

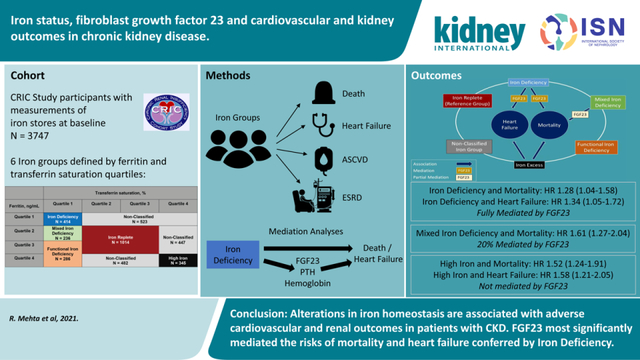
Abstract
Disordered iron and mineral homeostasis are interrelated complications of chronic kidney disease that may influence cardiovascular and kidney outcomes. In a prospective analysis of 3747 participants in the Chronic Renal Insufficiency Cohort Study, we investigated risks of mortality, heart failure, end-stage kidney disease (ESKD), and atherosclerotic cardiovascular disease according to iron status, and tested for mediation by C-terminal fibroblast growth factor 23 (FGF23), hemoglobin and parathyroid hormone. Study participants were agnostically categorized based on quartiles of transferrin saturation and ferritin as: “Iron Replete” (27.1% of participants; referent group for all outcomes analyses), “Iron Deficiency” (11.1%), “Functional Iron Deficiency” (7.6%), “Mixed Iron Deficiency” (iron indices between the Iron Deficiency and Functional Iron Deficiency groups; 6.3%), “High Iron” (9.2%), or “Non-Classified” (the remaining 38.8% of participants). In multivariable-adjusted Cox models, Iron Deficiency independently associated with mortality (hazard ratio 1.28, 95% confidence interval 1.04–1.58) and heart failure (1.34, 1.05–1.72). Mixed Iron Deficiency associated with mortality (1.61, 1.27–2.04) and ESKD (1.33, 1.02–1.73). High Iron associated with mortality (1.54, 1.24–1.91), heart failure (1.58, 1.21–2.05), and ESKD (1.41, 1.13–1.77). Functional Iron Deficiency did not significantly associate with any outcome, and no iron group significantly associated with atherosclerotic cardiovascular disease. Among the candidate facilitators, FGF23 most significantly mediated the risks of mortality and heart failure conferred by Iron Deficiency. Thus, alterations in iron homeostasis associated with adverse cardiovascular and kidney outcomes in patients with chronic kidney disease.
Keywords: chronic kidney disease, iron deficiency, iron excess, functional iron deficiency, fibroblast growth factor 23, mortality
Graphical Abstract
Introduction
Disordered iron homeostasis, which encompasses a spectrum of iron deficiency, functional iron deficiency and iron excess, is a common complication of chronic kidney disease (CKD) that worsens quality of life, increases health care costs and may influence clinical outcomes.1, 2, 3, 4 With the development of multiple new therapeutics in the area of iron and anemia management in CKD, further research is needed to determine how distinct phenotypes of disordered iron homeostasis influence clinical outcomes and what mechanisms underlie these risks.
Fibroblast growth factor 23 (FGF23) is a bone- and bone marrow-derived hormone that regulates phosphate and vitamin D homeostasis.5 Levels of FGF23 are elevated in CKD and associated with adverse clinical outcomes, most notably, mortality and heart failure, for which it might contribute mechanistically.6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Emerging data demonstrate that iron is an important regulator of FGF23 and that absolute and functional iron deficiency due to chronic inflammation induce FGF23 transcription.16, 17, 18 Unlike in individuals with preserved kidney function, iron deficiency-induced increases in FGF23 transcription in CKD result in increased circulating levels of full-length biologically active FGF23 due to impaired cleavage of nascent FGF23.18, 19
We investigated iron status and risks of mortality, heart failure, end-stage renal disease (ESRD), and atherosclerotic cardiovascular disease (ASCVD) in patients with moderate to severe CKD enrolled in the Chronic Renal Insufficiency Cohort (CRIC) Study. We hypothesized that more severe iron deficiency would associate with worse cardiovascular and renal outcomes, and that elevated FGF23 is a mediator of risks of mortality and heart failure that are attributable to iron deficiency.
Methods
Study Population
The CRIC Study is a multi-center prospective cohort study of risk factors for cardiovascular disease and progressive kidney disease among 3939 individuals with moderate to severe CKD at enrollment.20, 21 Participants aged 21–74 were recruited between June 2003 and September 2008 from 7 clinical centers across the United States.20, 21 Exclusion criteria included New York Heart Association class III or IV heart failure, multiple myeloma, polycystic kidney disease, renal cancer, HIV infection, organ transplantation, previous dialysis treatment for at least one month, recent chemotherapy or immunosuppressive therapy, cirrhosis, institutionalization, enrollment in other studies, pregnancy, or inability to provide informed consent.21 The study protocol was approved by the institutional review boards at each participating site and all participants provided written informed consent. Within the CRIC Study population, 3747 of 3939 (95.1%) participants had baseline samples available for measurement of iron, transferrin and ferritin.
Primary Exposure and Outcomes
Given the lack of evidence-based cut points for functional iron deficiency and iron excess in CKD, we agnostically defined categories of iron status based on quartiles of transferrin saturation (TSAT) and ferritin for the primary analyses, as has been done previously.3 In sensitivity analyses, we also investigated cut points used in clinical practice.22, 23 Initially, we categorized the study population into 5 exposure groups: “Iron Deficiency,” “Functional Iron Deficiency,” “Iron Replete,” “High Iron,” and “Non-Classified,” which included all remaining uncategorized participants (Figure 1). Upon investigation of the different subgroups that comprised the Non-Classified group, participants in the TSAT quartile 1–ferritin quartile 2 group (between the Iron Deficiency and Functional Iron Deficiency groups) were at significantly increased risk for certain clinical outcomes compared to the Iron Replete group, unlike all other Non-Classified subgroups. Therefore, we re-categorized this group as “Mixed Iron Deficiency,” and conducted all analyses using 6 exposure groups (Figure 1). Compared to the Iron Replete group, none of the other Non-classified subgroups were at significantly increased risk of any outcome (Supplemental Table 1).
Figure 1. Iron status groups.
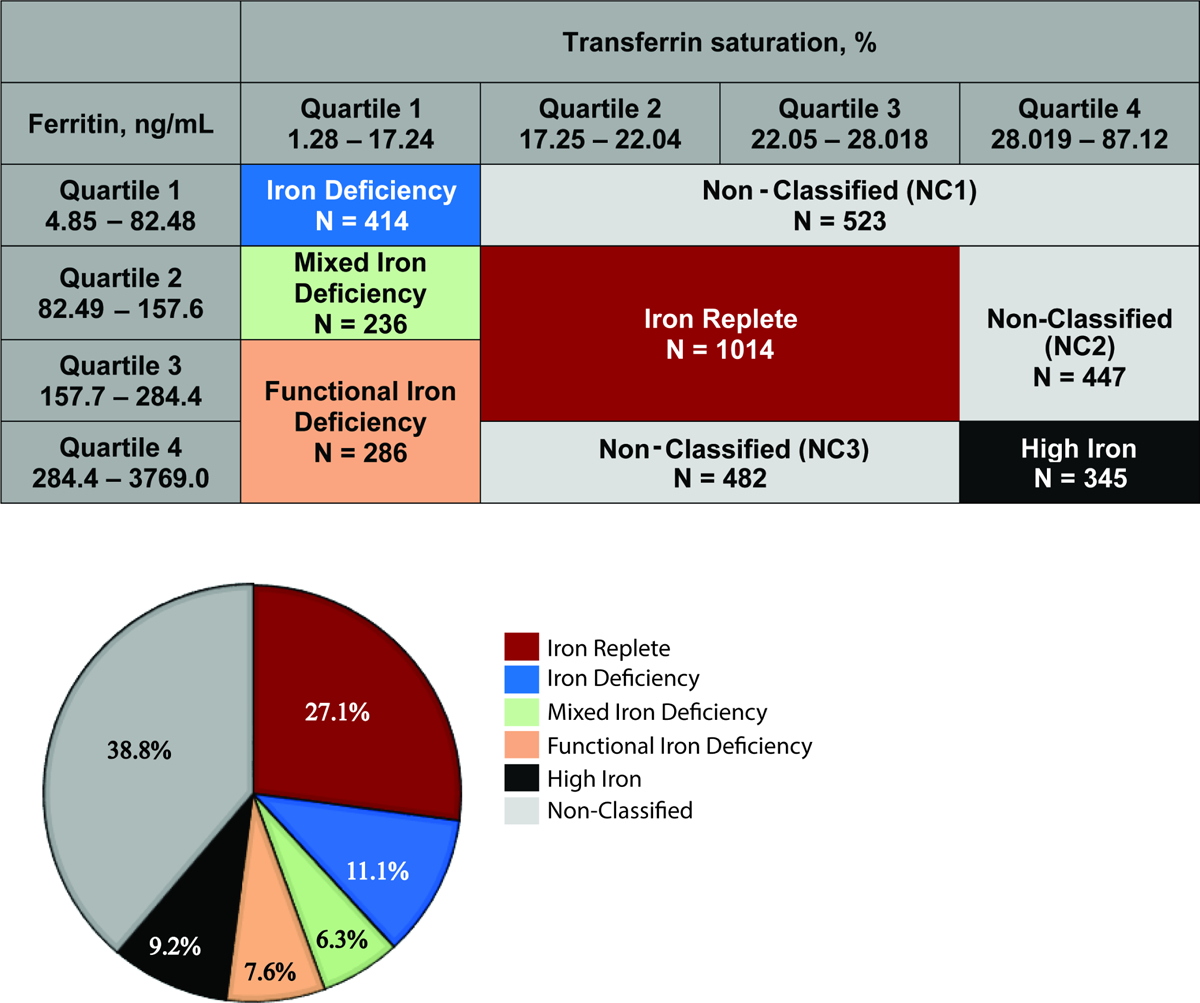
(A) Classification of iron groups according to quartiles of ferritin and transferrin saturation; (B) Proportions of each iron group within the Chronic Renal Insufficiency Cohort Study
The primary outcomes were mortality, heart failure events, progression to ESRD, and ASCVD events. Participants were followed until death, or censored for withdrawal from the study, loss to follow-up, or when the database was locked for analysis in 2013. The CRIC Study staff ascertained death through review of hospital records, reports from next of kin, retrieval of death certificates or obituaries, and linkage with the Social Security Death Index.24 All cardiovascular events were adjudicated by two independent physicians. Heart failure events were classified as definite, probable or possible using symptoms (dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea) in conjunction with either physical examination features (comprising >2 of pulmonary rales, S3 gallop, jugular venous distention > 5 centimeters, peripheral edema), radiographic findings (pulmonary edema, vascular congestion, pleural effusion), or invasive hemodynamic or echocardiographic findings of heart failure (pulmonary capillary wedge pressure > 18 mmHg, cardiac index < 2.0 L/min/m2, left ventricular ejection fraction ≤ 35%). For this study, we defined heart failure events only if they were considered definite or probable. Adjudicated ASCVD events included a composite of myocardial infarction (definite/probable/possible), stroke (definite/probable) and peripheral artery disease. ESRD, defined as initiation of chronic dialysis or receipt of a kidney transplant, was determined through self-report, by participants’ point of contact that was confirmed by CRIC Study staff, and by cross-linkage with the US Renal Data System.24
Covariates
Demographic and laboratory data and medications were collected from all participants at the baseline visit when iron status was ascertained. Self-reported history of cardiovascular disease was defined by prior coronary revascularization, heart failure, myocardial infarction, ischemic stroke, peripheral artery revascularization, or amputation. Any versus no iron supplementation was determined by dietary questionnaire; missing responses were considered no supplementation. Resting blood pressure was measured via standardized protocols.21
Assays
All analytes were measured by the CRIC Central Laboratory after a single thaw of frozen samples collected at the baseline CRIC study visit. Iron was measured on a Roche (c501) autoanalyzer; the inter-assay coefficients of variation (CV) were 2.1% and 1.1%. Transferrin was measured using an immunoturbidometric format (Roche Diagnostics); the inter-assay CV were 0.9% and 0.5%. Total iron binding capacity (μg/l) was estimated as transferrin (mg/l) × 1.43, and TSAT was calculated as serum iron concentration (μg/l) / total iron binding capacity (μg/l) × 100. Ferritin was measured using a sandwich immunoassay (Roche Diagnostics); the inter-assay CV was 6.7%. Hemoglobin, serum creatinine and phosphate, and urinary albumin-to-creatinine ratio (UACR) were measured using standard assays.20, 21 We calculated estimated glomerular filtration rate (eGFR) using the creatinine-based Chronic Kidney Disease Epidemiology Collaboration equation.25 As described previously, parathyroid hormone (PTH) was measured using a total PTH assay (Scantibodies), and C-terminal FGF23 was measured using a second-generation immunoassay (Immutopics).26
Statistical Analysis
We compared baseline characteristics and demographics across the 6 iron status groups. We used Cox proportional hazards models to analyze time to each outcome according to iron status. We adjusted for demographics (age, gender, race, ethnicity), and in the full model, further adjusted for renal risk factors (eGFR, UACR [< 300 mg/dl, ≥ 300 mg/dl, or missing (3.5% of the population)]), cardiovascular risk factors (prior cardiovascular disease, systolic blood pressure, diabetes, smoking, body mass index) and erythropoietin use. In secondary analyses, we further adjusted the full models for C-reactive protein (CRP) as a marker of inflammation, for oral iron use (yes or no), and for phosphate binder use (yes or no). All models included stratification terms to account for study site. In secondary analyses, we analyzed time-to-incident heart failure and time-to-incident ASCVD in the subpopulations without a history of heart failure or ASCVD at baseline. For all time-to-event analyses, we verified the proportional hazards assumption by visual inspection.
Mediation Analyses
Iron deficiency stimulates FGF23 production,5 and FGF23 excess is associated with heart failure and mortality.12, 13 To investigate potential mediation of the effects of iron status by FGF23, we first compared least square mean levels of FGF23 across the iron status groups, adjusted for eGFR, phosphate, CRP, and PTH. Next, we investigated the effects on the hazard ratios of the iron groups in Cox models that excluded and then included FGF23, focusing only on outcomes that were significantly associated with iron status. Finally, we used a counterfactual model-based approach for causal mediation analyses using a SAS macro that uses a Cox proportional hazards model as the outcomes model to calculate estimates for direct, indirect and total effects.27, 28 Direct effects refer to effects of iron stores on outcomes independent of FGF23. Indirect effects refer to effects of iron stores on outcomes that act through FGF23. Total effects refer to the product of the direct and indirect effects. In all mediation analysis models, we adjusted for the same covariates as in the primary analyses including center, age, sex, race, ethnicity, diabetes, smoking, systolic blood pressure, body mass index, prior cardiovascular disease, eGFR, urine albumin-to-creatinine ratio, and erythropoietin use. We computed the proportion mediated using a counterfactual model-based approach for causal mediation analyses using R package mediation based on a Weibull model which generates estimates, 95% confidence intervals (CI) and P values.29 To benchmark potential mediation effects of FGF23, we repeated the mediation analyses using PTH as another marker of mineral metabolism, and hemoglobin, which is on a causal pathway between iron deficiency and clinical outcomes.
Sensitivity Analyses
We investigated the multivariable-adjusted associations of TSAT and ferritin as continuous variables on each outcome. We re-categorized the study population according to clinical thresholds of iron status (Supplemental Figure 1), and repeated the multivariable and mediation modeling that we used in the primary analyses. We also repeated the primary analyses adjusting for UACR as a natural log-transformed continuous variable instead of a categorical variable.
All analyses were performed using SAS version 9.4 (Cary, NC, USA) and R package version 3.4.4. Two-sided P values <0.05 were considered statistically significant.
Results
Table 1 presents baseline characteristics according to the 6 iron exposure groups. The mean TSAT ranged between 12–15% in the Iron Deficiency, Mixed Iron Deficiency, and Functional Iron Deficiency groups. Individuals in the Iron Deficiency group had the lowest iron, TSAT, hemoglobin, and ferritin levels, and the highest total iron binding capacity. Individuals in the High Iron group had the highest iron, TSAT, hemoglobin and ferritin levels, and the lowest total iron binding capacity. Individuals in the Functional Iron Deficiency group had the highest median CRP and eGFR. Erythropoietin use was highest in the Functional Iron Deficiency and High Iron groups, whereas oral iron use was highest in the Iron Deficiency group. The Mixed Iron Deficiency group demonstrated features of both iron deficiency (low TSAT and ferritin), and functional iron deficiency (elevated CRP).
Table 1.
Baseline characteristics according to iron groups
| Iron replete N=1014 (27.1%) |
Iron deficiency N=414 (11.1%) |
Mixed iron deficiency N=236 (6.3%) |
Functional iron deficiency N=286 (7.6%) |
High Iron N=345 (9.2%) |
Non-Classified N=1452 (38.8%) |
|
|---|---|---|---|---|---|---|
| Age, years | 58.6 ± 10.7 | 56.3 ± 12.1 | 58.6 ± 10.2 | 58.4 ± 10.3 | 58.0 ± 10.7 | 57.1 ± 11.1 |
| Female, % | 44.8 | 66.2 | 53.4 | 45.5 | 27.8 | 42.2 |
| Black, % | 41.0 | 41.1 | 52.5 | 58.4 | 43.8 | 38.1 |
| Hispanic, % | 12.2 | 15.0 | 12.3 | 12.9 | 10.4 | 13.4 |
| Current smoking, % | 13.6 | 15.2 | 11.4 | 15.0 | 13.9 | 11.9 |
| BMI, kg/m2 | 32.3 ± 7.4 | 33.7 ± 9.5 | 33.9 ± 10.5 | 33.8 ± 8.4 | 30.5 ± 6.1 | 31.2 ± 7.0 |
| Systolic BP, mmHg | 128.6 ± 21.9 | 130.0 ± 23.5 | 133.1 ± 22.1 | 130.4 ± 22.5 | 127.1 ± 22.4 | 127.3 ± 21.8 |
| Hypertension, % | 88.7 | 87.7 | 85.6 | 90.6 | 85.8 | 83.5 |
| Diabetes, % | 50.5 | 51.9 | 54.2 | 59.4 | 42.0 | 45.1 |
| Heart failure, % | 9.4 | 11.8 | 9.8 | 10.5 | 10.1 | 8.3 |
| Stroke, % | 11.0 | 10.9 | 9.8 | 11.2 | 9.9 | 8.3 |
| CVD, % | 34.1 | 36.5 | 34.3 | 37.4 | 32.2 | 30.4 |
| Oral iron use, % | 34.8 | 40.1 | 34.3 | 31.8 | 33.6 | 38.1 |
| ESA, % | 2.9 | 3.9 | 2.1 | 6.7 | 6.1 | 3.8 |
| Hemoglobin, g/dL | 12.7 ± 1.7 | 11.7 ± 1.6 | 12.2 ± 1.8 | 11.8 ± 1.7 | 12.9 ± 1.9 | 12.9 ± 1.7 |
| Iron, ug/dL | 85.8 ± 25.6 | 53.9 ± 17.8 | 58.7 ± 17.6 | 57.4 ± 21.7 | 132.2 ± 44.2 | 104.3 ± 37.8 |
| TIBC, ug/L | 3871.9 ± 1096.1 | 4366.8 ± 1178.8 | 4101.9 ± 1024.8 | 3975.3 ± 1329.5 | 3754.6 ± 1158.3 | 3898.5 ± 1163.5 |
| TSAT, % | 22.3 ± 3.0 | 12.6 ± 3.2 | 14.3 ± 2.2 | 14.4 ± 2.5 | 35.7 ± 8.2 | 27.2 ± 7.5 |
| Ferritin, ng/mL | 159.8 (119.9, 209.3) |
42.7 (27.3, 62.3) |
116.1 (98.7, 137.2) |
248.7 (195.5, 355.0) |
449.0 (354.6, 654.7) |
158.3 (67.4, 330.5) |
| CRP, mg/L | 2.4 (1.1 – 5.6) |
3.8 (1.3 – 8.9) |
3.9 (1.6 – 8.9) |
6.8 (2.5 – 16.0) |
2.0 (0.9 – 4.3) |
2.0 (0.9 – 5.0) |
| eGFR, ml/min/1.73m2 | 43.6 ± 15.0 | 44.2 ± 15.7 | 43.4 ± 15.2 | 40.1 ± 13.3 | 43.2 ± 14.5 | 46.0 ± 14.9 |
| UACR, ug/mg | 58.3 (8.9, 445.1) |
66.8 (11.2, 441.5) |
68.3 (11.6, 454.1) |
82.3 (14.7, 568.3) |
45.6 (8.0, 610.2) |
40.0 (7.1, 442.3) |
| Serum albumin, g/dL | 4.0 ± 0.5 | 3.8 ± 0.4 | 3.9 ± 0.5 | 3.9 ± 0.5 | 4.0 ± 0.6 | 4.0 ± 0.5 |
| Phosphate, mg/dL | 3.7 ± 0.7 | 3.8 ± 0.7 | 3.8 ± 0.6 | 3.8 ± 0.6 | 3.7 ± 0.7 | 3.7 ± 0.7 |
| FGF23, RU/ml | 142.3 (98.4, 225.5) |
238.1 (149.2, 439.5) |
179.9 (110.8, 264.6) |
166.0 (108.5, 277.7) |
130.6 (87.1, 192.9) |
129.0 (87.6, 205.7) |
| PTH, pg/ml | 54.0 (35.0, 90.0) |
60.0 (35.1, 95.0) |
59.3 (36.3, 114.9) |
62.0 (41.4, 106.4) |
53.0 (35.4, 86.3) |
50.8 (33.3, 83.0) |
Results are reported as means ± standard deviations, medians with interquartile ranges or percentiles.
Abbreviations: N, number; BMI, body mass index; BP, blood pressure; CVD, cardiovascular disease; ESA, erythropoietin stimulating agents; TSAT, transferrin saturation; CRP, C-reactive protein; eGFR, estimated glomerular filtration rate; UACR, urine albumin-to-creatinine ratio; FGF23, fibroblast growth factor 23; PTH, parathyroid hormone
Iron Status and Mortality
During a median follow-up of 9.5 years, 1104 of the 3747 participants died. Compared to the Iron Replete group, the Iron Deficiency, Mixed Iron Deficiency and High Iron groups were each at significantly increased risk of death in analyses that adjusted for demographics, renal and cardiovascular risk factors, and erythropoietin use (Table 2). The results were qualitatively unchanged after further adjustment for CRP, oral iron use and phosphate binder use (Table 2). In contrast, the Functional Iron Deficiency group was not at significantly increased risk of death compared to the Iron Replete group in any analysis (Table 2).
Table 2.
Iron groups and risks of clinical outcomes
| Group | Incidence Density (95% CI) |
Demographics Model HR (95% CI) |
Full Model HR (95% CI) |
Full Model + oral iron use HR (95% CI) |
Full Model + CRP HR (95% CI) |
Full Model + Phosphate Binder Use HR (95% CI) |
|---|---|---|---|---|---|---|
| All-cause Mortality: 1104 events over median follow-up of 9.5 years | ||||||
| Iron Replete | 3.24 (2.88, 3.64) |
Reference | Reference | Reference | Reference | Reference |
| Iron Deficiency | 3.65 (3.04, 4.35) |
1.32 (1.07, 1.63) |
1.28 (1.04, 1.58) |
1.28 (1.03, 1.58) |
1.24 (1.00, 1.53) |
1.28 (1.04, 1.59) |
| Mixed Iron Deficiency | 4.89 (3.95, 5.99) |
1.48 (1.17, 1.87) |
1.61 (1.27, 2.04) |
1.61 (1.27, 2.05) |
1.53 (1.20, 1.94) |
1.61 (1.27, 2.04) |
| Functional Iron Deficiency | 4.23 (3.46, 5.13) |
1.19 (0.95, 1.49) |
1.16 (0.92, 1.46) |
1.16 (0.92, 1.46) |
1.02 (0.81, 1.29) |
1.16 (0.93, 1.46) |
| High Iron | 4.30 (3.57, 5.13) |
1.37 (1.11, 1.70) |
1.54 (1.24, 1.91) |
1.55 (1.25, 1.92) |
1.57 (1.27, 1.95) |
1.54 (1.24, 1.91) |
| Heart Failure: 728 events over median follow-up of 8.8 years | ||||||
| Iron Replete | 2.55 (2.20, 2.93) |
Reference | Reference | Reference | Reference | Reference |
| Iron Deficiency | 3.45 (2.80, 4.20) |
1.49 (1.17, 1.90) |
1.34 (1.05, 1.72) |
1.35 (1.05, 1.72) |
1.34 (1.04, 1.71) |
1.35 (1.05, 1.72) |
| Mixed Iron Deficiency | 3.54 (2.68, 4.58) |
1.28 (0.95, 1.73) |
1.35 (1.00, 1.82) |
1.35 (1.00, 1.82) |
1.33 (0.98, 1.79) |
1.36 (1.01, 1.84) |
| Functional Iron Deficiency | 3.55 (2.77, 4.47) |
1.23 (0.93, 1.61) |
1.18 (0.90, 1.55) |
1.18 (0.90, 1.55) |
1.12 (0.85, 1.47) |
1.19 (0.90, 1.56) |
| High Iron | 3.32 (2.63, 4.12) |
1.34 (1.03, 1.74) |
1.58 (1.21, 2.05) |
1.57 (1.21, 2.05) |
1.59 (1.22, 2.08) |
1.58 (1.21, 2.06) |
| End Stage Renal Disease: 1041 events over median follow-up of 8.8 years | ||||||
| Iron Replete | 3.98 (3.55, 4.46) |
Reference | Reference | Reference | Reference | Reference |
| Iron Deficiency | 3.93 (3.25, 4.72) |
1.00 (0.81, 1.25) |
1.00 (0.80, 1.24) |
1.00 (0.80, 1.24) |
1.00 (0.80, 1.25) |
1.00 (0.80, 1.25) |
| Mixed Iron Deficiency | 4.38 (3.42, 5.52) |
1.03 (0.79, 1.33) |
1.33 (1.02, 1.73) |
1.33 (1.02, 1.73) |
1.33 (1.02, 1.73) |
1.33 (1.02, 1.73) |
| Functional Iron Deficiency | 4.66 (3.77, 5.69) |
0.97 (0.77, 1.23) |
0.89 (0.70, 1.12) |
0.89 (0.70, 1.12) |
0.89 (0.71, 1.13) |
0.89 (0.70, 1.12) |
| High Iron | 4.65 (3.83, 5.61) |
1.14 (0.91, 1.42) |
1.41 (1.13, 1.77) |
1.41 (1.13, 1.77) |
1.41 (1.12, 1.77) |
1.41 (1.13, 1.77) |
| Atherosclerotic Cardiovascular Disease: 673 events over median follow-up of 8.8 years | ||||||
| Iron Replete | 2.75 (2.39, 3.15) |
Reference | Reference | Reference | Reference | Reference |
| Iron Deficiency | 2.42 (1.89, 3.05) |
1.00 (0.76, 1.31) |
0.93 (0.71, 1.22) |
0.93 (0.71, 1.22) |
0.91 (0.70, 1.20) |
0.93 (0.71, 1.22) |
| Mixed Iron Deficiency | 2.49 (1.79, 3.36) |
0.88 (0.63, 1.23) |
0.94 (0.67, 1.31) |
0.94 (0.67, 1.31) |
0.89 (0.64, 1.24) |
0.93 (0.67, 1.31) |
| Functional Iron Deficiency | 2.81 (2.14, 3.62) |
0.94 (0.70, 1.26) |
0.89 (0.67, 1.20) |
0.89 (0.67, 1.19) |
0.80 (0.60, 1.08) |
0.89 (0.67, 1.19) |
| High Iron | 2.76 (2.16, 3.49) |
0.99 (0.75, 1.29) |
1.05 (0.80, 1.39) |
1.05 (0.80, 1.39) |
1.07 (0.81, 1.41) |
1.05 (0.80, 1.39) |
Results are reported as hazard ratios compared to the referent group. The Non-classified group is not shown.
Demographics Model: Stratified by center, adjusted for age, sex, race, and ethnicity
Full Model: Demographics model plus diabetes, smoking, systolic blood pressure, body mass index, history of cardiovascular disease, estimated glomerular filtration rate, urinary albumin-to-creatinine ratio, and erythropoietin use
Abbreviations: HR, hazard ratio; CI, confidence interval; CRP, C-reactive protein
Iron Status and Heart Failure
During a median follow up of 8.8 years, 728 participants developed a heart failure event. Similar to the analyses of mortality, the Iron Deficiency and High Iron groups were at significantly higher risk of heart failure compared to the Iron Replete group in all analyses, whereas the Functional Iron Deficiency group was not at a significantly increased risk of heart failure in any analysis (Table 2). The Mixed Iron Deficiency group also was at increased risk of heart failure, but the association was no longer significant after adjustment for CRP (Table 2). Although fewer events reduced power, the results were qualitatively unchanged when restricted to incident heart failure events in participants with no prior history of heart failure (Supplemental Table 2).
Iron Status and ESRD
During a median follow up of 8.8 years, 1041 participants reached ESRD. Compared to the Iron Replete group, the High Iron and Mixed Iron Deficiency groups were at significantly increased risk of ESRD in the full model and after further adjustment for CRP, oral iron use and phosphate binder use (Table 2). Iron Deficiency and Functional Iron Deficiency were not significantly associated with increased risk of ESRD (Table 2).
Iron Status and Atherosclerotic Events
During a median follow up of 8.8 years, 673 participants developed an ASCVD event. No iron status group was at significantly increased risk of ASCVD events (Table 2). The results were unchanged when restricted to incident ASCVD events in participants with no prior history of ASCVD (Supplemental Table 2)
Mediation Analyses
To test the hypothesis that associations between iron status and clinical outcomes are mediated, in part, by FGF23, we first compared least square mean levels of FGF23 across the iron status groups. In an analysis parsimoniously adjusted for eGFR, CRP, phosphate, and PTH, FGF23 levels were inversely associated with iron stores: FGF23 was highest in the Iron Deficiency group and lowest in the High Iron group; the intermediate iron groups had intermediate FGF23 levels (Figure 2). In contrast, adjusted PTH levels were similar across all iron status groups (Figure 2).
Figure 2. FGF23 and PTH according to iron status.
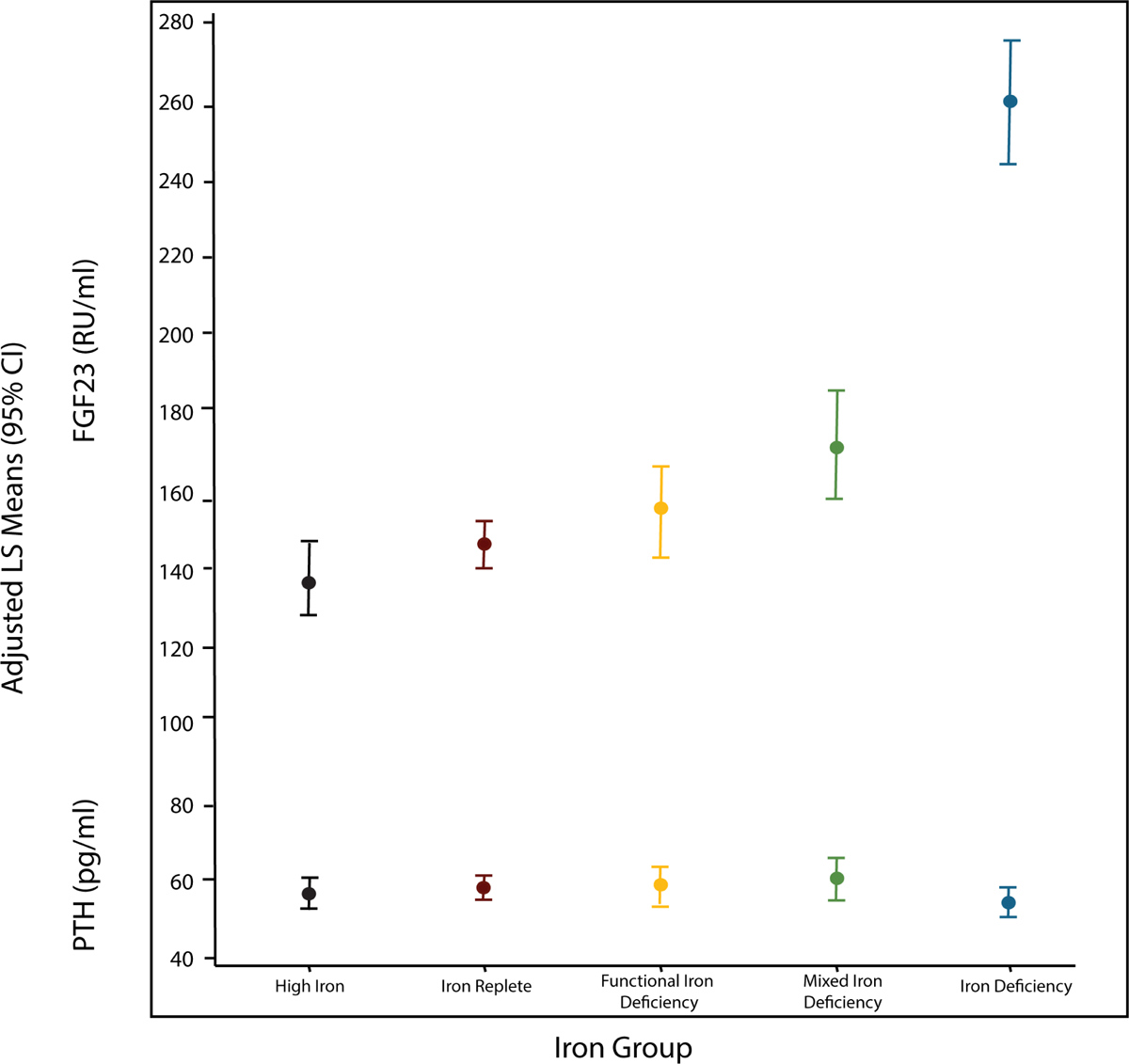
Least square means of FGF23 and PTH by iron group adjusted for eGFR, CRP, phosphate, and PTH or FGF23. Results are represented as mean values plus 95% confidence intervals.
Abbreviations: CRP, C-reactive protein; eGFR, estimated glomerular filtration rate; FGF23, fibroblast growth factor 23; PTH, parathyroid hormone
Next, we confirmed that higher FGF23 levels independently associated with increased risks of mortality and heart failure in fully adjusted models (Figure 3), as previously reported.11, 13 When FGF23 and the iron exposure groups were included together in fully adjusted models, the significant effects of FGF23 were unchanged, but the increased risks of mortality and heart failure in the Iron Deficiency group were fully attenuated (mortality: from 1.28 [95%CI 1.04, 1.58] to 0.97 [95%CI 0.77, 1.21]; heart failure: from 1.34 [95%CI 1.05, 1.72] to 0.98 [95%CI 0.75, 1.27]; Figure 3). Adjustment for FGF23 partially attenuated the effects of Mixed Iron Deficiency, but had no effect on the association of High Iron with risks of mortality or heart failure (Figure 3). Formal mediation analyses confirmed that the effects of Iron Deficiency on mortality and heart failure were fully accounted by FGF23, whereas FGF23 mediated 20% of the association between Mixed Iron Deficiency and death, and did not significantly mediate the effects of High Iron on either outcome (Table 3, Supplemental Figure 2).
Figure 3. Mediation analyses of iron status and risks of mortality and heart failure.
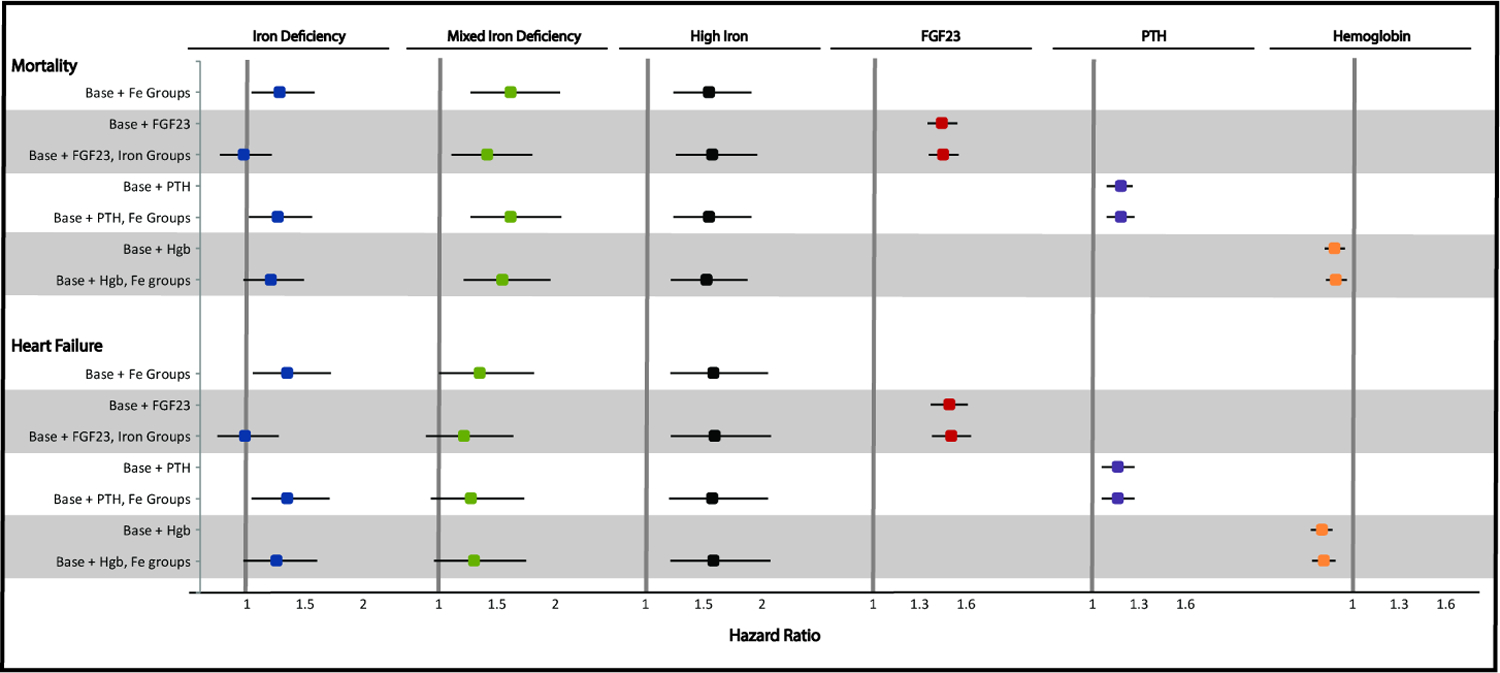
Results for the iron groups are reported as hazard ratios and 95% confidence intervals compared to the iron replete group. For the continuous variables, results are reported as hazard ratios per 1 standard deviation of natural log FGF23, hemoglobin, and natural log PTH. The full model was stratified by center, adjusted for age, sex, race, ethnicity, diabetes, smoking, systolic blood pressure, body mass index, prior cardiovascular disease, eGFR, urine albumin-to-creatinine ratio, and erythropoietin use.
Abbreviations: eGFR, estimated glomerular filtration rate; FGF23, fibroblast growth factor 23; Hgb, hemoglobin; PTH, parathyroid hormone
Table 3.
Mediation Analyses
| Direct Effect of Iron Stores (95%CI) P value |
Indirect Effect of Iron Stores through Mediator (95%CI) P Value |
Total Effect of Iron Stores (95%CI) P value |
*Proportion Mediated (%) (95%CI) P value |
|
|---|---|---|---|---|
| FGF23 as mediator | ||||
| Mortality | ||||
| Iron Deficiency | 0.96 (0.77, 1.20) 0.74 |
1.32 (1.24, 1.40) <0.001 |
1.27 (1.02, 1.57) 0.03 |
115% (50, 635) 0.04 |
| Mixed Iron Deficiency | 1.41 (1.11, 1.80) 0.005 |
1.10 (1.05, 1.15) < 0.001 |
1.56 (1.22, 1.99) <0.001 |
20% (10, 43) < 0.001 |
| High Iron | 1.57 (1.26, 1.96) < 0.001 |
0.98 (0.94, 1.02) 0.34 |
1.54 (1.24, 1.92) <0.001 |
−5% (−24, 5) 0.31 |
| Heart Failure | ||||
| Iron Deficiency | 0.98 (0.76, 1.28) 0.90 |
1.34 (1.25, 1.45) <0.001 |
1.32 (1.03, 1.70) 0.03 |
112% (51, 579) 0.03 |
| Mixed Iron Deficiency | 1.20 (0.89, 1.63) 0.23 |
1.11 (1.05, 1.17) <0.001 |
1.33 (0.98, 1.81) 0.07 |
35% (−70, 235) 0.06 |
| High Iron | 1.60 (1.23, 2.09) <0.001 |
0.98 (0.94, 1.02) 0.34 |
1.57 (1.20, 2.06) 0.001 |
−6% (−31, 5) 0.29 |
| PTH as mediator | ||||
| Mortality | ||||
| Iron Deficiency | 1.27 (1.02, 1.58) 0.03 |
1.00 (0.98, 1.01) 0.79 |
1.27 (1.02, 1.57) 0.03 |
2% (−12, 21) 0.55 |
| Mixed Iron Deficiency | 1.62 (1.27, 2.06) <0.001 |
1.01 (0.99, 1.02) 0.50 |
1.62 (1.28, 2.07) <0.001 |
0.6% (−5, 6) 0.80 |
| High Iron | 1.55 (1.24, 1.92) <0.001 |
0.99 (0.98, 1.01) 0.27 |
1.53 (1.23, 1.91) <0.001 |
−2% (−11, 3) 0.35 |
| Heart Failure | ||||
| Iron Deficiency | 1.36 (1.06, 1.74) 0.02 |
1.00 (0.99, 1.01) 0.79 |
1.35 (1.06, 1.73) 0.02 |
2% (−7, 16) 0.52 |
| Mixed Iron Deficiency | 1.28 (0.94, 1.74) 0.12 |
1.00 (0.99, 1.02) 0.51 |
1.28 (0.94, 1.74) 0.11 |
1% (−40, 40) 0.82 |
| High Iron | 1.59 (1.22, 2.08) <0.001 |
0.99 (0.98, 1.00) 0.30 |
1.58 (1.21, 2.07) <0.001 |
−2% (−12, 3) 0.35 |
| Hemoglobin as mediator | ||||
| Mortality | ||||
| Iron Deficiency | 1.19 (0.96, 1.48) 0.11 |
1.05 (1.02, 1.09) 0.003 |
1.26 (1.01, 1.56) 0.04 |
23% (4, 130) 0.04 |
| Mixed Iron Deficiency | 1.54 (1.21, 1.96) <0.001 |
1.02 (1.00, 1.04) 0.03 |
1.57 (1.24, 2.00) <0.001 |
5% (1, 13) 0.003 |
| High Iron | 1.51 (1.22, 1.88) <0.001 |
1.00 (0.99, 1.01) 0.77 |
1.52 (1.22, 1.88) <0.001 |
0.4% (−4, 5) 0.81 |
| Heart Failure | ||||
| Iron Deficiency | 1.26 (0.98, 1.62) 0.07 |
1.10 (1.05, 1.15) <0.001 |
1.38 (1.08, 1.77) 0.01 |
31% (13, 138) 0.02 |
| Mixed Iron Deficiency | 1.29 (0.95, 1.74) 0.10 |
1.04 (1.01, 1.07) 0.009 |
1.34 (0.99, 1.81) 0.06 |
14% (−69, 113) 0.09 |
| High Iron | 1.59 (1.22, 2.08) <0.001 |
1.00 (0.98, 1.02) 0.77 |
1.60 (1.23, 2.09) <0.001 |
0.6% (−6, 7) 0.80 |
Estimates for direct, indirect and total effects are hazard ratios. The direct effect refers to the effect of iron stores on the outcome independent of the mediator. The indirect effect refers to the effect of iron stores on the outcome that acts through the mediator. Total effects are computed as the product of the direct and indirect effects.
Proportion mediated is computed using R package mediation based on a Weibull model and presented as percentages.
Covariates included in the model: center, age, sex, race, ethnicity, diabetes, smoking, systolic blood pressure, body mass index, history of cardiovascular disease, estimated glomerular filtration rate, urinary albumin-to-creatinine ratio, and erythropoietin use.
Abbreviations: PTH, parathyroid hormone; FGF23, fibroblast growth factor 23
To benchmark the mediation effects of FGF23, we conducted similar analyses with PTH and hemoglobin. PTH and hemoglobin were each independently associated with mortality and heart failure in fully adjusted models (Figure 3), but when PTH and the iron exposure groups were included together in fully adjusted models, the effects of iron status were unchanged. In formal mediation analyses, PTH did not significantly mediate any of the associations of Iron Deficiency, High Iron, or Mixed Iron Deficiency with mortality or heart failure (Table 3). When hemoglobin and the iron exposure groups were included together in fully adjusted models, the point estimates of the Iron Deficiency and Mixed Iron Deficiency groups were attenuated, but to a lesser degree than in the parallel FGF23 analyses (Figure 3). In formal mediation analyses, hemoglobin accounted for 23% of the association of Iron Deficiency with mortality, 31% of the association of Iron Deficiency with heart failure, and 5% of the association of Mixed Iron Deficiency with mortality (Table 3).
Sensitivity Analyses
Multivariable-adjusted risk of mortality, HF, ESRD, and ASCVD across the spectra of continuous TSAT and ferritin values demonstrate non-linear relationships between ferritin, TSAT and outcomes that highlight the need to categorize patients into groups across the two dimensions of TSAT and ferritin (Supplemental Figures 3 and 4). When we re-categorized the iron exposure groups using clinical cut points (Supplemental Table 3), the results of the outcomes and mediation analyses were qualitatively similar to the primary analyses (Supplemental Table 4 and 5). Analyses that adjusted for UACR as a continuous variable yielded qualitatively similar results as the primary outcomes analyses (Supplemental Table 6). Likewise, adjusting for UACR as a continuous variable did not qualitatively change the results of the primary mediation analyses (Supplemental Table 7).
Discussion
In a diverse cohort of individuals with moderate to severe CKD, we found that iron deficiency associated with significantly increased risks of mortality and heart failure compared to normal iron stores, but did not associate with ESRD or ASCVD events. Analyses that used clinical rather than agnostic thresholds to define the iron exposure groups yielded qualitatively similar results. Overall, these results confirm prior reports of increased risk of mortality in association with iron deficiency in patients with CKD,3, 4 and extend them by comprehensively investigating iron deficiency and a range of important cardiovascular and renal outcomes, including heart failure, ASCVD events and ESRD.
Multiple plausible biological mechanisms may contribute to increased risks of mortality and heart failure among patients with CKD and iron deficiency. For example, iron deficiency reduces oxygen storage in myoglobin and tissue oxidative capacity, adversely effects cardiac mitochondrial energetics and alters immune responses.30, 31 Iron deficiency also increases FGF23 expression. FGF23 levels were progressively higher in groups with more severe iron deficiency, and FGF23 mediated the risks of mortality and heart failure associated with iron deficiency. In contrast, PTH did not mediate the effects of iron deficiency on mortality and heart failure, despite independent associations of PTH with these outcomes. These contrasting data suggest specific mediation effects of FGF23 rather than non-specific effects attributable to more severe alterations in mineral metabolism. Furthermore, hemoglobin only partially mediated the effects of iron deficiency on mortality and heart failure, despite anemia being an immediate downstream consequence of iron deficiency.
Together, these contrasts support FGF23 as a mediator on a potential causal pathway between iron deficiency, heart failure and mortality in CKD. Mechanistically, iron deficiency stimulates FGF23 transcription by stabilizing hypoxia-inducible factor 1α.18 Reduced FGF23 cleavage is a “second hit” that enables iron deficiency to increase full-length biologically active FGF23 specifically in CKD.18, 32 Higher FGF23 levels associate with increased risks of mortality and heart failure, and may contribute directly to cardiac toxicity by activating FGF receptor 4 (FGFR4), which promotes left ventricular hypertrophy in a klotho-independent manner.11, 12, 13, 14, 15, 33, 34, 35, 36, 37 The results of the current study suggest that increases in FGF23 induced by iron deficiency could magnify the cardiac toxicity of FGF23. Further studies are needed to determine whether correction of iron deficiency can improve clinical outcomes in CKD, as it does in patients with heart failure and iron deficiency, who also manifest elevated FGF23 levels that are independently associated with worse outcomes.30, 38, 39, 40, 41, 42, 43, 44, 45
Functional Iron Deficiency did not significantly increase risk of any outcome in the primary analyses. This finding was surprising given prior studies3 and because this group had the highest rates of hypertension, diabetes, and prior cardiovascular disease, the highest CRP and UACR, and the lowest eGFR. Some individuals in the current Mixed Iron Deficiency group likely would have been categorized as having functional iron deficiency in prior studies, which may have driven the associations between functional iron deficiency and poor outcomes in prior studies.3 Functional iron deficiency is primarily caused by chronic inflammation-induced increases in hepcidin, which reduces iron absorption and availability.46 Given that inflammation and erythropoietin also upregulate FGF23 transcription,18 we expected FGF23 elevation in patients with functional iron deficiency. Although FGF23 levels were higher in the Functional Iron Deficiency group than in the High Iron and Iron Replete groups, levels were not as high as in the Iron Deficiency or Mixed Iron Deficiency groups. Whether iron treatment will be beneficial or harmful in patients with Functional Iron Deficiency is unknown.
Another unexpected finding was the emergence of the Mixed Iron Deficiency group, which had an iron phenotype intermediate between the Iron Deficiency and Functional Iron Deficiency groups, but clinical outcomes most similar to the Iron Deficiency group. Likewise, FGF23 levels were elevated in this group and partially mediated its association with increased risks of heart failure and mortality. Additional research is needed to better understand the Mixed Iron Deficiency phenotype, to determine whether there would be clinical utility in separating Mixed Iron Deficiency from its current inclusion in clinically-defined Functional Iron Deficiency, and whether this group would benefit from iron repletion.
High Iron significantly increased risks of mortality and heart failure regardless of whether it was defined by agnostic or clinical criteria. High Iron also increased risk of ESRD, as reported previously.3, 47 Iron excess may increase catalytic iron that is not bound to transferrin and is associated with acute kidney injury and mortality.48, 49 Excess systemic iron may also increase intracellular iron, which can cause lipid peroxidation that leads to mitochondrial and cardiac dysfunction.50 Further studies are needed to investigate the causes and consequences of High Iron.
This study has limitations. Since we only measured iron parameters at a single time-point at baseline, variability of ferritin and TSAT over time may have caused misclassification of the primary exposure. Although we found that FGF23 mediated the associations between iron deficiency and clinical outcomes, it is undoubtedly only one of many factors that contribute to the adverse effects of iron deficiency on outcomes. Residual confounding by unknown or unmeasured factors that were not included in the analysis cannot be excluded. Additionally, despite our use of established methods to investigate mediation by FGF23, observational studies cannot prove causality. Since we measured FGF23 with a C-terminal assay that detects full-length FGF23 and its C-terminal fragments, but not an intact assay that exclusively measures the full-length peptide, we cannot determine which FGF23 peptides account for the effects we observed. Although full-length FGF23 is the biologically active peptide for phosphate homeostasis, C-terminal peptides (or perhaps the equimolar amounts of N-terminal fragments that are generated by FGF23 cleavage) might exert toxic but currently unknown effects that contribute to adverse outcomes.4, 51 We also did not have data on the duration of prior cardiovascular disease or vitamin D levels. Finally, for all analyses, the true association between iron status and outcomes may fall anywhere within the confidence interval, and in many cases, this lower limit of the confidence interval is close to 1.00. Interpretation of point estimates should recognize the inherent limitations of confidence intervals.
Lack of data on intravenous iron therapy is another limitation, but during the study period of 2008–2013, intravenous iron was uncommon in non-dialysis-dependent CKD.52 Since then, highly bioavailable oral iron preparations and intravenous iron formulations that enable rapid correction of iron deficiency became available. Reports that these treatments may improve clinical outcomes in CKD and heart failure populations,43, 44, 45, 53, 54 along with the current and recent results linking iron deficiency and FGF23 excess to each other and to adverse outcomes in CKD,4 justify randomized clinical outcomes trials of iron replacement in select patients with non-dialysis-dependent CKD.
Supplementary Material
Supplemental Table 1. Risks of outcomes in the subgroups of the Non-Classified Group versus the Iron Replete Group
Supplemental Table 2. Iron stores and incident heart failure and atherosclerotic cardiovascular disease
Supplemental Table 3. Baseline characteristics according to iron groups using clinical thresholds
Supplemental Table 4. Iron groups defined by clinical thresholds and risks of mortality and cardiovascular events
Supplemental Table 5. Iron groups defined by clinical thresholds and risks of mortality and cardiovascular events with adjustment for FGF23, Hemoglobin and PTH
Supplemental Table 6. Iron groups and risks of mortality and cardiovascular events adjusting for UACR as a continuous variable
Supplemental Table 7. Iron groups and risks of mortality and cardiovascular events adjusting for UACR as a continuous variable and additionally adjusting for potential mediators: FGF23, hemoglobin and PTH
Supplemental Figure 1. Classification of iron groups according clinical thresholds of ferritin and transferrin saturation
Supplemental Figure 2. Direct acyclic graph detailing exposure, confounders, mediators and outcomes
Supplemental Figure 3. Transferrin saturation and mortality and cardiovascular events
Supplemental Figure 4. Ferritin and mortality and cardiovascular events
Acknowledgements
The authors thank the participants, investigators, and staff of the CRIC study for their time and commitment. The authors take responsibility for decision to submit the manuscript for publication. Drs. Mehta and Wolf had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This study was supported by grants P30DK114857, K23HL150236 (RM), R01DK081374 (MW), K24DK093723 (MW), R01DK102438 (TI), R01DK111952 (JS), R03 HL146788 (SLS), CX001566-01A1 (MC), National Kidney Foundation of Illinois Young Investigator Grant (RM), and a Strategically Focused Research Network Center Grant from the American Heart Association (MW). Research reported in this publication was also supported, in part, by the National Institutes of Health’s National Center for Advancing Translational Sciences, Grant Number KL2TR001424 and by the National Institutes of Health’s National Center for Advancing Translational Sciences, Grant Number UL1TR001422. Funding for the CRIC Study was obtained under a cooperative agreement from National Institute of Diabetes and Digestive and Kidney Diseases (U01DK060990, U01DK060984, U01DK061022, U01DK061021, U01DK061028, U01DK060980, U01DK060963, and U01DK060902). In addition, this work was supported in part by: the Perelman School of Medicine at the University of Pennsylvania Clinical and Translational Science Award NIH/NCATS UL1TR000003, Johns Hopkins University UL1TR-000424, University of Maryland GCRC M01 RR-16500, Clinical and Translational Science Collaborative of Cleveland, UL1TR000439 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research, Michigan Institute for Clinical and Health Research (MICHR) UL1TR000433, University of Illinois at Chicago CTSA UL1RR029879, Tulane COBRE for Clinical and Translational Research in Cardiometabolic Diseases P20 GM109036, Kaiser Permanente NIH/NCRR UCSF-CTSI UL1 RR-024131. The funders had no role in the study design, data collection, analysis, reporting or decision to submit for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Disclosure
RM has stock ownership in AbbVie, Inc. and Teva Pharmaceuticals Industries Ltd and consultant/honoraria fees from Akebia/Oksuba and AstraZeneca. SLS has received research support or consultant fees from Pfizer, Novartis, and Global Blood Therapeutics. TI has received honoraria from Kyowa Kirin and LifeSci Capital. MW has received research support, honoraria or consultant fees from Akebia, Ardelyx, AstraZeneca, Bayer, Jnana, Pharmacosmos, Unicycive, and Walden Biosciences and has equity interests in Akebia, Unicycive and Walden Biosciences. The remaining authors declare that they have no relevant financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary information is available on Kidney International’s website.
References
- 1.Batchelor EK, Kapitsinou P, Pergola PE, et al. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. Journal of the American Society of Nephrology. 2020;31(3):456–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wish JB, Aronoff GR, Bacon BR, et al. Positive Iron Balance in Chronic Kidney Disease: How Much is Too Much and How to Tell? American journal of nephrology. 2018;47(2):72–83 [DOI] [PubMed] [Google Scholar]
- 3.Cho ME, Hansen JL, Peters CB, et al. An increased mortality risk is associated with abnormal iron status in diabetic and non-diabetic Veterans with predialysis chronic kidney disease. Kidney international. 2019;96(3):750–760 [DOI] [PubMed] [Google Scholar]
- 4.Eisenga MF, van Londen M, Leaf DE, et al. C-Terminal Fibroblast Growth Factor 23, Iron Deficiency, and Mortality in Renal Transplant Recipients. Journal of the American Society of Nephrology : JASN. 2017;28(12):3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf M, White KE. Coupling fibroblast growth factor 23 production and cleavage: iron deficiency, rickets, and kidney disease. Current opinion in nephrology and hypertension. 2014;23(4):411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew JS, Sachs MC, Katz R, et al. Fibroblast Growth Factor-23 and Incident Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation. 2014;130(4):298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kestenbaum B, Sachs MC, Hoofnagle AN, et al. Fibroblast growth factor-23 and cardiovascular disease in the general population: the Multi-Ethnic Study of Atherosclerosis. Circulation Heart failure. 2014;7(3):409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta R, Cai X, Lee J, et al. Association of Fibroblast Growth Factor 23 With Atrial Fibrillation in Chronic Kidney Disease, From the Chronic Renal Insufficiency Cohort Study. JAMA cardiology. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ix JH, Katz R, Kestenbaum BR, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). Journal of the American College of Cardiology. 2012;60(3):200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehta R, Cai X, Lee J, et al. Serial Fibroblast Growth Factor 23 Measurements and Risk of Requirement for Kidney Replacement Therapy: The CRIC (Chronic Renal Insufficiency Cohort) Study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA : the journal of the American Medical Association. 2011;305(23):2432–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isakova T, Cai X, Lee J, et al. Longitudinal FGF23 Trajectories and Mortality in Patients with CKD. Journal of the American Society of Nephrology : JASN. 2018;29(2):579–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scialla JJ, Xie H, Rahman M, et al. Fibroblast growth factor-23 and cardiovascular events in CKD. Journal of the American Society of Nephrology : JASN. 2014;25(2):349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jovanovich A, Ix JH, Gottdiener J, et al. Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis. 2013;231(1):114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutierrez OM, Januzzi JL, Isakova T, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119(19):2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munoz Mendoza J, Isakova T, Ricardo AC, et al. Fibroblast growth factor 23 and Inflammation in CKD. Clinical journal of the American Society of Nephrology : CJASN. 2012;7(7):1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28(8):1793–1803 [DOI] [PubMed] [Google Scholar]
- 18.David V, Martin A, Isakova T, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney international. 2016;89(1):135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nature Reviews Nephrology. 2020;16(1):7–19 [DOI] [PubMed] [Google Scholar]
- 20.Lash JP, Go AS, Appel LJ, et al. Chronic Renal Insufficiency Cohort (CRIC) Study: baseline characteristics and associations with kidney function. Clinical journal of the American Society of Nephrology : CJASN. 2009;4(8):1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldman HI. The Chronic Renal Insufficiency Cohort (CRIC) Study: Design and Methods. Journal of the American Society of Nephrology : JASN. 2003;14(90002):148S–153 [DOI] [PubMed] [Google Scholar]
- 22.Locatelli F, Barany P, Covic A, et al. Kidney Disease: Improving Global Outcomes guidelines on anaemia management in chronic kidney disease: a European Renal Best Practice position statement. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28(6):1346–1359 [DOI] [PubMed] [Google Scholar]
- 23.Cappellini MD, Comin-Colet J, de Francisco A, et al. Iron deficiency across chronic inflammatory conditions: International expert opinion on definition, diagnosis, and management. American journal of hematology. 2017;92(10):1068–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ku E, Yang W, McCulloch CE, et al. Race and Mortality in CKD and Dialysis: Findings From the Chronic Renal Insufficiency Cohort (CRIC) Study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2020;75(3):394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Annals of internal medicine. 2009;150(9):604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international. 2011;79(12):1370–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valeri L, VanderWeele TJ. SAS macro for causal mediation analysis with survival data. Epidemiology. 2015;26(2):e23–24 [DOI] [PubMed] [Google Scholar]
- 28.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychological methods. 2013;18(2):137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imai K, Keele L, Tingley D. A general approach to causal mediation analysis. Psychological methods. 2010;15(4):309–334 [DOI] [PubMed] [Google Scholar]
- 30.Anand IS, Gupta P. Anemia and Iron Deficiency in Heart Failure. Circulation. 2018;138(1):80–98 [DOI] [PubMed] [Google Scholar]
- 31.Lill R, Hoffmann B, Molik S, et al. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823(9):1491–1508 [DOI] [PubMed] [Google Scholar]
- 32.Clinkenbeard EL, Farrow EG, Summers LJ, et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2014;29(2):361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. The New England journal of medicine. 2008;359(6):584–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grabner A, Schramm K, Silswal N, et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Scientific reports. 2017;7(1):1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation. 2011;121(11):4393–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X, Cai C, Xiao Z, et al. FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. Journal of molecular and cellular cardiology. 2020;138:66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grabner A, Amaral A, Schramm K, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metabolism. 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Udell JA, Morrow DA, Jarolim P, et al. Fibroblast growth factor-23, cardiovascular prognosis, and benefit of angiotensin-converting enzyme inhibition in stable ischemic heart disease. Journal of the American College of Cardiology. 2014;63(22):2421–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rousseau MF, Gruson D, Lepoutre T, et al. FGF23 IS A STRONG PREDICTOR OF SURVIVAL IN CONGESTIVE HEART FAILURE. Journal of the American College of Cardiology. 2012;59(13 Supplement):E878 [Google Scholar]
- 40.Almahmoud MF, Soliman EZ, Bertoni AG, et al. Fibroblast Growth Factor-23 and Heart Failure With Reduced Versus Preserved Ejection Fraction: MESA. Journal of the American Heart Association. 2018;7(18):e008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anker SD, Comin Colet J, Filippatos G, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. The New England journal of medicine. 2009;361(25):2436–2448 [DOI] [PubMed] [Google Scholar]
- 42.Bolger AP, Bartlett FR, Penston HS, et al. Intravenous iron alone for the treatment of anemia in patients with chronic heart failure. Journal of the American College of Cardiology. 2006;48(6):1225–1227 [DOI] [PubMed] [Google Scholar]
- 43.Toblli JE, Lombrana A, Duarte P, et al. Intravenous iron reduces NT-pro-brain natriuretic peptide in anemic patients with chronic heart failure and renal insufficiency. Journal of the American College of Cardiology. 2007;50(17):1657–1665 [DOI] [PubMed] [Google Scholar]
- 44.Okonko DO, Grzeslo A, Witkowski T, et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron deficiency FERRIC-HF: a randomized, controlled, observer-blinded trial. Journal of the American College of Cardiology. 2008;51(2):103–112 [DOI] [PubMed] [Google Scholar]
- 45.van Veldhuisen DJ, Ponikowski P, van der Meer P, et al. Effect of Ferric Carboxymaltose on Exercise Capacity in Patients With Chronic Heart Failure and Iron Deficiency. Circulation. 2017;136(15):1374–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010;55(4):726–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salonen JT, Nyyssönen K, Korpela H, et al. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803–811 [DOI] [PubMed] [Google Scholar]
- 48.Leaf DE, Rajapurkar M, Lele SS, et al. Iron, Hepcidin, and Death in Human AKI. Journal of the American Society of Nephrology. 2019;30(3):493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leaf DE, Rajapurkar M, Lele SS, et al. Plasma Catalytic Iron, AKI, and Death among Critically Ill Patients. Clin J Am Soc Nephro. 2014;9(11):1849–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang H-C, Shapiro JS, Ardehali H. Getting to the “Heart” of Cardiac Disease by Decreasing Mitochondrial Iron. Circulation research. 2016;119(11):1164–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goetz R, Nakada Y, Hu M, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney inter., Suppl 2012; 2: 279–335. [Google Scholar]
- 53.Block GA, Block MS, Smits G, et al. A Pilot Randomized Trial of Ferric Citrate Coordination Complex for the Treatment of Advanced CKD. Journal of the American Society of Nephrology : JASN. 2019;30(8):1495–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Macdougall IC, White C, Anker SD, et al. Intravenous Iron in Patients Undergoing Maintenance Hemodialysis. New England Journal of Medicine. 2018;380(5):447–458 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Risks of outcomes in the subgroups of the Non-Classified Group versus the Iron Replete Group
Supplemental Table 2. Iron stores and incident heart failure and atherosclerotic cardiovascular disease
Supplemental Table 3. Baseline characteristics according to iron groups using clinical thresholds
Supplemental Table 4. Iron groups defined by clinical thresholds and risks of mortality and cardiovascular events
Supplemental Table 5. Iron groups defined by clinical thresholds and risks of mortality and cardiovascular events with adjustment for FGF23, Hemoglobin and PTH
Supplemental Table 6. Iron groups and risks of mortality and cardiovascular events adjusting for UACR as a continuous variable
Supplemental Table 7. Iron groups and risks of mortality and cardiovascular events adjusting for UACR as a continuous variable and additionally adjusting for potential mediators: FGF23, hemoglobin and PTH
Supplemental Figure 1. Classification of iron groups according clinical thresholds of ferritin and transferrin saturation
Supplemental Figure 2. Direct acyclic graph detailing exposure, confounders, mediators and outcomes
Supplemental Figure 3. Transferrin saturation and mortality and cardiovascular events
Supplemental Figure 4. Ferritin and mortality and cardiovascular events