

Abstract
Alzheimer’s disease (AD) is a major contributor of dementia leading to the degeneration of neurons in the brain with major symptoms like loss of memory and learning. Many evidences suggest the involvement of neuroinflammation in the pathology of AD. Cytokines including TNF-α and IL-6 are also found increasing the BACE1 activity and expression of NFκB resulting in generation of Aβ in AD brain. Following the interaction of Aβ with microglia and astrocytes, other inflammatory molecules also get translocated to the site of inflammation by chemotaxis and exaggerate neuroinflammation. Various pathways like NFκB, p38 MAPK, Akt/mTOR, caspase, nitric oxide and COX trigger microglia to release inflammatory cytokines. PPARγ agonists like pioglitazone increases the phagocytosis of Aβ and reduces inflammatory cytokine IL-1β. Celecoxib and roficoxib like selective COX-2 inhibitors also ameliorate neuroinflammation. Non-selective COX inhibitor indomethacin is also potent inhibitor of inflammatory mediators released from microglia. Mitophagy process is considered quite helpful in reducing inflammation due to microglia as it promotes the phagocytosis of over activated microglial cells and other inflammatory cells. Mitophagy induction is also beneficial in the removal of damaged mitochondria and reduction of infiltration of inflammatory molecules at the site of accumulation of the damaged mitochondria. Targeting these pathways and eventually ameliorating the activation of microglia can mitigate neuroinflammation and come out as a better therapeutic option for the treatment of Alzheimer’s disease.
Keywords: Alzheimer’s disease, Neuroinflammation, Molecular pathways, Therapeutics
Introduction
Multiple factors are involved in the development of Alzheimer’s disease which makes it difficult to find the complete cure for the disease. Biomarkers related pathologies have also been observed (Sweeney et al. 2019). The major attributes of Alzheimer’s disease are senile plaques or Aβ plaques generated by the abnormal cleavage of Amyloid Precursor Protein (APP) and neurofibrillary tangles produced by hyperphosphorylation of tau. Brain regions mostly affected by the disease are hippocampus, neocortex and amygdala. Alzheimer’s disease leads to dementia which accounts for over 131.5 million people worldwide (Sengoku 2020). Out of all dementia cases across the globe AD accounts for 90% of the whole. Behavioral and psychological symptoms are common in AD (Kales et al. 2019). It is believed that Alzheimer’s disease symptoms arise after 20 years of acquiring the disease. The nerve cells involved in thinking, memory and learning get damaged due to which symptoms like memory loss and language difficulties arise. This makes the patient unable to perform his routine works. Gradually the person becomes bedridden and ultimately leads to death of the Alzheimer’s Association (2019). Vascular diseases, age, head trauma, family history, low physical activity, environmental factors and smoking are major risk elements of the disease. Cerebrospinal fluid analysis and amyloid Positron Emission Tomography (PET) are helpful in the diagnosis of the disease. Magnetic Resonance Imaging (MRI), brain biopsy and Retinal Optical coherence tomography (OCT) imaging are other diagnostic methods among which brain biopsy is considered to be the most definitive one (Atri 2019).
Out of the multiple factors responsible for the disease neuroinflammation has a vital role (Fig. 1). Studies suggest the involvement of over-activation of microglial cells and astrocytes along with inflammatory molecules in the pathogenesis of the disease (Uddin et al. 2020). Neuroinflammation acts as double edged sword for the brain, as it clears deposited Aβ and produces cytotoxic substances which aggravate the deposition of Aβ and cause neurodegeneration. Many experiments proved high accumulation of inflammatory mediators around NFTs and amyloid plaques which makes the involvement of neuroinflammation in AD quite evident (Ahmad et al. 2019).
Fig. 1.
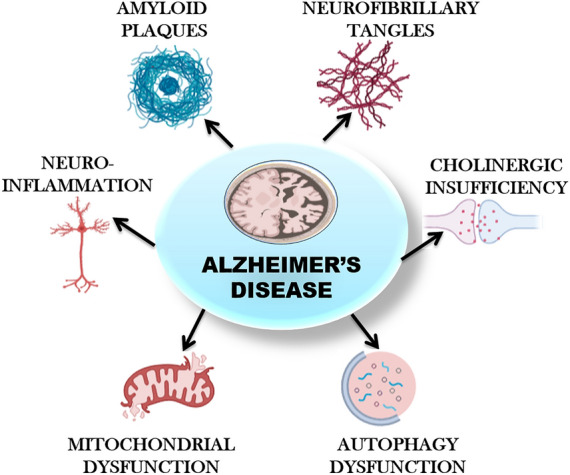
Pathophysiology of Alzheimer’s disease. Figure 1 depicts the multiple factors responsible for the progression of Alzheimer’s disease. Amyloid plaques and hyperphosphorylated tau are the major ones. Extracellular amyloid β deposition leads to the generation of senile plaques. Hyperphosphorylated tau leads to the disassembly of microtubules and damages the cytoskeleton and signal transduction processes in neuronal cells. Other factors like neuroinflammation, oxidative stress, cholinergic insufficiency, mitochondrial dysfunction and autophagy dysfunction also play major role in the disease progression
Major inflammatory cells in the CNS are microglia and astrocytes. Microglia are macrophages which cover 10% of the total population of cells in the CNS. It is involved in the neuronal plasticity and neurogenesis and acts as first line of defense in any injury of the brain. Major Cytokines found in AD brains are Tumor necrosis factor-α (TNF-α) and Interleukin-6 (IL-6). The activity of β-site APP cleaving enzyme (BACE1) and nuclear factor κB (NFκB) is increased by these cytokines resulting in increased generation of Aβ (Calsolaro and Edison 2016). Aging has profound effect in the pathogenesis of AD, it also affects innate immunity of the body. Inflammasomes are the structures responsible for the release of interleukins but during aging there is marked reduction of inflammasomes leading to reduction in cytokines (Cao and Zheng 2018). Due to the degeneration of neurons there is infiltration of inflammatory mediators, activation of innate immune system and glial cells at the accumulation site. It is believed that this occurs at initial stages of the disease and contributes in the progression of the disease (Deardorff and Grossberg 2017).
Mild activation of microglia is beneficial as it clears the cell debris, damaged neurons and promotes survival of cells in the brain. But if there is chronic inflammation of neurons the microglia and other inflammatory cells get hyperactivated which can be toxic to the neuronal cells (Domingues et al. 2017). When microglia becomes hyperactive, a protein known as Translocator protein 18 kDa (TSPO) is also highly expressed which serves as a biomarker and helps in imaging of inflammation in the brain and TSPO levels are monitored by employing PET techniques (Knezevic and Mizrahi 2018). Reduction in the infiltration of inflammatory molecules and removal of damaged mitochondria is stimulated by an innate system of the body known as mitophagy (Lautrup et al. 2019).
Neuroinflammation and AD
Inflammation is a response produced towards any injury, trauma or infection to the cells and tissues. It is believed that brain and immune system has a biochemical link. Inflammation that occurs in the brain is known as neuroinflammation. Neuroinflammation is sometimes beneficial and sometimes destructing to the neuronal cells as over-activation of the inflammatory molecules can cause damage to the cells of the brain (Fig. 2) (Shabab et al. 2017). Neuroinflammation leads to activation of inflammatory cells in the brain including microglia and astrocytes. The brain is regarded as an "immunological privileged organ" because peripheral immune cells are expected not to enter the blood–brain barrier. Rather, glial cells—microglia and astrocytes, are the primary component of the committed neuroimmune system, and interrelate with peripheral immunity but not clearly justified. The glial cells provide favorable and anti-inflammatory actions under normal and pathological conditions, counting phagocytosis, steroid release, free radical depletion, and repairing of cells. Release of cytokines and generation of free radicals can cause neuronal cell death and synaptic dysfunction. So if there is imbalance between the regulations of proinflammatory anti-inflammatory function, it can cause brain injury (Schain and Kreisl 2017).
Fig. 2.

Neuroinflammation in Alzheimer’s disease. Figure 2 shows the hyperactive glial cells including astrocytes and microglia upon interaction between Aβ and tau. Microglia and astrocytes are the major cells in the brain responsible for inflammatory responses. Due to the activation of glial cells various pro-inflammatory mediators are released which direct more inflammatory molecules at the site of injury leading to exaggerated inflammatory response in brain called neuroinflammation
Microglia and astrocytes are the two main constituents of the immune system of brain and they have a key function in the neuroinflammation process. In normal conditions microglia have a phagocytic action which removes damaged neurons thus encourages the repair of tissue at the site of invasion by foreign molecules or pathogens where the immune functions of the cells are initiated. Astrocytes remove the debris from the cerebrospinal fluid and play a neuroprotective role. In Alzheimer’s disease there is accretion of these cells around the NFTs and senile plaques. In activated state these cells release cytokines, interleukins and chemokines which act as pro-inflammatory components and potentiate the neuroinflammatory process in the brain contributing to AD (Walters et al. 2016).
ApoE is the risk element for Alzheimer disease that possesses immunomodulatory action. This capacity of ApoE is conjugated with triggering receptor expressed on myeloid cells 2 (TREM2), which is mediated by microglia in the CNS (Shi et al. 2018). Elevated levels of cytokines in the brain are closely linked with AD. The interleukins and TNF α along with Cox-2 are the major cytokines considered to be involved. Cytokines generation in the brain mediated by Aβ also promotes microglia mediated free radical generation. In general IL-1 is involved in the progression of AD which is released from neurons, astrocytes and microglia. IL-1 enhances the abnormal processing of APP and generates Aβ. There is an increased concentration of IL-1 in the brain of AD patients. Aβ is generally considered to increase the interleukin levels. In contrast, it is seen that interleukins are responsible to generate Aβ from neurons and astrocytes. IL-1 also induces the release of other cytokines and increase NOS activity which results in toxicity of neurons (Sawikr et al. 2017).
Role of inflammatory molecules in AD
Interleukins are secreted by leukocytes and activated microglia. Interleukins promote the activation of T-lymphocytes mediated toxicity and increase the activity of macrophages and neutrophils. It has various receptors including IL-6 receptors which in normal concentrations are helpful in neuronal cell survival but increased activation of IL-6 may lead to neurodegeneration. Mutations in IL-6 may potentiate the risk of AD. Other receptors are IL-1β, IL-1α, IL-9, IL-17, IL-15, IL-16, IL-12, IL-18, IL-4 and IL-10. IL-4 decreases the clearance of Aβ and thus enhances deposition of Aβ leading to the formation of senile plaques. IL-10 suppresses the activity of several pro-inflammatory mediators and thus acts as anti-inflammatory molecule. IL-18 activates the production of interferon-γ. These all somehow are related to the pathogenesis of AD either indirectly by activating inflammatory mediators leading to neuroinflammation or directly by causing neurotoxicity (Bagyinszky et al. 2017).
Neuroinflammation in AD is believed to be basically determined by microglial cells. Aβ oligomers and fibrils are equipped for preparing microglial cells through communications with different receptors, which improve the generation of provocative cytokines and chemokines [interleukin-1, interleukin-6, tumor necrosis factor α (TNF-α), C1q etc.] and make microglia progressively vulnerable to auxiliary boosts, stimulating microglial activation. In AD, conglomeration of Aβ brings about interminable stimulation of microglia, improving generation of inflammatory cytokines and chemokines, which further enhances the levels of activated microglia (Marttinen et al. 2018).
Inflammatory cytokines are released from glial cells and are of two types, pro-inflammatory and anti-inflammatory. As the name suggests pro-inflammatory cytokines like IL-1, IL-6 and TNF-α enhance inflammation by mediating microglial activation and increasing Aβ thus contributing to inflammation mediated progression of AD (Cai et al. 2014).
Chemokines are chemotactic cytokines which act as mediator of AD by stimulating chemotaxis in the brain. Chemokines are comprised of four families: CXC, CC, CX3C and C, according to the number and cysteine buildups in the N-terminal, otherwise called α, β, γ and δ chemokines, respectively. Microglia, astrocytes and neurons are the key cells responsible for the synthesis of chemokines and their receptors. Under inflammatory and diseased condition the chemokines produced by CNS are CCL2, CXCL8, CXCL10, CCL5 and CCL3. Chemokines direct microglia and astrocytes to the site of neuroinflammation aggravating local inflammation (Domingues et al. 2017).
In AD patients the Cd11b integrin was found over-expressed in neutrophils present peripherally which potentiates the adhesion and translocation of neutrophils. Some studies also suggested the increased level of free radicals in peripheral neutrophils. It was found that neutrophils are hyperactive in AD brains. Lymphocyte function associated antigen -1 (LFA-1) is an integrin present on neutrophils whose activity is increased by Aβ proteins. It is believed that the infilteration of neutrophils is highly regulated by LFA-1 and depletion in the LFA-1 levels reduces the migration of neutrophils towards the brain and this leads to decrease in the levels of phosphorylated tau and improved cognitive functions in AD patients (Stock et al. 2018).
Various pathways playing role in neuroinflammation
The toll-like receptors are responsible for the activation of NFκB pathways. It is a transcription factor specific to B-lymphocytes. Activation of NFκB initiates phagocytosis mediated by microglia, release cytokines and activates molecules which are required for the function of adaptive immune response. IκBα regulates the activity of NFκB along with its translocation in nucleus. Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α (IκBα) is an inhibitory molecule which when removed from NFκB, leads to its activation. TNF-α and IL-1β also activate the NFκB pathway. Ubiquitin inhibits NF-κB, degrades IκB, processing of precursors of NFκB and activation of IKK (Shabab et al. 2017).
In brain, NFκB signaling occurs mainly in microglia, to some extent in astrocytes and oligodendrocytes but neurons are devoid of this signaling. Nuclear factor erythroid 2-related factor-2 (Nrf2) is another transcription factor which regulates various anti-oxidant pathways. Due to inflammation in AD there occurs oxidative stress and increased activation of microglia and astrocytes which leads to increased NFκB activity and deformed activity of Nrf2. It is believed on the basis of various studies that NRf2 by increasing anti-oxidant activity can mitigate NFκB activity and hence play a neuroprotective role. Redox status of the cells regulate the expression of NFκB, which in turn is regulated by Nrf2 (Liddell 2017).
Activation of microglia also leads to the induction of other signaling pathway like PI3K/Akt pathway which is involved in apoptosis and regulation of inflammatory responses. NFκB translocation is also promoted by activation of PI3K. In turn PI3K is activated by Akt by undergoing various downstream pathways. The mammalian target of rapamycin (mTOR) also belongs to PI3 kinase family. Activation of PI3K leads to activation of Akt which subsequently activates mTOR. Microglia activation takes place as soon as there is initiation of phosphorylation of mTOR. mTOR further regulates the activity of NFκB proportionally. Increase in mTOR activation eventually increases the activity of NFκB which leads to stimulation of inducible nitric oxide synthase (iNOS) and Cox-2 (Shabab et al. 2017).
Normally inflammation in brain is associated with two types of cycloxygenase enzymes counting cox-1 and cox-2. Cox-1 is present in neurons of cerebral cortex and hippocampus whereas cox-2 is over-expressed due to inflammation in both activated microglia and neurons. Inflammatory mediators in neuroinflammation activate cox-2 which synthesizes and releases prostaglandins via arachidonic pathway and this prostaglandin further stimulates microglia. Prostaglandin E2 (PGE2) is mostly involved in microglia and macrophages and its receptors are predominantly found in microglia (Sil and Ghosh 2016).
AMP-activated protein kinase (AMPK) is believed to play an advantageous role in neuroinflammation by modulation of signal transducer and activator of transcription (STAT1) and blocking the expression of interferon γ (IFNγ), iNOS and TNF-α. AMPK also protects neurons from glutamate excitotoxicity. AMPK activators in some studies also showed attenuated NFκB activity. But the activation of AMPK is found detrimental according to some studies, therefore, the exact role of AMPK is still unclear (Peixoto et al. 2017).
Lipopolysaccharides also induce mitogen activated protein kinase (MAPK) pathway along with NFκB pathway. c-Jun N-terminal kinase (JNK), extracellular-signal-regulated kinase1/2 (ERK1/2) and p38 are the major signaling pathways underlying MAPKs. MAPK pathways are majorly involved in regulating immune response, oxidative stress and apoptosis. Anti-inflammatory molecules like neurotropin reduce the activation of p38, ERK and JNK pathways by ameliorating the phosphorylation of these, thus inhibiting the MAPK pathway which leads to induction of inflammation (Zheng et al. 2018).
Interferon γ binds to the interferon γ receptors and activates the M1 state of microglia via JAK/STAT pathway leading to its activation followed by release of various cytokines and chemokines. Phosphorylation of interferon regulatory factor-3 (IRF3) leads to the generation of interferon type 1 (IFN-β) which binds to its receptors and activates janus kinase family which further phosphorylates and activates STAT. This STAT then enters into the nucleus and activates interferon stimulation genes which results in the release of various inflammatory mediators (Thawkar and Kaur 2019).
Nitric oxide is a signaling molecule which is synthesized by three forms of nitric oxide synthase enzymes counting neuronal NOS, endothelial NOS and inducible NOS. Inducible NOS is activated only in pathological conditions of AD in brain particularly in microglia. Lipopolysaccharides, TNFα, interleukins and IFNγ are capable of stimulating iNOS. Increased levels of NO can induce nitration of several proteins in the brain leading to neuronal apoptosis. Further increase in the level of iNOS and NO the production of free radicals is also increased which significantly contributes in increasing the cytotoxic effects in the neuronal cells. Under normal levels NO is helpful in cell proliferation (Shabab et al. 2017).
Therapeutic strategies targeting neuroinflammation in AD
As pro-inflammatory cytokines and their receptors are major contributors of neuroinflammation in Alzheimer’s disease, inhibiting cytokines gene expression and blocking or binding their receptors can come out to be a better therapeutic option for treatment of AD. Antibodies are used to counteract the effects of IL1-β and TNF-α along with its receptors. Anti-inflammatory molecules like minocycline are also employed for reducing Aβ and tau pathologies by mitigating the release of pro-inflammatory cytokines from glial cells. Inhibition of iNOS and cox-2 is also considered effective in ameliorating neuroinflammation in AD patients (Bronzuoli et al. 2016).
Mitophagy is a process which is involved in the clearance of damaged mitochondria which is also emerging out as a good option in mitigating neuroinflammation in AD. In AD, there is decrease in mitophagy process which enhances neuroinflammation due to reduced phagocytosis and excessive stimulation of microglia which leads to increased accumulation of Aβ plaques and hyperphosphorylated tau. Damaged mitochondria accretion also leads to the activation of damage-associated molecular patterns (DAMPs) which on gaining entry into the cytoplasm act as pro-inflammatory molecules and stimulate NLR family pyrin domain containing 3 (NLRP3) inflammasomes, IL-1β activation and caspase-1 activation. Urolithin A and actinonin mitigate neuroinflammation and enhance microglial phagocytosis by increasing mitophagy resulting in decreased tau and amyloid β accumulation. IL-10 which is an anti-inflammatory cytokine is also responsible for mitigating inflammation by phosphorylating AMPK and activating STAT-3/ DNA damage inducible transcript 4 (DDIT4) pathway and inhibiting mTOR which potentiates the mitophagy process (Lautrup et al. 2019). M1 state of microglia can be modulated into M2 state which has neuroprotective role. Therefore, microglial polarization can be a potential target. A second generation tetracycline known as minocycline is selective for inhibiting M1 state of microglia and resulting in anti-neuroinflammatory effects in brain of AD patients (Regen et al. 2017).
At present there are various drugs under investigation which can prevent neuroinflammatory pathways contributing to AD (Fig. 3). Phytochemicals found naturally have anti-inflammatory and anti-oxidant activities. Polyphenolic compounds are one in this broad category. Some of the compounds include vitamin E, vitamin C, Curcumin, catechin and resveratrol. It has been seen in various studies that nutraceuticals containing these compounds act as pro-oxidants rather than anti-oxidants. So the exact actions of these nutraceuticals are still unclear and investigations are still going on (Sawikr et al. 2017).
Fig. 3.
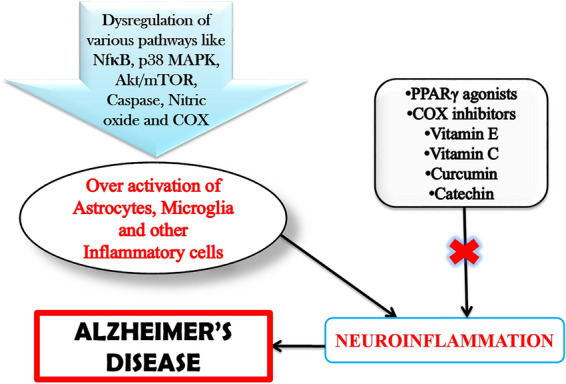
Neuroinflammatory pathways and drugs targeting neuroinflammation. Figure 3 shows that due to dysregulation of various pathways in brain and periphery leads to hyperactivation of Glial cells and other inflammatory cells in the brain which contributes to neuroinflammatory pathway of AD. Drugs like PPARγ agonists, COX inhibitors, vitamin E, vitamin C, curcumin and catechin act via various mechanisms to reduce the neuroinflammatory pathways thus prevent the progression of AD
Anti-oxidant nutrients repress the synthesis of TNF-α, IL-6 and NO by the activated microglia in a fixation subordinate way. Vitamin E gives neuro-protection by lessening TNF-α along with NO production and it decreases the LPS-instigated increment in free radicals and IL-6 in microglia. Polyphenolic compounds like flavonoids and Vitamin C may hinder age-related neurodegenerative diseases. Flavonoids, that are available in day to day dietary products, ensure survival and wellbeing of neuronal cells by decreasing oxidation of proteins, restraining of JNK and p38 pathways and forestalling production of free radicals (Shabab et al. 2017).
Pioglitazone acts as Peroxisome proliferator-activated receptor γ (PPARγ) agonist; it changes the M1 state of microglia to M2 state which increases the phagocytosis of deposited Aβ. PPARγ agonists also reduce the synthesis of IL-1β inflammatory cytokines. Transhinone I is an inhibitor of IL-6, IL-1β, TNF-α and nitric oxide and subsequently inhibits NFκB pathway in microglia. Gastrodin inhibits MAPK pathway and NFκB resulting in reduction of pro-inflammatory mediators, cytokines and interleukins. Resveratrol also inhibits NFκB, cAMP response element binding protein (CREB) and MAPK by mTOR pathway. Trans-cinnamaldehyde inhibits and degrades iNOS which inhibits ERK1/2/MEK pathways and reduce production of NO. Glatiramer acetate reduces the release of TNF-α from microglial cells but it does not have any profound effect in the synthesis of NO (Thawkar and Kaur 2019) (Table.1).
Table 1.
Drugs in clinical trial targeting neuroinflammation for the treatment of AD
| S. No. | Drug name | Structure | Mechanism of action | NCT No. | Route of adminis-tration | Status |
|---|---|---|---|---|---|---|
| 1 | Indomethacin |
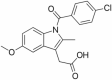
|
COX1/2 inhibitor | NCT00432081 | Oral | Completed |
| 2 |
VX-745 (Neflamapimod) |
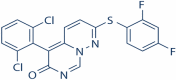
|
P38-α MAPK inhibitor | NCT03435861 | Oral | Recruiting |
| 3 | PTI-125 (Simufilam) |
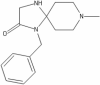
|
Binds to Filamin A (FLNa) Anti-neuro-inflammatory | NCT04388254 | Oral | Active, Not recruiting |
| 4 | Candesartan |
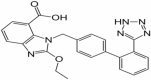
|
Reduces NO, TNF-α and TGF-β1 levels | NCT02646982 | Oral | Completed |
| 5 | Florbetaben |
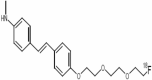
|
Targets amyloid β | NCT03744312 | i.v | Enrolling by invitation |
| 6 | Minocycline |
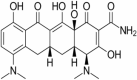
|
eIF2α Inhibition | NCT01463384 | Oral | Completed |
| 7 | Pioglitazone |
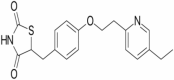
|
PPAR γ agonist | NCT00982202 | Oral | Completed |
| 8 | Simvastatin |
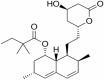
|
Immuno-modulatory and anti-inflammatory | NCT00486044 | Oral | Completed |
| 9 | Atomoxetine |
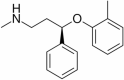
|
Reduces NF-κB expression | NCT01522404 | Oral | Completed |
| 10 | Dexmedetomidine |
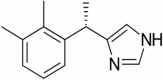
|
MAPK/ERK1/2 inhibitor | NCT04205539 | I.V. infusion | Suspended (Pending Covid-19 pandemic) |
| 11 | NP001 | Immuno-modulator and macrophage modulator | NCT03179501 | i.v | Terminated | |
| 12 | Celecoxib |
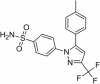
|
COX-2 inhibitor | NCT00065169 | Oral | Completed |
| 13 | Entanercept |
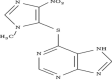
|
TNF-α inhibitor | NCT00203359 | Perispinal injection | Completed |
| 14 | Memantine |
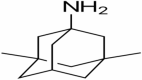
|
Reduces release of pro-inflammatory factors, NMDA antagonist | NCT03918616 | Oral | Completed |
| 15 | Cyclophosphamate |
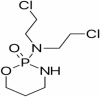
|
Immuno-suppressor and alkylating agent | NCT00013650 | i.v/oral | Completed |
Drugs in various phases of clinical trials for the treatment of Alzheimer’s disease targeting neuroinflammation
Indomethacin
Indomethacin is in phase 3 of clinical trial with the main objective of treatment of Alzheimer’s disease. The actual enrollment for the study is of 160 participants. The study design is randomized with parallel assignment model and double masking. The recruitment status of the study is completed. In preclinical trials on rat models indomethacin reduced the levels of inflammatory molecules like TNFα and IL1β in the hippocampus region of the brain (Rawat et al. 2019). Indomethacin is a non-steroidal anti-inflammatory drug which acts as PPARγ activator that results in decreased activity of BACE1 reducing the production of Aβ from APP (Deardorff and Grossberg, 2017). The results are although negative in some studies as this drug failed to slow down the progression of AD (de Jong et al. 2008) however some recent studies revealed neuroprotective action of indomethacin and improvement in cognitive decline in AD (Ali et al. 2019). More research needs to be done to establish a definitive role of indomethacin in AD treatment.
VX-745
VX-745 (Neflamapimod) is in phase 2 of clinical trials with an objective of treatment of Alzheimer’s disease. The study design is randomized, parallel assignment with quadruple masking. It is a double blind and placebo-controlled study. Estimated enrollment is of 40 participants. The recruitment status of the study is under recruiting. In AD transgenic mice models VX-745 has found to be effective in slowing down the progression of AD by acting as a p38 MAPK inhibitor (Dong et al. 2019). It is an oral drug with selective p38α blocking activity. p38 is involved in the activation of microglia and release of pro-inflammatory cytokines (Scheltens et al. 2018).
PTI-125
PTI-125 (Simufilam) is under phase 2 of clinical trial. The primary purpose of the study is treatment of the disease. The study is open label with single group assignment and multi-centric. The estimated enrollment for the study is of 200 participants. The recruitment status of the study is active, not recruiting. In animal models simufilam has been proven to reduce inflammation and tau phosphorylation via binding filamin thus preventing binding of Aβ42 α7 nicotinic acetylcholine receptors (Pleen and Townley 2021). PTI-125 binds to altered filamin A (FLNA) and restores its normal structure, improves receptor and synaptic functions and reduces its linkage with α7nAChR/TLR4 downstream pathologies which leads to increase in hyperphosphorylated tau levels and increased release of inflammatory mediators respectively (Wang et al. 2017).
Candesartan
Candesartan is in phase 2 of clinical trials for treatment of Alzheimer’s disease. The study design is randomized having quadruple masking with parallel assignment model. The estimated enrollment for the study is of 77 participants. The recruitment status of the study is completed. Candesartan being an angiotensin receptor blocker has a potential to prevent the AT2R binding. Stress stimuli such as cold, predator exposure activate release of RAS and Ang II. In familial AD mice models candesartan reduced nitric oxide synthase and cyclooxygenase levels. It also reduced the hyperactivation of microglia in the hippocampal region of AD mice brains (Torika et al. 2018). Candesartan potentially decrease the activity of angiotensin I receptor which contributes in the modulation of inflammation in AD (Trofimiuk et al. 2018).
Florbetaben
Florbetaben is in phase 2 of clinical trial with the objective of diagnosis of the disease. The study design is non-randomized, open label with parallel assignment model. Estimated enrollment for the study is of 50 participants. The recruitment status of the study is under recruiting. Florbetaben is used in PET scanning using 18F labeling. It is non-invasive method to detect amyloid β levels. It has been used in APP-Swe mice to detect amyloidogenesis progression in the mice brain (Rominger et al. 2013). It is frequently used to check the amyloid β decreasing ability of drugs (Stenzel et al. 2018).
Minocycline
Minocycline is in phase 2 of clinical trials for the objective of treatment of Alzheimer’s disease. The design of the study is open label with single group assignment model. Actual enrollment is of 13 participants. The recruitment status of the study is completed. It is a second generation tetracycline antibiotic. In various neurodegenerative animal models minocycline has shown neuroprotective effects. It reduced the levels of inflammatory markers like IL6 and the microglial activation in the brains of AD models (Cheng et al. 2015). Minocycline reduces TNF-α and IL-1β levels and other inflammatory cytokines leading to the anti-inflammatory action (Amani et al. 2019).
Pioglitazone
Pioglitazone is in phase 2 of clinical trial with the primary motive of treatment of the disease. The design of the study is randomized, parallel assignment model with quadruple masking. Actual enrollment for the study is of 25 participants. The recruitment status of the study is completed. Pioglitazone has been found to reduce Glial inflammation and Aβ levels in AD transgenic mice models (Galimberti and Scarpini 2017). Pioglitazone acts as PPARγ agonist; it changes the M1 state of microglia to M2 state which increases the phagocytosis of deposited Aβ. PPARγ agonists also reduce the synthesis of IL-1β inflammatory cytokines (Thawkar and Kaur 2019).
Simvastatin
Simvastatin is in phase 2 clinical trial for the primary aim of prevention of the disease. The study design is randomized, parallel assignment with triple masking. The actual enrollment for the study is of 103 participants. The recruitment status of the study is completed. Preclinical studies in mice models have shown that simvastatin reduces neuroinflammation via modulation of NFκB/p65 pathway (Yu-Ben et al. 2019). Simvastatin reduces the apoptosis and release of inflammatory mediators and cytokines and increase the rate of survival of neuronal cells. Statins also acts as HMG CoA reductase inhibitor. Simvastatin also inhibits miR-106b thus prevents neuronal death and inhibits inflammatory response in brain of AD patients (Huang et al. 2017).
Atomoxetine
Atomoxetine is in phase 2 of clinical trial. The actual enrollment for the study is of 39 participants. The design of the study is randomized and crossover-assignment with quadruple masking. The recruitment status of the study is completed. It acts as non-adrenaline reuptake inhibitor. Atomoxetine efficiently reduced microglial activation and decreased the expression of inflammatory cytokine TNFα in experimental models (Yssel et al. 2018). It increases the neurotrophic effects and decreases the expression of TNF-α by suppressing the activation of microglia. In addition atomoxetine increases tyrosine hydroxylase activity and improves motor activities (Yssel et al. 2018).
Dexmedetomidine
Dexmedetomidine is in phase 1 of clinical trials for the objective of treatment of Alzheimer’s disease. The design of the study is open label with single group assignment. Estimated enrollment for the study is of 50 participants. The recruitment status of the study is enrolling by invitation. In rat models dexmedetomidine reduces the expression of TLR-4 mRNA in the hippocampus region thus inhibiting inflammatory pathways and cognitive dysfunction (Wang et al. 2018). It is a selective α2 adrenoreceptor agonist; it inhibits the excessive production of TNF inflammatory mediator and mitigates microglial activation thus acting as an anti-inflammatory agent (Clark and Vissel 2018).
NP001
NP001 is in phase 1 of clinical trial. The study design is randomized, double blind and placebo-controlled with parallel assignment model. The recruitment status of the study is terminated due to poor recruitment. Actual enrollment for the study is of four participants. It belongs to anti-inflammatory drugs. NP001 has shown immunomodulatory effects, it reduces the activation of abnormal inflammatory macrophages in preclinical studies (Bucchia et al. 2015). It acts as immune regulator of inflammatory monocytes or macrophages and reduces the release of inflammatory mediators and ameliorates neuroinflammation (Cummings et al. 2018).
Celecoxib
Celecoxib is in clinical trial for the prevention of Alzheimer’s disease. The study design is randomized, double blind with parallel assignment model. The actual enrollment for the study is of 138 participants. In Aβ induced rat models celecoxib reduced neuroinflammation by reducing Cox-2 protein expression in the brain of rat models (Mhillaj et al. 2018). It is a non-steroidal anti-inflammatory drug (NSAID) which selectively blocks Cox-2 activity thus reducing the over-activation of inflammatory mediators causing neuroinflammation. NSAIDs are also efficient in modulating PPARγ and processing APP (Deardorff and Grossberg, 2017).
Etanercept
Etanercept is in phase 1 clinical trial for treatment of the disease. The design of the study is open label and non-randomized with single group assignment model. The actual enrollment for the study is of 15 participants. The recruitment status of the study is completed. It acts as TNF-α inhibitor. Being a highly specific antagonist of TNF etanercept have shown efficacy in reducing inflammation via reduction of Il-6 levels in the brain of animal models (Galinsky et al. 2020). TNFR1 and TNFR2 are the two types of receptors for TNF which mediate pro-apoptotic pathway along with inflammation and neuroprotection, respectively. It reduces the levels of TNF in hippocampus region of brain and improves memory functions (Ortí-Casañ et al. 2019).
Memantine
This study involving Memantine is an observational study with case control model. Actual enrollment for the study is of 50 participants. The recruitment status of the study is completed. Preclinical studies of Memantine were performed in Tg2576 and triple- transgenic mice, concluding that Memantine reduced tau pathology and levels of Aβ along with reduction in inflammation and improved behavioral activities (Scearce-Levie et al. 2020). Memantine blocks NMDA, a glutamate receptors sub-family, which has very important role in the functioning of brain. The recruitment status of the study is terminated (Khalaf et al. 2019).
Conclusion
AD is a rapidly progressing disorder contributing to neurodegeneration. Many hypotheses have been made regarding the pathophysiology of the disease including mainly tau hypothesis and amyloid β hypothesis. But due to the involvement of several factors in the pathogenesis of the disease complete cure is still on the urge of development. Many evidences support the involvement of brain inflammation in the development of the disease. The major cells involved in the neuroinflammation process in the brain are glial cells including microglia and astrocytes. Other cells involved in neuroinflammation are oligodendrocytes, pericytes and neurons. In AD brains these cells become hyperactive and potentiate the release of inflammatory mediators like cytokines, chemokines, interferons and interleukins which get accumulated around senile plaques in the brain and contribute to neuroinflammation.
Various inflammatory molecules are involved in the progression of AD. Interleukins promotes the activation of T-lymphocytes mediated toxicity, increase the activity of macrophages and neutrophils. Inflammatory cytokines are released from glial cells and are of two types, pro-inflammatory and anti-inflammatory. Chemokines may powerfully direct microglial relocation and enlistment of astrocytes to the site of neuroinflammation, preferring the degree of local aggravation of inflammation. Other inflammatory molecules are neutrophils and interferons.
Pathways like NFκB, MAPK, Akt/mTOR, p38, COX and nitric oxide pathways are involved in the release of inflammatory molecules from microglia by activating it or potentiating M1 state of microglia. AMPK pathway generally ameliorates the inflammatory response by inhibiting mTOR pathway. Anti-inflammatory molecules like minocycline are employed for reducing Aβ and tau pathologies by mitigating the release of pro-inflammatory cytokines from glial cells. Inhibition of iNOS and Cox-2 is also considered effective in ameliorating neuroinflammation in AD patients. Other anti-inflammatory molecules are curcumin, catechin, Vitamin E and Vitamin C. Celecoxib a cox-2 selective inhibitor, also ameliorates inflammation in brain. Pioglitazone a PPARγ agonist, changes the M1 state of microglia to M2 state which increases the phagocytosis of deposited Aβ. PPARγ agonists also reduce the synthesis of IL-1β inflammatory cytokines. Many drugs targeting neuroinflammation inducing pathways are under different phases of clinical trials. As there is a need of development of complete cure for Alzheimer’s disease, focusing neuroinflammation pathways can come out as effective treatment for the disease.
Author contributions
RD wrote the manuscript, PS wrote the manuscript, BM design and edited manuscript, AB edited manuscript, DHR design, wrote and edited Manuscript.
Funding
None.
Availability of data and material
Not applicable.
Declarations
Conflicts of interest
No conflicts of interest.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ahmad MH, Fatima M, Mondal AC. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci. 2019;59:6–11. doi: 10.1016/j.jocn.2018.10.034. [DOI] [PubMed] [Google Scholar]
- Ali MM, Ghouri RG, Ans AH, et al. Recommendations for anti-inflammatory treatments in Alzheimer’s disease: a comprehensive review of the literature. Cureus. 2019 doi: 10.7759/CUREUS.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019;15:321–387. doi: 10.1016/j.jalz.2019.01.010. [DOI] [Google Scholar]
- Amani M, Shokouhi G, Salari AA. Minocycline prevents the development of depression-like behavior and hippocampal inflammation in a rat model of Alzheimer’s disease. Psychopharmacology. 2019;236:1281–1292. doi: 10.1007/s00213-018-5137-8. [DOI] [PubMed] [Google Scholar]
- Atri A. The Alzheimer’s disease clinical spectrum: diagnosis and management. Med Clin North Am. 2019;103:263–293. doi: 10.1016/j.mcna.2018.10.009. [DOI] [PubMed] [Google Scholar]
- Bagyinszky E, Van GV, Shim K, et al. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J Neurol Sci. 2017;376:242–254. doi: 10.1016/j.jns.2017.03.031. [DOI] [PubMed] [Google Scholar]
- Bronzuoli MR, Iacomino A, Steardo L, Scuderi C. Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res. 2016;9:199–208. doi: 10.2147/JIR.S86958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucchia M, Ramirez A, Parente V, et al. Therapeutic development in amyotrophic lateral sclerosis. Clin Ther. 2015;37:668–680. doi: 10.1016/J.CLINTHERA.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–321. doi: 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimer’s Dement. 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Cao W, Zheng H. Peripheral immune system in aging and Alzheimer’s disease 11 medical and health sciences 1109 neurosciences. Mol Neurodegener. 2018;13:1–17. doi: 10.1186/s13024-018-0284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Hou J, Zhang C, et al. Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci Reports. 2015;51(5):1–14. doi: 10.1038/srep10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Vissel B. The inflammatory nature of post-surgical delirium predicts benefit of agents with anti-TNF effects, such as dexmedetomidine. Front Neurosci. 2018;12:257. doi: 10.3389/fnins.2018.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement Transl Res Clin Interv. 2018;4:195–214. doi: 10.1016/j.trci.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong D, Jansen R, Hoefnagels W, et al. No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS ONE. 2008 doi: 10.1371/JOURNAL.PONE.0001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff WJ, Grossberg GT. Targeting neuroinflammation in Alzheimer’s disease: evidence for NSAIDs and novel therapeutics. Expert Rev Neurother. 2017;17:17–32. doi: 10.1080/14737175.2016.1200972. [DOI] [PubMed] [Google Scholar]
- Domingues C, da Cruz e Silva OAB, Henriques AG. Impact of cytokines and chemokines on Alzheimer’s disease neuropathological hallmarks. CAR. 2017 doi: 10.2174/1567205014666170317113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Li X, Cheng J, Hou L. Drug development for Alzheimer’s disease: microglia induced neuroinflammation as a target? Int J Mol Sci. 2019 doi: 10.3390/IJMS20030558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Scarpini E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs. 2017;26:97–101. doi: 10.1080/13543784.2017.1265504. [DOI] [PubMed] [Google Scholar]
- Galinsky R, Dhillon SK, Dean JM, et al. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J Neuroinflammation. 2020;171(17):1–16. doi: 10.1186/S12974-020-01769-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li Z, Zhao L, Zhao W. Simvastatin ameliorate memory deficits and inflammation in clinical and mouse model of Alzheimer’s disease via modulating the expression of miR-106b. Biomed Pharmacother. 2017;92:46–57. doi: 10.1016/j.biopha.2017.05.060. [DOI] [PubMed] [Google Scholar]
- Kales HC, Lyketsos CG, Miller EM, Ballard C. Management of behavioral and psychological symptoms in people with Alzheimer’s disease: an international Delphi consensus. Int Psychogeriatrics. 2019;31:83–90. doi: 10.1017/S1041610218000534. [DOI] [PubMed] [Google Scholar]
- Khalaf SS, Hafez MM, Mehanna ET, et al. Combined vildagliptin and memantine treatment downregulates expression of amyloid precursor protein, and total and phosphorylated tau in a rat model of combined Alzheimer’s disease and type 2 diabetes. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:685–695. doi: 10.1007/s00210-019-01616-3. [DOI] [PubMed] [Google Scholar]
- Knezevic D, Mizrahi R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog Neuro-Psychopharmacol Biol Psychiatry. 2018;80:123–131. doi: 10.1016/j.pnpbp.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Lautrup S, Lou G, Aman Y, et al. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem Int. 2019;129:104469. doi: 10.1016/j.neuint.2019.104469. [DOI] [PubMed] [Google Scholar]
- Liddell JR. Interplay between Nrf2 and NF-κB in neuroinflammatory diseases. J Clin Cell Immunol. 2017 doi: 10.4172/2155-9899.1000489. [DOI] [Google Scholar]
- Marttinen M, Takalo M, Natunen T, et al. Molecular mechanisms of synaptotoxicity and neuroinflammation in Alzheimer’s disease. Front Neurosci. 2018;12:963. doi: 10.3389/fnins.2018.00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhillaj E, Morgese MG, Tucci P, et al. Celecoxib prevents cognitive impairment and neuroinflammation in soluble amyloid β-treated rats. Neuroscience. 2018;372:58–73. doi: 10.1016/J.NEUROSCIENCE.2017.12.046. [DOI] [PubMed] [Google Scholar]
- Ortí-Casañ N, Wu Y, Naudé PJW, et al. Targeting TNFR2 as a novel therapeutic strategy for Alzheimer’s disease. Front Neurosci. 2019;13:49. doi: 10.3389/FNINS.2019.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto CA, de Oliveira WH, da Araújo SM, R, Nunes AKS, AMPK activation: role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol. 2017;298:31–41. doi: 10.1016/j.expneurol.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Pleen J, Townley R. Alzheimer’s disease clinical trial update 2019–2021. J Neurol. 2021;1:1–14. doi: 10.1007/S00415-021-10790-5. [DOI] [PubMed] [Google Scholar]
- Rawat C, Kukal S, Dahiya UR, Kukreti R. Cyclooxygenase-2 (COX-2) inhibitors: future therapeutic strategies for epilepsy management. J Neuroinflammation. 2019;161:1–15. doi: 10.1186/S12974-019-1592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regen F, Hellmann-Regen J, Costantini E, Reale M. Neuroinflammation and Alzheimer’s disease: implications for microglial activation. Curr Alzheimer Res. 2017 doi: 10.2174/1567205014666170203141717. [DOI] [PubMed] [Google Scholar]
- Rominger A, Brendel M, Burgold S, et al. Longitudinal assessment of cerebral β-Amyloid deposition in mice overexpressing Swedish mutant β-Amyloid precursor protein using 18F-Florbetaben PET. J Nucl Med. 2013;54:1127–1134. doi: 10.2967/JNUMED.112.114660. [DOI] [PubMed] [Google Scholar]
- Sawikr Y, Yarla NS, Peluso I, et al. Neuroinflammation in Alzheimer’s disease: the preventive and therapeutic potential of polyphenolic nutraceuticals. Adv Protein Chem Struct Biol. 2017 doi: 10.1016/bs.apcsb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Scearce-Levie K, Sanchez PE, Lewcock JW, et al. Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat Rev Drug Discov. 2020;19:447–462. doi: 10.1038/s41573-020-0065-9. [DOI] [PubMed] [Google Scholar]
- Schain M, Kreisl WC. Neuroinflammation in neurodegenerative disorders—a review. Curr Neurol Neurosci Rep. 2017;17:25. doi: 10.1007/s11910-017-0733-2. [DOI] [PubMed] [Google Scholar]
- Scheltens P, Prins N, Lammertsma A, et al. An exploratory clinical study of p38 α kinase inhibition in Alzheimer’s disease. Ann Clin Transl Neurol. 2018;5:464–473. doi: 10.1002/acn3.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengoku R. Aging and Alzheimer’s disease pathology. Neuropathology. 2020;40:22–29. doi: 10.1111/neup.12626. [DOI] [PubMed] [Google Scholar]
- Shabab T, Khanabdali R, Moghadamtousi SZ, et al. Neuroinflammation pathways: a general review. Int J Neurosci. 2017;127:624–633. doi: 10.1080/00207454.2016.1212854. [DOI] [PubMed] [Google Scholar]
- Shi Y, Holtzman DM, Immunology DH-NR, 2018 U Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18:759–772. doi: 10.1038/s41577-018-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sil S, Ghosh T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s disease. J Neuroimmunol. 2016;291:115–124. doi: 10.1016/j.jneuroim.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Stenzel J, Rühlmann C, Lindner T, et al. [18 F]-florbetaben PET/CT Imaging in the Alzheimer’s disease Mouse Model APPswe/PS1dE9. Curr Alzheimer Res. 2018;16:49–55. doi: 10.2174/1567205015666181022095904. [DOI] [PubMed] [Google Scholar]
- Stock AJ, Kasus-Jacobi A, Pereira HA. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J Neuroinflammation. 2018;15:1–15. doi: 10.1186/s12974-018-1284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Montagne A, Sagare AP, et al. Vascular dysfunction—the disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019;15:158–167. doi: 10.1016/j.jalz.2018.07.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thawkar BS, Kaur G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol. 2019;326:62–74. doi: 10.1016/j.jneuroim.2018.11.010. [DOI] [PubMed] [Google Scholar]
- Torika N, Asraf K, Apte RN, Fleisher-Berkovich S. Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci Ther. 2018;24:231–242. doi: 10.1111/CNS.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofimiuk E, Wielgat P, Braszko JJ. Candesartan, angiotensin II type 1 receptor blocker is able to relieve age-related cognitive impairment. Pharmacol Rep. 2018;70:87–92. doi: 10.1016/j.pharep.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Uddin MS, Kabir MT, Al MA, et al. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int Immunopharmacol. 2020;84:106479. doi: 10.1016/j.intimp.2020.106479. [DOI] [PubMed] [Google Scholar]
- Walters A, Phillips E, Zheng R, et al. Evidence for neuroinflammation in Alzheimer’s disease. Prog Neurol Psychiatry. 2016;20:25–31. doi: 10.1002/pnp.444. [DOI] [Google Scholar]
- Wang HY, Lee KC, Pei Z, et al. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol Aging. 2017;55:99–114. doi: 10.1016/j.neurobiolaging.2017.03.016. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhou W, Dong H, et al. Dexmedetomidine pretreatment inhibits cerebral ischemia/reperfusion-induced euroinflammation via activation of AMPK. Mol Med Rep. 2018;18:3957–3964. doi: 10.3892/MMR.2018.9349. [DOI] [PubMed] [Google Scholar]
- Yu-Ben X, Zhang HN, Dai Y, et al. Simvastatin prevents and ameliorates depressive behaviors via neuroinflammatory regulation in mice. J Affect Disord. 2019;245:939–949. doi: 10.1016/J.JAD.2018.11.086. [DOI] [PubMed] [Google Scholar]
- Yssel JD, O’Neill E, Nolan YM, et al. Treatment with the noradrenaline re-uptake inhibitor atomoxetine alone and in combination with the α2-adrenoceptor antagonist idazoxan attenuates loss of dopamine and associated motor deficits in the LPS inflammatory rat model of Parkinson’s disease. Brain Behav Immun. 2018;69:456–469. doi: 10.1016/j.bbi.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Fang W, Fan S, et al. Neurotropin inhibits neuroinflammation via suppressing NF-κB and MAPKs signaling pathways in lipopolysaccharide-stimulated BV2 cells. J Pharmacol Sci. 2018;136:242–248. doi: 10.1016/j.jphs.2018.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.