

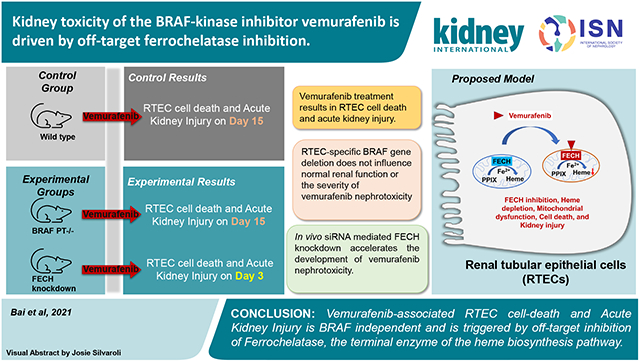
Abstract
A multitude of disease and therapy related factors drive the frequent development of kidney disorders in cancer patients. Along with chemotherapy, the newer targeted therapeutics can also cause kidney dysfunction through on and off-target mechanisms. Interestingly, among the small molecule inhibitors approved for the treatment of cancers that harbor BRAF-kinase activating mutations, vemurafenib can trigger tubular damage and acute kidney injury. BRAF is a proto-oncogene involved in cell growth. To investigate the underlying mechanisms, we developed cell culture and mouse models of vemurafenib kidney toxicity. At clinically relevant concentrations vemurafenib induces cell-death in transformed and primary mouse and human kidney tubular epithelial cells. In mice, two weeks of daily vemurafenib treatment causes moderate acute kidney injury with histopathological characteristics of kidney tubular epithelial cells injury. Importantly, kidney tubular epithelial cell-specific BRAF gene deletion did not influence kidney function under normal conditions or alter the severity of vemurafenib-associated kidney impairment. Instead, we found that inhibition of ferrochelatase, an enzyme involved in heme biosynthesis contributes to vemurafenib kidney toxicity. Ferrochelatase overexpression protected kidney tubular epithelial cells and conversely ferrochelatase knockdown increased the sensitivity to vemurafenib-induced kidney toxicity. Thus, our studies suggest that vemurafenib-associated kidney tubular epithelial cell dysfunction and kidney toxicity is BRAF-independent and caused, in part, by off-target ferrochelatase inhibition.
Keywords: Onconephrology, Acute kidney injury, BRAF kinase, Protein kinase inhibitors, Ferrochelatase, Renal tubular epithelial cells
Graphical Abstract
Introduction
A broad array of factors increase the risk of renal dysfunction in cancer patients1-3. While certain malignancies can directly affect kidney function4, cancer therapy can also trigger fluid and electrolyte disorders5, acute kidney injury (AKI)6, and chronic kidney disease (CKD)7. Renal disorders jeopardize the continuation of cancer therapy and contribute to the development of debilitating short and long-term adverse sequelae. Along with chemotherapeutics, targeted antibodies8 and small-molecule inhibitors9 are also associated with renal impairment. Thus, one of the challenges in the emerging field of onconephrology10-13 is to determine the mechanisms associated with these toxicities in order to develop mitigating strategies.
Dysregulation of the mitogen-activated protein kinase (MAPK) pathway is a major driver of oncogenesis14. The Raf (rapidly accelerated fibrosarcoma) family of serine/threonine kinases act as a conduit between upstream Ras signaling and downstream MAPK kinase activation, relaying signaling cues from the extracellular environment and directing cell proliferation, differentiation, migration and survival15. Mammals possess three RAF proteins: RAF1 (CRAF), ARAF and BRAF, which play essential and distinct physiological roles. Importantly, somatic activating mutations in BRAF are frequent in hairy-cell leukemia (100%), melanoma (50%–60%), and thyroid cancer (40%– 60%)15,16. The recent development of orally bioavailable BRAF-inhibitors, namely vemurafenib17,18 and dabrafenib19 have brought exceptional clinical benefits, especially in melanoma patients.
Dermatological toxicities are the major side effects related to vemurafenib treatment20. While renal toxicities were not observed in the clinical trials21, a growing body of literature suggests that a significant percentage of patients treated with vemurafenib can develop AKI22-24,24-26. Acute tubular necrosis, electrolyte disorders, and subnephrotic-range proteinuria have been reported in a subset of vemurafenib-treated patients22,25. These studies have shown that patients treated with vemurafenib as compared to dabrafenib have a higher incidence of AKI and analysis of kidney biopsies have established tubular injury as the major histopathological lesion22.
Due to paucity of experimental models, the mechanisms underlying vemurafenib nephrotoxicity remain unclear. Furthermore, it is unknown if BRAF is essential for renal tubular function. It is also unknown whether vemurafenib nephrotoxicity is BRAF-dependent. To address these key questions, here we have developed a renal tubule-specific BRAF knockout mice and established in vitro and in vivo models of vemurafenib nephrotoxicity. Our studies suggest that BRAF knockout in renal tubules does not trigger renal impairment and vemurafenib-associated RTEC dysfunction and AKI is BRAF-independent.
Material and Methods
Animal Experiments.
Mice were housed and handled in accordance with the Institutional Animal Care and Use Committee of the Ohio State University and the University of Tennessee Health Science Center. Vemurafenib (20 mg/kg, p.o., b.i.d) was administered for 15-20 days and blood urea nitrogen, serum creatinine, Ngal expression, and H&E based histological examinations were used to assess the extent of renal injury. Details regarding animal procedures are provided in Supplementary methods.
Cell culture.
BUMPT, HK-2, and primary murine RTECs were cultured according to previously described methods27,28. Cell viability and cell-death was monitored by trypan blue, MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide), and caspase activity measurements. Further information regarding the experimental procedures are provided in Supplementary methods.
Vemurafenib pharmacokinetic analysis.
Plasma levels of vemurafenib were measured using previously described methods29,30 and as detailed in the Supplementary methods.
Ferrochelatase assay and intracellular heme measurement.
Ferrochelatase activity was measured by enzymatic formation of zinc-protoporphyrin IX (Zn-PpIX) using a well-established method31. For determination of total intracellular heme levels, cells were homogenized in 1%Triton-X100 in TBS followed by heme quantification based on a previously described method32. These procedures are described in detail in the Supplementary methods.
Protein and Gene expression analysis.
For protein analysis, whole cell lysates were prepared using a modified RIPA buffer supplemented with 1% SDS. Invitrogen Bis-tris gradient midi-gels were used for western blot analysis and the antibodies used are described in the Supplementary methods. For gene expression analysis, we performed quantitative polymerase chain reaction (qPCR) as described in our previous work33 and in the Supplementary methods.
Statistical Analysis.
Data are presented as mean with SD. P < 0.05 was considered statistically significant. The GraphPad Prism software was used for statistical analysis. Additional details are provided in supplementary methods.
Results
Vemurafenib induces cell-death in cultured RTECs.
We initially sought to determine if vemurafenib could cause direct toxicity in cultured RTECs. To this end, we treated transformed tubular epithelial cells of murine (BUMPT) and human origin (HK-2) with vemurafenib and tested its effect on cellular survival. In these experiments, we also included cisplatin, a well-studied nephrotoxic drug34, as well as BRAF inhibitor dabrafenib, and the multi-kinase and CRAF-inhibitor sorafenib35. Similar to cisplatin, vemurafenib treatment reduced cellular viability as measured by trypan blue staining (Fig. 1A-B) in both BUMPT and HK-2 cells (IC50=~50μM). Notably, pharmacokinetic studies in humans have shown that plasma levels of vemurafenib can range from 50-100 μM21. Confirmatory experiments with MTT assays showed that at 50 μM concentration, vemurafenib can cause ~50% reduction in cellular viability within 48 hours (Suppl. Fig.1A-B), with a parallel increase in caspase activation (Fig. 1C-D).
Figure 1: Vemurafenib induces cell death in murine and human tubular epithelial cell lines.

Tubular epithelial cell lines of murine (BUMPT) and human (HK-2) origin were treated with vehicle, cisplatin or kinase inhibitors including vemurafenib followed by assessment of cell viability and cell death. (A-B) Dose response experiments (0-100 μM) and trypan blue based cellular viability assays at 48 hours post treatment showed that vemurafenib can induce cell death in RTEC cell lines with IC50 values of approximately 50 μM. Survival data was normalized to vehicle group and are presented as mean (n = 5 biologically independent samples), from one out of three independent experiments, all producing similar results. (C-D) BUMPT and HK-2 cells were treated with vehicle or indicated drugs at 50 μM concentration, followed by measurement of caspase activity at 48 hours. The results show that similar to cisplatin, vemurafenib can reduce RTEC viability. Data are presented as individual data points (n = 8 biologically independent samples from three independent experiments). (E) RNAi mediated Braf knockdown in BUMPT cells did not influence vemurafenib associated cell death (50 μM for 48 hours) as assessed by trypan blue based viability assay. Data are presented as individual data points (n = 6 biologically independent samples from three independent experiments). A representative immunoblot (right panel) shows the successful knockdown of Braf gene. (F) CRISPR/Cas9 mediated Braf knockout in HK-2 cells did not influence vemurafenib associated cell death (50 μM for 48 hours) as assessed by trypan blue based viability assay. Data are presented as individual data points (n = 8 biologically independent samples from three independent experiments). A representative immunoblot (right panel) shows the successful knockout of Braf gene. In all the graphs (n=5-8 biologically independent samples), experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Dunnett’s was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Intriguingly, kinase assays showed that at 50 μM concentration vemurafenib, dabrafenib, and sorafenib inhibited BRAF-kinase activity to similar levels (Suppl. Fig.1C-D), however, under these conditions only vemurafenib triggered RTEC cell-death. We next questioned whether BRAF kinase is essential for RTEC survival and a causal factor in vemurafenib-induced cell-death. To address this, we carried out RNAi-mediated knockdown and CRISPR/Cas9-mediated BRAF knockout in BUMPT and HK-2 cells respectively (Fig. 1E-F). BRAF knockdown or knockout had no impact on cellular viability under normal conditions and neither did it influence vemurafenib-induced cell-death (Fig. 1E-F and Suppl. Fig. 2). These results suggest that vemurafenib can trigger cell-death in cultured RTECs, seemingly through a BRAF-independent mechanism.
Establishment of a mouse model of vemurafenib nephrotoxicity.
To establish a murine model of vemurafenib nephrotoxicity, we initially performed a pharmacokinetic study in C57B6/J mice. In concordance with previous work36, we found that at 20 mg/kg dose, plasma drug (~50μM) concentrations (Suppl. Fig. 3) were similar to those observed in humans21. We then determined the effect of 20 mg/kg twice daily vemurafenib administration on kidney function (Fig. 2A). We noticed a significant increase in blood urea nitrogen (Fig. 2B) and serum creatinine (Fig. 2C) levels after 2 weeks of b.i.d. dosing. In concordance with studies in humans, our histological examination (Fig. 2D-F and Suppl. Fig. 4) revealed tubular epithelial injury and cell-death as the major pathological lesion. In C57B6/J mice, several injury37, repair38 and inflammatory genes39-42 are upregulated during ischemia, cisplatin, and rhabdomyolysis associated AKI43. We found a similar increase in injury, repair, and inflammation-related genes during vemurafenib nephrotoxicity (Fig. 2G and Suppl. Fig. 5). Similar to humans22,25, we also found that treatment discontinuation in mice reverses vemurafenib-associated renal impairment (day 20 versus 22).
Figure 2: Development of a mouse model of vemurafenib nephrotoxicity.

(A) Schematic representation of dosing strategy. Age-matched, 8-12 weeks old male C57BL/6J mice were treated with either vehicle or 20 mg/kg vemurafenib (p.o, b.i.d.) for 20 days followed by cessation of drug administration for 2 days and subsequent endpoint analysis of renal function. (B) Analysis of blood urea nitrogen levels showed that vemurafenib can induce AKI after 2 weeks of continuous treatment and cessation of drug administration reversed the increase in BUN levels. (C) Vemurafenib treatment also resulted in increased serum creatinine levels indicating significant renal impairment (D-E) Histological analysis of renal tissues showed that vemurafenib treated mice had clear tubular epithelial injury and cell death. Representative H&E staining depicting renal tubular damage (indicated by an asterisk) linked with vemurafenib-associated AKI. (D & F) TUNEL staining of renal tissues revealed significant tubular epithelial cell death in the vemurafenib treated mice. (G) Renal Ngal gene expression analysis further confirmed significant renal damage in vemurafenib treated mice. In all the bar graphs (n=5-7 biologically independent samples), experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. Student’s t-test or non-parametric Mann–Whitney U test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar (D): 100 μm.
Vemurafenib nephrotoxicity is BRAF-independent.
To evaluate the RTEC-specific role of BRAF kinase, we generated conditional knockout mice (BRAFPT−/−) by crossing the BRAF floxed44 mice with the Ggt1-Cre mice45. We found that BRAF deficiency (Figure 3A) did not influence normal renal function (Suppl. Fig. 6). Moreover, when the control and BRAFPT−/− littermates were challenged with vemurafenib, the extent of renal damage as evaluated by blood urea nitrogen, serum creatinine, as well as histological damage and injury biomarker analysis was found to be similar in both groups (Figure 3B-F). To corroborate the in vivo findings, we isolated primary RTECs from BRAF floxed mice and carried out in vitro Cre-mediated deletion (Suppl. Fig. 7A). Vemurafenib treatment induced cell-death in primary RTECs and BRAF gene-deletion did not influence cell-death under these conditions (Suppl. Fig. 7B). These findings support the notion that BRAF-kinase inhibition is unlikely to be the underlying cause of vemurafenib-associated AKI.
Figure 3: Vemurafenib nephrotoxicity is not influenced by RTEC-specific Braf gene deletion.

To generate mice with renal tubule-specific Braf knockout, Ggt1-Cre mice were crossed with Braf-floxed mice. (A-B) Representative genotyping and immunoblots showing successful knockout in the renal tissues. Littermate control and Braf conditional knockout mice (indicated by BrafPT−/−) were used to study the role of Braf in vemurafenib nephrotoxicity. Age-matched, 8-12 weeks old male littermate mice were treated with either vehicle or 20 mg/kg vemurafenib (p.o, b.i.d.) for 20 days followed by subsequent endpoint analysis of renal function. Blood urea nitrogen (C), serum creatinine (D), and histological analysis (E) were performed to examine renal function and damage. The extent of functional renal impairment and damage was similar between the control and Braf deficient mice. (F) TUNEL staining also showed similar amount of renal epithelial cell death in the control and knockout tissues. (G) Renal Ngal gene expression analysis. In all the bar graphs (n=8 biologically independent samples), experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test or non-parametric Mann–Whitney U test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Identification of molecular targets associated with vemurafenib toxicity.
Small-molecule kinase inhibitors target the ATP-binding pocket; however, the high conservation of the ATP binding site within the kinase families poses a significant challenge in developing highly specific kinase inhibitors46. Previous studies47,48 have systematically profiled the promiscuity of kinase inhibitors including vemurafenib. To ascertain if inhibition of these kinases contribute to vemurafenib cytotoxicity in RTECs, we used a chemical genetics approach49. BUMPT cells were transfected with plasmids encoding wild-type or inhibitor-resistant kinase genes and their effect on cellular survival was monitored (Suppl. Fig. 8). Surprisingly, inhibitor-resistant kinase overexpression did not rescue cytotoxicity in BUMPT cells (Suppl. Fig. 8). Furthermore, vemurafenib is frequently used in conjunction with cobimetinib, a MEK kinase inhibitor, and this combination therapy has been suggested50 to possibly reduce vemurafenib nephrotoxicity. However, our in vivo and in vitro combination studies at clinically relevant concentrations51 (Suppl. Fig. 9 and 10) showed that concomitant treatment with cobimetinib does not influence the severity or prevalence of vemurafenib nephrotoxicity. These results raised the possibility of the involvement of a non-kinase related mechanism in vemurafenib nephrotoxicity.
Interestingly, chemical proteomics studies52,53 have shown that vemurafenib but not dabrafenib can inhibit ferrochelatase (FECH). FECH is a mitochondrial protein that catalyzes the insertion of ferrous iron into protoporphyrin IX, and is the terminal enzyme involved in heme biosynthesis54. To ascertain if FECH inhibition causes vemurafenib-associated RTEC cell-death, we initially quantified FECH activity in BUMPT and HK-2 cells. We found that vemurafenib significantly inhibited FECH activity in RTEC cell lines (Fig. 4A-B) and caused a reduction in intracellular heme levels (Suppl. Fig. 11). Importantly, intracellular heme serves as a prosthetic group for several proteins that constitute the complexes of mitochondrial electron transport chain55. Therefore, heme plays a pivotal role in oxidative phosphorylation and oxygen consumption. Using, seahorse flux analyzer-based assay, a well-established technique for the measurement of oxygen consumption rate (OCR) and mitochondrial respiration42,56, we found that mitochondrial dysfunction is triggered at early time points after vemurafenib treatment(Suppl. Fig. 12). To establish the functional role of FECH in vemurafenib-mediated RTEC cell death, we performed overexpression studies. We found that overexpression of wild-type FECH protected BUMPT and HK-2 from vemurafenib associated cell-death (Fig. 4C-G). Overexpression of an inactive FECH mutant (FECHmut) did not influence vemurafenib associated cell-death. These results indicate that FECH inhibition might contribute to RTEC cell-death under in vitro conditions.
Figure 4: Ferrochelatase inhibition contributes to Vemurafenib mediated RTEC cell death.

Tubular epithelial cell lines of murine (BUMPT) and human (HK-2) origin were treated with vehicle, cisplatin or kinase inhibitors including vemurafenib at 50 μM concentration, followed by assessment of ferrochelatase activity at 24 hours. (A-B) Ferrochelatase activity was inhibited by vemurafenib in BUMPT and HK-2 cells. (B) Representative immunoblot showing overexpression of FLAG-tagged wild type and mutant FECH. Blots are representative of three independent experiments. (D-G) Empty vector, wild type FECH, or FECH mutant (M98K) was overexpressed in BUMPT (transient transfection) and HK-2 (lentiviral transduction) cells followed by treatment with either vehicle or 50 μM vemurafenib for 48 hours. Trypan blue based survival assays and caspase assays showed that wild type FECH overexpression can protect BUMPT and HK-2 cells from vemurafenib-associated cell death. In all the bar graphs (n=5-6 biologically independent samples) from one out of three independent experiments, all producing similar results, and experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
RTEC-specific inhibition of FECH during vemurafenib nephrotoxicity.
Since vemurafenib nephrotoxicity is associated with RTEC injury and cell-death, we examined FECH expression in these and other renal cells. To this end, analysis of single cell RNAseq data57 (Suppl. Fig. 13) showed that Fech is expressed in multiple renal cell lines, with a subset of proximal tubular cells showing particularly high expression. Immunofluorescence studies showed that FECH is expressed in the cortical tubular epithelial cells, the major site of nephrotoxic injury (Suppl. Fig. 14A). Time-course studies showed that while FECH protein levels remained unaltered, a progressive decline in FECH activity occurred during vemurafenib nephrotoxicity (Suppl. Fig. 14B-C). Reduction in FECH activity was vemurafenib-specific and not a generalized consequence of AKI (Suppl. Fig. 14D). We next investigated whether the decrease in cortical renal FECH activity is RTEC specific. To label and isolate RTECs from murine kidneys, we crossed the ROSAmT/mG strain with the RTEC-specific Ggt1-Cre mice to generate transgenic mice that express membrane-localized green fluorescent protein (GFP) in the tubular epithelial cells (Fig. 5A). As described in our recent work28,43, we then isolated GFP-positive cells (RTECs) and GFP-negative (other cell types) from the kidneys of untreated and vemurafenib-treated mice (Fig. 5B). Initial examination of cells from untreated mice, showed that FECH protein expression is relatively higher in RTECs as compared to other cell types (Fig. 5C-D). Similar to C57B6/J mice, upon vemurafenib treatment, these reporter mice also exhibited similar extent of nephrotoxicity (Suppl. Fig. 15). Remarkably, when we isolated GFP positive and negative cells from vehicle and vemurafenib treated mice, we found that FECH activity (Fig. 5E) and intracellular heme levels (Fig. 5F) were specifically reduced in RTECs. Finally, we observed robust caspase activation specifically in RTECs from vemurafenib treated mice (Fig. 5G). These results suggest that RTECs are specifically sensitive to vemurafenib-associated FECH inhibition, heme depletion, and cell-death.
Figure 5: Ferrochelatase expression and inhibition in RTECs during vemurafenib nephrotoxicity.

To label, isolate, and examine RTECs, we crossed the Ggt1-Cre mice with ROSAmT/mG mice. The resulting transgenic mice express membrane localized EGFP in renal tubular epithelial cells, while the other cells type express membrane localized tdTomato. (A) A representative image showing EGFP expression in renal tubular cells, while the cells within the glomerulus are TdTomato positive. (B) Schematic representation of the methods used to isolate EGFP-positive renal epithelial cells. (C-D) Kidneys from control mice were used to isolate GFP positive (RTECs) and negative cells, followed by immunoblot and densitometric analysis. FECH expression was found to be higher in RTECs as compare to other cell types. As shown in the representative blot, the effectiveness of the cell isolation method was monitored by immunoblot analysis of RTEC (GFP and SLC22A2) and non-RTEC (tdTomato) specific markers. (E-G) Age-matched, 8-12 weeks old male transgenic mice were treated with either vehicle or 20 mg/kg vemurafenib (p.o, b.i.d.) for 15 days followed by isolation of GFP positive and negative cells. These isolated cells were then used to examine FECH activity, intracellular heme levels, and caspase activity. The results show that in the vemurafenib treated mice, there is an RTEC-specific decrease in FECH activity, heme depletion, and caspase activation. In all the bar graphs (n=8-10 biologically independent samples) are shown from three independent experiments and experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. Student’s t-test or One-way ANOVA followed by Tukey’s multiple-comparison test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
In vivo FECH knockdown hastens vemurafenib nephrotoxicity.
Given that a progressive decline in FECH activity preceded the development of AKI at 2 weeks, we questioned if FECH knockdown would influence vemurafenib nephrotoxicity. Using hydrodynamic siRNA injection approach29,33, we identified a specific siRNA that reduced FECH protein expression by ~70% (Fig. 6A-B). FECH knockdown did not influence normal renal function, however strikingly, mice with FECH knockdown developed vemurafenib nephrotoxicity within three days in contrast to 2 weeks in control mice (Fig. 6C-F). This increased sensitivity was vemurafenib-specific because no difference in the extent of renal impairment was observed when the mice were challenged with cisplatin (Suppl. Fig. 16). We also carried out these in vivo siRNA experiments in the reporter mice with RTEC-specific GFP expression. Immunoblot analysis of isolated GFP positive cells from these mice confirmed siRNA mediated FECH knockdown in RTECs (Suppl. Fig. 17A-B). Similar to C57B6/J mice, siRNA mediated FECH knockdown also hastened the development of vemurafenib nephrotoxicity in these reporter mice (Suppl. Fig. 17C-F). These results suggest that FECH knockdown can remarkably accelerate the development of vemurafenib-associated AKI.
Figure 6: In vivo siRNA mediated FECH knockdown hastens the development of vemurafenib nephrotoxicity.

Age-matched male (8-12 weeks) C57BL/6 mice were administered with three once-daily intravenous injections of control (non-specific) or FECH targeting siRNAs (25 μg in 0.5 ml of PBS). In one group (mock) 0.5 ml of PBS was injected. One day later mice were treated with either vehicle or 20 mg/kg vemurafenib (p.o, b.i.d.) for 3 days followed by endpoint analysis of renal function. (A) Representative blots and (B) densitometric analysis showed that the targeted siRNA was able to knock-down FECH proteins levels by approximately 75%. Blots are representative of three independent experiments, all producing similar results. Blood urea nitrogen (C), serum creatinine (D), and histological analysis (E) Renal Ngal gene expression analysis (F) showed that the FECH knockdown mice developed vemurafenib nephrotoxicity within 3 days of treatment, while the control group demonstrated no obvious renal injury or damage. In all the bar graphs (n=6-10 biologically independent samples), experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test or non-parametric Mann–Whitney U test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Accelerated development of vemurafenib nephrotoxicity in FECH mutant mice.
In humans, mutations associated with reduced ferrochelatase activity can cause erythropoietic protoporphyria (EPP), a disease characterized by cutaneous photosensitivity and liver damage. In the mice with homozygous (fch/fch) mutations that reduce the FECH activity to 2.7-6.3%, photosensitivity and hepatic dysfunction is observed54. On the other hand, the heterozygotes (+/fch) mice have 45-65% of normal FECH activity and do not display any skin or liver damage. Congruent with a previous study54, we found that the heterozygous mutant mice had approximately 50% reduction in renal FECH activity (Fig. 7A). Next, we studied the consequence of reduced FECH activity on the development of vemurafenib nephrotoxicity. In comparison to control littermates, the heterozygous mice developed vemurafenib nephrotoxicity in an accelerated manner. At 7 days of vemurafenib treatment, when the control mice had no apparent renal impairment, BUN, serum creatinine, histological, and gene expression analysis (Fig. 7B-E) showed a precipitous decline in renal function in the heterozygous mice. Given that vemurafenib inhibits FECH activity in RTECs and genetic reduction of FECH activity accelerates the development of AKI, we propose that inhibition of renal FECH function contributes to vemurafenib nephrotoxicity.
Figure 7: Accelerated development of vemurafenib nephrotoxicity in FECH mutant mice.

(A) Renal tissues from wild type and heterozygous (Fch/+) mutant mice (littermates) were used to evaluate FECH activity using an ezymmatic assay. The result show an approximately 50% reduction in FECH activity in the heterozygous mice. The wild type and heterozygous mice were then challenged with either vehicle or 20 mg/kg vemurafenib (p.o, b.i.d.) for 7 days followed by endpoint analysis of renal function. Blood urea nitrogen (B), serum creatinine (C), and histological analysis (D) Renal Ngal gene expression analysis (E) showed that the FECH heterozygous mutant mice developed vemurafenib nephrotoxicity within 7 days of treatment, at a time-point when the wild type group demonstrated no obvious renal injury or damage. In all the bar graphs (n=6-8 biologically independent samples), experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Tukey’s multiple-comparison test or non-parametric Mann–Whitney U test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
CRISPR/Cas9 mediated FECH knockout triggers cell-death in HK-2 cells.
We questioned if genetic FECH inhibition would be sufficient to trigger cell-death in cultured renal tubular epithelial cells. To address this, we utilized a doxycycline-inducible CRISPR/Cas9 system, wherein we generated stable HK-2 cells that express either a control or FECH-targeted sgRNA, along with doxycycline-inducible Cas9. As shown in Fig. 8A-B, doxycycline treatment resulted in decline in FECH protein expression in the stable cells expressing the FECH targeted sgRNA. When we further monitored these cells for 24-72 hours post doxycycline induction, we noticed a clear decline in cellular viability in the FECH knockout cells as measured by trypan blue staining (Fig. 8C). At 72 hours, MTT and caspase activity assays confirmed reduced viability and increased cell-death in the FECH knockout cells (Fig. 8D-E). The observation that genetic FECH deficiency impairs RTEC viability supports the hypothesis that FECH inhibition could trigger RTEC cell-death and dysfunction.
Figure 8: CRISPR/Cas9 mediated FECH knockout induces cell death in HK-2 cells.

Stable HK-2 cells were generated with either a control or FECH targeted sgRNA. In these stable cells lines, Cas9 expression can be induced by doxycycline treatment. (A) In the presence of doxycycline, FECH gene deletion occurs in the cells that express FECH targed sgRNA. Representative immunoblot depicting successful gene knockout. Blots are representative of three independent experiments. (B) Densitometric analysis of FECH protein after 72 hours of doxycycline treatment. (C) Control (vector) and FECH sgRNA (FECHDox-KO) stable cells growing in normal media, were treated with doxycycline and trypan blue based survival assays were performed at 24-72 hours. The results show that FECH knockdown resulted in significant reduction in cell survival. (D-E) MTT and caspase assays at 72 hrs confirmed that FECH gene depletion results in cell death activation. (In all the bar graphs (n=5-6 biologically independent samples) from one out of three independent experiments, all producing similar results, and experimental values are presented as mean ± s.d. The height of error bar = 1 s.d. and p < 0.05 was indicated as statistically significant. One-way ANOVA followed by Dunnett’s test was carried out and statistical significance is indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In the current study, we have established cell culture and murine models of vemurafenib nephrotoxicity that have provided important mechanistic insights. Our studies reveal that vemurafenib triggers RTEC dysfunction and nephrotoxicity through a BRAF-independent and ferrochelatase-dependent manner.
Similar to most protein kinase inhibitors, vemurafenib is mainly excreted via feces (94%), with urinary excretion (1%) playing an insignificant role21,58. Based on our in vitro and in vivo studies, we propose that vemurafenib causes direct toxicity to tubular epithelial cells. Several lines of evidence suggest that this toxicity is independent of BRAF-kinase inhibition. Firstly, both vemurafenib and dabrafenib treatment resulted in BRAF inhibition, however only vemurafenib triggered RTEC cell-death. Secondly, BRAF deletion in murine and human RTECs did not influence cell survival under normal conditions. BRAF deletion or knockdown also did not influence vemurafenib-induced cell-death and AKI. This also eliminates the possibility that paradoxical MAPK activation initiated by inhibitor-induced wild type BRAF dimerization59 might contribute to nephrotoxicity. Thirdly, chemical genetic studies with transfection of vemurafenib-resistant BRAF gatekeeper mutants did not confer protection from vemurafenib-induced cell-death. While genetic compensation remains a possibility, collectively our results suggest that BRAF-kinase is not an essential gene in RTECs and vemurafenib nephrotoxicity is BRAF-independent. Though it is important to note that AKI is a multifaceted disease2 that results from a complex interplay between epithelial38,60,61, immune40, and endothelial62,63 cells. Thus, notwithstanding our studies with RTECs, further work is required to discern the role of immune and endothelial cells in vemurafenib nephrotoxicity.
We propose that vemurafenib inhibits ferrochelatase, which likely contributes to RTEC cell-death and AKI. Ferrochelatase is the terminal enzyme of the heme biosynthesis pathway54. FECH is a mitochondrial protein that catalyzes the insertion of the reduced form of iron (Fe2+) into protoporphyrin. In humans, mutations associated with reduced FECH activity are associated with erythropoetic protoporphyria (EPP) that is characterized by cutaneous photosensitivity and progressive liver damage54. Proteomic profiling of leukemia cells previously revealed that vemurafenib but not dabrafenib is a potent FECH inhibitor52,53. It was thus speculated that FECH inhibition might contribute to photosensitivity and skin toxicities associated with vemurafenib treatment. Our in vivo studies show that vemurafenib treatment results in a progressive decline in FECH activity and development of AKI two weeks post treatment initiation. Importantly, RNAi mediated knockdown or FECH loss-of-function mutations, cause a striking acceleration in the development and onset of vemurafenib-associated AKI (3-7 days versus 15 days). Furthermore, FECH overexpression in cultured RTECs provided significant protection from vemurafenib-induced cell-death.
In contrast with targeted anti-angiogenic therapeutics64 where the entire class of drugs cause hypertension due to ‘on-target’ effects, not all BRAF inhibitors seem to cause AKI22. This is supported by the observation that patients treated with vemurafenib as compared to dabrafenib have a higher incidence of AKI22,25. We propose that this is not a consequence of differential BRAF-inhibition but is dependent on the unique property of vemurafenib to inhibit FECH. In support, BRAF gene deletion in RTECs did not result in renal impairment in vivo. While it is unknown if BRAF is an essential gene in other renal cell types, it is interesting that BRAF inhibition has been suggested as a therapeutic option to prevent podocyte injury65,66. Currently, it is difficult to predict which patients are more likely to develop vemurafenib nephrotoxicity. It would be interesting to examine if genetic polymorphisms or physiological conditions associated with altered renal FECH activity play a contributing role. Altered iron homeostasis is a known feature of AKI67-69; however, the role of FECH in RTEC function and AKI have not been previously explored.
We currently do not know if RTECs in particular proximal tubular segments have differential FECH expression and or are more sensitive to vemurafenib toxicity. However, our cell labelling experiments clearly show that as compared to other cell types, vemurafenib treatment results in RTEC-specific FECH inhibition, intracellular heme depletion, and cell-death activation. So, why are RTECs particularly sensitive to vemurafenib-mediated FECH inhibition and cell-death? We propose that this could be due to two unique functional and physiological properties of RTECs. Firstly, due to the higher expression of transport systems, it is likely that vemurafenib might accumulate at higher levels in RTECs than other cell types. While future studies are required to identify the transport mechanisms, this differential uptake could explain why FECH inhibition only occurs in RTECs. Secondly, while liver and the bone marrow are the key sites for systemic heme synthesis, kidneys also have significant heme biosynthetic capacity, which is interestingly localized mainly within the cells of the proximal convoluted tubule67,70. Hence, within the kidneys FECH might have a particularly critical role in heme synthesis and cellular viability in RTECs. Given the critical role of mitochondria in RTEC function61 and the critical role of heme in mitochondrial function, it is not surprising that FECH is an essential gene in these cells. Indeed, our studies show that both pharmacological and genetic FECH inhibition in cultured RTECs results in cell-death induction. Studies with conditional knockout mice could further clarify if FECH is an essential gene in vivo in RTECs and other renal cells.
Why does FECH inhibition result in RTEC cell-death? FECH is a mitochondrial enzyme that is involved in biosynthesis of heme, an iron-containing cyclic tetrapyrrole. Heme is a co-factor for protein complexes involved in oxygen transport, mitochondrial respiration, redox reactions, circadian rhythm, transcription and translation55,71. Importantly, heme is a co-factor for several electron transport chain (ETC) components, where it mediates electron transfer reactions that are coupled to formation of the mitochondrial proton gradient. One possible indirect consequence of vemurafenib-mediated FECH inhibition could be the accumulation of by-products of heme biosynthesis53 within the mitochondria, resulting in increased oxidative stress, mitochondrial dysfunction, and direct cellular toxicity. On the other hand the direct consequence of vemurafenib-mediated FECH inhibition could be heme depletion, loss of mitochondrial function, and RTEC cell-death. In support of these possibilities, our studies show that vemurafenib treatment causes a significant decrease in intracellular heme levels in RTECs, which correlates with a precipitous decline in mitochondrial function, and cell-death induction. Future in-depth studies are however warranted to tease out exactly how vemurafenib-mediated FECH inhibition influences the ETC and triggers mitochondrial dysfunction. Additionally, while genetic FECH knockout triggers RTEC cell-death, it is unlikely that FECH inhibition in RTECs is solely responsible for vemurafenib nephrotoxicity. FECH-dependent and independent mechanisms in RTECs and other renal cells including endothelial and immune cells might play a role in this toxicity.
Due to the availability of BRAF inhibitors such as dabrafenib and the moderate nature of vemurafenib nephrotoxicity, currently there is no dire need to develop therapeutic strategies to prevent vemurafenib-associated AKI. However, several approved, experimental, and investigational kinase inhibitors have recently been demonstrated to inhibit FECH activity, through mechanisms that remain elusive53. For instance, an ALK (anaplastic lymphoma kinase) inhibitor alcetinib can inhibit FECH52 and is linked with renal dysfunction72, although the causality remains to be investigated. Since, FECH inhibition is not limited to vemurafenib, but has emerged as a relatively common feature of several protein kinase inhibitors, future genetic and pharmacological studies are required to understand the role of this enzyme in renal function and AKI.
In summary, our work suggests that vemurafenib-associated AKI is an off-target toxicity that can be partly attributed to FECH inhibition in RTECs. By identifying the underlying mechanisms, our study reveals that drugs with FECH-inhibiting ability might cause RTEC dysfunction and nephrotoxicity.
Supplementary Material
Translational Statement.
BRAF is the most frequently mutated protein kinase and a critical oncogenic driver in human cancers. In melanoma and other cancers with BRAF activating mutations, BRAF targeted small-molecule therapeutics such as vemurafenib, and dabrafenib have shown remarkable clinical benefits. However, recent clinical studies have shown that a significant number of patients that receive vemurafenib develop AKI through mechanisms that remain unknown. The present study describes the development of novel experimental models of vemurafenib nephrotoxicity and reveals the underlying off-target mechanisms that contribute to renal injury.
Acknowledgements
We thank Dr. Shuiying Hu for assistance with pharmacokinetic analysis. This work was supported by funds from the OSU Cancer Center (N.S.P.), National Institute of Health grants R01CA215802 (A.S), and R01DK117183 (A.B.). Y.B. was supported by postdoctoral fellowships from the Pelotonia foundation and the American Heart Association.
Sources of support:
OSU Cancer Center (N.S.P.), National Institute of Health grants R01CA215802 (A.S), and R01DK117183 (A.B.).
Footnotes
Disclosure Statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary information is available on Kidney International's website.
References
- 1.Malyszko J, Tesarova P, Capasso G, Capasso A. The link between kidney disease and cancer: complications and treatment. The Lancet. 2020;396(10246):277–287. doi: 10.1016/S0140-6736(20)30540-7 [DOI] [PubMed] [Google Scholar]
- 2.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380(9843):756–766. doi: 10.1016/S0140-6736(11)61454-2 [DOI] [PubMed] [Google Scholar]
- 3.Iff S, Craig JC, Turner R, et al. Reduced estimated GFR and cancer mortality. Am J Kidney Dis. 2014;63(1):23–30. doi: 10.1053/j.ajkd.2013.07.008 [DOI] [PubMed] [Google Scholar]
- 4.Schwartz WB, Bennett W, Curelop S, Bartter FC. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. Am J Med. 1957;23(4):529–542. doi: 10.1016/0002-9343(57)90224-3 [DOI] [PubMed] [Google Scholar]
- 5.Uppal NN, Wanchoo R, Barnett R, Sinha A, Jhaveri KD. Hyponatremia in a Patient With Cancer. Am J Kidney Dis. 2020;75(1):A15–A18. doi: 10.1053/j.ajkd.2019.09.005 [DOI] [PubMed] [Google Scholar]
- 6.Rosner MH, Perazella MA. Acute Kidney Injury in Patients with Cancer. New England Journal of Medicine. 2017;376(18):1770–1781. doi: 10.1056/NEJMra1613984 [DOI] [PubMed] [Google Scholar]
- 7.Launay-Vacher V Epidemiology of chronic kidney disease in cancer patients: lessons from the IRMA study group. Semin Nephrol. 2010;30(6):548–556. doi: 10.1016/j.semnephrol.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 8.Seethapathy H, Zhao S, Chute DF, et al. The Incidence, Causes, and Risk Factors of Acute Kidney Injury in Patients Receiving Immune Checkpoint Inhibitors. Clin J Am Soc Nephrol. 2019;14(12):1692–1700. doi: 10.2215/CJN.00990119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jhaveri KD, Sakhiya V, Wanchoo R, Ross D, Fishbane S. Renal effects of novel anticancer targeted therapies: a review of the Food and Drug Administration Adverse Event Reporting System. Kidney Int. 2016;90(3):706–707. doi: 10.1016/j.kint.2016.06.027 [DOI] [PubMed] [Google Scholar]
- 10.Lam AQ, Humphreys BD. Onco-Nephrology: AKI in the Cancer Patient. Clin J Am Soc Nephrol. 2012;7(10):1692–1700. doi: 10.2215/CJN.03140312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salahudeen AK, Bonventre JV. Onconephrology: The Latest Frontier in the War against Kidney Disease. JASN. 2013;24(1):26–30. doi: 10.1681/ASN.2012070690 [DOI] [PubMed] [Google Scholar]
- 12.Rosner MH, Jhaveri KD, McMahon BA, Perazella MA. Onconephrology: The intersections between the kidney and cancer. CA Cancer J Clin. Published online August 27, 2020. doi: 10.3322/caac.21636 [DOI] [PubMed] [Google Scholar]
- 13.Cambier J-F, Ronco P. Onco-nephrology: glomerular diseases with cancer. Clin J Am Soc Nephrol. 2012;7(10):1701–1712. doi: 10.2215/CJN.03770412 [DOI] [PubMed] [Google Scholar]
- 14.Yaeger R, Corcoran RB. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019;9(3):329–341. doi: 10.1158/2159-8290.CD-18-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nature Reviews Cancer. 2014;14(7):455–467. doi: 10.1038/nrc3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 17.Flaherty KT, Yasothan U, Kirkpatrick P. Vemurafenib. Nature Reviews Drug Discovery. 2011;10(11):811–812. doi: 10.1038/nrd3579 [DOI] [PubMed] [Google Scholar]
- 18.Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF -mutant cancer. Nature Reviews Drug Discovery. 2012;11(11):873–886. doi: 10.1038/nrd3847 [DOI] [PubMed] [Google Scholar]
- 19.Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. doi: 10.1016/S0140-6736(12)60868-X [DOI] [PubMed] [Google Scholar]
- 20.Lacouture ME, Duvic M, Hauschild A, et al. Analysis of Dermatologic Events in Vemurafenib-Treated Patients With Melanoma. Oncologist. 2013;18(3):314–322. doi: 10.1634/theoncologist.2012-0333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jhaveri KD, Sakhiya V, Fishbane S. Nephrotoxicity of the BRAF Inhibitors Vemurafenib and Dabrafenib. JAMA Oncol. 2015;1(8):1133–1134. doi: 10.1001/jamaoncol.2015.1713 [DOI] [PubMed] [Google Scholar]
- 23.Uthurriague C, Thellier S, Ribes D, Rostaing L, Paul C, Meyer N. Vemurafenib significantly decreases glomerular filtration rate. J Eur Acad Dermatol Venereol. 2014;28(7):978–979. doi: 10.1111/jdv.12322 [DOI] [PubMed] [Google Scholar]
- 24.Regnier-Rosencher E, Lazareth H, Gressier L, Avril MF, Thervet E, Dupin N. Acute kidney injury in patients with severe rash on vemurafenib treatment for metastatic melanomas. Br J Dermatol. 2013;169(4):934–938. doi: 10.1111/bjd.12555 [DOI] [PubMed] [Google Scholar]
- 25.Teuma C, Perier-Muzet M, Pelletier S, et al. New insights into renal toxicity of the B-RAF inhibitor, vemurafenib, in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2016;78(2):419–426. doi: 10.1007/s00280-016-3086-7 [DOI] [PubMed] [Google Scholar]
- 26.Hurabielle C, Pillebout E, Stehlé T, et al. Mechanisms Underpinning Increased Plasma Creatinine Levels in Patients Receiving Vemurafenib for Advanced Melanoma. PLoS One. 2016;11(3):e0149873. doi: 10.1371/journal.pone.0149873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pabla N, Bhatt K, Dong Z. Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chk1 that regulates cell cycle and DNA damage checkpoints. Proc Natl Acad Sci USA. 2012;109(1):197–202. doi: 10.1073/pnas.1104767109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JY, Bai Y, Jayne LA, et al. A kinome-wide screen identifies a CDKL5-SOX9 regulatory axis in epithelial cell death and kidney injury. Nat Commun. 2020;11(1):1924. doi: 10.1038/s41467-020-15638-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sprowl JA, Ong SS, Gibson AA, et al. A phosphotyrosine switch regulates organic cation transporters. Nat Commun. 2016;7:10880. doi: 10.1038/ncomms10880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pabla N, Gibson AA, Buege M, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A. 2015;112(16):5231–5236. doi: 10.1073/pnas.1424313112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshioka E, Chelakkot VS, Licursi M, et al. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics. 2018;8(8):2134–2146. doi: 10.7150/thno.22641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khechaduri A, Bayeva M, Chang H-C, Ardehali H. Heme Levels are Increased in Human Failing Hearts. J Am Coll Cardiol. 2013;61(18):1884–1893. doi: 10.1016/j.jacc.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JY, Jayne LA, Bai Y, et al. Ribociclib mitigates cisplatin-associated kidney injury through retinoblastoma-1 dependent mechanisms. Biochem Pharmacol. 2020;177:113939. doi: 10.1016/j.bcp.2020.113939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73(9):994–1007. doi: 10.1038/sj.ki.5002786 [DOI] [PubMed] [Google Scholar]
- 35.Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nature Reviews Drug Discovery. 2006;5(10):835–844. doi: 10.1038/nrd2130 [DOI] [PubMed] [Google Scholar]
- 36.Yang H, Higgins B, Kolinsky K, et al. Antitumor Activity of BRAF Inhibitor Vemurafenib in Preclinical Models of BRAF-Mutant Colorectal Cancer. Cancer Res. 2012;72(3):779–789. doi: 10.1158/0008-5472.CAN-11-2941 [DOI] [PubMed] [Google Scholar]
- 37.Ichimura T, Bonventre JV, Bailly V, et al. Kidney Injury Molecule-1 (KIM-1), a Putative Epithelial Cell Adhesion Molecule Containing a Novel Immunoglobulin Domain, Is Upregulated in Renal Cells after Injury. J Biol Chem. 1998;273(7):4135–4142. doi: 10.1074/jbc.273.7.4135 [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Liu J, Pang P, et al. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep. 2015;12(8):1325–1338. doi: 10.1016/j.celrep.2015.07.034 [DOI] [PubMed] [Google Scholar]
- 39.Okusa MD. The inflammatory cascade in acute ischemic renal failure. Nephron. 2002;90(2):133–138. doi: 10.1159/000049032 [DOI] [PubMed] [Google Scholar]
- 40.Bajwa A, Kinsey GR, Okusa MD. Immune mechanisms and novel pharmacological therapies of acute kidney injury. Curr Drug Targets. 2009;10(12):1196–1204. doi: 10.2174/138945009789753174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110(6):835–842. doi: 10.1172/JCI15606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rousselle TV, Kuscu C, Kuscu C, et al. FTY720 Regulates Mitochondria Biogenesis in Dendritic Cells to Prevent Kidney Ischemic Reperfusion Injury. Front Immunol. 2020;11:1278. doi: 10.3389/fimmu.2020.01278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JY, Bai Y, Jayne LA, et al. SOX9 promotes stress-responsive transcription of VGF nerve growth factor inducible gene in renal tubular epithelial cells. J Biol Chem. 2020;295(48):16328–16341. doi: 10.1074/jbc.RA120.015110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen AP, Ohno M, Giese KP, Kühn R, Chen RL, Silva AJ. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J Neurosci Res. 2006;83(1):28–38. doi: 10.1002/jnr.20703 [DOI] [PubMed] [Google Scholar]
- 45.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–350. doi: 10.1172/JCI15518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125(5):1780–1789. doi: 10.1172/JCI76094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–1051. doi: 10.1038/nbt.1990 [DOI] [PubMed] [Google Scholar]
- 48.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10(2):130–137. doi: 10.1038/nrc2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Rix U, Fang B, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6(4):291–299. doi: 10.1038/nchembio.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teuma C, Pelletier S, Amini-Adl M, et al. Adjunction of a MEK inhibitor to Vemurafenib in the treatment of metastatic melanoma results in a 60% reduction of acute kidney injury. Cancer Chemother Pharmacol. 2017;79(5):1043–1049. doi: 10.1007/s00280-017-3300-2 [DOI] [PubMed] [Google Scholar]
- 51.Liston DR, Davis M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin Cancer Res. 2017;23(14):3489–3498. doi: 10.1158/1078-0432.CCR-16-3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savitski MM, Reinhard FBM, Franken H, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205). doi: 10.1126/science.1255784 [DOI] [PubMed] [Google Scholar]
- 53.Klaeger S, Gohlke B, Perrin J, et al. Chemical Proteomics Reveals Ferrochelatase as a Common Off-target of Kinase Inhibitors. ACS Chem Biol. 2016;11(5):1245–1254. doi: 10.1021/acschembio.5b01063 [DOI] [PubMed] [Google Scholar]
- 54.Bloomer J, Bruzzone C, Zhu L, Scarlett Y, Magness S, Brenner D. Molecular defects in ferrochelatase in patients with protoporphyria requiring liver transplantation. J Clin Invest. 1998;102(1):107–114. doi: 10.1172/JCI1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ponka P Cell biology of heme. Am J Med Sci. 1999;318(4):241–256. doi: 10.1097/00000441-199910000-00004 [DOI] [PubMed] [Google Scholar]
- 56.Namwanje M, Bisunke B, Rousselle TV, et al. Rapamycin Alternatively Modifies Mitochondrial Dynamics in Dendritic Cells to Reduce Kidney Ischemic Reperfusion Injury. Int J Mol Sci. 2021;22(10):5386. doi: 10.3390/ijms22105386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dhillon P, Park J, Hurtado Del Pozo C, et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. 2021;33(2):379–394.e8. doi: 10.1016/j.cmet.2020.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldinger SM, Rinderknecht J, Dummer R, et al. A single-dose mass balance and metabolite-profiling study of vemurafenib in patients with metastatic melanoma. Pharmacol Res Perspect. 2015;3(2). doi: 10.1002/prp2.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–435. doi: 10.1038/nature08833 [DOI] [PubMed] [Google Scholar]
- 60.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13(10):629–646. doi: 10.1038/nrneph.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72(2):151–156. doi: 10.1038/sj.ki.5002312 [DOI] [PubMed] [Google Scholar]
- 63.Bullen A, Liu ZZ, Hepokoski M, Li Y, Singh P. Renal Oxygenation and Hemodynamics in Kidney Injury. Nephron. 2017;137(4):260–263. doi: 10.1159/000477830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Meiracker AH, Danser AHJ. Mechanisms of Hypertension and Renal Injury During Vascular Endothelial Growth Factor Signaling Inhibition. Hypertension. 2016;68(1):17–23. doi: 10.1161/HYPERTENSIONAHA.116.07618 [DOI] [PubMed] [Google Scholar]
- 65.Sieber J, Wieder N, Clark A, et al. GDC-0879, a BRAFV600E Inhibitor, Protects Kidney Podocytes from Death. Cell Chem Biol. 2018;25(2):175–184.e4. doi: 10.1016/j.chembiol.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bryer JS, Susztak K. Screening Drugs for Kidney Disease: Targeting the Podocyte. Cell Chem Biol. 2018;25(2):126–127. doi: 10.1016/j.chembiol.2018.01.018 [DOI] [PubMed] [Google Scholar]
- 67.Tracz MJ, Alam J, Nath KA. Physiology and Pathophysiology of Heme: Implications for Kidney Disease. JASN. 2007;18(2):414–420. doi: 10.1681/ASN.2006080894 [DOI] [PubMed] [Google Scholar]
- 68.Walker VJ, Agarwal A. Targeting Iron Homeostasis in Acute Kidney Injury. Semin Nephrol. 2016;36(1):62–70. doi: 10.1016/j.semnephrol.2016.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scindia Y, Leeds Md J, Swaminathan Md S. Iron Homeostasis in Healthy Kidney and its Role in Acute Kidney Injury. Semin Nephrol. 2019;39(1):76–84. doi: 10.1016/j.semnephrol.2018.10.006 [DOI] [PubMed] [Google Scholar]
- 70.Woods JS. Regulation of porphyrin and heme metabolism in the kidney. Semin Hematol. 1988;25(4):336–348. [PubMed] [Google Scholar]
- 71.Li Y, Ivica NA, Dong T, et al. MFSD7C switches mitochondrial ATP synthesis to thermogenesis in response to heme. Nat Commun. 2020;11(1):4837. doi: 10.1038/s41467-020-18607-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Izzedine H, El-Fekih RK, Perazella MA. The renal effects of ALK inhibitors. Invest New Drugs. 2016;34(5):643–649. doi: 10.1007/s10637-016-0379-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.