. designed the study. S.L. and J.Z. collected the data. C.L. and C.H. analyzed the data. M.B. and X.C. analyzed the IHC staining. C.M., X.C., and X.Z. searched the literature. Y.C. and J.T. edited the manuscript. B.L. acquired the funding and supervised the whole study.
Declaration of interests
The authors declare no competing interests.
References
- 1.Cedeno J.D., Light D.E., Leslie S.W. StatPearls. StatPearls Publishing; 2020. Testicular Seminoma; pp. 1–2. [Google Scholar]
- 2.Woldu S.L., Bagrodia A. Update on epidemiologic considerations and treatment trends in testicular cancer. Curr. Opin. Urol. 2018;28:440–447. doi: 10.1097/MOU.0000000000000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Bray F., Richiardi L., Ekbom A., Pukkala E., Cuninkova M., Møller H. Trends in testicular cancer incidence and mortality in 22 European countries: continuing increases in incidence and declines in mortality. Int. J. Cancer. 2006;118:3099–3111. doi: 10.1002/ijc.21747. [DOI] [PubMed] [Google Scholar]
- 5.Znaor A., Lortet-Tieulent J., Jemal A., Bray F. International variations and trends in testicular cancer incidence and mortality. Eur. Urol. 2014;65:1095–1106. doi: 10.1016/j.eururo.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Pierorazio P.M., Albers P., Black P.C., Tandstad T., Heidenreich A., Nicolai N., Nichols C. Non-risk-adapted surveillance for stage I testicular cancer: critical review and summary. Eur. Urol. 2018;73:899–907. doi: 10.1016/j.eururo.2017.12.030. [DOI] [PubMed] [Google Scholar]
- 7.Cheng L., Albers P., Berney D.M., Feldman D.R., Daugaard G., Gilligan T., Looijenga L.H.J. Testicular cancer. Nat. Rev. Dis. Primers. 2018;4:29. doi: 10.1038/s41572-018-0029-0. [DOI] [PubMed] [Google Scholar]
- 8.International germ cell consensus classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J. Clin. Oncol. 1997;15:594–603. doi: 10.1200/JCO.1997.15.2.594. [DOI] [PubMed] [Google Scholar]
- 9.de Wit R., Stoter G., Sleijfer D.T., Neijt J.P., ten Bokkel Huinink W.W., de Prijck L., Collette L., Sylvester R. Four cycles of BEP vs four cycles of VIP in patients with intermediate-prognosis metastatic testicular non-seminoma: a randomized study of the EORTC Genitourinary Tract Cancer Cooperative Group. European Organization for Research and Treatment of Cancer. Br. J. Cancer. 1998;78:828–832. doi: 10.1038/bjc.1998.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calero M., Rostagno A., Frangione B., Ghiso J. Clusterin and Alzheimer's disease. Subcell. Biochem. 2005;38:273–298. [PubMed] [Google Scholar]
- 11.Albers P., Albrecht W., Algaba F., Bokemeyer C., Cohn-Cedermark G., Fizazi K., Horwich A., Laguna M.P., Nicolai N., Oldenburg J. Guidelines on testicular cancer: 2015 update. Eur. Urol. 2015;68:1054–1068. doi: 10.1016/j.eururo.2015.07.044. [DOI] [PubMed] [Google Scholar]
- 12.Miyake H., Hara I., Gleave M.E. Antisense oligodeoxynucleotide therapy targeting clusterin gene for prostate cancer: Vancouver experience from discovery to clinic. Int. J. Urol. 2005;12:785–794. doi: 10.1111/j.1442-2042.2005.01173.x. [DOI] [PubMed] [Google Scholar]
- 13.Praharaj P.P., Patra S., Panigrahi D.P., Patra S.K., Bhutia S.K. Clusterin as modulator of carcinogenesis: a potential avenue for targeted cancer therapy. Biochim. Biophys. Acta Rev. Cancer. 2020;1875:188500. doi: 10.1016/j.bbcan.2020.188500. [DOI] [PubMed] [Google Scholar]
- 14.Blaschuk O., Burdzy K., Fritz I.B. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 1983;258:7714–7720. [PubMed] [Google Scholar]
- 15.Sala A., Bettuzzi S., Pucci S., Chayka O., Dews M., Thomas-Tikhonenko A. Regulation of CLU gene expression by oncogenes and epigenetic factors implications for tumorigenesis. Adv. Cancer Res. 2009;105:115–132. doi: 10.1016/S0065-230X(09)05007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Fan Z., Dou X., Zhou Q., Zeng G., Liu L., Chen W., Lan R., Liu W., Ru G., et al. Inactivation of tumor suppressor gene clusterin leads to hyperactivation of TAK1-NF-κB signaling axis in lung cancer cells and denotes a therapeutic opportunity. Theranostics. 2020;10:11520–11534. doi: 10.7150/thno.44829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B., Han M.T., Zhang J., Lu P., Li J., Song N., Wang Z., Yin C., Zhang W. Downregulation of clusterin expression in human testicular seminoma. Cell Physiol. Biochem. 2013;32:1117–1123. doi: 10.1159/000354511. [DOI] [PubMed] [Google Scholar]
- 18.Tang M., Li J., Liu B., Song N., Wang Z., Yin C. Clusterin expression and human testicular seminoma. Med. Hypotheses. 2013;81:635–637. doi: 10.1016/j.mehy.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Kier M.G., Lauritsen J., Mortensen M.S., Bandak M., Andersen K.K., Hansen M.K., Agerbaek M., Holm N.V., Dalton S.O., Johansen C., et al. Prognostic factors and treatment results after bleomycin, etoposide, and cisplatin in germ cell cancer: a population-based study. Eur. Urol. 2017;71:290–298. doi: 10.1016/j.eururo.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Clementz A.G., Mutolo M.J., Leir S.H., Morris K.J., Kucybala K., Harris H., Harris A. Collagen XV inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS One. 2013;8:e72250. doi: 10.1371/journal.pone.0072250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martins V.L., Vyas J.J., Chen M., Purdie K., Mein C.A., South A.P., Storey A., McGrath J.A., O'Toole E.A. Increased invasive behaviour in cutaneous squamous cell carcinoma with loss of basement-membrane type VII collagen. J. Cell Sci. 2009;122:1788–1799. doi: 10.1242/jcs.042895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shintani Y., Fukumoto Y., Chaika N., Svoboda R., Wheelock M.J., Johnson K.R. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Biol. 2008;180:1277–1289. doi: 10.1083/jcb.200708137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandrashekar D.S., Bashel B., Balasubramanya S.A.H., Creighton C.J., Ponce-Rodriguez I., Chakravarthi B., Varambally S. UALCAN: a portal for Facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adra N., Einhorn L.H. Testicular cancer update. Clin. Adv. Hematol. Oncol. 2017;15:386–396. [PubMed] [Google Scholar]
- 25.Walker C., Mojares E., Del Río Hernández A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018;19:3028. doi: 10.3390/ijms19103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geiger B., Yamada K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harbor Perspect. Biol. 2011;3:a005033. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang M., Yuan J., Peng C., Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014;35:2871–2882. doi: 10.1007/s13277-013-1511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provenzano P.P., Eliceiri K.W., Campbell J.M., Inman D.R., White J.G., Keely P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers J.C., Dion A.S., Abraham V., Amenta P.S. Type XV collagen exhibits a widespread distribution in human tissues but a distinct localization in basement membrane zones. Cell Tissue Res. 1996;286:493–505. doi: 10.1007/s004410050719. [DOI] [PubMed] [Google Scholar]
- 30.Harris H. Is collagen XV a tumor suppressor? DNA Cel. Biol. 2003;22:225–226. doi: 10.1089/104454903321908601. [DOI] [PubMed] [Google Scholar]
- 31.Harris A., Harris H., Hollingsworth M.A. Complete suppression of tumor formation by high levels of basement membrane collagen. Mol. Cancer Res. 2007;5:1241–1245. doi: 10.1158/1541-7786.MCR-07-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amenta P.S., Hadad S., Lee M.T., Barnard N., Li D., Myers J.C. Loss of types XV and XIX collagen precedes basement membrane invasion in ductal carcinoma of the female breast. J. Pathol. 2003;199:298–308. doi: 10.1002/path.1303. [DOI] [PubMed] [Google Scholar]
- 33.Amenta P.S., Briggs K., Xu K., Gamboa E., Jukkola A.F., Li D., Myers J.C. Type XV collagen in human colonic adenocarcinomas has a different distribution than other basement membrane zone proteins. Hum. Pathol. 2000;31:359–366. doi: 10.1016/s0046-8177(00)80251-8. [DOI] [PubMed] [Google Scholar]
- 34.Fukushige T., Kanekura T., Ohuchi E., Shinya T., Kanzaki T. Immunohistochemical studies comparing the localization of type XV collagen in normal human skin and skin tumors with that of type IV collagen. J. Dermatol. 2005;32:74–83. [PubMed] [Google Scholar]
- 35.Reiter F., Wienerroither S., Stark A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017;43:73–81. doi: 10.1016/j.gde.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Rhee H.S., Pugh B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Giorgio E., Franforte E., Cefalù S., Rossi S., Dei Tos A.P., Brenca M., Polano M., Maestro R., Paluvai H., Picco R., et al. The co-existence of transcriptional activator and transcriptional repressor MEF2 complexes influences tumor aggressiveness. PLoS Genet. 2017;13:e1006752. doi: 10.1371/journal.pgen.1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui Y., Miao C., Hou C., Wang Z., Liu B. Apolipoprotein C1 (APOC1): a novel diagnostic and prognostic biomarker for clear cell renal cell carcinoma. Front. Oncol. 2020;10:1436. doi: 10.3389/fonc.2020.01436. [DOI] [PMC free article] [PubMed] [Google Scholar]