

Abstract
The sodium-dependent taurocholate co-transporting polypeptide (NTCP)-S267F variant is known to be associated with a reduced risk of hepatitis B virus (HBV) infection and disease progression. The NTCP-S267F variant displays diminished function in mediating HBV entry, but its function in HBV infection has not been fully established in more biologically relevant models. We introduced the NTCP-S267F variant and tested infectivity by HBV in genetically edited hepatic cells. HepG2-NTCP clones with both homozygous and heterozygous variants were identified after CRISPR base editing. NTCP-S267F homozygous clones did not support HBV infection. The heterozygote clones behaved similarly to wild-type clones. We generated genetically edited human stem cells with the NTCP-S267F variant, which differentiated equally well as wild-type into hepatocyte-like cells (HLCs) expressing high levels of hepatocyte differentiation markers. We confirmed that HLCs with homozygous variant did not support HBV infection, and heterozygous variant clones were infected with HBV equally as well as the wild-type cells. In conclusion, we successfully introduced the S267F variant by CRISPR base editing into the NTCP/SLC10A gene of hepatocytes, and showed that the variant is a loss-of-function mutation. This technology of studying genetic variants and their pathogenesis in a natural context is potentially valuable for therapeutic intervention against HBV.
Keywords: HBV, NTCP, S267F polymorphism, CRISPR base editing, stem cell-derived hepatocyte
Graphical abstract
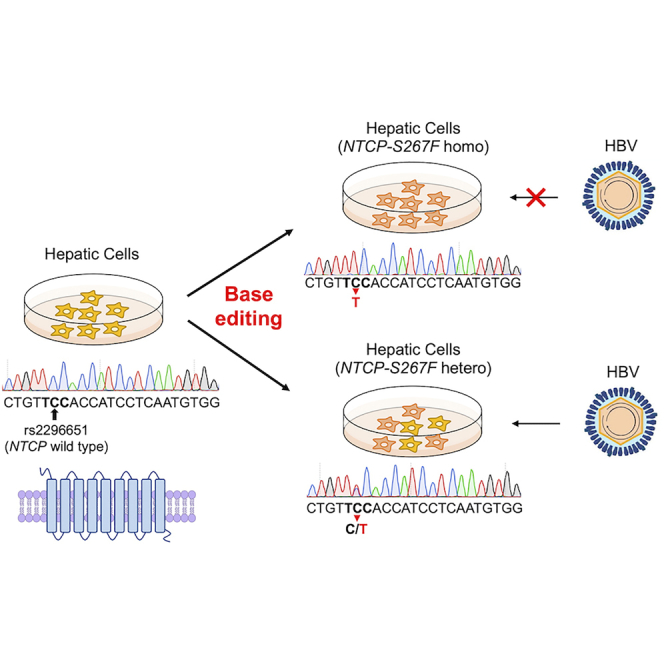
The authors introduced the S267F polymorphism into the HBV receptor NTCP gene of human hepatic cells using CRISPR base editing. They characterized the functional effect of homozygous or heterozygous NTCP-S267F variant on HBV infection and showed that the variant is a loss-of-function mutation. The findings have implications in the pathogenesis and therapy of HBV.
Introduction
Hepatitis B virus (HBV) is a partially double-stranded hepatotropic DNA virus and a major cause of chronic liver disease worldwide. Although the incidence of HBV infection is decreasing due to the utilization of antiviral therapy and vaccination, 3.5% of the global population is still chronically infected with HBV. Chronic HBV infection leads to liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) in 25%–40% of HBV carriers.1
The sodium-dependent taurocholate co-transporting polypeptide (NTCP) was identified as a cellular receptor for HBV and hepatitis delta virus. NTCP specifically interacts with the large surface protein of HBV and mediates viral entry.2,3 NTCP is expressed specifically on the hepatic basolateral membranes and functions as co-transporter of bile acids and sodium ions into hepatocytes.4 Overexpression of NTCP gene into non-susceptible HCC cell lines permits infection with HBV.2,3,5 Identification of NTCP as the HBV receptor not only provides a major advance for the establishment of in vitro and in vivo HBV models but also serves as an attractive target for HBV drug development.6
NTCP is encoded by the SLC10A gene, a member of the solute carrier family. Several single-nucleotide polymorphisms (SNPs) that may affect the function of NTCP have been identified so far.7,8 A nonsynonymous variant, Ser267Phe (S267F, c.800C>T, rs2296651 SNP) in NTCP is found only in East Asia and is absent in African and European populations.8,9 The S267F variant has been shown to diminish the HBV receptor function of NTCP and results in reduced HBV entry and infection in a transfection cell-culture system.9,10 However, the function of this variant in HBV infection has not been clearly established in more biologically relevant models. The rs2296651 (S267F) variant has also been associated with a reduced risk of HBV and HDV infection11, 12, 13, 14, 15, 16, 17 as well as a slow disease progression in chronic HBV-infected patients.11,12,15, 16, 17
CRISPR/Cas9 is a well-established method for gene editing and relies on the initial introduction of double-stranded DNA breaks (DSBs), resulting in insertions, deletions, and translocations at the target genome.18 The efficiency of single-base editing, however, is low using this system. Most of the distribution of human pathogenic genetic variants consist of point mutations.19 Therefore, installing and correcting pathogenic SNPs efficiently is of great interest for the treatment of genetic disorders and the study of pathogenic mechanisms of diseases. Base editing based on the CRISPR platform is a novel genome-editing strategy that can convert one target genome DNA base to another without inducing DSBs. Two kinds of base editor, cytosine base editor and adenine base editor, allow the introduction of all four possible transition mutations (C to T, A to G, T to C, and G to A). Base editing was initially developed by fusing rat APOBEC1 to catalytically inactive Cas9, followed by various further modifications leading to enhanced editing efficiency and specificity.19 Genome-wide off-target editing effects induced by the CRISPR-guided DNA base editors can be substantially diminished by the introduction of various mutations, such as R33A and K34A mutations in the APOBEC1 portion of the base editors.20 In addition, another base editor that can efficiently induce targeted C-to-G base transversion has been reported.21 CRISPR base-editing technology thus can correct or introduce disease-associated or pathogenic mutations with relative ease and safety.
In this study, we introduced the rs2296651 (S267F) variant onto the endogenous NTCP/SLC10A gene by a CRISPR base editor into the human hepatoma cell line and stem cells that are subsequently differentiated into hepatocyte-like cells (HLCs) and tested their infectivity by HBV.
Results
Establishment of base editor-expressing HepG2-NTCP cells
We first generated an FNLS base editor construct with the APOBEC1-R33A mutation that confers a lower off-target effect and similar base-editing efficacy.20 The FNLS base editor carries a rat APOBEC cytidine deaminase at the N terminus of optimized Cas9n (Cas9D10A) and a uracil glycosylase inhibitor domain at the C terminus followed by a nuclear-localization signal (NLS). It also carries NLS sequences at the N terminus of the base editor with an FLAG epitope tag. In addition, we also used a doxycycline (Dox)-inducible system to control target editing and reduce undesirable base editing. The NanoLuc luciferase (Nluc) gene was fused to the base editor with an intervening autocleavage site (P2A) and served as a convenient means to measure base editor expression (Figure 1A). The FNLS(R33A) construct was transduced into HepG2-NTCP cells, and HepG2-NTCP/FNLS(R33A) cells were selected with G418. The selected cell line was treated with Dox to assess the induction of the base editor. Dose-dependent Nluc activity was observed and Cas9 protein expression by Dox was confirmed in the HepG2-NTCP/FNLS(R33A) cells (Figures 1B and 1C).
Figure 1.

Base editing of NTCP(S267F) in HepG2-NTCP cell and generation of HepG2-NTCP(S267F) cells
(A) Schematic representation of FNLS with APOBEC(R33A) mutation. The FNLS base editor carries a rat APOBEC cytidine deaminase at the N terminus of optimized Cas9n and a uracil glycosylase inhibitor (UGI) domain at the C terminus. Nuclear-localization signal (NLS) sequence is located at both the N terminus (with an FLAG epitope tag) and the C terminus. (B) Western blotting of Cas9 in HepG2-NTCP/FNLS(R33A) cells treated with Dox (500 ng/mL) for 1 day or untreated (0 day). (C) Nluc assay in HepG2-NTCP/FNLS(R33A) in different Dox concentrations. (D) Base editing on target site after Dox treatment in HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA. Sequence around the SLC10A-sgRNA rs2296651 is shown below. Bold letters represent the amino acid codon for S267F. Red arrow and blue arrow show C6 (rs2296651) and C7 position with editing to T, respectively. (E) Frequency (%) of C-to-T conversion on C6 and C7 in HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA after Dox treatment. (F) Sequence of selected homozygous and heterozygous clone of NTCP(S267F). (G) NTCP mRNA expression in selected HepG2-NTCP(S267F) clones. Non-edited (NE) cell is HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA without Dox treatment. (H) Immunostaining of NTCP in HepG2-NTCP(WT) and HepG2-NTCP(S267F) shows expression and membrane location of NTCP in HepG2-NTCP(WT) and -NTCP(S267F). (I) RNA was extracted from NE cells, homozygous and heterozygous clones, and subjected to RT-PCR and sequence determination. Quantitative data are shown as means ± standard deviation of triplicates.
The construct expressing the SLC10A-single guide RNA (sgRNA) was transduced into the HepG2-NTCP/FNLS(R33A) cells and selected with puromycin. HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA cells were then treated with Dox for 9 days. Sanger sequencing showed base editing at both the sixth nucleotide (C6) and seventh nucleotide (C7) positions of the SLC10A gene in HepG2-NTCP cell line in a treatment-period-dependent manner (Figure 1D). The C6 position represents the location of the rs2296651 SNP in the SLC10A gene. Editing at the C7 position leads to a silent mutation. Using the EditR assay, we observed 80%, 89%, and 94% of target C-to-T conversion at C6 after 3, 6, and 9 days, respectively, after Dox treatment (Figure 1E). Less efficient base editing was observed at the C7 compared with C6 position (Figure 1E). This editing profile is consistent with the behavior of the FNLS base editor.22
To select single clones with edited NTCP-S267F variant, we treated HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA cells with Dox for different periods: 1–2 days for heterozygous and 6 days for homozygous S267F clone selection. After Dox treatment, the cells were seeded at low density to select single clones. Selected clones were expanded and genotyped for the NTCP-S267F variant; clones with both homozygous and heterozygous variants were identified (Figure 1F). All clones expressed similar levels of NTCP mRNA (Figure 1G). NTCP protein expression and membrane localization in NTCP-S267F variant were similar to that of wild-type cells by immunofluorescence (Figure 1H).
The HepG2-NTCP cells harbor both endogenous and transduced NTCP genes, although the endogenous genes are not transcriptionally active. We designed qPCR primers for NTCP mRNA with primers spanning an intron in the endogenous NTCP gene and measured exogenous NTCP expression. As expected, NTCP expression of HepG2-NTCP was predominantly from the transduced NTCP gene (Figure S1). To precisely determine the homozygosity or heterozygosity of the targeted base-editing sites on the transduced NTCP gene would be difficult by DNA sequencing. Thus, we harvested NTCP mRNA from homozygous and heterozygous clones and parental HepG2-NTCP cells and performed RT-PCR to determine the sequences. The sequencing data confirmed the homozygous and heterozygous nature of this variant in various clones (Figure 1I).
NTCP-S267F does not support HBV infection in edited HepG2-NTCPcells
We next tested HBV infectivity in the various HepG2-NTCP(S267F) clones. Four homozygous HepG2-NTCP(S267F) were infected with cell-culture-derived HBV genotype D (HBVcc). Parental HepG2-NTCP cells with or without Myrcludex B (MyrB) were infected as a control. Medium hepatitis B e antigen (HBeAg), hepatitis B surface antigen (HBsAg), and HBV RNA in the cells were determined 7 days after infection. As expected, HepG2-NTCP cells showed high levels of HBV markers that were much reduced in MyrB-treated cells. In contrast, NTCP-S267F homozygous clones showed very low levels of HBV markers that were not different from samples with MyrB treatment (Figure 2A), suggesting that the NTCP-S267F variant does not support HBV infection.
Figure 2.

HBV infection in HepG2-NTCP(S267F) cells
Non-edited (NE) cell is HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA without Dox treatment. HBV RNA (left panel), HBeAg (middle panel), and HBsAg (right panel) are shown. (A) HBV infection in homozygous of NTCP(S267F). (B) HBV infection in clones with either wild-type or heterozygous. “Homo” indicates HepG2-NTCP(S267F) homozygous clone #1 as a negative control for infection experiment. (C) HBV time-of-addition assay. The experimental scheme is shown on the left. (−/−) denotes no MyrB treatment all the time. (−/+) denotes no MyrB at 4°C for 4 h and MyrB added after 4 h when the incubation temperature is raised to 37°C. (+/+) denotes with MyrB all the time. Quantitative data are shown as means ± standard deviation of triplicates. Unpaired t test was used for comparisons: for (A), comparing all the clones with NE cells; for (C), compared with NE cells (−/−). ∗p < 0.05.
We next tested clones with either wild-type or heterozygous sequence for HBV infection. In general, they all showed similar high HBV markers (Figure 2B), indicating that the heterozygotes with a single copy of the wild-type NTCP gene is sufficient to support HBV infection, like the non-edited cells.
To assess the mechanism of NTCP-S267F variant in affecting HBV infection, we performed a time-of-addition experiment as a means to determine the step of HBV entry mediated by NTCP. Addition of MyrB after HBV binding at 4°C was effective in blocking HBV entry in wild-type cells, indicating that NTCP mediates post-binding viral entry (Figure 2C). This finding is consistent with previous reports that heparan sulfate proteoglycan mediates the initial attachment of HBV to cells followed by NTCP-mediated entry.23,24 The homozygous clone showed no entry activity under all the conditions, suggesting that NTCP-S267F confers resistance to HBV infection during a post-binding entry step.
To determine whether the NTCP-S267F affects infection of other HBV genotypes, we infected homozygous and heterozygous clones with either HBV genotype A or E. They infected wild-type and heterozygous clone equally well (Figure S2), whereas homozygous clone was not infected by either genotype. The two genotypes tested here had somewhat different infectivity in wild-type cells (genotype E > genotype A), which had been shown previously25 and probably relates to a difference in the amount of infectious HBV particles in the preparation.
We also tested HepG2-NTCP(S267F) homozygous clones with the C7 mutation. They expressed similar levels of NTCP RNA (Figure 3A) and were also not infected by HBV (Figure 3B). To ensure no other untoward mutations that may impair HBV infection were introduced during the base-editing step, we transduced wild-type NTCP fused with GFP into a HepG2-NTCP(S267F) homozygous clone. The transduced cells showed high GFP positivity (Figure 3C) and could be infected with HBV. These cells showed complete restoration of HBV infectivity (Figure 3D).
Figure 3.

HBV infection in HepG2-NTCP(S267F) cells with C7 mutation and restoration of HBV infectivity in HepG2-NTCP(S267F) cells
Non-edited (NE) cell is HepG2-NTCP/FNLS(R33A)/SLC10A-sgRNA without Dox treatment. #1 is homozygous clone of rs2296651(S267F). (A) NTCP mRNA expression in HepG2-NTCP(S267F) clones. (B) HBV infection in HepG2-NTCP(S267F) cells with C7 mutation. (C) GFP expression in HepG2-NTCP(S267F) after the transduction of NTCP-P2A-GFP. (D) Restoration of HBV infectivity in HepG2-NTCP(S267F) after NTCP-GFP transduction. Quantitative data are shown as means ± standard deviation of triplicates. Unpaired t test was used for comparisons: for (B), comparing all the clones with NE cells; for (D), comparing mock with NTCP-GFP. ∗p < 0.05
Generation of NTCP-S267F variant-containing human stem cells
To validate the functional effect of the S267F variant in cells more similar to primary human hepatocytes, we edited human stem cells, either embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), for this variant, differentiated them into HLCs, and tested them for infection with HBV. This system permits the direct assessment of the effect of this variant in a genetically identical background of hepatocytes. We selected H9hESC and a human iPSC line, SWiPSC, generated in our laboratory.26, 27, 28 We first tried to generate FNLS(R33A)-expressing stem cells using the same approach as used for the HepG2-NTCP cells. However, we could not detect enough Cas9 protein expression and base editing in FNLS(R33A)-expressing H9ESCs and SWiPSCs. It is possible that the use of the hALB minimal promoter in constructing the Dox-inducible expression plasmid was only weakly active in human stem cells.
Next, we used a newer version of the base editor, AncBE4max, which may have a more efficient base-editing activity.29 We generated pLenti-TRE-EFS-AncBE4max(R33A)-P2A-Nluc-neo and transduced it into H9ESCs and SWiPSCs. Again, no Cas9 expression and base editing were observed, consistent with our above reasoning. Thus, we switched to a cytomegalovirus (CMV) promoter-driven construct and transient transfection system. The pCMV-AncBE4max-P2A-GFP or pCMV-AncBE4max(R33A)-P2A-GFP construct together with the SLC10A-sgRNA-expressing plasmid was transiently transfected into H9ESCs and SWiPSCs. GFP+ cells were sorted using a fluorescence-activated cell sorter 2 days after transfection and expanded for a week. A C-to-T conversion on target site was detected in GFP-sorted cells in both H9ESCs and SWiPSCs. The APOBEC-R33A mutation resulted in a similar conversion effect on C6 (the NTCP-S267F variant) compared with the unmutated AncBE4max. On the other hand, AncBE4max(R33A) showed less conversion on C7 compared with AncBE4max (Figure 4A). Thus, AncBE4max(R33A) seemed to function more specifically at the C6 position compared with the wild-type protein.
Figure 4.

Base editing of NTCP(S267F) into human stem cells
(A) Base editing on target site in GFP+-H9ESCs and -SWiPSCs. Sequence around the SLC10A-sgRNA rs2296651 is shown below. Bold letters represent the amino acid codon for S267F. Red arrow and blue arrow show C6 (rs2296651) and C7 position with editing to T, respectively. (B) Western blotting of Cas9 in GFP+-H9ESCs and -SWiPSCs. Stem cell-derived HLCs were generated through a three-step HLC differentiation protocol.28 (C) Microscopic images of HLCs derived from either wild-type or genetically edited H9ESCs. Scale bars, 200 μm. Secreted hALB level in supernatant (D) and expression of HNF4a and NTCP mRNA (E) after HLC differentiation. (F) Intracellular HBV RNA levels in genetically edited H9ESC-derived HLC with or without MyrB treatment. Quantitative data are shown as means ± standard deviation of triplicates. Unpaired t test was used for comparisons: comparing the homo and hetero clones without MyrB with the parental H9 clone without MyrB. ∗p < 0.05.
We could not detect AncBE4max protein in the GFP-sorted H9ESCs and SWiPSCs at 10 days after expansion (Figure 4B). However, it is possible that a low level of the AncBE4max protein was expressed early and was sufficient to edit the genome before being silenced in the transfected stem cells. We thus selected single clones from the GFP-sorted cells and screened them for the S267F variant. We were able to identify several clones with homozygous and heterozygous variants from both stem cell lines (Figure S3). To assess whether the editing affected the phenotype of the stem cell lines, we assessed these clones for their abilities to differentiate into HLCs. HLCs were obtained from H9ESCs through a three-step differentiation protocol. Like their precursor stem cells, HLCs derived from genetically edited cells exhibited characteristic hepatocyte morphology: cuboidal shape, distinctive round small nuclei, and compact cell-cell contacts (Figure 4C). They differentiated equally well into HLCs that expressed high levels of hepatocyte differentiation markers (Figures 4D and 4E). The NTCP mRNA levels were comparable among the various clones and the unedited stem cell-derived HLCs (Figure 4E).
Stem cell-derived HLCs were then infected with HBV with or without MyrB. In HLC-infected culture, much input virus tends to obscure accurate determination of de novo HBV infection by measuring HBV markers in the supernatant (little difference between samples with or without MyrB). Intracellular HBV RNA was a better marker to monitor HBV infection. HBV-infected NTCP-S267F homozygous clones showed levels of HBV RNA similar to those of cells with MyrB treatment control. On the other hand, heterozygous clones, similar to that of wild-type cells, showed high HBV RNA levels compared with samples under MyrB treatment. These results suggest that, similar to HepG2-NTCP cell lines, HLCs with homozygous variant clones were resistant to HBV infection, and heterozygous variant clones were infected with HBV equally as well as the wild-type cells in stem cell-derived HLCs (Figure 4F).
Discussion
Previous clinical studies have reported an association of rs2296651 (S267F) variant in the NTCP/SLC10A gene with infection rate, disease severity, and progression in chronic HBV-infected patients.11,12,15, 16, 17 The S267F variant is also associated with treatment response.30 It has been reported that the S267F variant abolishes the function of NTCP,9 and yet another report shows that NTCP-267F variant is a functional but less efficient receptor for HBV entry.10 The 267F variant apparently affects the same domain on the NTCP protein that interacts with bile acid (its natural substrate) and the HBV pre-S1 sequence.7, 8, 9 Despite the deleterious effect of this variant on the bile-acid transport function of NTCP, individuals carrying this variant do not have any overt phenotype, probably reflecting compensatory action by other transporters.7,8,17 This association is similar to the phenotype for the null delta32 allele of CCR5 (the major cellular receptor for HIV), which protects carriers from HIV infection and progression.31 Finally, the NTCP-267F variant (in heterozygotes) may exert a dominant negative effect on the wild-type protein because various genetic association studies have reported a difference in the disease phenotype of heterozygotes from that of wild type. Thus, more biologically relevant models are necessary to fully define the functions of NTCP-S267F variant in HBV infection.
CRISPR/Cas9 base editing can efficiently convert a single nucleotide to another with ease and reduce the off-target effect. This novel technique has profoundly changed the approach to the establishment of disease models. In this study, we successfully edited NTCP-267F variant into HepG2-NTCP and human stem cells. Both NTCP-S267F homozygous and heterozygous clones were generated and studied. The off-target effects of base editing were also minimized by using an inducible (HepG2-NTCP) or transient transfection (human stem cell) system. Homozygous base editing to the NTCP-S267F variant resulted in a near-complete abrogation of HBV infection, supporting that this variant represents a null mutation for HBV infection. It is possible that this variant may not be totally nonfunctional in supporting HBV infection, as HBV infection does occur in individuals with homozygous alleles.11,12,17 On the other hand, it is intriguing to speculate that there may be a minor, alternative entry pathway for HBV. It is well known that viruses can use different ways to enter cells.32
Our study also clearly shows that heterozygous clones of HepG2-NTCP(S267F) are fully susceptible to HBV infectivity, indicating that the NTCP-267F variant does not have a dominant negative effect on the wild-type protein. At present, based on clinical association studies it is not clear why heterozygous individuals with this variant have a slower HBV disease progression. It is possible that this variant may have an additional effect on HBV disease unrelated to viral entry.
In conclusion, we successfully introduced the NTCP-S267F SNP into hepatic cells by gene editing with CRISPR/Cas9 technology and clarified the function of this variant in HBV infection. Our study also supports the use of MyrB, an inhibitor of NTCP, in the treatment of HBV and HDV,33,34 as homozygous hepatic cells of this variant appear fully functional as hepatocytes other than resistance to infection by HBV. Further study is needed to define any subtle biological effect of this variant on hepatocytes. As the CRISPR/Cas9-mediated gene-editing technology advances and becomes a more widely accepted therapeutic modality, it would be intriguing to consider conferring an HBV-resistant phenotype to HBV-infected individuals as a means to cure HBV infection.
Materials and methods
Plasmid
For base editor constructs, pLenti-FNLS-PGK-Puro was a gift from Lukas Dow (Addgene plasmid #110844; http://n2t.net/addgene:110844; RRID:Addgene_110844)22 and pCMV_AncBE4max_P2A_GFP was a gift from David Liu (Addgene plasmid #112100; http://n2t.net/addgene:112100; RRID:Addgene_112100).29 APOBEC1-R33A mutations were generated using a QuikChange Lightning site-directed mutagenesis kit (Agilent, Santa Clara, CA). pLenti-TRE-hAlb-FNLS(R33A)-P2A-Nluc-neo and pLenti-TRE-EFS-AncBE4max(R33A)-P2A-Nluc-neo were generated using an In-Fusion HD cloning kit (TakaraBio, Shiga, Japan).
For the sgRNA construct, the target sequence of SLC10A (5′-CTG TTC CAC CAT CCT CAA TG-3′) was inserted into lentiGuide-Puro (lentiGuide-SLC10A-sgRNA-Puro) according to the protocol.35 lentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid #52963; http://n2t.net/addgene:52963; RRID: Addgene_52963). pLenti-CMV-NTCP-P2A-GFP was generated from pLenti CMV Blast empty (w263-1) as a backbone also using an infusion cloning method. pLenti CMV Blast empty (w263-1) was a gift from Eric Campeau and Paul Kaufman (Addgene plasmid #17486; http://n2t.net/addgene:17486; RRID: Addgene_17486).
Cell culture
HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS)/1% penicillin-streptomycin (PS), at 37°C and 5% CO2. HepG2-NTCP cells (provided by Ulrike Protzer, University of Munich)36 were maintained in DMEM with 10% FBS/1% PS/1% non-essential amino acids (NEAA), and 30 μg/mL blasticidin (Gibco) using collagen-coated plates. PXB cells,37 which are human hepatocytes passaged serially and expanded in cDNA-uPA/SCID mice, were maintained as described previously.25
H9 human ESCs and SW human iPSCs were maintained on the plates previously coated with high-concentration (0.4 mg/mL) Matrigel matrix (Corning, Corning, NY) in mTeSR1 (STEMCELL Technologies, Vancouver, Canada). Rock Inhibitor Y-27632 (10 μM) (STEMCELL Technologies) was added to mTeSR1 at the passaging as described previously.26,27 SW iPSCs were kindly provided by Dr. Cynthia E. Dunbar (NIH, Bethesda, MD).
Transfection and transduction
For lentivirus production, 3 million HEK293T cells were plated on a T-75 flask. One day after plating, cells were transfected with a prepared mix of DMEM containing 5 μg of base editor construct or lentiGuide-SLC10A-Puro with 3.75 μg of psPAX2 (Addgene #12260) and 2.5 μg of pMD2.G (Addgene #12259) using FuGENE 6 transfection reagent (Promega, Madison, WI). 24 h after transfection, the medium was replaced with DMEM with 10% BSA, and supernatants were harvested every day up to 3 days after transfection. Supernatants containing lentivirus were concentrated by ultracentrifugation (25,000 rpm, 4°C, 90 min). The concentrated lentivirus was resuspended in 200 μL of PBS and stored at −80°C until infection.
For transduction, cells were plated on 6-well plates on the day before transduction and transduced with concentrated lentivirus. Two days after transduction, cells were selected in G418 (0.5–0.75 mg/mL) or puromycin (1.5 μg/mL for HepG2-NTCP and 5 μg/mL for stem cells). For the transfection into stem cells, cells were transfected with 2 μg of base editor construct and 1.5 μg of lentiGuide-SLC10A-Puro on 6-well plates using FuGENE 6 transfection reagent (Promega).
Nanoluciferase assay
Cas9 expressing cells were plated on 96-well white microplates. After 24 h of treatment with 0.5 μg/mL of Dox (Sigma-Aldrich, St. Louis, MO, cat. #D9891), base editor protein expression level was quantified by Nluc activity using a Nano-Glo Luciferase Assay System (Promega, Cat. #N1120).
Protein analysis
Cell pellets from 6-well plates were resuspended in 200 μL of Pierce RIPA Buffer (Thermo Fisher Scientific, Waltham, MA) with protease inhibitor (Roche, Basel, Switzerland). After 10 min of incubation at room temperature, cell lysates were centrifuged at 14,000 rpm and 4°C for 10 min to collect protein lysates. After determination of protein concentration using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), automated quantitative western blotting (Wes assay) was performed on a Wes instrument (ProteinSimple, San Jose, CA) according to the manufacturer's instructions. Antibodies to the following proteins were used for the analysis: Cas9 (1:100 dilution; Novus Biologicals, Centennial, CO, cat. #NBP2-36440) and actin (1:100 dilution; Abcam, Cambridge, UK, cat. #ab8227).
Sequencing analysis and genotyping of NTCP-S267F
DNA was extracted using a DNAeasy blood and tissue kit (Qiagen), and target DNA was amplified using LAtaq (TakaraBio) according to the protocol provided by the manufacturer. For analysis of the transcript, RNA was extracted using an ISOLATE II RNA Mini Kit (Meridian Bioscience, Cincinnati, OH). Extracted RNA was reverse transcribed using a Maxima First Strand cDNA Synthesis Kit for qRT-PCR (Thermo Fisher Scientific). Primers for the amplification of SLC10A target site are listed in Table S1. Sanger sequencing was performed after gel purification using NucleoSpin gel and a PCR Clean-up kit (Macherey-Nagel, Dueren, Germany). Base editing efficacy was estimated using EditR (http://baseeditr.com/), an algorithm for predicting potential editing in a guide RNA region from a single Sanger sequencing run.38 For the genotyping assay, the S267F variant (rs2296651) was genotyped using a commercial TaqMan SNP Genotyping Assay (Assay ID: C__16,184,554_10; Applied Biosystems, Foster City, CA) with TaqMan Genotyping Master Mix (Applied Biosystems) on the ViiA 7 Real-Time PCR System (Applied Biosystems) according to the manufacturer's protocol.
Quantitative real-time PCR
For mRNA quantification, RNA was extracted using an ISOLATE II RNA Mini Kit (Meridian Bioscience). qRT-PCR was performed using a qScript One-Step RT-qPCR Kit (Quantabio, Beverly, MA). A relative unit is defined as the expression ratio of target gene against β-actin (ACTB). All qRT-PCR was performed on a ViiA 7 Real-Time PCR System (Applied Biosystems). Primers used for qRT-PCR in this study are listed in Table S1.
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 min. Slides were blocked with 3% BSA in PBS for 15 min, followed by incubation with the primary antibody diluted in 1% BSA/0.1% Triton X-100 in PBS overnight at 4°C. After extensive washing, cells were incubated with the secondary antibody in 1% BSA/0.1% Triton X-100 in PBS for 1 h at room temperature in darkness. Samples were incubated with 4′,6-diamidino-2-phenylindole and mounted. Images were captured with CellSens (Olympus, Tokyo, Japan) or LSM 700 confocal microscope (Zeiss, Oberkochen, Germany). Antibody for NTCP (kindly provided by Bruno Stieger) in 1:100 dilution was used for staining and followed by Alexa 488-conjugated goat anti-rabbit immunoglobulin G.
Hepatocyte-like cell differentiation
Monolayer type iPSCs or ESCs were resuspended using Accutase, and 2 million cells were seeded into one well in 6-well plates previously coated with high-concentration (0.4 mg/mL) growth factor reduced Matrigel matrix (Corning). From the next day, the cells were then cultured with STEMdiff Definitive Endoderm Kit (STEMCELL Technologies) for definitive endoderm induction. For the first day, the cells were cultured in STEMdiff definitive endoderm basal medium with supplements A and B followed by the basal medium with only supplement B for 3 days, with daily change of medium.
Differentiation medium contains high-glucose DMEM, F12, 10% KnockOut Serum Replacement (Gibco), 1% NEAA, 1% glutamine, and 1% PS. For hepatic specification, definitive endoderm cells were resuspended using Accutase and seeded with 79,000 cells/cm2 concentration on new plates coated with low-concentration (0.125 mg/mL) growth factor reduced Matrigel matrix (Corning) in differentiation medium containing 100 ng/mL human growth factor (HGF; PeproTech, Cranbury, NJ), 1% dimethyl sulfoxide (DMSO; Sigma-Aldrich), and 10 μM Rock Inhibitor Y-27632 (STEMCELL Technologies). The cells were cultured for a further 7 days in differentiation medium containing 100 ng/mL HGF and 1% DMSO, with daily change of medium. After the hepatic specification stage, the hepatoblast-like cells were matured in differentiation medium containing 10−7 M dexamethasone (Sigma-Aldrich) for 3 days. Differentiated HLCs were then maintained in hepatocyte maintenance medium, which is William's E Medium containing 10% FBS, 0.17 μM human Insulin (Sigma-Aldrich), 10 μM hydrocortisone 21-hemisuccinate (Sigma-Aldrich), 1.8% DMSO, and 1% PS. Medium was changed every 2–3 days after the differentiation.
HBV infection
HBVcc was concentrated from the supernatant of HepAD38 cells using a centrifugal filter device (Centricon Plus-70, Biomax 100.000; Millipore, Bedford, MA) and titered by HBV-DNA-qPCR. The virus stock was divided into aliquots and stored at −80°C until use. For genotype A and E infection, we used the serum obtained from humanized mice infected with HBVcc (HBVmp).25 HBV-infected mouse serum was stored at −80°C until use.
For infection, HepG2-NTCP cells were seeded on collagen-coated 24-well plates 1 day before HBV infection. The number of stem cell-derived HLCs were estimated at 1 million per well on 24-well plates after the differentiation. Cells were infected with HBVcc at a multiplicity of infection (MOI) of 200, HBVmp genotype A at an MOI of 100, and HBVmp genotype E at an MOI of 20 in cell-culture medium containing 5% polyethylene glycol. At the end of 24 h incubation after HBV inoculation, cells were washed with PBS five times and cultured in DMEM/2% DMSO or hepatocyte maintenance medium in HepG2-NTCP or stem cell-derived HLCs, respectively. Cells and supernatants were harvested 7 days after HBV inoculation.
For the time-of-addition assay, HepG2-NTCP cells were seeded 1 day before and exposed to HBV at 4°C with or without MyrB for 4 h. After the incubation, cells were washed with PBS five times and raised to 37°C in the presence of MyrB. Supernatant was harvested and assessed for HBeAg 3 days after HBV infection.
ELISA
Secreted HBeAg and HBsAg were determined using a Human HBeAg Elisa Kit (CD BioScience, Shirley, NY) and a Human HBsAg Elisa Kit (CD BioScience), respectively. Signal-to-noise ratio (S/N) is presented in the figures. The hALB level in the supernatant was determined using a Human Albumin ELISA Kit (Bethyl Laboratories, Montgomery, TX).
Acknowledgments
This work was supported by the Intramural Research program of the National Institute of Diabetes and Digestive and Kidney Diseases and National Institutes of Health and partially supported by research funding from the Research Program on Hepatitis from the Japan Agency for Medical Research and Development, AMED (grant number 21fk0310109).
Author contributions
Concept and design: T.U., S.B.P., and T.J.L. Experiments, procedures, and data analysis: T.U., S.B.P., M.Z., T.I., J.N.A., and T.J.L. Writing of manuscript: T.U. and T.J.L. Supervision and critical revision of manuscript: K.C. and T.J.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.11.002.
Supplemental information
and Table S1
References
- 1.Liang T.J., Block T.M., Mcmahon B.J., Ghany M.G., Urban S., Guo J.T., Locarnini S., Zoulim F., Chang K.M., Lok A.S. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ni Y., Lempp F.A., Mehrle S., Nkongolo S., Kaufman C., Fälth M., Stindt J., Königer C., Nassal M., Kubitz R., et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology. 2014;146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 4.Seeger C., Mason W.S. Sodium-dependent taurocholic cotransporting polypeptide: a candidate receptor for human hepatitis B virus. Gut. 2013;62:1093–1095. doi: 10.1136/gutjnl-2013-304594. [DOI] [PubMed] [Google Scholar]
- 5.Iwamoto M., Watashi K., Tsukuda S., Aly H.H., Fukasawa M., Fujimoto A., Suzuki R., Aizaki H., Ito T., Koiwai O., et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014;443:808–813. doi: 10.1016/j.bbrc.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 6.Li W., Urban S. Entry of hepatitis B and hepatitis D virus into hepatocytes: basic insights and clinical implications. J. Hepatol. 2016;64:S32–S40. doi: 10.1016/j.jhep.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho R.H., Leake B.F., Roberts R.L., Lee W., Kim R.B. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid Substrate recognition. J. Biol. Chem. 2004;279:7213–7222. doi: 10.1074/jbc.M305782200. [DOI] [PubMed] [Google Scholar]
- 8.Pan W., Song I.S., Shin H.J., Kim M.H., Choi Y.L., Lim S.J., Kim W.Y., Lee S.S., Shin J.G. Genetic polymorphisms in Na+-taurocholate co-transporting polypeptide (NTCP) and ileal apical sodium-dependent bile acid transporter (ASBT) and ethnic comparisons of functional variants of NTCP among Asian populations. Xenobiotica. 2011;41:501–510. doi: 10.3109/00498254.2011.555567. [DOI] [PubMed] [Google Scholar]
- 9.Yan H., Peng B., Liu Y., Xu G., He W., Ren B., Jing Z., Sui J., Li W. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J. Virol. 2014;88:3273–3284. doi: 10.1128/JVI.03478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C., Xu G., Gao Z., Zhou Z., Guo G., Li D., Jing Z., Sui J., Li W. The p.Ser267Phe variant of sodium taurocholate cotransporting polypeptide (NTCP) supports HBV infection with a low efficiency. Virology. 2018;522:168–176. doi: 10.1016/j.virol.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 11.An P., Zeng Z., Winkler C.A. The loss-of-function S267F variant in HBV receptor NTCP reduces human risk for HBV infection and disease progression. J. Infect. Dis. 2018;218:1404–1410. doi: 10.1093/infdis/jiy355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu H.-H., Liu J., Lin Y.-L., Luo W.-S., Chu Y.-J., Chang C.-L., Jen C.-L., Lee M.-H., Lu S.-N., Wang L.-Y., et al. The rs2296651 (S267F) variant on NTCP (SLC10A1) is inversely associated with chronic hepatitis B and progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B. Gut. 2016;65:1514–1521. doi: 10.1136/gutjnl-2015-310686. [DOI] [PubMed] [Google Scholar]
- 13.Li N., Zhang P., Yang C., Zhu Q., Li Z., Li F., Han Q., Wang Y., Lv Y., Wei P., et al. Association of genetic variation of sodium taurocholate cotransporting polypeptide with chronic hepatitis B virus infection. Genet. Test. Mol. Biomarkers. 2014;18:425–429. doi: 10.1089/gtmb.2013.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu W., Zeng Y., Lin J., Wu Y., Chen T., Xun Z., Ou Q. Genetic variants in NTCP exon gene are associated with HBV infection status in a Chinese Han population. Hepatol. Res. 2018;48:364–372. doi: 10.1111/hepr.13007. [DOI] [PubMed] [Google Scholar]
- 15.Binh M.T., Hoan N.X., Van Tong H., Sy B.T., Trung N.T., Bock C.-T., Toan N.L., Song L.H., Bang M.H., Meyer C.G., et al. NTCP S267F variant associates with decreased susceptibility to HBV and HDV infection and decelerated progression of related liver diseases. Int. J. Infect. Dis. 2019;80:147–152. doi: 10.1016/j.ijid.2019.01.038. [DOI] [PubMed] [Google Scholar]
- 16.Lee H.W., Park H.J., Jin B., Dezhbord M., Kim D.Y., Han K.-H., Ryu W.-S., Kim S., Ahn S.H. Effect of S267F variant of NTCP on the patients with chronic hepatitis B. Sci. Rep. 2017;7:17634. doi: 10.1038/s41598-017-17959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng L., Zhao Q., Li Q., Li M., Li C., Xu T., Jing X., Zhu X., Wang Y., Li F., et al. The p.Ser267Phe variant in SLC10A1 is associated with resistance to chronic hepatitis B. Hepatology. 2015;61:1251–1260. doi: 10.1002/hep.27608. [DOI] [PubMed] [Google Scholar]
- 18.Cohen J. “Base editors” open new way to fix mutations. Science. 2017;358:432–433. doi: 10.1126/science.358.6362.432. [DOI] [PubMed] [Google Scholar]
- 19.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurt I.C., Zhou R., Iyer S., Garcia S.P., Miller B.R., Langner L.M., Grünewald J., Joung J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021;39:41–46. doi: 10.1038/s41587-020-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E., et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–896. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulze A., Gripon P., Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46:1759–1768. doi: 10.1002/hep.21896. [DOI] [PubMed] [Google Scholar]
- 24.Somiya M., Liu Q., Yoshimoto N., Iijima M., Tatematsu K., Nakai T., Okajima T., Kuroki K., Ueda K., Kuroda S. Cellular uptake of hepatitis B virus envelope L particles is independent of sodium taurocholate cotransporting polypeptide, but dependent on heparan sulfate proteoglycan. Virology. 2016;497:23–32. doi: 10.1016/j.virol.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M., Zhang Z., Imamura M., Osawa M., Teraoka Y., Piotrowski J., Ishida Y., Sozzi V., Revill P.A., Saito T., et al. Infection courses, virological features and IFN-α responses of HBV genotypes in cell culture and animal models. J. Hepatol. 2021 doi: 10.1016/j.jhep.2021.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpentier A., Tesfaye A., Chu V., Nimgaonkar I., Zhang F., Lee S.B., Thorgeirsson S.S., Feinstone S.M., Liang T.J. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J. Clin. Invest. 2014;124:4953–4964. doi: 10.1172/JCI75456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carpentier A., Nimgaonkar I., Chu V., Xia Y., Hu Z., Liang T.J. Hepatic differentiation of human pluripotent stem cells in miniaturized format suitable for high-throughput screen. Stem Cell Res. 2016;16:640–650. doi: 10.1016/j.scr.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Y., Carpentier A., Cheng X., Block P.D., Zhao Y., Zhang Z., Protzer U., Liang T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017;66:494–503. doi: 10.1016/j.jhep.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–848. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thanapirom K., Suksawatamnuay S., Sukeepaisarnjaroen W., Treeprasertsuk S., Tanwandee T., Charatcharoenwitthaya P., Thongsawat S., Leerapun A., Piratvisuth T., Boonsirichan R., et al. Association of the S267F variant on NTCP gene and treatment response to pegylated interferon in patients with chronic hepatitis B: a multicentre study. Antivir. Ther. 2018;23:67–75. doi: 10.3851/IMP3179. [DOI] [PubMed] [Google Scholar]
- 31.An P., Winkler C.A. Host genes associated with HIV/AIDS: advances in gene discovery. Trends Genet. 2010;26:119–131. doi: 10.1016/j.tig.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helenius A. Virus entry: looking back and moving forward. J. Mol. Biol. 2018;430:1853–1862. doi: 10.1016/j.jmb.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loglio A., Ferenci P., Uceda Renteria S.C., Tham C.Y.L., van Bömmel F., Borghi M., Holzmann H., Perbellini R., Trombetta E., Giovanelli S., et al. Excellent safety and effectiveness of high-dose myrcludex-B monotherapy administered for 48 weeks in HDV-related compensated cirrhosis: a case report of 3 patients. J. Hepatol. 2019;71:834–839. doi: 10.1016/j.jhep.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Blank A., Markert C., Hohmann N., Carls A., Mikus G., Lehr T., Alexandrov A., Haag M., Schwab M., Urban S., et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016;65:483–489. doi: 10.1016/j.jhep.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko C., Chakraborty A., Chou W.M., Hasreiter J., Wettengel J.M., Stadler D., Bester R., Asen T., Zhang K., Wisskirchen K., et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018;69:1231–1241. doi: 10.1016/j.jhep.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishida Y., Yamasaki C., Yanagi A., Yoshizane Y., Fujikawa K., Watashi K., Abe H., Wakita T., Hayes C.N., Chayama K., et al. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. Am. J. Pathol. 2015;185:1275–1285. doi: 10.1016/j.ajpath.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 38.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: a method to quantify base editing from Sanger sequencing. Cris. J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and Table S1