

Abstract
Aims:
Neuronal cytoplasmic inclusions of TDP-43 are a pathological hallmark of diverse neurodegenerative disorders, yet the processes that mediate their formation and their functional significance remain incompletely understood. Both dysfunction in autophagy and neuroinflammation have been linked to TDP-43 mislocalisation. Here, we investigate TDP-43 proteinopathy in Niemann-Pick type C disease (NPC), an autosomal recessive lysosomal storage disease (LSD) distinguished by the accumulation of unesterified cholesterol within late endosomes and lysosomes. NPC is characterized by neurodegeneration, neuroinflammation and multifocal disruption of the autophagy pathway.
Methods:
We utilized immunohistochemistry, confocal microscopy, electron microscopy, and biochemical and gene expression studies to characterize TDP-43 pathology and autophagic substrate accumulation in Npc1 deficient mice.
Results:
In the NPC brain, cytoplasmic TDP-43 mislocalisation was independent of autophagic substrate accumulation. These pathologies occurred in distinct neuronal subtypes, as brainstem cholinergic neurons were more susceptible to TDP-43 mislocalisation while glutamatergic neurons exhibited hallmarks of autophagic dysfunction. Furthermore, TDP-43 mislocalisation did not co-localise with markers of stress granules or progress to ubiquitinated aggregates over months in vivo, indicating a stable, early stage in the aggregation process. Neither microgliosis nor neuroinflammation were sufficient to drive TDP-43 proteinopathy in the NPC brain. Notably, cytoplasmic TDP-43 co-localised with the nuclear import factor importin α, and TDP-43 mislocalised neurons demonstrated nuclear membrane abnormalities and disruption of nucleocytoplasmic transport.
Conclusion:
Our findings highlight the relationship between LSDs and TDP-43 proteinopathy, define its functional importance in NPC by triggering nuclear dysfunction, and expand the spectrum of TDP-43 pathology in the diseased brain.
Keywords: Niemann-Pick type C, TDP-43, lysosomal diseases, autophagy, nucleocytoplasmic transport
Introduction
A pathological hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) is nuclear depletion and neuronal cytoplasmic inclusions of TAR-DNA binding protein of 43 kDa (TDP-43) (1, 2). Although ALS and FTD are biochemically, genetically and clinically heterogeneous, pathological TDP-43 cytoplasmic inclusions are observed in over 90% of ALS and over 50% of FTD patients, highlighting its role in disease pathogenesis (3). TDP-43 is a nuclear RNA-binding protein that functions in RNA processing, including splicing, translation and degradation, and its expression levels and localisation are critical for normal cellular function. TDP-43 knockout and overexpression cause neurodegeneration (4-7), and cytoplasmic mislocalisation of TDP-43 is sufficient to drive neuron death (8). Despite increased understanding of both gain- and loss-of-function pathological mechanisms of TDP-43 mislocalisation (9), the processes that mediate the formation of these pathological aggregates remain incompletely understood.
TDP-43 contains a C-terminal low-complexity domain which renders it aggregation prone (10). Nearly all ALS-associated TDP-43 mutations are concentrated in this region, but contribute to only 2%-5% of ALS cases (9, 10). Increasingly, dysfunction in the clearance of TDP-43 aggregates by autophagy has been linked to disease pathogenesis (11-13), and autophagy induction enhances neuronal survival in ALS models (14). Consistent with this idea, multiple ALS- and FTD-associated genes are linked to autophagy and the lysosomal pathway (15), including C9ORF72, TBK1, UBQLN2, VCP, SQSTM1, and OPTN, and mutations in these genes result in TDP-43 pathology (16-21). Furthermore, mouse and patient brains with progranulin mutations that result in FTD accumulate TDP-43 cytoplasmic inclusions and exhibit pathologic hallmarks of the lysosomal storage disease (LSD) neuronal ceroid lipofuscinosis (22), highlighting a link between lysosomal dysfunction and TDP-43 proteinopathy.
Here, we investigate TDP-43 proteinopathy in Niemann-Pick type C disease (NPC), an autosomal recessive LSD characterized by the accumulation of unesterified cholesterol in lysosomes and late endosomes (23). Most cases of NPC (~95%) are due to mutations in the NPC1 gene (24), although a small subset (~5%) is due to mutations in NPC2. NPC1 and NPC2 function in concert to export cholesterol from lysosomes (25-28). Loss-of-function mutations in either of these proteins lead to cholesterol accumulation, liver disease, severe neurodegeneration, and early death, often in childhood (29). Several general principles have emerged that guide our understanding of NPC pathogenesis. The importance of lipid storage in the disease phenotype is apparent, as reduction of cholesterol storage by hydroxypropyl-beta-cyclodextrin dampens neurodegeneration and prolongs lifespan in Npc1 mutant mice and cats (30-32); similarly, peripheral administration of synthetic HDL nanoparticles to NPC mice mobilizes cholesterol and rescues liver dysfunction and body weight (33). Lipid storage has also been linked to lysosomal membrane permeabilization in NPC (34-36), highlighting the importance of functional lysophagy to mitigate cytotoxicity (36). Furthermore, the significance of neurons in disease has been established. Conditional deletion of Npc1 in neurons leads to age-dependent, cell autonomous neuron loss without evidence of a critical window for neuronal vulnerability (37) as well as non-cell autonomous dysmyelination (38). Conversely, transgenic over-expression of Npc1 in neurons of null mice ameliorates neurodegeneration (39). Notably, as lysosomal function is essential for autophagy, LSDs can also be viewed primarily as “autophagy disorders” (40), and NPC models exhibit multifocal disruption of macroautophagy (41-48).
With the association between dysregulated autophagy and TDP-43 proteinopathy, we sought to characterize neuronal TDP-43 pathology in NPC, examine mechanisms that drive TDP-43 mislocalisation, and explore functional consequences in the NPC brain. We show that Npc1−/− neurons accumulate cytoplasmic mislocalised TDP-43. Unexpectedly, this TDP-43 pathology occurs independently of autophagic substrate accumulation and does not progress to ubiquitinated protein aggregates over months in vivo. Moreover, we find that alternative factors implicated in TDP-43 pathology, including microgliosis and neuroinflammation are insufficient to drive TDP-43 mislocalisation in the NPC brain. We also present evidence of nuclear pathology and disruption in nucleocytoplasmic transport in Npc1−/− neurons with mislocalised TDP-43. Our findings highlight the relationship between TDP-43 pathology and LSDs, expand the spectrum of TDP-43 pathology in the diseased brain, and provide evidence of nuclear dysfunction in an LSD.
Materials and Methods
Primary antibodies (antigen, dilution, vendor)
Neurofilament, 1:200, Millipore MAB5266; Neurofilament, 1:1000, Abcam ab8135; NeuN, 1:500, Millipore ABN78; NeuN, 1:500, Millipore ABN90; Synapsin-1, 1:200, Cell Signaling D12G5; GFAP, 1:500, Dako Z0334; Tom20, 1:500, Santa Cruz Biotechnology Inc. sc-11415; Ubiquitin, 1:400, Cell Signaling 3933S; phospho-Ubiquitin (Ser65), 1:100, Millipore ABS1513-I; SQSTM1, 1:500, Abnova H00008878-MO1; LC3B, 1:500, Novus NB100-2220; TDP-43, 1:500, Proteintech 10782-2-AP; karyopherin α2, 1:150, Santa Cruz Biotechnology Inc. sc-55538; TDP43 Phospho (Ser409/410), 1:100 Biolegend 829901; TIA-1, 1:200, Abcam ab40693; G3BP1, 1:500 Proteintech 103057-2-AP; Nup62, 1:500 BD 610497; Lamin B1, 1:1000, abcam ab16048; LMP2, 1:1000, abcam ab3328; Iba1, 1:500, Wako 019-19741; Iba1, 1:100 abcam ab5076, LMP7, 1:500, abcam ab3329; PSMB10, 1:1000, abcam ab183506; β-Actin, 1:2000, Sigma-Aldrich A5441; Vinculin, 1:2000, Sigma-Aldrich V9131; p62/SQSTM1, 1:1000, Sigma-Aldrich P0067; VDAC1, 1:500, abcam ab14734; VGLUT1, 1:500, Synaptic Systems 135303; VGAT cytoplasmic domain, 1:100, Synaptic Systems 131008; Choline Acetyltransferase, 1:100, Sigma-Aldrich AB144P; FUS, 1:500, Sigma-Aldrich HPA008784; hnRNPA2/B1, 1:500, Santa Cruz Biotechnology Inc. sc-374053
Secondary antibodies
Alexa Fluor™ 594 donkey anti-goat IgG (H+L), 1:500, Invitrogen A11058; Alexa Fluor™ 594 Fab’2 fragment of goat anti-rabbit IgG (H+L), 1:500, Invitrogen A11072; Alexa Fluor™ 594 Fab’2 fragment of goat anti-mouse IgG (H+L), 1:500, Invitrogen A11020; Alexa Fluor™ 488 goat anti-rabbit IgG (H+L), 1:500, Invitrogen A11008; Alexa Fluor™ 594 goat anti guinea pig IgG (H+L), Invitrogen A1076; Alexa Fluor™ 488 goat anti-mouse IgG (H+L), Invitrogen A11029; Alexa Fluor™ 488 rabbit anti mouse IgG (H+L), 1:500, Invitrogen A11059; Alexa Fluor 488™ Tyramide SuperBoost™ Kit, goat anti-mouse IgG, Invitrogen B40941; Goat anti-mouse IgG (H+L)-HRP conjugate, 1:2000, Bio-Rad 170-6516; Goat anti-rabbit IgG (H+L)-HRP conjugate, 1:2000, Bio-Rad 170-6515
Mice
Npc1nih Balb/cJ mice were obtained from Jackson Laboratories (#003092) and backcrossed to C57BL6/J (≥10 generations). Npc1−/− mice were F1 hybrids between Npc1+/− mice on the C57BL6/J and Balb/cJ backgrounds, respectively, to restore the Mendelian frequency of the Npc1−/− genotype (41). Npc1flox/flox were generated as previously described (49). Syn1-Cre mice were obtained from Jackson Laboratories (#003474) and backcrossed to C57BL6/J (≥10 generations). Npc1-I1061T mice (50) were a gift from Daniel Ory (Washington University in St Louis) and backcrossed to C57BL/6 (≥10 generations).
Western blot
For tissue preparation, mice were perfused with saline before tissue was collected and flash frozen in liquid nitrogen. Tissue was homogenized and sonicated in RIPA buffer (Teknova) with complete protease inhibitor (Thermo Scientific 11836153001) and 0.625mg/ml N-ethylmaleimide (Sigma E3876). Protein concentrations were determined by DC™-protein assay (Bio-Rad) and normalized. For preparation of soluble and insoluble fractions, total lysates were spun at 20,000xg for 10 min. The supernatant was run as the soluble fraction, and the pellet was resuspended and run as the insoluble fraction. Proteins were separated on NuPAGE™ 4-12% Bis-Tris Protein Gels (Thermo Scientific NP0336BOX) and transferred to Immobilon-P 0.45um PVDF (Merck Millipore). Immunoreactivity was detected with ECL (Thermo Scientific) or SuperSignal™ West Pico PLUS Chemiluminscent Substrate (Thermo Scientific) and an iBright (Thermo Fisher Scientific). Quantification was performed using Image Studio. Band intensity was normalized to the indicated loading control.
RT-qPCR
RNA was collected using TRIzol® (Thermo Fisher) according to manufacturer’s instructions and converted to cDNA using the High Capacity Reverse Transcription kit (Applied Biosystems 4368814). Quantitative real-time PCR was performed using 10ng cDNA, FastStart Taqman Probe Master Mix (Roche), and gene-specific FAM-labelled TaqMan probes (Thermo Scientific) for mouse C3 (Mm01232779_m1), C1qa (Mm07295529_m1), PSMB8 (Mm00440207_m1), PSMB9 (Mm00479004_m1), PSMB10 (Mm00479052_g1), PSME1 (Mm00650858_g1), PSME2 (Mm01702832_g1), PSMB5 (Mm01615821_g1), and PSMB7 (Mm01327044_m1). Gene expression was normalized to mouse Cpsf2-Vic (Mm00489754) multiplexed within the same well. RT-qPCR was performed using an ABI 7900HT Sequence Detection System and relative expression calculated by the 2^(−ΔΔCt) method.
Immunofluorescence staining
For tissue preparation, mice were perfused with saline and 4% PFA. Then, tissue was removed and post-fixed in 4% PFA overnight at 4°C. Brains were bisected; one hemisphere processed for paraffin embedding, and the other half was incubated in 30% sucrose for 48hr at 4°C and frozen in O.C.T. (Tissue-Tek). Paraffin-embedded tissues were cut on a Reichert-Jung 2030 microtome into 5μm sections and placed on Fisher Scientific Superfrost Plus microscope slides. Sections were adhered onto slides in an oven at 55-60°C for 1 hr. Frozen sections were prepared at 16μm in a cryostat.
For staining of paraffin embedded sections:
Samples were deparaffinized and antigen retrieval was performed by boiling in 10mM sodium citrate (pH 6.0) for 10 min and incubating in hot citrate solution for an additional 20 min, then washed 3x in deionized water. For staining, slides were incubated in a solution with 0.1% Triton, 10% goat or donkey serum and 1% BSA in PBS for 20 min. Then, slides were placed in blocking solution (10% goat or donkey serum, 1% BSA in PBS) before incubating in primary antibody diluted in blocking solution overnight at 4°C. Slides were washed 3x in PBS and incubated for 1 hr with secondary antibody diluted in blocking solution. Slides were then washed 3x in PBS and mounted with Vectashield + DAPI (Vector Laboratories).
For all studies, at least 3 mice/genotype and age were stained with each antibody. Images are representative of consistent findings across these cohorts.
Filipin staining
For filipin staining of tissue, frozen sections were stained for immunofluorescence as described above (without deparaffinization). Following the washes after secondary antibody incubation, sections were incubated in filipin staining solution (4mL FBS + 40uL filipin solution (1mg filipin [Sigma, F9765] + 40μL DMSO) + 16mL PBS) for 2 hrs at room temperature. Slides were washed 3x in PBS and mounted with Vectashield (Vector Laboratories).
RNA Fluorescence In Situ Hybridization with Immunofluorescence
Immunofluorescence staining was performed on paraffin embedded tissue sections as described above. Following the washes after secondary antibody incubation, slides were washed in Stellaris® RNA FISH Wash Buffer A (Biosearch Technologies) with 10% formamide for 2-5 min. Then, slides were incubated with a solution containing 125 nM oligo-d(T) probes conjugated to CAL Fluor Red 610 (Biosearch Technologies) in Stellaris® RNA FISH Hybridization Buffer (Biosearch Technologies) with 10% formamide for 4 hrs at 37°C. Slides were then washed in Stellaris® RNA FISH Wash Buffer A (Biosearch Technologies) for 30 min at 37°C, Stellaris® RNA FISH Wash Buffer B (Biosearch Technologies) for 2-5 min, then 3x in PBS. Finally, slides were mounted with Vectashield + DAPI (Vector Laboratories).
Proteasome activity
To measure the activity of immunoproteasome subunits, Me4BodipyFL-Ahx3Leu3VS was used as previously described (51). Briefly, tissue was homogenized and sonicated in HR buffer (50mM Tris-HCl [pH 7.4], 5mM MgCl2, 250mM sucrose, 1mM DTT, 2mM ATP). Protein concentrations were determined by DC™-protein assay (Bio-Rad) and incubated with 1μM Me4BodipyFL-Ahx3Leu3VS for 1 hr at 37°C. Samples were run on NuPAGE™ 12% Bis-Tris Protein Gels (Thermo Scientific) at 140V for 2 hrs and imaged using a Typhoon Trio Plus Scanner (Amersham Bioscience).
Electron Microscopy
Mice were perfused with 0.9% normal saline followed by 3% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M Sorensen's buffer. The brainstem was removed and post-fixed in perfusion solution overnight, followed by fixation in 1% osmium tetroxide solution for 1 h at room temperature. After dehydration, tissues were embedded in epoxy resin. For transmission electron microscopy, ultrathin sections were cut, and images were captured on a Philips CM-100 imaging system.
Microscopy
Confocal images were collected using a Nikon A-1 confocal microscope with diode-based laser system. Filipin images were captured on a Zeiss Axioplan 2 imaging system. For quantification of the nuclear-to-cytoplasmic (N:C) ratio of poly(A) RNA, the pixel intensity in the nucleus and cytoplasm was measured by ImageJ. Quantification of circularity of Lamin B1 staining was performed by ImageJ. Co-localisation was determined using CellProfiler Analyst Software.
Phenotype analysis
Balance beam:
The balance beam consists of a 12mm diameter circular beam (thick beam) and a 5mm wide square beam (thin beam) suspended at 50cm. Mice were trained at 4 weeks of age to cross the beam and then tested every other week starting at 5 weeks. For testing, mice were run 3 times across the beam, and the average time was taken. Maximum time was set at 20 sec and falls were scored as 20 sec.
Rotarod:
After acclimating to the testing room for 30 min, mice were gently placed on a moving (4 RPM) rotarod for 30 sec. Then, over a period of 235 sec, rotarod speed was increased to 40 RPM. The trial ended if mice stopped walking for two revolutions or dropped onto the paddle. Mice were trained three times per day with a 30 min interval between each training session over a period of 3 days. The following week, mice were tested on a single day using the training protocol.
Statistics
Graphpad Prism 7.0 was used to determine significance (P<0.05), F (F-statistic) and t (T-statistic) values. Unpaired Student’s t-test (two-tailed) and one-way or two-way ANOVA were used as indicated in the figure legends. A P value less than 0.05 was considered significant. All error bars are s.e.m.
Results
Npc1 deficiency triggers age-dependent accumulation of swollen axons containing autophagic cargo
As NPC1 deficiency causes autophagic impairment, we sought to characterize the accumulation of autophagic substrates in mutant mouse brain, initially focusing on swollen axons (so-called spheroids) that are a prominent neuropathologic feature in the brainstem of diseased mice. Axonal spheroids, as identified by neurofilament staining, appeared as NPC1 deficient mice aged (Supplementary Figure 1). In 4-week Npc1−/− mice, before the onset of behavioural phenotypes (Supplementary Figure 2), axonal spheroids were sparse; as mice progressed in the disease course to 11 weeks, the numbers of axonal spheroids significantly increased (Figure 1a, Supplementary Figure 1a). Similar pathology occurs in the brainstem of mice deficient for Npc1 only in neurons (37). To further characterize the cell autonomous development of axonal spheroids, we utilized Npc1flox, Syn1-Cre+ mice, which harbour a conditional null allele of the Npc1 gene (Npc1flox) (49) and express Cre recombinase as a transgene under the control of the Synapsin1 promoter (Syn1-Cre+). In these mice, Npc1 is deleted in neurons throughout the brain during late embryonic development (52). As with global Npc1−/− mice, axonal spheroids increased in an age-dependent manner in Npc1flox/−, Syn1-Cre+ mice from 7 to 16 weeks (Figure 1a, Supplementary Figure 1b).
Figure 1. Npc1 deficiency triggers age-dependent accumulation of swollen axons containing autophagic cargo.

(a) Axonal spheroids in the brainstem, quantified from 4-5 fields, N=3 mice per genotype and age. (b) Electron micrographs of the brainstem taken from 16-week Npc1flox/−, Syn-Cre+ mice. Top: An axonal spheroid surrounded by normal sized axons. Scale bar: 2 μm. Bottom: Higher magnification shows accumulation of vesicular cargo. Scale bar: 500 nm. (c) Brain sections from 16-week Npc1flox/+, Syn-Cre+ and Npc1flox/−, Syn-Cre+ mice were stained with the indicated markers and imaged by confocal microscopy. Scale bar: 10 μm. Data are shown as mean ± s.e.m. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001 by (a) one-way ANOVA with Tukey’s multiple comparisons (a) F=10.04 (top); F=566.1 (bottom)
Axonal spheroids may reflect impaired autophagy, as autophagosomes are formed at the distal ends of axons and must be transported in a retrograde manner towards the cell soma for autolysosome formation and degradation; disruption of this process can cause accumulation of cargo and dysfunctional organelles within swollen axons (53-55). Accordingly, deleting essential autophagy genes in Purkinje cells causes axonal swellings (56, 57), and swollen axons in mouse models of Alzheimer’s disease contain autophagic cargo (58-60). In agreement with these observations, electron micrographs of 16-week Npc1flox/−, Syn1-Cre+ mice revealed striking enlargement of axons in the brainstem with the accumulation of polymorphous vesicular structures, including vesicles consistent with double-membraned autophagosomes and mitochondria (Figure 1b). To confirm the presence of autophagic and organellar cargo, we performed immunofluorescent staining on the brainstem of Npc1flox/−, Syn1-Cre+ mice. Both ubiquitinated cargo and autophagosomes were visualized within axonal spheroids (Figure 1c). Furthermore, Tom20 staining identified mitochondria (Figure 1c). As impaired neuronal autophagy leads to accumulation of dysfunctional mitochondria (61), and mitochondrial abnormalities occur in NPC (46, 62, 63), we sought to determine if damaged mitochondria accumulated within swollen axons. Indeed, staining for pS65-Ub, a marker of damaged mitochondria (64), indicated the presence of mitochondria targeted for degradation by autophagy (Figure 1c). Supporting this conclusion, staining for pS65-Ub co-localised with VDAC1 (Mander’s coefficient = 0.84) (Supplementary Figure 3). Altogether, these findings demonstrated an accumulation of autophagic and organellar cargo within the swollen axons of Npc1-deficient mice.
Independent accumulation of autophagic cargo and cytoplasmic TDP-43 mislocalisation
Defective autophagy is linked to the development of ALS/FTD and to the cytoplasmic mislocalisation and aggregation of TDP-43 (11, 13-15, 65). As autophagic impairment is prominent in NPC (41-48) and manifests with the accumulation of autophagic cargo (Figure 1), we sought to determine whether TDP-43 proteinopathy was present in Npc1-deficient mice. Interestingly, in 4-week Npc1−/− mice, we observed cytoplasmic mislocalisation of TDP-43 primarily within brainstem neurons (Figure 2a). This occurred at an early age, before the onset of behavioural phenotypes (Supplementary Figure 2). With the described connection between deficient autophagy and TDP-43 mislocalisation, we determined whether TDP-43 mislocalised neurons also accumulated autophagic cargo. As a marker of autophagic substrate accumulation, we stained for p62, showing accumulation in neuronal cell bodies of Npc1-deficient mice, but not at synapses or in astrocytes (Supplementary Figure 4). Unexpectedly, we observed that neurons accumulating p62 did not accumulate cytoplasmic TDP-43 and vice versa (Figure 2b, 2d). This indicated that Npc1−/− mice harbour distinct subpopulations of neurons: those that accumulate autophagic cargo and those that accumulate mislocalised TDP-43. Notably, filipin staining showed that both subpopulations of neurons accumulated unesterified cholesterol (Supplementary Figure 5), suggesting divergent pathologic responses following lipid storage.
Figure 2. Independent accumulation of autophagic cargo and cytoplasmic TDP-43 mislocalisation.

(a-c) The brainstem from 4-week Npc1+/+ and Npc1−/− mice was stained for the indicated markers and imaged by confocal microscopy. Scale bar: 10 μm. (d) Left: The percentage of TDP-43 mislocalised cells co-localised with importin α or p62. Right: The percentage of p62+ cells co-localised with cytoplasmic TDP-43. Twenty to thirty TDP-43+ or p62+ cells were quantified per mouse, N=3 mice. Data are shown as mean ± s.e.m.
Given its role as a ubiquitin binding protein, lack of p62 co-localisation indicated that cytoplasmic TDP-43 was not present in ubiquitinated aggregates, but instead resembled TDP-43 de-mixing (the formation of cytoplasmic liquid droplets) that has been reported to occur in cell culture at an early stage of progressive TDP-43 aggregation (66). We investigated whether this cytoplasmic TDP-43 co-localised with other proteins shown to de-mix with it in vitro, including nuclear transport factors (66). Indeed, the nuclear import factor importin α co-localised with TDP-43 in affected neurons (Figure 2c-d), suggesting the occurrence of impaired nucleocytoplasmic transport with TDP-43 mislocalisation. Unlike ALS/FTD, we did not observe coincident nuclear clearance of TDP-43 (Figure 2a), a hallmark of TDP-43 aggregation (2) that has been shown to promote neurodegeneration (67). Our findings support an early stage of TDP-43 proteinopathy in 4-week Npc1−/− mice.
TDP-43 cytoplasmic mislocalisation persists in vivo for months, up to the end-stage in Npc1−/− mice
With prolonged stress, cytoplasmic TDP-43 droplets progress to aggregates and clear TDP-43 from the nucleus in cell culture (66). As aberrant phase transitions are suggested to drive the formation of pathological inclusions of RNA-binding proteins (10, 68, 69), we sought to determine if cytoplasmic mislocalised TDP-43 in 4-week Npc1−/− mice progressed to aggregates in Npc1−/− mice at the end-stage of disease. Thus, we examined 11-week Npc1−/− mice, which have developed significant weight loss and motor impairment (Supplementary Figure 2). Unexpectedly, TDP-43 pathology at this stage was indistinguishable from that present at 4-weeks of age (Figure 2). We focused our analysis on the brainstem, where TDP-43 and importin α mislocalised neurons were prominent. We did not observe similar findings in the cerebellum (Supplementary Figure 6).
TDP-43 mislocalised to the cytoplasm of 11-week Npc1−/− neurons, with continued localisation in the nucleus (Figure 3a). Furthermore, neurons that accumulated mislocalised TDP-43 did not accumulate p62, but co-localised with importin α (Figure 3b-d). Similar findings were observed in 16-week Npc1flox, Syn1-Cre+ mice following conditional deletion of Npc1 in neurons (Figure 3e, Supplementary Figure 7). Therefore, cytoplasmic mislocalised TDP-43 in Npc1-deficient neurons persisted in vivo for months without progression to ubiquitinated aggregates or nuclear depletion and remained independent of autophagic substrate accumulation.
Figure 3. TDP-43 cytoplasmic mislocalisation persists in vivo for months, up to the end-stage in Npc1−/− mice.

(a-c) The brainstem from 11-week Npc1+/+ and Npc1−/− mice was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 10 μm. (d-e) Left: The percentage of TDP-43 mislocalised cells co-localised with importin α or p62. Right: The percentage of p62+ cells co-localised with cytoplasmic TDP-43. Twenty to thirty TDP-43+ or p62+ cells were quantified per mouse, N=3 mice per genotype. (f) Top: The brainstem from 11-week Npc1−/− mice was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 5 μm. Bottom: The percentage of importin α+ or p62+ that were ChAT+. Fifteen to twenty-five cells were quantified per mouse, N=3 mice per genotype. (g) Left: The brainstem from 11-week Npc1−/− mice was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 5 μm. Right: The percentage of VGLUT1+ cells that were p62+ or importin α+. Fifteen to twenty-five cells were quantified per mouse, N=3 mice per genotype. (h) Brainstem from 11-week Npc1−/− mice was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 5 μm. Data are shown as mean ± s.e.m. **P ≤ 0.01 by (f-g) Student’s t-test (f) t=5.388 (g) t=7.429
Moreover, as TDP-43 can be recruited to stress granules, which may increase its propensity to form pathological aggregates (70-72), we stained for stress granule markers G3BP1 and TIA-1 but found no co-localisation (Supplementary Figure 8a-b). We conclude that TDP-43 mislocalisation occurred independently of stress granule formation, in agreement with prior observations of TDP-43 de-mixing (66, 73).
We sought to determine whether other hallmarks of TDP-43 proteinopathy were present in Npc1-deficient neurons, including phosphorylated TDP-43 and the accumulation of C-terminal fragments (2). Immunofluorescence and western blot analysis were negative for these pathologies, and TDP-43 protein levels were unchanged in both total brain lysates and in soluble and insoluble fractions (Supplementary Figure 8c, Supplementary Figure 9). Furthermore, despite co-localisation with importin α (Figure 2c, Figure 3c), we did not observe mislocalisation of the nucleoporin Nup62 (Supplementary Figure 8d), which has been associated with TDP-43 pathology in vitro (66, 74). Similarly, we did not observe cytoplasmic mislocalisation of FUS or hnRNPA2/B1, proteins that also contain low-complexity domains and are mutated in some ALS/FTD patients (Supplemental Figure 8e-f).
To examine neuron subtypes involved by these pathologies, we first co-stained for choline acetyltransferase (ChAT) to identify brainstem cholinergic neurons. Interestingly, almost 50% of neurons that were importin α+, but not p62+, co-stained with ChAT (Figure 3f), indicating a preferential susceptibility to TDP-43 pathology among brainstem cholinergic neurons. Conversely, p62 but not importin α accumulated in VGLUT1+ cells (Figure 3g), indicating that brainstem glutamatergic neurons preferentially accumulated autophagic substrates over mislocalised TDP-43. VGAT expressing neurons have been shown to form inhibitory synapses on excitatory neurons (75). Correspondingly, we found VGAT+ synapses surrounding the soma of p62+ but not importin α+ neurons (Figure 3h), revealing differential inhibitory signalling and connectivity between these neuron populations.
Progressive accumulation of autophagic cargo without TDP-43 cytoplasmic mislocalisation in Npc1-I1061T mice
To further characterize the relationship between autophagic dysfunction and TDP-43 pathology we studied Npc1-I1061T knock-in mice (50), which harbour the most common disease-causing allele in patients of Western European ancestry (76). In contrast to null models, Npc1-I1061T mice express mutant NPC1 protein that misfolds in the ER and is largely degraded, while a small fraction of protein that escapes degradation can traffic to the lysosome and function in cholesterol efflux (77, 78). Therefore, Npc1-I1061T mice exhibit a similar although milder phenotype than Npc1−/− mice. We first assessed autophagic impairment in this model. Npc1-I1061T mice developed stored cholesterol as early as 3.7 weeks of age and showed progressive accumulation of ubiquitinated substrates, axonal spheroids and p62 by 7 weeks of age (Figure 4, Supplementary Figure 10). However, despite the progressive accumulation of autophagic cargo, Npc1-I1061T mice did not accumulate either cytoplasmic TDP-43 or importin α in neurons (Figure 4e). This observation further supported the conclusion that autophagic impairment is not sufficient to drive TDP-43 mislocalisation, and provided a model system in which to test the contribution of alternative factors that have been implicated as underlying TDP-43 pathology.
Figure 4. Progressive accumulation of autophagic cargo in Npc1-I1061T mice.

(a-c) The brainstem from Npc1-I1061T mice at 3.7, 7 and 10-weeks of age was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 10 μm. (d) The timeline summarises the age-dependent accumulation of cholesterol and autophagic substrates in I1061T mice. (e) Importin α+ cells in the brainstem, quantified from N=3 mice per genotype. Data are shown as mean ± s.e.m.
Recent studies have focused on the role of microglia in neurodegenerative disease (79, 80) because of their production of pro-inflammatory cytokines and other factors that underlie neuroinflammation (81). Neurotoxic microglia and complement activation promote TDP-43 proteinopathy in progranulin deficient neurons (82). Thus, we quantified the number of Iba1+ microglia in the brainstem of 4-week and 11-week Npc1+/+ and Npc1−/− mice and compared them with 11-week WT and Npc1-I1061T mice, which display a milder phenotype than 11-week Npc1−/− mice but which have progressed in disease with neuropathological and behavioural deficits (50). We found that 11-week Npc1-I1061T mice exhibited intermediate levels of microgliosis compared to 4- and 11-week Npc1−/− mice (Figure 5a). Despite microgliosis at greater levels than that of 4-week Npc1−/− mice, TDP-43 was not mislocalised in Npc1-I1061T neurons (Figure 4e). Similarly, expression of genes encoding complement factors C1qa and C3 in 11-week Npc1-I1061T mice was intermediate to that of 4- and 11-week Npc1−/− mice (Figure 5b). Thus, induction of microgliosis and expression of complement components were not sufficient to account for TDP-43 mislocalisation in NPC1 deficient neurons.
Figure 5. Microgliosis and complement activation in NPC mice.

(a) The brainstem from 4-week Npc1+/+ and Npc1−/− mice, 11-week WT and Npc1-I1061T mice, and 11-week Npc1+/+ and Npc1−/− mice was stained with Iba-1 and imaged by confocal microscopy. Scale bar: 10 μm. Right: Iba1+ cells per field, quantified from 4-5 fields, N=3 mice per genotype. (b) Relative expression of C1qa and C3 mRNA in brainstem by qPCR. N=4-5 mice per genotype. Data are shown as mean ± s.e.m. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001 by (a-b) one-way ANOVA with Tukey’s multiple comparisons (a) F=64.48; (b) F=40.57 (C1qa) and F=22.51 (C3)
To more broadly assess neuroinflammation in the NPC brain, we examined induction of the immunoproteasome (83). While the constitutive proteasome is a key effector of proteolysis, the immunoproteasome plays an important role in antigen processing and is induced in response to oxidative stress and proinflammatory cytokines in neurodegenerative diseases (84, 85). The constitutive proteasome is composed of a 20S cylindrical core containing four stacked α and β rings, with the β rings containing three catalytic β subunits (PSMB5-7). In contrast, the immunoproteasome incorporates three alternative β subunits (PSMB8-10) that replace the constitutive catalytic subunits as well as distinct 11S lid components (PSME1-2) (86). In Npc1-I1061T mice, expression of these immunoproteasome subunits was increased in the cerebellum and brainstem in an age-dependent manner (Figure 6a-b, Supplementary Figure 11a-b). This induction was specific to immunoproteasome subunits as expression of constitutive proteasome β subunits was unchanged (Supplementary Figure 11c). Correspondingly, protein levels (Figure 6c) and activity (Supplementary Figure 11d) of immunoproteasome subunits were increased, and immunofluorescence localised the immunoproteasome to both microglia and axonal spheroids (Supplementary Figure 11f). Notably, immunoproteasome subunit expression was increased to comparable levels in the brainstem of 11-week Npc1−/− mice (Figure 6d), while immunoproteasome subunit expression was less robustly increased in 4-week Npc1−/− mice (Supplementary Figure 11e). Thus, neuroinflammation as reflected by microgliosis, complement expression, and immunoproteasome induction occurred similarly in Npc1−/− and I1061T mice. Our analyses suggest a role for alterative stressors in the occurrence of TDP-43 pathology in Npc1−/− neurons.
Figure 6. The immunoproteasome is induced in NPC mice.
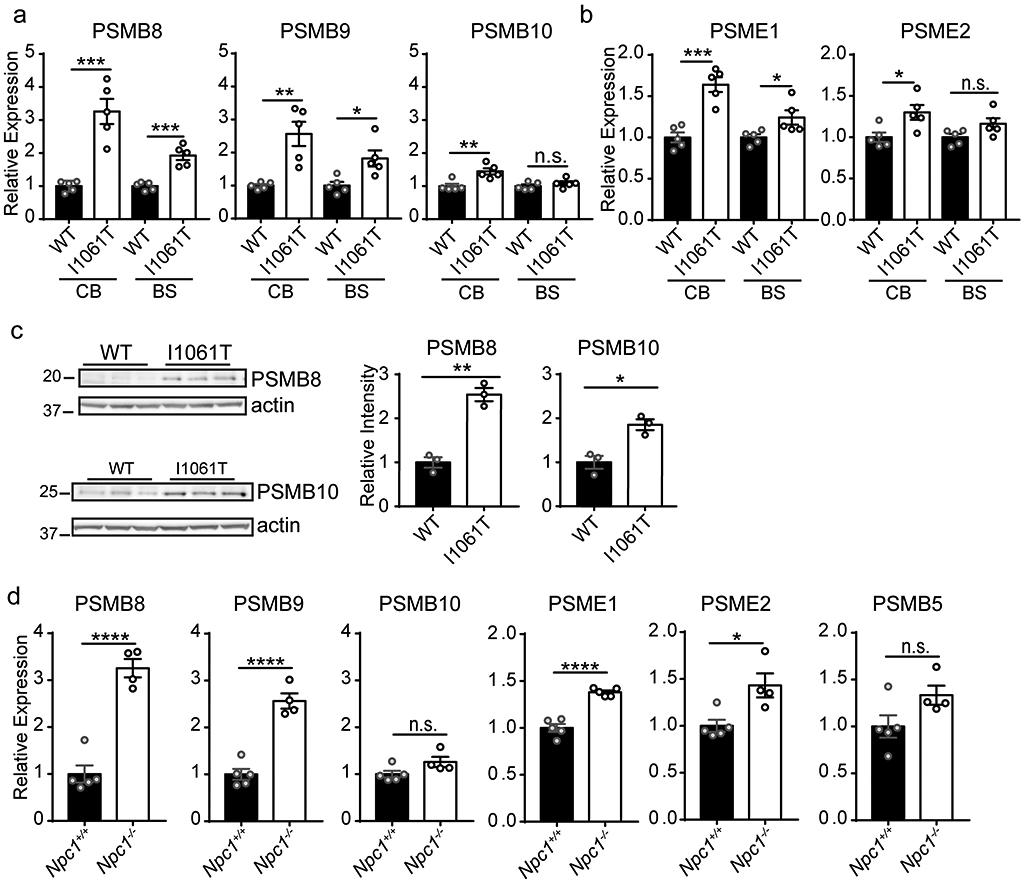
(a-b) Relative expression of immunoproteasome 20S core subunits (a) and alternative lid (b) was determined in the cerebellum (CB) and brainstem (BS) of 12-week WT and Npc1-I1061T mice by qPCR. N=5 mice per genotype. (c) The relative abundance of immunoproteasome subunits in the cerebellum of 12-week WT and Npc1-I1061T mice was determined by western blot. N=3 mice per genotype. Quantified at right. (d) Relative expression of immunoproteasome, alternative lid and constitutive proteasome subunits was determined by qPCR in the brainstem of 11-week Npc1+/+ and Npc1−/− mice. N=4 mice per genotype. Data are shown as mean ± s.e.m. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, **** P ≤ 0.0001 by (a-d) Student’s t-test (a) t=5.833 (CB PSMB8); t=6.27 (BS PSMB8); t=4.23 (CB PSMB9); t=3.088 (BS PSMB9); t=3.864 (CB PSMB10); t=1.083 (BS PSMB10) (b) t=5.893 (CB PSME1); t=2.556 (BS PSME1); t=2.853 (CB PSME2); t=2.077 (BS PSME2) (c) t=8.039 (PSMB8); t=4.454 (PSMB10) (d) t=8.351 (PSMB8); t=8.067 (PSMB9); t=2.077 (PSMB10); t=8.281 (PSME1); t=3.216 (PSME2); t=2.05 (PSMB5)
Importin α mislocalisation marks neurons with disrupted poly(A) RNA export and nuclear membrane morphology
With persistent cytoplasmic TDP-43 mislocalisation in Npc1−/− mice, we sought to characterize the functional consequences of this pathology. As TDP-43 proteinopathy has been associated with mislocalisation or aggregation of nucleocytoplasmic transport factors and disruption of the nuclear membrane and nuclear pore complexes (66, 74, 87, 88), we hypothesized that nucleocytoplasmic transport was disrupted in TDP-43 mislocalised neurons. To assess this hypothesis, we performed RNA fluorescence in situ hybridization (FISH) to quantify the N:C ratio of poly(A) RNA in Npc1−/− neurons. We focused on the subpopulations of neurons that accumulated p62 versus those that accumulated importin α. We detected a significantly increased N:C ratio in importin α+ neurons (Figure 7a), indicating nuclear retention and a defect in poly(A) RNA export. Similar findings have been observed in models of TDP-43 proteinopathy (74, 87, 89). Further indication of nuclear pathology in TDP-43 mislocalised neurons came from our observation of altered nuclear membrane morphology. Lamin B1 staining demonstrated significantly decreased circularity of the nuclear membrane in importin α+ neurons compared to p62+ neurons (Figure 7b). These results indicated that TDP-43 mislocalisation in Npc1−/− neurons compromises nucleocytoplasmic transport and nuclear membrane integrity, providing the first evidence of nuclear pathology in an LSD.
Figure 7. Importin α mislocalisation marks neurons with disrupted poly(A) RNA export and nuclear membrane morphology.

(a) The brainstem from 11-week Npc1−/− mice was stained with p62 or importin α. Poly(A) RNA was detected by RNA fluorescence in situ hybridization (FISH) with an oligo(dT) probe and imaged by confocal microscopy. Scale bar: 5 μm. Poly(A) RNA N:C ratio quantified from 20-30 p62+ or importin α+ cells per mouse, with N=6 mice. (b) The brainstem from 11-week Npc1−/− mice was stained with the indicated markers and imaged by confocal microscopy. Scale bar: 5 μm. Circularity of Lamin B1 staining was quantified from 20-30 p62+ or importin α+ cells per mouse, with N=6 mice. Data are shown as mean ± s.e.m. *P ≤ 0.05, **P ≤ 0.01, by (a-b) Student’s t-test (a) t=2.603 (b) t=4.53
Discussion
We demonstrate novel aspects of degeneration in the NPC brain that are mediated by TDP-43 proteinopathy. NPC1-deficient neurons exhibit mislocalisation of TDP-43 that is characterized by hallmarks of cytoplasmic de-mixing, an early stage in the aggregation process that was recently described in stressed neurons in cell culture (66). Cytoplasmic TDP-43 in Npc1−/− neurons co-localises with the nuclear import factor importin α, but not with markers of stress granules or ubiquitinated protein aggregates, nor is it associated with loss of nuclear TDP-43 or the generation of C-terminal cleavage fragments. This pathology is remarkably stable in the brainstem of Npc1 null mice, appearing as early as 4 weeks of age, before the onset of behavioural phenotypes, and remaining constant through end stage disease, almost two months later. Similarly long-lasting TDP-43 de-mixing has been documented to occur in cell culture (66).
Prior work has linked TDP-43 proteinopathy to LSDs in the context of progranulin mutations (22) and has suggested the occurrence of slightly diminished nuclear TDP-43 in cerebellar neurons of Npc1−/− mice (90). Our findings strengthen this connection with LSDs and expand the spectrum of TDP-43 proteinopathy in neurodegenerative brains by providing a first in vivo demonstration of stable cytoplasmic mislocalisation that does not progress to ubiquitinated aggregates, even over several months. These observations indicate that while certain stressors are sufficient to induce TDP-43 mislocalisation, distinct stressors are needed to promote its cytoplasmic aggregation. This is in line with data from in vitro models where exposure of neurons to fragmented amyloid-like fibrils promotes TDP-43 de-mixing while additional sodium arsenite-induced stress converts these liquid droplets to gels/solids that later recruit p62 (66).
TDP-43 mislocalisation occurs independently from the accumulation of autophagic cargo in the NPC brain. This conclusion is based on finding that these pathologies occur in distinct populations of neurons in Npc1−/− mice and that Npc1-I1061T mice accumulate autophagic cargo but not mislocalised TDP-43. This was unexpected as autophagic dysfunction has been postulated to mediate TDP-43 proteinopathy (11-13), as mutations in autophagy genes cause ALS/FTD (15) and as inhibiting autophagy induces cytoplasmic accumulation of pathologic TDP-43 (65). Furthermore, we demonstrate the occurrence of preferential susceptible to either TDP-43 proteinopathy (cholinergic) or autophagic dysfunction (glutamatergic) in distinct subpopulations of neurons, highlighting differential contributions to disease pathogenesis. Notably, neuroinflammation, as reflected by microgliosis, complement expression and immunoproteasome induction, is insufficient to account for TDP-43 mislocalisation in the NPC brain. Identifying the biological stressors in NPC that induce TDP-43 mislocalisation without progression to insoluble aggregates emerges as an important unknown to be addressed by future research.
One intriguing hypothesis is that the observed cytoplasmic TDP-43 mislocalisation represents a physiological response to neuronal injury. While TDP-43 cytoplasmic mislocalisation without concomitant nuclear depletion and progression to aggregates is an unusual finding, it has been detected in motor neurons following axotomy. In these injured neurons, TDP-43 expression was upregulated and showed prominent cytosolic localisation, findings that were restored following neuron recovery (91). This was proposed to be due in part to TDP-43’s involvement in neurofilament mRNA metabolism and transport to facilitate axonal repair (92). Thus, investigating the relationship between neuronal injury and TDP-43 cytoplasmic mislocalisation could provide important insights into the physiologic and/or pathologic roles of TDP-43 mislocalisation in NPC.
Finally, our studies also provide the first demonstration of nuclear pathology in an LSD. Importin α mislocalises with cytoplasmic TDP-43 in Npc1−/− neurons. These neurons also exhibit impaired nuclear export of RNA and abnormal nuclear membrane morphology. Similar disruption of nucleocytoplasmic transport and abnormalities of nuclear membrane architecture have been documented in other disorders with TDP-43 pathology (66, 74, 87, 88). Our observations suggest that shared mechanisms of lysosomal dysfunction and TDP-43 proteinopathy impair neuronal function in diverse degenerative disorders, including both LSDs and ALS/FTD. These shared disease mechanisms may provide future opportunities for therapeutic interventions that will benefit patients affected by these relentlessly progressive disorders.
Supplementary Material
Acknowledgments
Funding
This work was supported by the U.S. National Institutes of Health [R01 NS063967 to A.P.L.; T32-GM007863, T32-GM007315 to E.A.L.].
List of abbreviations
- ALS
Amyotrophic lateral sclerosis
- ChAT
Choline acetyltransferase
- FTD
Frontotemporal degeneration
- LSD
Lysosomal storage disease
- NPC
Niemann-Pick type C disease
Footnotes
Study Approval
All procedures involving mice were approved by the University of Michigan Committee on Use and Care of Animals (PRO00008133) and conducted in accordance with institutional and federal guidelines.
The authors declare that they have no competing interests
Data Availability Statement
Data available on request from the authors.
References
- 1.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11. [DOI] [PubMed] [Google Scholar]
- 2.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. [DOI] [PubMed] [Google Scholar]
- 3.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iguchi Y, Katsuno M, Niwa J, Takagi S, Ishigaki S, Ikenaka K, et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain. 2013;136(Pt 5):1371–82. [DOI] [PubMed] [Google Scholar]
- 5.Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(Pt 9):2610–26. [DOI] [PubMed] [Google Scholar]
- 7.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30(2):639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2011;13(1):38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127(Pt 6):1263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urushitani M, Sato T, Bamba H, Hisa Y, Tooyama I. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J Neurosci Res. 2010;88(4):784–97. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469(1):112–6. [DOI] [PubMed] [Google Scholar]
- 14.Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng Z, Sheehan P, Chen S, Yue Z. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol Neurodegener. 2017;12(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–6. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–6. [DOI] [PubMed] [Google Scholar]
- 20.Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122(6):673–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Mossevelde S, van der Zee J, Gijselinck I, Engelborghs S, Sieben A, Van Langenhove T, et al. Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain. 2016;139(Pt 2):452–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014;127(6):845–60. [DOI] [PubMed] [Google Scholar]
- 23.Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A. 1985;82(23):8247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–31. [DOI] [PubMed] [Google Scholar]
- 25.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105(40):15287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. Proc Natl Acad Sci U S A. 2016;113(29):8212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, et al. Structural Basis of Low-pH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell. 2020;182(1):98–111 e18. [DOI] [PubMed] [Google Scholar]
- 29.Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64(4):269–81. [DOI] [PubMed] [Google Scholar]
- 30.Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, et al. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4(9):e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc Natl Acad Sci U S A. 2009;106(7):2377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med. 2015;7(276):276ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz ML, Fawaz MV, Azaria RD, Hollon TC, Liu EA, Kunkel TJ, et al. Synthetic high-density lipoprotein nanoparticles for the treatment of Niemann-Pick diseases. BMC Med. 2019;17(1):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung C, Puthanveetil P, Ory DS, Lieberman AP. Genetic and pharmacological evidence implicates cathepsins in Niemann-Pick C cerebellar degeneration. Hum Mol Genet. 2016;25(7):1434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis OB, Shin HR, Lim CY, Wu EY, Kukurugya M, Maher CF, et al. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev Cell. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu EA, Schultz ML, Mochida C, Chung C, Paulson HL, Lieberman AP. Fbxo2 mediates clearance of damaged lysosomes and modifies neurodegeneration in the Niemann-Pick C brain. JCI Insight. 2020;5(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu T, Shakkottai VG, Chung C, Lieberman AP. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum Mol Genet. 2011;20(22):4440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu T, Lieberman AP. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013;9(4):e1003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez ME, Klein AD, Dimbil UJ, Scott MP. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci. 2011;31(12):4367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8(5):719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet. 2012;21(22):4876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1(1):81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A, et al. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 −/− mouse brain. Am J Pathol. 2007;171(3):962–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AW, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports. 2014;2(6):866–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meske V, Erz J, Priesnitz T, Ohm TG. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol Dis. 2014;64:88–97. [DOI] [PubMed] [Google Scholar]
- 46.Ordonez MP, Roberts EA, Kidwell CU, Yuan SH, Plaisted WC, Goldstein LS. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum Mol Genet. 2012;21(12):2651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16(12):1495–503. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5(5):1302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elrick MJ, Pacheco CD, Yu T, Dadgar N, Shakkottai VG, Ware C, et al. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet. 2010;19(5):837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Praggastis M, Tortelli B, Zhang J, Fujiwara H, Sidhu R, Chacko A, et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J Neurosci. 2015;35(21):8091–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Jong A, Schuurman KG, Rodenko B, Ovaa H, Berkers CR. Fluorescence-based proteasome activity profiling. Methods Mol Biol. 2012;803:183–204. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15(7):859–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maday S Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016;1649(Pt B):143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30(1):71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196(4):407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr., Iwata J, Kominami E, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104(36):14489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3(6):591–6. [DOI] [PubMed] [Google Scholar]
- 58.Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112(28):E3699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–22. [DOI] [PubMed] [Google Scholar]
- 60.Tammineni P, Jeong YY, Feng T, Aikal D, Cai Q. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer's disease neurons. Hum Mol Genet. 2017;26(22):4352–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron. 2015;87(2):355–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kennedy BE, Madreiter CT, Vishnu N, Malli R, Graier WF, Karten B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J Biol Chem. 2014;289(23):16278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wos M, Szczepanowska J, Pikula S, Tylki-Szymanska A, Zablocki K, Bandorowicz-Pikula J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch Biochem Biophys. 2016;593:50–9. [DOI] [PubMed] [Google Scholar]
- 64.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang MC, Srinivasan K, Friedman BA, Suto E, Modrusan Z, Lee WP, et al. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J Exp Med. 2017;214(9):2611–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, et al. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron. 2019;102(2):339–57 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137(1):27–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 2016;24(9):1537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474(8):1417–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–61. [DOI] [PubMed] [Google Scholar]
- 71.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, et al. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int. 2012;60(4):415–24. [DOI] [PubMed] [Google Scholar]
- 73.Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron. 2019;102(2):321–38 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin TW, Tan Z, Barik A, Yin DM, Brudvik E, Wang H, et al. Regulation of Synapse Development by Vgat Deletion from ErbB4-Positive Interneurons. J Neurosci. 2018;38(10):2533–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gelsthorpe ME, Baumann N, Millard E, Gale SE, Langmade SJ, Schaffer JE, et al. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J Biol Chem. 2008;283(13):8229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, et al. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun. 2018;9(1):3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. [DOI] [PubMed] [Google Scholar]
- 80.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21(10):1359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94. [DOI] [PubMed] [Google Scholar]
- 82.Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. 2020;588(7838):459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moritz KE, McCormack NM, Abera MB, Viollet C, Yauger YJ, Sukumar G, et al. The role of the immunoproteasome in interferon-gamma-mediated microglial activation. Sci Rep. 2017;7(1):9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orre M, Kamphuis W, Dooves S, Kooijman L, Chan ET, Kirk CJ, et al. Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain. 2013;136(Pt 5):1415–31. [DOI] [PubMed] [Google Scholar]
- 85.Wagner LK, Gilling KE, Schormann E, Kloetzel PM, Heppner FL, Kruger E, et al. Immunoproteasome deficiency alters microglial cytokine response and improves cognitive deficits in Alzheimer's disease-like APPPS1 mice. Acta Neuropathol Commun. 2017;5(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci. 2012;109:75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351(6269):173–6. [DOI] [PubMed] [Google Scholar]
- 90.Dardis A, Zampieri S, Canterini S, Newell KL, Stuani C, Murrell JR, et al. Altered localization and functionality of TAR DNA Binding Protein 43 (TDP-43) in niemann- pick disease type C. Acta Neuropathol Commun. 2016;4(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–11. [DOI] [PubMed] [Google Scholar]
- 92.Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ. Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1296:176–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request from the authors.